DOI:
10.1039/C9RA09376A
(Paper)
RSC Adv., 2020,
10, 3646-3658
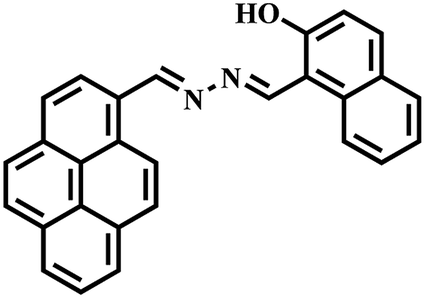
Pyrene-appended bipyridine hydrazone ligand as a turn-on sensor for Cu2+ and its bioimaging application†
Received
11th November 2019
, Accepted 5th January 2020
First published on 22nd January 2020
Abstract
A pyrene-appended bipyridine hydrazone-based ligand, HL, was synthesized and characterized by spectroscopic methods. Upon complexation with Cu(II), HL formed a hexanuclear paddlewheel metal–organic macrocycle (MOM) via self-assembly with a high association constant with the molecular formula of [Cu6L6(NO3)6]. Intermolecular and intramolecular π–π interactions were demonstrated in this hexanuclear Cu(II) complex. Further, it was observed that HL had the potential to detect a trace level of Cu(II) ion selectively among a wide range of biologically relevant metal ions in aqueous medium at physiological pH. Using HL, it was feasible to sense copper(II) ions in living cells due to its good cell permeability and high solubility under physiological conditions along with its high IC50 value. The low detection limit, high sensitivity and good reproducibility make this Cu–sensor very promising. The complex (MOM) formed between the ligand and Cu(II) was found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 on the basis of fluorescence titrations and was confirmed by ESI-MS. Moreover, single-crystal study of the hexanuclear self-assembled fluorescent species provided better insight into its chemistry, e.g. coordination environment and binding mode, unlike most of the metal sensors due to the lack of a single-crystal structure of the metal sensor complex. Cytotoxicity assay and bioimaging were performed in living cells (Vero cells), giving green fluorescent images. Fluorescence lifetime measurements and theoretical calculations were carried out. The morphology and topographic details on the surface of the metal–organic macrocycle (MOM) were studied by field-emission scanning electron microscopy (FESEM).
:1 on the basis of fluorescence titrations and was confirmed by ESI-MS. Moreover, single-crystal study of the hexanuclear self-assembled fluorescent species provided better insight into its chemistry, e.g. coordination environment and binding mode, unlike most of the metal sensors due to the lack of a single-crystal structure of the metal sensor complex. Cytotoxicity assay and bioimaging were performed in living cells (Vero cells), giving green fluorescent images. Fluorescence lifetime measurements and theoretical calculations were carried out. The morphology and topographic details on the surface of the metal–organic macrocycle (MOM) were studied by field-emission scanning electron microscopy (FESEM).
Introduction
Copper is the third-most abundant ubiquitous metal found in nature, where it occurs in the form of rock, water, soil and sediments. It is an essential element not only for human beings but also for almost all living organisms. In vivo, the proper functioning of organs and metabolic processes are ensured by trace concentrations of copper. It is an obligatory nutrient-element for metabolism in the physiological cycle as well as in the ecological cycle.1,2 Moreover, Cu2+ ions represent special trace ions that have both beneficial and cytotoxic effects in various enzymatic processes and they also act as a cofactor for many fundamental biological processes and catalyse the production of highly reactive oxygen species.3 However, the accumulation of unregulated and excess amounts over the permitted limit of copper even for a short time-span can cause gastrointestinal dysfunction, while the accumulation of the same for a longer period of time can damage the liver or kidney4 and lead to the increased possibility of long-term disease, such as Wilson's,5 Alzheimer's,6 Menke's,7 Prion's8 disease and Indian childhood cirrhosis.9 On the other hand, haematological and neurological syndromes can occur due to an insufficiency of copper concentration in the human body.10,11 An increased concentration of Cu2+ may be harmful for the environment also as it affects the self-purification capability of water in the sea or rivers and can ruin the water for aquatic life.12 So, the precise identification and detection of Cu2+ in aqueous solution is essential for monitoring its concentration level in the environment and biological systems, ensuring the regular functioning of the ecosystem, health, and humanity. The detection of Cu2+ ions at the trace level (ppm–ppb level) is not an easy task. At present, some instrumentation techniques do exist but either they require high-end instruments or difficult sample preparation methods, which limit the easy and fast detection of Cu2+ ions. Keeping these difficulties in mind, the role of a sensitive chemosensor becomes very important as it can sense trace amounts of the ions by showing a significant change in its electric, magnetic or optical properties. Fluorescent chemosensors have added advantages in terms of their ease of synthesis, sensitivity, selectivity and versatility, allowing economical and real-time monitoring with a fast response time.13–15 Recently, tremendous efforts have been made to design and develop a chemical sensor to overcome the difficulties mentioned above. For example, enormous progress has been made in the design, isolation and application of chemiluminescent MOFs towards gas storage,16 separation,17 chemical sensing,18 catalysis19 and biomedicines20 in general and for sensing important targets, such as cations and anions,13–15 small molecules,21 gas,22 pesticides23 and explosives,24,25 in particular. A metal–organic framework (MOF) signifies a new class of hybrid material built through the careful design or selection of organic bridging ligands and a variety of metal ions with a well-defined coordination geometry. Along similar lines, here we incorporated a metalladiazamacrocycle and termed it a metal–organic macrocycle (MOM),26 which could be interesting because of its fascinating architecture. In such MOM molecules, if there exist multiple luminescent centres and the tuneable coordination of metal centres with organic linkers, those could potentially generate “turn-on” luminescence.27–30 Because of their various applications in different scientific fields, such as biochemistry, clinical and medical sciences, cell biology, analytical chemistry and environmental sciences, the development of metal ion chemosensors that could functions in aqueous solution is of great importance. However, most of the reported chemosensors for Cu2+ ions are based on the quenching of the fluorescence intensity due to the high binding ability and high negative CFSE energy of paramagnetic Cu2+-ion, which leads to a “turn-off”31–34 fluorescence sensor. Now, the design and synthesis of a Cu2+-induced “turn-on”14,35–37 fluorescent chemosensor is attracting increasing interest. The essential requirements for the development of efficient optical chemosensors are high selectivity and sensitivity, quick response time, high complex stability, low detection limit and reusability.
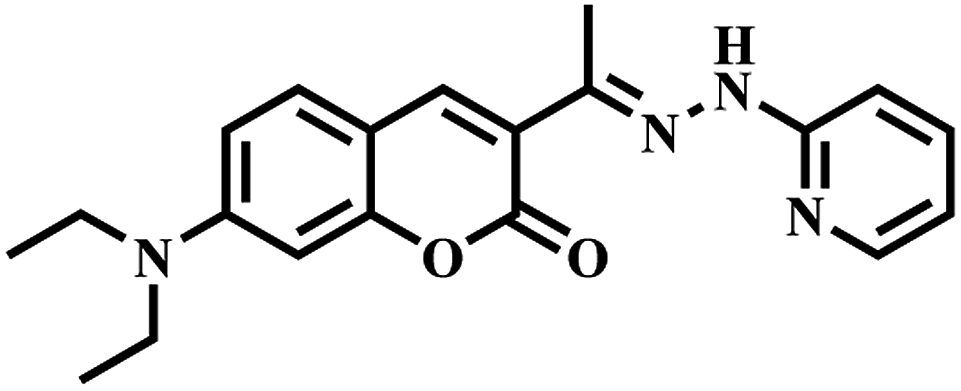
Keeping the above-mentioned points in mind, herein we report a novel pyrene derivative probe, HL, containing a bipyridinehydrazone moiety for copper ion detection. Bipyridine is well known for its good chelating capability. Its hydrazone moiety was introduced as a binding unit and pyrene was purposefully attached with it as a fluorophore unit. HL was synthesized in pure form by the condensation of pyrene aldehyde with bipyridine hydrazone in methanol. It was fully characterized by spectroscopic techniques, such as NMR, IR and ESI-MS. When HL was reacted with a Cu2+ precursor in a 1:1 stoichiometry, it formed a fluorescent hexanuclear self-assembled MOM. Fluorescence lifetime measurements and theoretical calculations were also carried out. Further, the electronic effect of organic molecules on this chemosensor was studied. It is worth mentioning that most of the fluorescent chemosensors for Cu2+ reported to date are “turn-off”; however, this MOM joins a relatively rare family of “turn-on” chemosensors for Cu2+. It showed highly selective turn-on fluorescence at physiological pH for copper ions over other metal ions with a low detection limit in a (8:2, v/v) water–MeCN solvent mixture. HL, being water soluble, gave us the opportunity to study its sensing ability and biological applications in aqueous media, such as cytotoxicity assay and bioimaging in living cells.
Experimental
Materials and instrumentation
All solvents (analytical grade and spectroscopic grade) were obtained from Finar (India) and Spectrochem (India) and the solvents were purified using standard literature methods. Deuterated solvents for NMR were bought from Sigma-Aldrich (India) and used as received. All metal salts and other chemicals were obtained from Alfa-Aesar (India), Spectrochem (India) and Merck (India). Melting points were measured using a BUCHI M-500 instrument. Fourier transform infrared (FT-IR) spectra were recorded on a BRUKER ALPHA-T FT-IR spectrometer using KBr pellets. A PerkinElmer model Lambda 650 UV-vis spectrophotometer was used for recording the absorption spectra. Emission spectra were recorded on a Fluoromax 4P Spectro-fluorometer (Horiba-Jobin-Mayer, Edison, NJ, USA). The bioimaging experiments were carried out using a Leica TCS SP5 confocal microscope and the fluorescence lifetime was measured using a time-correlated single-photon counting (TCSPC) spectrometer (Edinburgh, OB920) instrument. 1H and 13C NMR spectra were recorded on a Bruker AVANCE 400 NMR spectrometer. A Bruker microTOF-Q II mass spectrometer was used for electrospray ionization mass spectrometry (ESI-MS) analysis of the compounds. X-ray data were collected using a Rigaku Oxford diffractometer with graphite-monochromated Cu-Kα (λ = 1.54178 Å) radiation at 110(2) K.
Synthesis of HL
[2,2′]-Bipyridinyl-3-carboxylic acid hydrazide38 (0.535 g, 2.5 mmol) was taken in a round-bottom flask and dissolved in 20 ml dry MeOH. Pyrene aldehyde (0.575 mg, 2.5 mmol) was dissolved in 30 ml dry MeOH in another reagent bottle. The second solution was dropwise added to the first solution. Then the mixture was heated to reflux for 12 h. A dark yellow precipitate appeared. Then it was cooled for complete precipitation. After, it was filtered, washed with MeOH and dried over a high vacuum to give a deep-yellow solid ligand (HL). Yield: 0.543 g, 51%. Mp: 255–257 °C; ESI-MS m/z ([HL + H]+): calcd, 427.16; found, 427.15. 1H NMR (400 MHz, DMSO-d6) δ (ppm) = 12.20 (s, 1H), 9.85 (s, 1H), 8.98 (d, 1H), 8.92 (d, 1H), 8.76 (d, 1H) 8.67 (m, 2H), 8.38 (m, 4H), 8.24 (m, 4H), 8.13 (m, 2H), 7.55 (m, 1H). FTIR (KBr, cm−1); 3440.72 (N–H), 1688.38 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1593.16 and 1581.61 (CN). UV-vis (5 × 10−5 M, MeCN): λ [nm] (ε [L mol−1 cm−1]) 240.5 (49254); 276 (36683) and 343 (20867).
O), 1593.16 and 1581.61 (CN). UV-vis (5 × 10−5 M, MeCN): λ [nm] (ε [L mol−1 cm−1]) 240.5 (49254); 276 (36683) and 343 (20867).
Synthesis of Cu2+ complex of HL; [Cu6L6(NO3)6]
The ligand HL (0.100 g, 0.23 mmol) was added to a hot solution of Cu(NO3)2·3H2O (0.085 g, 0.35 mmol) in a (1:1, v/v) methanol–chloroform mixture (14 ml) with constant stirring and heating. The resulting clear green solution was filtered to remove any undissolved solid and left at room temperature for slow evaporation. After 2 days, dark green crystals suitable for X-ray diffraction were isolated, washed first with methanol, then diethyl ether and collected. Yield: 0.070 g, 55%. ESI–MS m/z: ([Cu6L6]6+) calcd, 488.07; found, 488.07. IR (KBr) ν (cm−1): 1631.07 (CO), and 1553.46 (CN). UV-vis (5 × 10−5 M, MeCN): λ [nm] (ε [L mol−1 cm−1]) 243 (33003); 287 (28603); 361 (17804) and 394 (16647).
Optical detection of Cu2+ using HL
Stock solution of the ligand (HL) was prepared in acetonitrile and of the metal salts in water. All the metal solutions were prepared by using their nitrate salts. All fluorescence data were collected from 400 to 600 nm at ambient temperature in a (8:2, v/v) water–MeCN solvent mixture. The excitation and maximum emission wavelengths were λex = 365 nm and λem = 466 nm, respectively. In all the experiments, 40 mM HEPES buffer solution was used to maintain the pH at 7.4 during the solution preparation.
Crystallography
CIF table, selected bond lengths and bond angles of the Cu2+–L complex are given in Tables 2, S1 and S2,† respectively. Single-crystal X-ray data were collected using a Rigaku Oxford diffractometer with graphite-monochromated Cu-Kα (λ = 1.54178 Å) radiation at 110(2) K. The “Bruker Saint Plus” program39 was used for data reduction. Data were corrected for Lorentz and polarization effects; an empirical absorption correction (SADABS) was applied.39 The structure was solved by direct methods with SIR-97 and refined by full-matrix least squares methods based on F2 by using SHELXL-97,40 incorporated in the WinGX 2014.1 crystallographic package.41 All nonhydrogen atoms were refined with anisotropic thermal parameters. The positions of the hydrogen atoms were calculated assuming ideal geometries, but were not refined.
Cell line and cell culture
The Vero cells obtained from Dr M. M. Parida (DRDE, Gwalior, India) and maintained in Dulbecco's modified Eagle's medium (DMEM, Pan Biotech) containing 10% (v/v) fetal bovine serum (PAN Biotech), penicillin and streptomycin (100 μg ml−1) (Sigma Aldrich) in a CO2 incubator according to the method described earlier.42 Cells were initially propagated in 75 cm2 polystyrene, filter-capped tissue culture flask under an atmosphere of 5% CO2 and 95% air at 37 °C in a CO2 incubator. When the cells reached 90% confluency, the cells were split into 35 mm dishes (TPP) with cover slips containing 0.7 × 106 cells per dish. After adhesion, the cells were treated according to the experimental requirement.
Cytotoxicity assay
The cytotoxic effects of the Cu2+–L complex were determined by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay following the protocol described earlier.43 Vero cells were cultured in 96-well plates (104 cells per well) for 24 h. After overnight incubation, the medium was removed, and various concentrations of Cu2+–L complex (0, 5, 10, 15, 20, 25, 30, 35, 40, 45 and 50 μM) made in DMEM were added to the cells, followed by incubation for 24 h. Control experiments were set with DMSO; cells without any treatment and cell-free medium were also included in the study. Following incubation, the growth medium was removed and fresh DMEM containing MTT solution was added. The plate was incubated for 3–4 h at 37 °C. Subsequently, the supernatant was removed, the insoluble coloured formazan product was solubilized in DMSO and its absorbance was measured using a microplate reader (PerkinElmer) at 570 nm. The assay was performed in triplicate for each concentration of Cu2+–L. Cell viability was calculated by the following formula:
| Cell viability = (mean OD in treated wells/mean OD in control wells) × 100 |
Cell imaging study
For the confocal imaging studies, Vero cells in 35 mm dishes were washed twice with 1 × PBS. Cells were then treated with Cu(NO3)2 in DMEM and incubated at 37 °C in a CO2 incubator for 2 h. After 2 h, the cells were again washed twice with 1 × PBS and incubated with 25 μM HL in DMEM at 37 °C for 30 min in a CO2 incubator. The cells were then washed twice with 1 × phosphate buffered saline (PBS) (pH 7.4) and fixed with 4% paraformaldehyde (w/v) for 15 min at room temperature. Subsequently, the cells on the coverslips were washed twice with 1 × PBS and the coverslips containing fixed cells were mounted onto a glass slide with a mounting medium containing antifade (Invitrogen) to reduce photobleaching. Fluorescence microscopic images were obtained using a Leica TCS SP5 confocal microscope using a 40× objective at λex = 405 and λem = 466 nm. All the images were analyzed using Leica Application Suite Advanced Fluorescence software (LASAF).44
Theoretical calculations
Quantum chemical calculations were carried out using the Gaussian 09 program.45 The possible ground state structures of both the gaseous and aqueous phases were optimized with density functional theory (DFT) at the B3LYP/6-31G(d) and LANL2DZ level. The effect of water was considered using a polarized continuum model (PCM).46 The GaussView 5 program47 was used for the visualization of the studied systems.
Results and discussion
The pyrene hydrazone and bipyridine-based Schiff base probe HL was synthesized by coupling [2,2′]-bipyridinyl-6-carboxylic acid hydrazide and 1-pyrenealdehyde in dry methanol (Scheme 1 and details in the Experimental section). The photophysical properties of this chemosensor were analytically examined using absorbance (UV-visible), fluorescence and time-resolved spectroscopic methods.
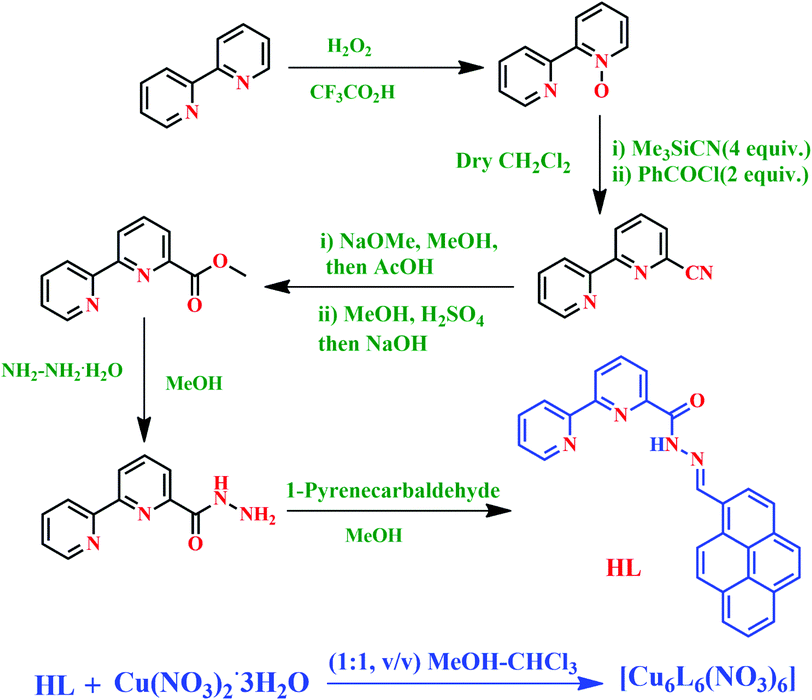 |
| | Scheme 1 Synthesis of the probe HL and its corresponding Cu2+ complex. | |
Solvent selectivity
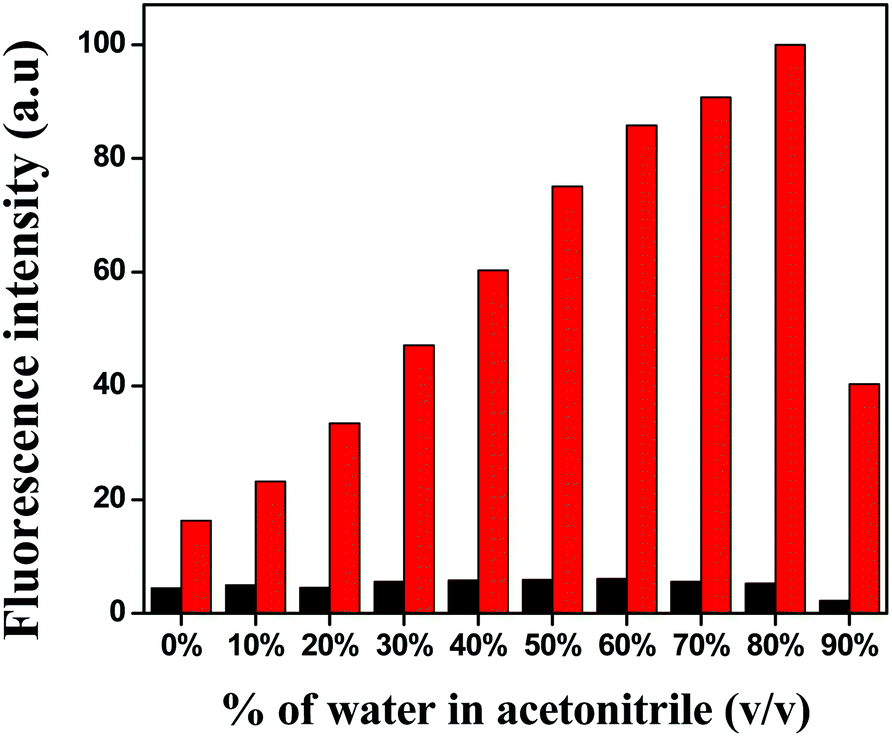
At first, the solvent selectivity of the fluorescence improvement for Cu2+ ions was measured. The maximum emission was observed in the case of MeCN over THF, DMSO, DMF, MeOH and EtOH (Fig. S5†). Next, the effect of water on the emission intensity was studied at the same excitation and emission wavelength and it was clearly seen that up to 80% of water addition resulted in an increase in the fluorescence intensity (Fig. 1), which proved the water–MeCN (8:2, v/v) solvent mixture was the best composition of solvents for the fluorescence study.
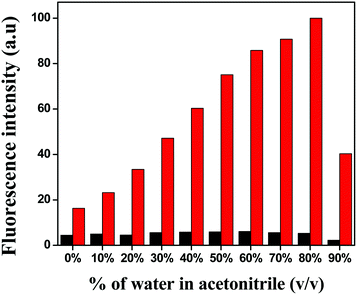 |
| | Fig. 1 Bar graph of the relative normalised emission intensity of HL (black) and L–Cu2+ (red). | |
Selectivity and sensitivity of HL
At first, the fluorescence behaviour of HL was studied upon treatment with different metal ions. HL alone did not show any prominent fluorescence when excited at 365 nm. However, upon the addition of Cu2+, the emission intensity of HL was enhanced at a wavelength of 466 nm. On the other hand, the addition of other relevant metal ions, such as Na+, K+, Mg2+, Ca2+, Ag+, Mn2+, Fe3+, Co2+, Ni2+, Zn2+ and Al3+, caused almost no fluorescence increment (Fig. 2).
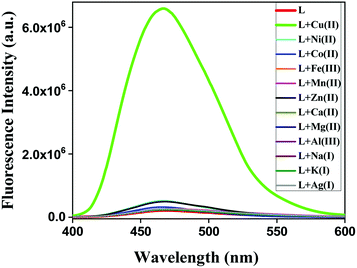 |
| | Fig. 2 Fluorescence spectra of HL (10 μM) before and after the addition of different metal salts at λem = 466 nm in a water–MeCN (8:2, v/v) solvent mixture. | |
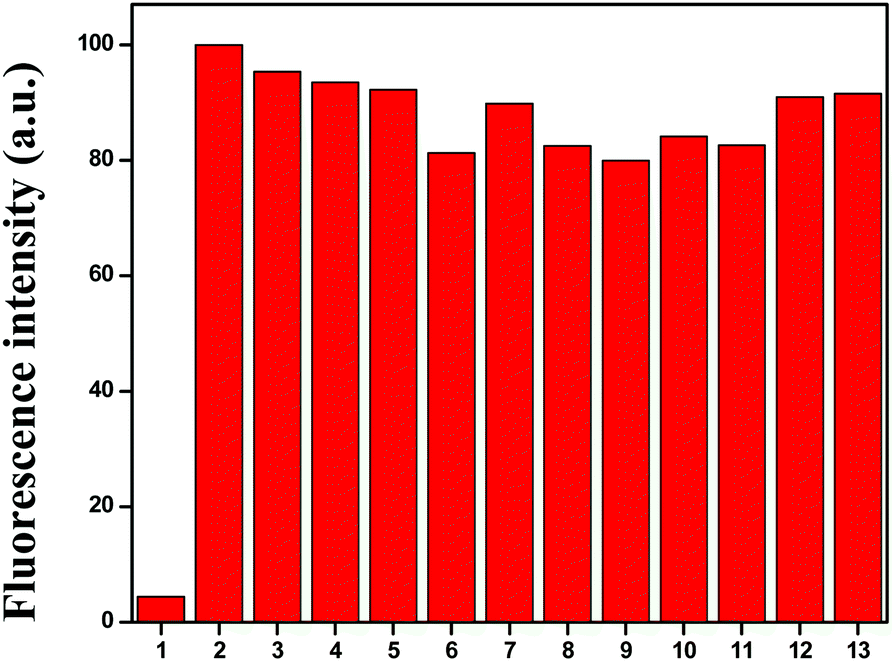
To understand the better selectivity, complex formation ability and stability of HL with Cu2+ ions over other metals, a 10 μM ligand (HL) and 10 μM Cu2+ mixture was treated with 1 equiv. of different competitive metal ions (Mn2+, Fe3+, Co2+, Ni2+, Zn2+, Mg2+, Ca2+, Al3+, Na+, K+ and Ag+) in a mixed aqueous solution (water–MeCN, 8:2; v/v). The recorded fluorescence spectra at 466 nm did not show any significant variation in the fluorescence intensity of the L–Cu2+ complex in the presence of other metal ions (Fig. 3). These results demonstrated that the response of the chemosensor to Cu2+ ions was unaffected by the presence of other metal ions.
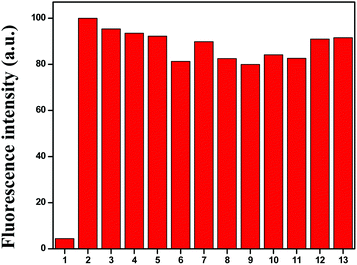 |
| | Fig. 3 Fluorescence response of Cu2+–L complex in the presence of 1 equiv. of competing metal ions: (1) HL, (2) blank (L + Cu2+), (3) blank + Ni2+, (4) blank + Co2+, (5) blank + Fe3+, (6) blank + Mn2+, (7) blank + Zn2+, (8) blank + Ca2+, (8) blank + Ag+, (9) blank + Mg2+, (10) blank + Al3+ (11) blank + Na+, (12) blank + K+ and (13) blank + Ag+ at λem 466 nm. | |
pH effect
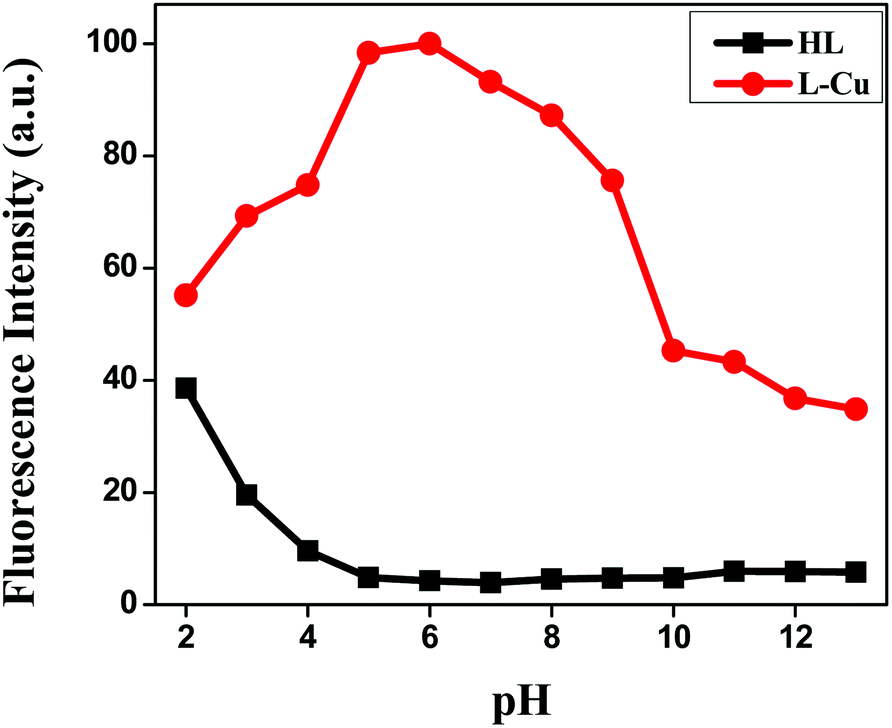
For better applicability, the pH effect was also examined to normalize the suitable pH range for Cu2+ sensing in the absence and presence of Cu2+ ions (Fig. 4). The overall emission intensity of Cu2+–L remained higher than that of the free receptor HL at 466 nm. For the biological application of the fluorescent sensor, the sensing should function in an extensive range of pH. The experimental results clearly demonstrated the ability of HL to detect Cu2+ ions in aqueous solution at a suitable pH range (4–9) to show its practicality, whereby the fluorescence “on–off” could be operated by copper-ion binding.
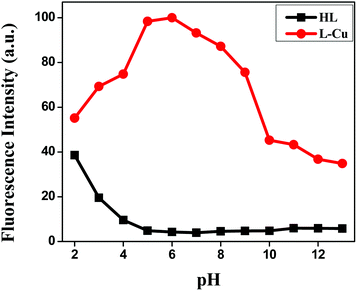 |
| | Fig. 4 Effect of pH on the fluorescence intensities of HL and the Cu2+–L complex. | |
Detection limit
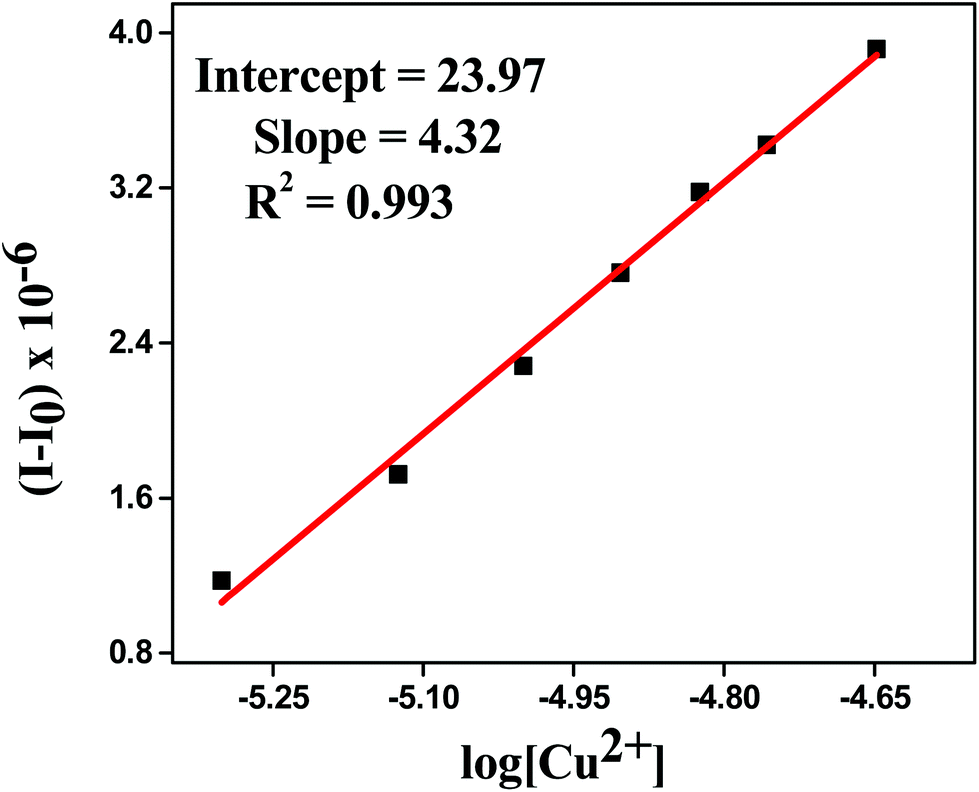
The detection limit was calculated by determining the slope and intercept of the plot between the fluorescence intensity versus the concentration of metal ion from titration experiments.48,49 It was found to be 66 × 10−8 M (Fig. 5), which confirmed that HL can be used for even less than the micro molar level detection of Cu2+ in the environment as well as in biological systems. The detection limit was calculated by using the following equation:
| Detection limit = 10−[slope/intercept] |
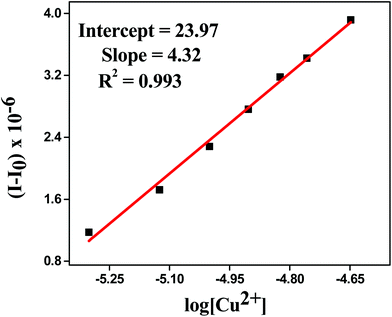 |
| | Fig. 5 Detection limit plot of HL for Cu2+ ions. | |
A state-of-the-art comparison of this Cu sensor (type, detection limit and structure of Cu–sensor complex) with some of the relevant reported Cu–sensors is given in (Table 1).14,32,35,36,50–55 It is worth mentioning here that this sensor showed outstanding performance among all the relevant reported Cu sensors. It was evident that it had the best detection limit among all the structurally characterized turn-on Cu–sensors.
Table 1 State-of-the-art comparison of this Cu sensor (type, detection limit and structure of Cu–sensor complex) with some of the relevant reported Cu–sensors
| Cu2+ recognition probe |
Chemosensor type |
Detection limit |
Cu–sensor complex structurally characterized or not? |
Reference |
 |
“Turn-off” Cu2+ sensor |
7.11 nM |
No |
50 |
 |
“Turn-on” Cu2+ sensor |
20 μM |
Yes |
51 |
 |
“Turn-off” Cu2+ sensor |
1.62 nM |
No |
52 |
 |
“Turn-on” Cu2+ sensor |
30 μM |
No |
14 |
 |
“Turn-off” Cu2+ sensor |
29.6 μM |
Yes |
53 |
 |
“Turn-on” Cu2+ sensor |
1.53 μM |
No |
35 |
 |
“Turn-on” Cu2+ sensor |
10.1 μM |
No |
54 |
 |
“Turn-on” Cu2+ sensor |
0.04 μM |
No |
36 |
 |
“Turn-on” Cu2+ sensor |
Not mentioned |
No |
55 |
 |
“Turn-off” Cu2+ sensor |
0.5 μM |
Yes |
32 |
| HL (this article) |
“Turn-on” Cu2+ sensor |
0.66 μM |
Yes |
XX |
Table 2 Crystallographic details for the Cu-complex
| Parameters |
Cu-complex |
| Empirical formula |
C168H102Cu6N32O24 |
| Formula weight |
3334.05 |
| Temperature (K) |
110(2) |
| Wavelength (Å) |
1.54178 |
| Crystal system, space gr. |
Trigonal, R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
| Unit cell dimensions |
a = 24.322(3) Å α = 90° |
| b = 24.322(3) Å β = 90° |
| c = 21.069(3) Å γ = 120° |
| Volume (Å3) |
10794(3) |
| Z, calculated density (g cm−3) |
3, 1.539 |
| Absorption coefficient (mm−1) |
1.690 |
| F(000) |
5100 |
| Crystal size (mm3) |
0.3 × 0.2 × 0.2 |
| Theta range for data collection |
2.966 to 88.356° |
| Limiting indices |
−30 ≤ h ≤ 30, −30 ≤ k ≤ 29, −11 ≤ l ≤ 26 |
| Reflections collected/unique |
11485 |
| Completeness to theta |
99.1% |
| Absorption correction |
Empirical |
| Refinement method |
Full-matrix least-squares on F2 |
| Data/parameters |
5026/373 |
| Goodness-of-fit on F2 |
1.013 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0760, wR2 = 0.1951 |
| R indices (all data) |
R1 = 0.1435, wR2 = 0.2487 |
| Largest diff. peak and hole (e Å−3) |
0.651 and −0.370 e Å−3 |
Stoichiometry and binding property of the complex
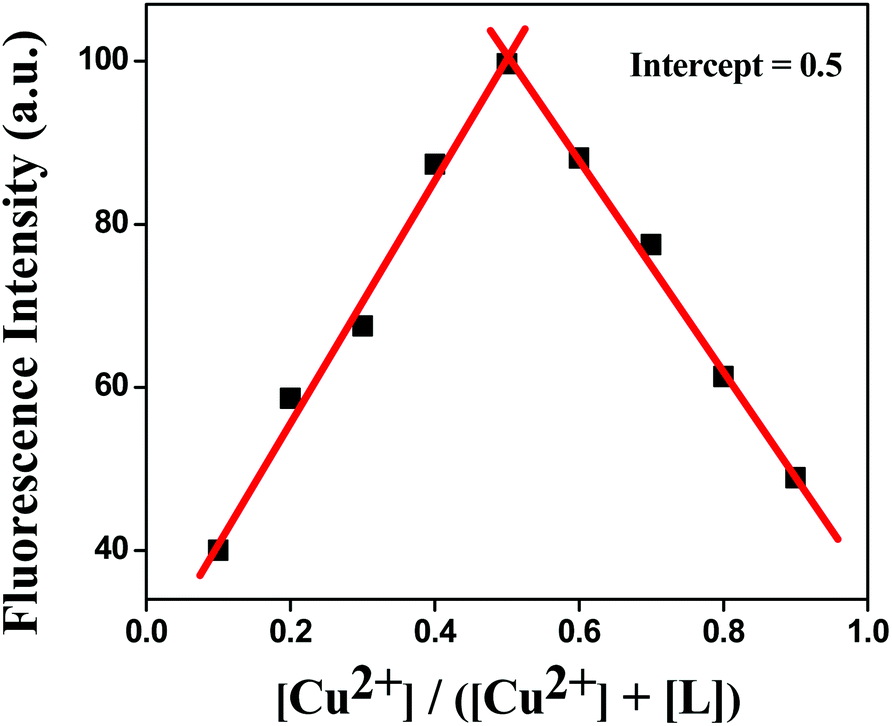
The stoichiometry and binding property of the ligand with Cu2+ were confirmed by absorbance spectra, ESI-mass, Job's plot and single-crystal X-ray. From the Job's plot, it could be observed that the L:M stoichiometry was 1:1 (Fig. 6). The absorbance spectra of the chemosensor HL was measured in the range of 230 to 450 nm in the absence and presence of Cu2+ ions, in acetonitrile. Three absorption peaks were found for free HL at 240, 276 and 343 nm. After the addition of copper to the ligand solution, the above-mentioned peaks were shifted to 243, 287 and 361 nm and two new peaks were generated at 373 and 394 nm, which indicated the formation of the Cu2+–L complex (Fig. S6†). This was further confirmed by ESI-MS, in which a sharp intense peak was found at m/z 488.07 (calcd, 488.07), indicating the formation and high stability of a 1:1 [L + Cu2+] complex (Fig. S4†) in the solution phase as well.
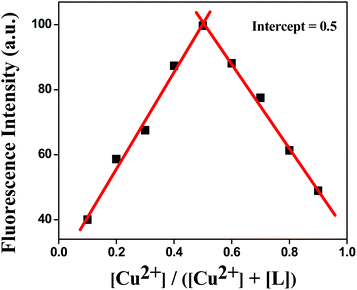 |
| | Fig. 6 Job's plot for L–Cu2+ complexes in 80% H2O/MeCN (v/v) solution. | |
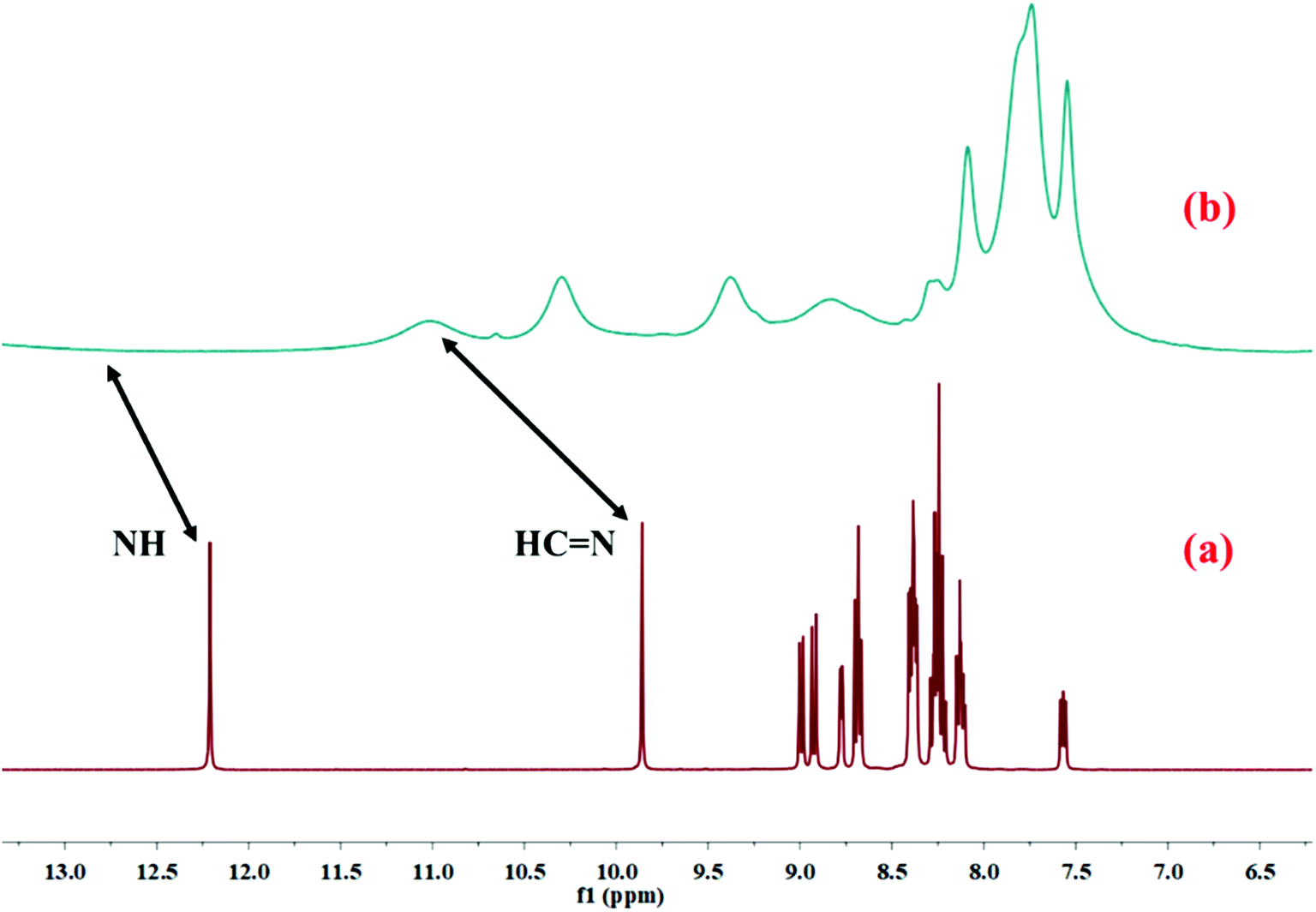
The binding mode of HL with Cu2+ in the solution phase was further confirmed by 1H NMR study. The 1H NMR of HL was taken before and after the addition of Cu2+–salt (Fig. 7). After the addition of copper nitrate, all the 1H NMR peaks were broadened and the proton signal for amide hydrogen (NH) at 12.20 ppm disappeared completely due to the coordination of Cu2+ with HL. The intensity of the proton (CHN) signal at 9.85 ppm decreased and was shifted to 11.02 ppm. Moreover, IR analysis of free HL and Cu–MOM established the binding property. The bands at 1688 and 1581 cm−1 were associated with double-bonds (CO and CN) and were shifted to 1631 and 1553 cm−1 after metalation.
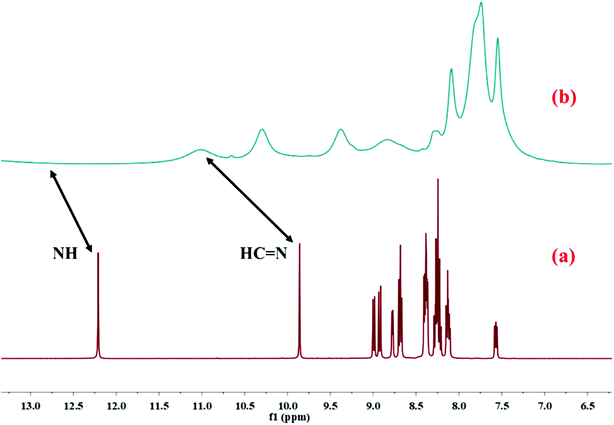 |
| | Fig. 7 1H NMR spectra of (a) HL only and (b) Cu2+–L complex in DMSO-d6. | |
Electronic effect on fluorescence
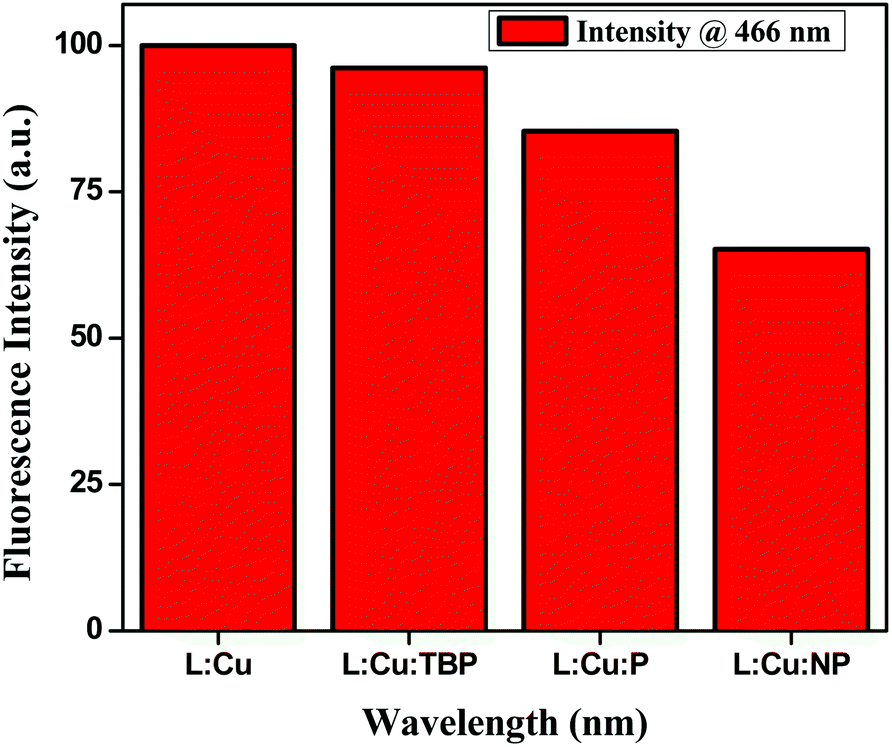
We measured the quenching effect of various substituted aromatics on the hexanuclear L–Cu2+ MOM complex. This MOM complex emits at 466 nm. When exposed to a range of aromatic analytes (phenols), it was found that the phenol with more electron-withdrawing substituent, e.g. nitrophenol, quenched the fluorescence maximum (Fig. 8). The quenching efficiency increased in the order of nirophenol < phenol < tbutyl-phenol ≈ MOM. Also, we found that this MOM complex selectively recognized the catechol colourimetrically from its three positional isomers of benzenediol (catechol, resorcinol and quinol). It changed its colour from greenish yellow to reddish brown upon reaction with catechol, whereas with the other two isomers no appreciable colour change was observed (Fig. S7†).
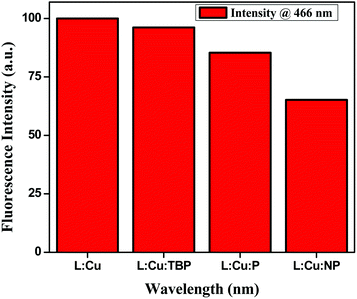 |
| | Fig. 8 Change in fluorescence intensity in the presence of phenol derivatives having different electronic properties. | |
Topographic studies on the surface of the metal–organic macrocycle (MOM)
The morphology and topographic details on the surface of the metal–organic macrocycle (MOM) was studied by field-emission scanning electron microscopy (FESEM). It clearly demonstrated a nice cubic block-like structure with the dimensions of 2 micron (Fig. 9).
 |
| | Fig. 9 FESEM images of Cu–MOM. | |
Crystal structure
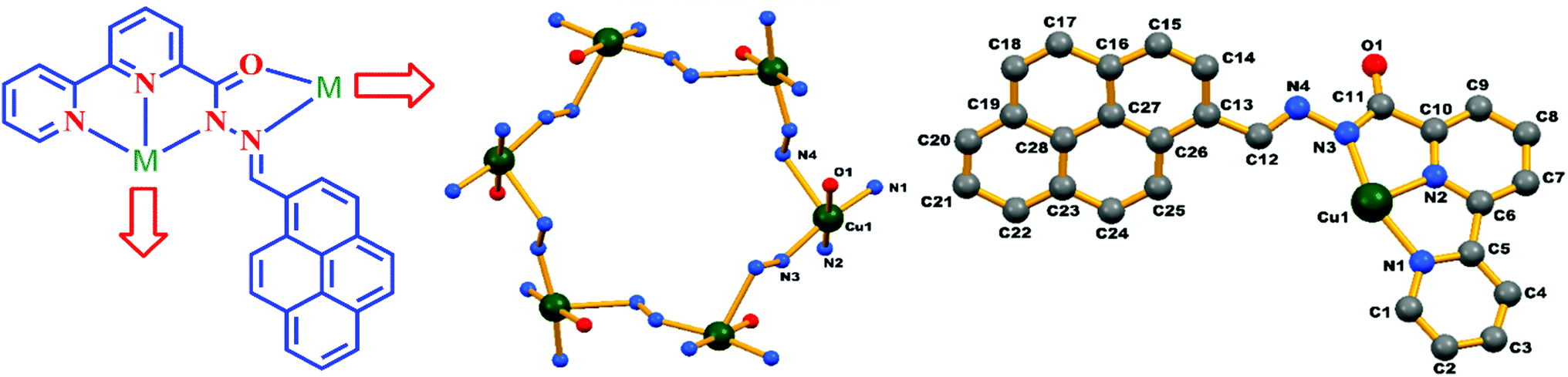
From the literature available, we observed that most of the reported chemosensors and their interaction with the particular metal ion that they sense are based on spectroscopic studies and analysis. Here we report the chemosensor–Cu complex, (MOM), which we not only fully spectroscopically characterized but also X-ray structurally characterized. Single-crystal X-ray diffraction studies confirmed the reaction of HL with copper(II) in a 6:6 stoichiometric ratio to form a hexanuclear metal–organic supramolecular scaffold. Green-coloured single crystals of the L–Cu2+ complex were obtained from the slow evaporation of methanol–chloroform mixture. The complex crystallized in the rhombohedral space group R. The binding mode and the core structure framework are shown in Fig. 10. Further the overall crystal packing is shown in Fig. S8.† The cyclic repeating units from metal ions and the multidentate bridging ligands are –[M–N–N]– in this MOM, which generated an 18 membered hexanuclear copper diazamacrocycle with the S6 symmetry. Two donor atoms of the ligand are coordinated with one metal ion in a bidentate mode on the one side, whereas the other three donor atoms are coordinated with another metal ion on another side in the tridentate mode. It is to be noted that the configuration around each metal centre was chiral; however, the overall configuration of the system was achiral because of the cyclic alternation of the chiral configurations. The charge balance for each Cu centre was ensured by each deprotonated monoanionic ligand and axially coordinated nitrate ion to make the MOM neutral. In this MOM, each copper centre retained its coordination number 6 by coordinating with two stable ligands in bidentate/tridentate chelation modes in five coordination sites, while the 6th coordination for the Jahn–Teller Cu2+ ion was fulfilled by an axially ligated disordered nitrate ion.
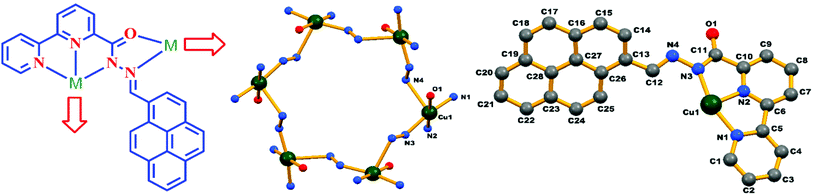 |
| | Fig. 10 Schematic diagram of the coordination modes of the ligand HL in the complex (left), core structure and binding atoms of the complex in an asymmetric unit (right). Hydrogen atoms and disordered nitrate molecules are omitted for clarity. | |
Each individual unit in Cu–MOM was stabilized through intramolecular C–H⋯O hydrogen bonds [C(12)–H(12)⋯O(1): 2.33 Å; C(12)–H(12): 0.95 Å, C(12)⋯O(1): 3.038(9) Å and C(12)–H(12)⋯O(1): 130°] involving an O-atom [O(1)] of a –CO group and H-atom [C(12)–H(12)] of an imine –CHN– group of the ligand (as shown in Fig. 11). Interestingly, two sets of three pyrene rings located on either side of the cyclic molecule were involved in weak intramolecular C–H⋯π interactions [C(15)–H(15)–Cg(3): 2.75 Å, 142.87° and C(17)–H(17)–Cg(5): 2.95 Å, 131.28°; Cg(3): C(13)–C(14)–C(15)–C(16)–C(27)–C(26) and Cg(5): C(19)–C(20)–C(21)–C(22)–C(23)–C(28)] as shown in Fig. 12, where two consecutive pyrene planes formed a dihedral angle of ca. 60°.
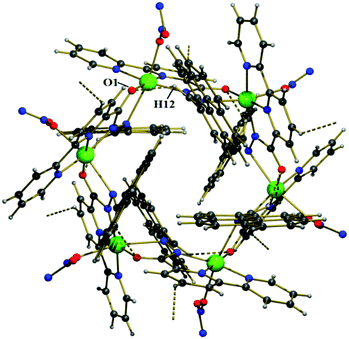 |
| | Fig. 11 A view of the intramolecular C–H⋯O hydrogen bonding interactions in the Cu2+–L complex. | |
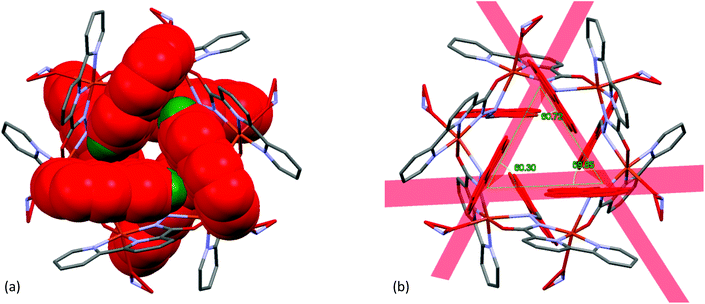 |
| | Fig. 12 (a) A view of the intramolecular C–H⋯π interactions in the Cu2+–L complex (pyrene rings are shown in red and hydrogen atoms in green) and (b) the planes of three pyrene rings with their respective dihedral angles. | |
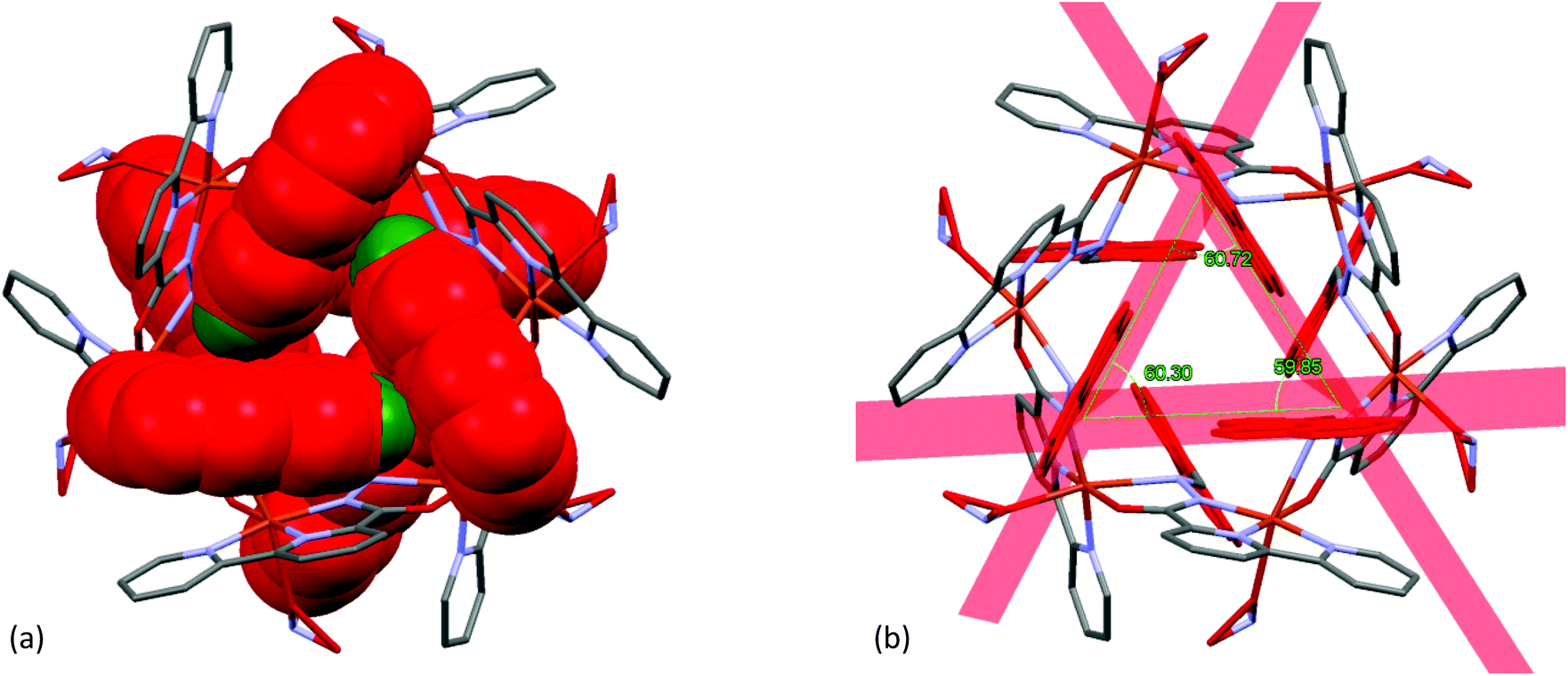
It is clearly evident from Fig. 13(a), that one of the pyridine ring [Cg(2): N(2)–C(6)–C(7)–C(8)–C(9)–C(10)] was engaged in intramolecular π–π stacking interactions [Cg(3)–Cg(2) distance 3.653(5) Å, dihedral angle between two planes (α) 0° and slippage 1.812 Å] with a fused phenyl ring [Cg(3): C(13)–C(14)–C(15)–C(16)–C(27)–C(28)] of the closest pyrene moiety.
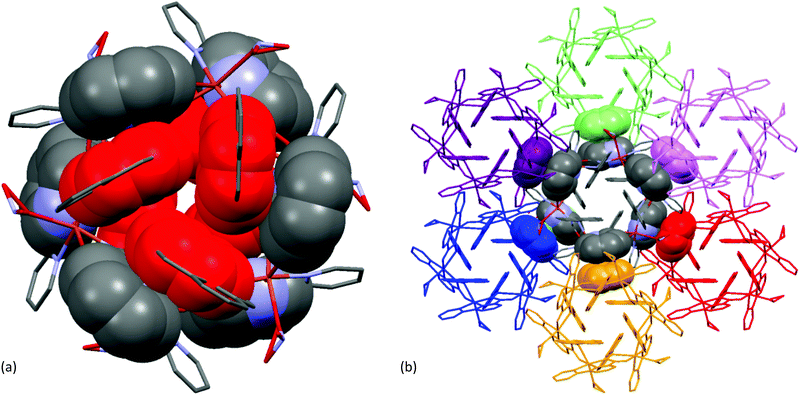 |
| | Fig. 13 A view of: (a) intramolecular π–π stacking interactions in the Cu2+–L complex and (b) intermolecular π–π stacking interactions among six neighbouring molecules. | |
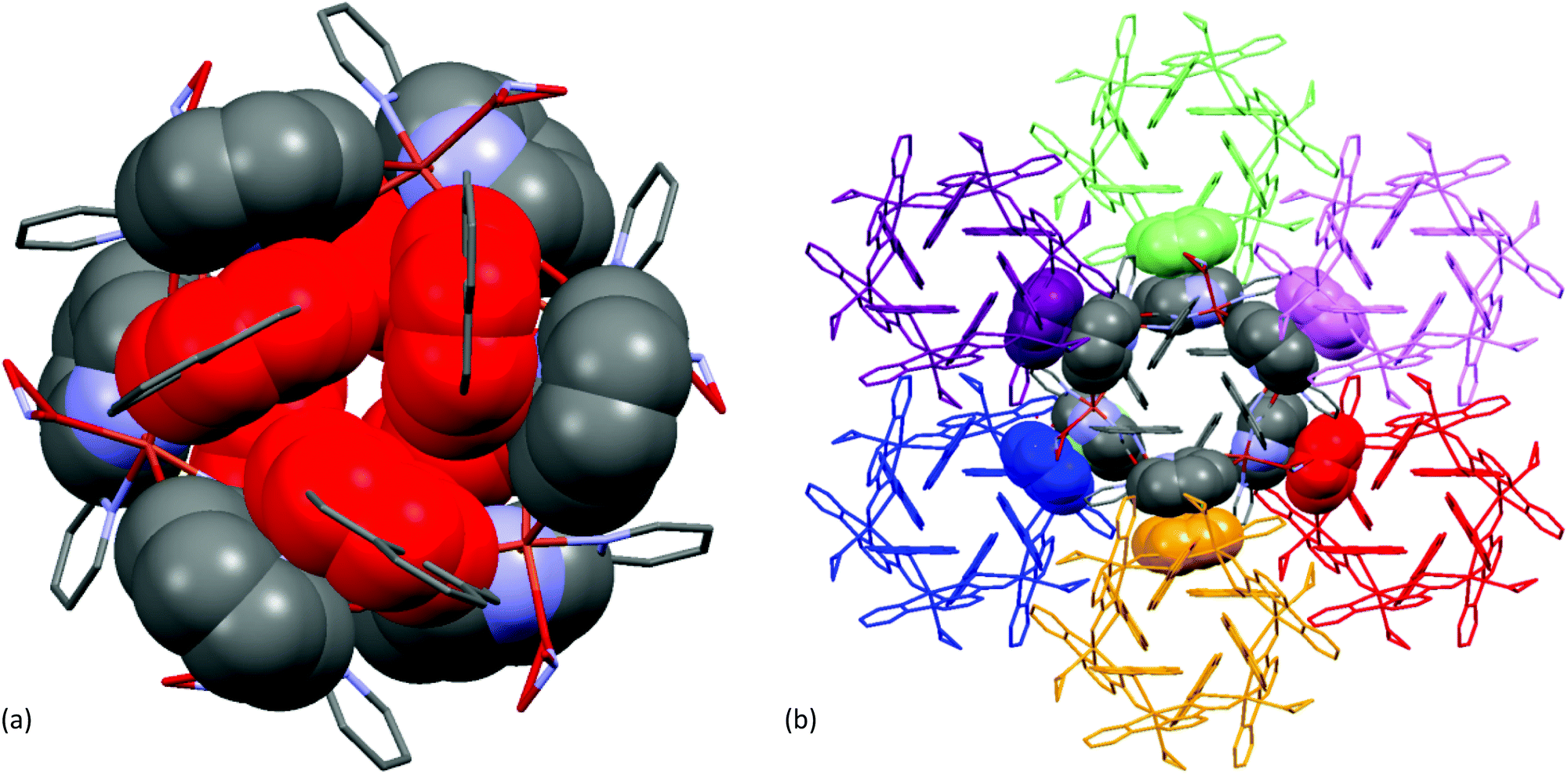
In the crystalline state, each hexa-nuclear molecule was linked to its six neighbouring molecules (as shown in Fig. 13(b)) through intermolecular π–π stacking interactions involving the aromatic pyridine ring [Cg(2): N2–C6–C7–C8–C9–C10; Cg(2)–Cg(2) distance 3.442(5) Å, dihedral angle between two planes (α) 0° and slippage 0.945 Å] forming a 3D network structure.
Fluorescence lifetime measurement
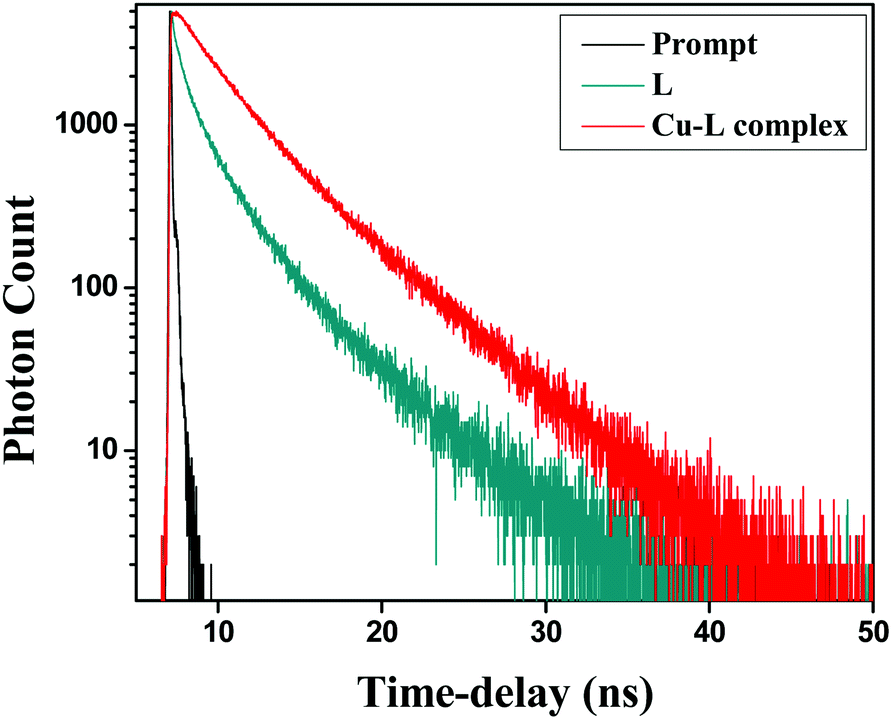
To gain a better insight into the excited state behaviour of a fluorophore, fluorescence lifetime measurement is a significant tool and gives important information. In this regard, the fluorescence lifetime was monitored at (λex = 375 nm) in a water–acetonitrile mixture (8:2 v/v) at λem = 466 nm (Fig. 14). The average lifetimes of the free HL and Cu2+–L complex were calculated from the fitted triple exponential data. The average lifetimes were 0.855 and 3.252 ns, respectively, for 10 μM ligand and the complex in the same solvent mixture.
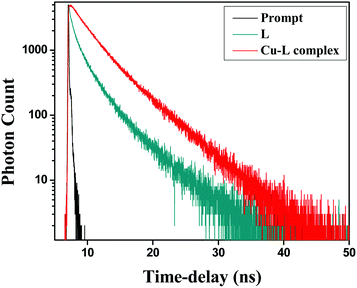 |
| | Fig. 14 Fluorescence decay profiles of HL and Cu2+–L; λex = 375 nm, λem (max) = 366 nm. | |
Biological studies (MTT assay and bioimaging)
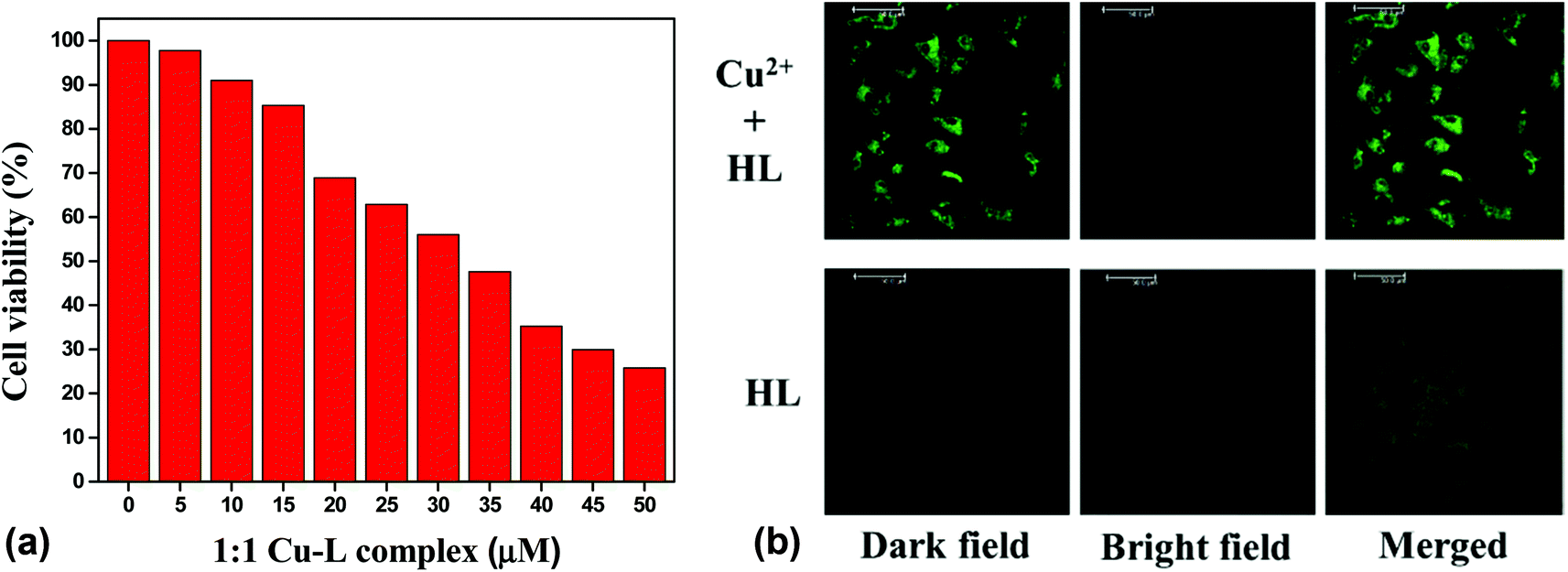
The potential biological application of the chemosensor was studied for imaging Cu2+ in living cells. First, an MTT assay with the Vero cell line was used to determine the cytotoxicity of the chemosensor. It was found that the cellular viability was around 90% after 24 h of treatment of the 10 μM of chemosensor. The IC50 value was found to be approximately 35 μM (Fig. 15(a)), which clearly indicated the low cytotoxicity of the chemosensor. Cell images were further obtained using a confocal fluorescence microscope at below IC50 value, 25 μM of both copper and HL in the mixed aqueous solution (water–MeCN, 8:2; v/v). No significant fluorescence was found in the free ligand, but bright green fluorescence was observed for the Cu2+–L complex. An overlay of the fluorescence and bright-field images is shown in Fig. 15(b).
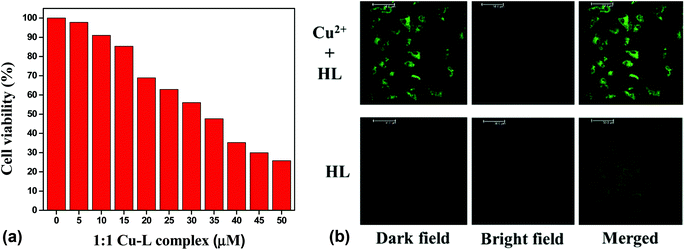 |
| | Fig. 15 (a) Cell viability values (%) estimated by MTT assay versus the incubation concentrations of HL (left) and (b) confocal fluorescence images of 25 μM HL in the presence and absence of 1 equiv. Cu2+ ion (right). | |
Computational studies
In order to have a better understanding of the host–guest interactions, the possible ground state structures of HL and the Cu2+–L complex (asymmetric unit for simplification) in both states (gaseous and aqueous) were optimized (Fig. S9†) using density functional theory DFT at the B3LYP/6-31G(d) and LANL2DZ levels. The effect of water was considered using a polarized continuum model (PCM).
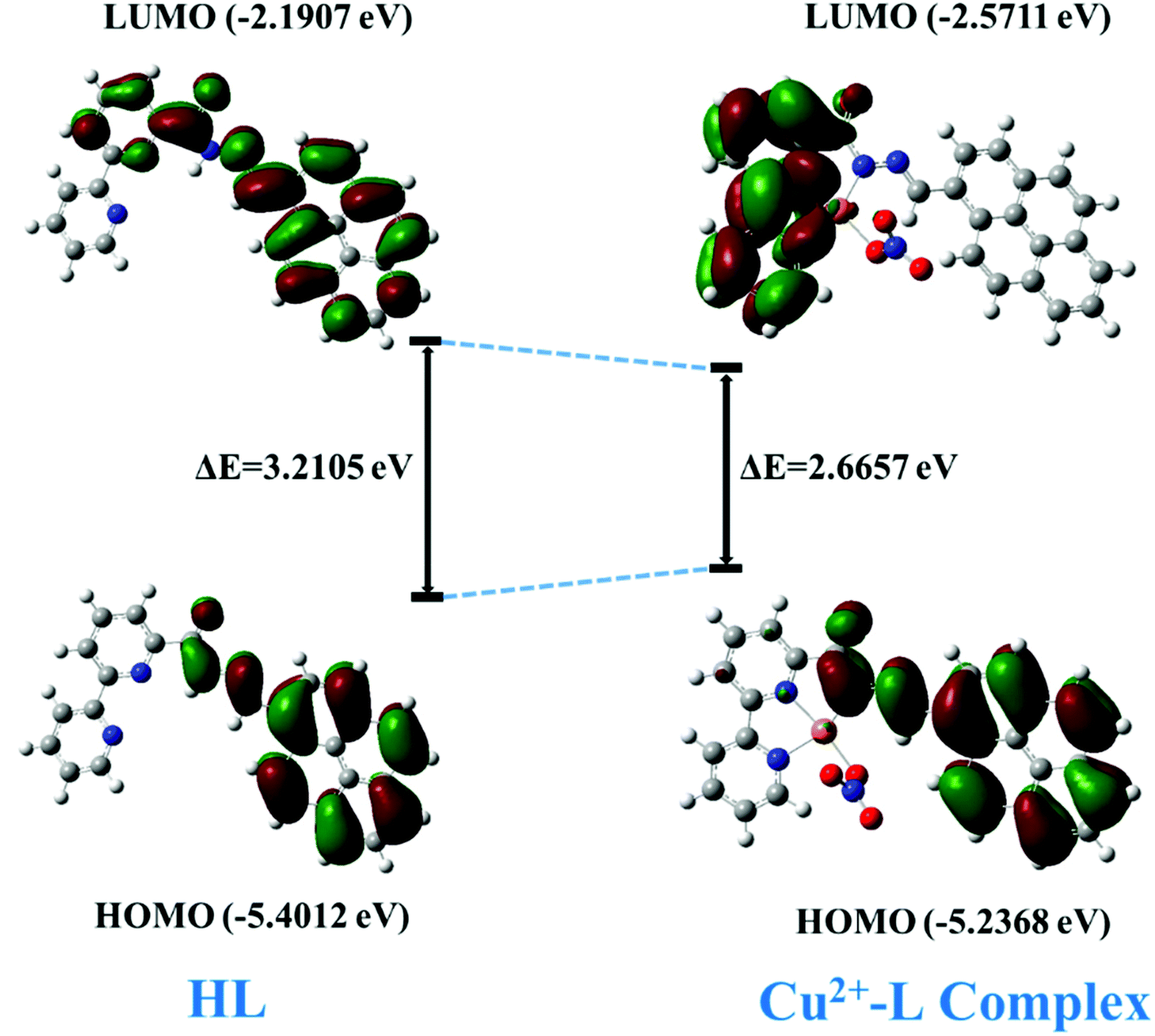
The DFT calculation revealed a sharp decrease in the HOMO–LUMO energy gap, which is in agreement with the subsequent generation of the new red-shifted absorbance peak upon the addition of Cu2+ to HL. Upon coordination of the metal ion with the HL, the energy level of the HOMO increased a little bit, but the energy of the LUMO decreased quite a bit relative to those of free HL. That led to a decrease in the HOMO–LUMO gap by 0.5448 eV (HOMO–LUMO gap was 3.2105 eV for the free ligand and 2.6657 eV for its Cu-complex) (Fig. 16) and resulted in a red-shift of the absorbance spectrum and a change of the photophysical property upon coordination with Cu2+ ion.
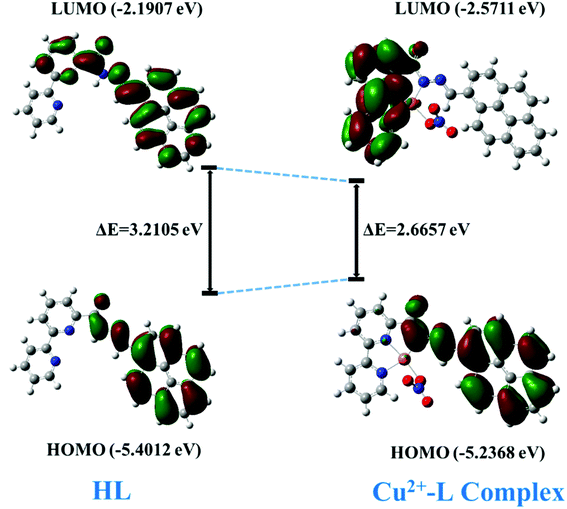 |
| | Fig. 16 The HOMO–LUMO energy gap of HL and its Cu2+ complex. | |
Conclusion
In conclusion, a novel “turn-on” chemosensor for Cu2+ was synthesized and well characterized. It readily functioned in aqueous medium at physiological pH. The low value of its cytotoxicity (high IC50 value ≈ 35 μM) encouraged us to study it by confocal fluorescence microscopy for bioimaging using the Vero cell line to get a green fluorescent image. The X-ray structure of the hexanuclear Cu2+–L complex (MOM) gave us opportunity to understand the exact binding nature of the sensor with the metal ion. Thus this MOM joins the family of very few structurally characterized M-sensor complexes with a turn-on sensing capability. This MOM complex also recognized the catechol moiety selectively among its positional isomers of benzenediol. The electronic effect on the fluorescent MOM confirmed that the phenol derivative having the most electron-withdrawing group quenched the fluorescence intensity maximum.
Conflicts of interest
Authors declare no conflicts of interest.
Acknowledgements
AKS acknowledges IIT Bhubaneswar for infrastructure and Dr K. Bhar, Central University of Rajasthan for providing help in crystal structure of Cu2+–L complex. SMH acknowledges IIT Bhubaneswar for fellowship and PM & SC acknowledge ILS Bhubaneswar.
References
- P. Li, X. Duan, Z. Chen, Y. Liu, T. Xie, L. Fang, X. Li, M. Yin and B. Tang, Chem. Commun., 2011, 47, 7755 RSC.
- N. J. Robinson and D. R. Winge, Biochemistry, 2010, 79, 537 CrossRef CAS PubMed.
- B. Halliwell and J. M. Gutteridge, Biochem. J., 1984, 219, 1 CrossRef CAS.
- P. G. Georgopoulos, A. Roy, M. J. Yonone-Lioy, R. E. Opiekun and P. J. Lioy, J. Toxicol. Environ. Health, Part B, 2001, 4, 341 CAS.
- P. C. Bull, G. R. Thomas, J. M. Rommens, J. R. Forbes and D. W. Cox, Nat. Genet., 1993, 5, 327 CrossRef CAS.
- Y. Hung, A. Bush and R. Cherny, J. Biol. Inorg Chem., 2010, 15, 61 CrossRef CAS PubMed.
- C. Vulpe, B. Levinson, S. Whitney, S. Packman and J. Gitschier, Nat. Genet., 1993, 3, 7 CrossRef CAS.
- D. R. Brown, Brain Res. Bull., 2001, 55, 165 CrossRef CAS.
- S. H. Hahn, M. S. Tanner, D. M. Danke and W. A. Gahl, Biochem. Mol. Med., 1995, 54, 142 CrossRef CAS PubMed.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995 CrossRef CAS.
- G. Multhaup, A. Schlicksupp, L. Hesse, D. Beher, T. Ruppert, C. L. Masters and K. Beyreuther, Science, 1996, 271, 1406 CrossRef CAS.
- Z. Han, Q. Yang, L. Liang and X. Zhang, Adv. Mater. Phys. Chem., 2013, 3, 314 CrossRef.
- L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Coord. Chem. Rev., 2000, 205, 59 CrossRef CAS.
- Y. Zhao, X. B. Zhang, Z. X. Han, L. Qiao, C. Y. Li, L. X. Jian, G. L. Shen and R. Q. Yu, Anal. Chem., 2009, 81, 7022 CrossRef CAS.
- M. Saleem and K. H. Lee, RSC Adv., 2015, 5, 72150 RSC.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC.
- J. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed.
- A. Corma, H. García and F. X. L. Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed.
- J. D. Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957 CrossRef.
- Y. Kang, X. J. Zheng and L. P. Jin, J. Colloid Interface Sci., 2016, 471, 1 CrossRef CAS PubMed.
- V. Pentyala, P. Davydovskaya, M. Ade, R. Pohle and G. Urban, Sens. Actuators, B, 2016, 222, 904 CrossRef CAS.
- L. L. Wen, F. Wang, X. K. Leng, C. G. Wang, L. Y. Wang, J. M. Gong and D. F. Li, Cryst. Growth Des., 2010, 10, 2835 CrossRef CAS.
- L. Sun, H. Xing, J. Xu, Z. Liang, J. Yua and R. Xu, Dalton Trans., 2013, 42, 5508 RSC.
- C. Zhang, L. Sun, Y. Yan, J. Li, X. Song, Y. Liua and Z. Liang, Dalton Trans., 2015, 44, 230 RSC.
- M. J. Prakash and M. S. Lah, Chem. Commun., 2009, 3326 RSC.
- M. J. Dong, M. Zhao, S. Ou, C. Zou and C. D. Wu, Angew. Chem., Int. Ed., 2014, 53, 1575 CrossRef CAS PubMed.
- N. Yanai, T. Uemura, M. Inoue, R. Matsuda, T. Fukushima, M. Tsujimoto, S. Isoda and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 4501 CrossRef CAS PubMed.
- J. Yu, Y. Cui, H. Xu, Y. Yang, Z. Wang, B. Chen and G. Qian, Nat. Commun., 2013, 4, 2719 CrossRef PubMed.
- M. Ma, A. Gross, D. Zacher, A. Pinto, H. Noei, Y. Wang, R. A. Fischer and N. M. Nolte, CrystEngComm, 2011, 13, 2828 RSC.
- J. T. Yeh, W. C. Chen, S. R. Liu and S. P. Wu, New J. Chem., 2014, 38, 4434 RSC.
- H. S. Jung, P. S. Kwon, J. W. Lee, J. I. Kim, C. S. Hong, J. W. Kim, S. Yan, J. Y. Lee, J. H. Lee, T. Joo and J. S. Kim, J. Am. Chem. Soc., 2009, 131, 2008 CrossRef CAS PubMed.
- F. Ye, Q. Chai, X. M. Liang, M. Q. Li, Z. Q. Wang and Y. Fu, Molecules, 2017, 22, 1741 CrossRef PubMed.
- S. Anbu, R. Ravishankaran, M. F. C. G. da Silva, A. A. Karande and A. J. L. Pombeiro, Inorg. Chem., 2014, 53, 6655 CrossRef CAS PubMed.
- A. K. Mahapatra, S. Mondal, S. K. Manna, K. Maiti, R. Maji, M. R. Uddin, S. Mandal, D. Sarkar, T. K. Mondal and D. K. Maiti, Dalton Trans., 2015, 44, 6490 RSC.
- P. Venkatesan and S. P. Wu, RSC Adv., 2015, 5, 42591 RSC.
- M. Koneswaran and R. Narayanaswamy, Sens. Actuators, B, 2009, 139, 104 CrossRef CAS.
- K. V. Shuvaev, L. N. Dawe and L. K. Thompson, Eur. J. Inorg. Chem., 2010, 4583 CrossRef CAS.
- APEX2 and SAINT, Bruker AXS Inc., Madison, WI, 2004 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- A. Kumar, P. Mamidi, I. Das, T. K. Nayak, S. Kumar, J. Chhatai, S. Chattopadhyay, A. R. Suryawanshi and S. Chattopadhyay, PLoS One, 2014, 9, e85714 CrossRef PubMed.
- G. He, J. Li, Z. Wang, C. Liu, X. Liu, L. Ji, C. Xie and Q. Wang, Tetrahedron, 2017, 73, 272 CrossRef CAS.
- S. Chattopadhyay, A. Kumar, P. Mamidi, T. K. Nayak, I. Das, J. Chhatai, I. Basantray, U. Bramha, P. K. Maiti, S. Singh, A. R. Suryawanshi and S. Chattopadhyay, J. Virol. Methods, 2014, 199, 86 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- S. M. Hossain, K. Singh, A. Lakma, R. N. Pradhan and A. K. Singh, Sens. Actuators, B, 2017, 239, 1109 CrossRef CAS.
- S. M. Hossain, A. Lakma, R. N. Pradhan, A. Chakraborty, A. Biswas and A. K. Singh, RSC Adv., 2015, 5, 63338 RSC.
- B. Guo, X. Pan, Y. Liu, L. Nie, H. Zhao, Y. Liu, J. Jing and X. Zhang, Sens. Actuators, B, 2018, 256, 632 CrossRef CAS.
- V. Chandrasekhar, S. Das, R. Yadav, S. Hossain, R. Parihar, G. Subramaniam and P. Sen, Inorg. Chem., 2012, 51, 8664 CrossRef CAS PubMed.
- A. Paul, S. Anbu, G. Sharma, M. L. Kuznetsov, M. F. C. G. da Silva, B. Koch and A. J. L. Pombeiro, Dalton Trans., 2015, 44, 16953 RSC.
- D. Bansal and R. Gupta, Dalton Trans., 2016, 45, 502 RSC.
- S. Ghosh, A. Ganguly, M. R. Uddin, S. Mandal, M. A. Alam and N. Guchhait, Dalton Trans., 2016, 45, 11042 RSC.
- Y. Xiang, A. Tong, P. Jin and Y. Ju, Org. Lett., 2006, 6, 2863 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic information file (CIF) for X-ray crystallographic data, crystal structure, bond length bond angles, 1H and 13C NMR of HL in DMSO-d6, ESI mass of HL and its corresponding Cu2+ complex, UV-vis spectra, etc. CCDC 1836452. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra09376a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Soma Chattopadhyayb and
Akhilesh Kumar Singh
b,
Soma Chattopadhyayb and
Akhilesh Kumar Singh