
Engineering proteinosomes with renewable predatory behaviour towards living organisms†
Chunyu
Zhao
,
Mei
Zhu
,
Ye
Fang
,
Xiaoman
Liu
*,
Lei
Wang
 ,
Dafa
Chen
and
Xin
Huang
*
,
Dafa
Chen
and
Xin
Huang
*
MIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin, 150001, China. E-mail: liuxiaoman@hit.edu.cn; xinhuang@hit.edu.cn
First published on 9th August 2019
Abstract
Communication is a fundamental feature of life, and all living organisms interact actively with their surroundings to better their chance of survival through the coordination of group behavior. To develop mutual interactions between an artificial cell and living organism, we constructed a positively charged thermo-sensitive proteinosome with loaded L-arginine modified chitosan oligosaccharide as an antimicrobial in the hydrogel-based core domain, and then by controlling the temperature and ionic strength we demonstrate that changes in both the hydrophobicity and the electrostatic attraction allowed the constructed proteinosomes to show a programmed interaction with the living organism E. coli, including capture, aggregation induced self-suicide, release, and then the renewal of the proteinosomes. It is anticipated that our results will contribute to the development of the communication behaviours within artificial cell and living organism hybrid microsystems, and lay a foundation for further engineering artificial cell models towards biology inspired materials.
New conceptsFor the first time, we developed a biology-inspired proteinosome-based material which could show programmed mutual interaction with the living organism E. coli, including capture, aggregation induced self-suicide, and release of dead E. coli. By the incorporation of L-arginine grafted chitosan oligosaccharides into the hydrogel matrix of the proteinosome, the E. coli aggregation-induced self-suicide behaviours allowed the designed proteinosomes to show a smart and highly efficient bactericidal behaviour. Significantly, the activity of the proteinosomes could then be recovered by adding salt and cooling down the temperature. In general, it is anticipated that the proposed concept such as capture-killing-release could play an important role in bridging the gap design between biology and materials science. In particular, given that bacterial contamination and the associated risk of infection are serious issues in a variety of areas including public health, medical equipment and water purification etc., the studied interaction behaviours between the constructed proteinosomes and E. coli should offer a potential possibility for the development of proteinosome-based smart antibacterial chasis. In addition, taking the designed proteinosome as a type of protocell, our results should also illustrate a way to engineer synthetic protocells towards advanced programmed interaction behaviors in protocell and living organism hybrid microsystems. We think that this work will be of interest to a wide community of scientists engaged in research based around bio-inspired materials, protocells and soft matter. |
Introduction
The spontaneous self-assembly and spatial integration of molecular and nanoscale building blocks into semi-permeable aqueous micro-compartments is providing a new approach to the design and construction of rudimentary artificial cell-like constructs with biomimetic functions.1,2 Examples of artificial cells include liposomes,3–5 polymersomes,6–14 dendrimersomes,15,16 inorganic nanoparticle-stabilized colloidosomes,17 layer-by-layer microcapsules of counter-charged polyelectrolytes,18,19 surfactants or proteins stabilized oil-in-water emulsions,20,21 and membrane-free coacervates.22,23 Recently, based on the interfacial assembly of protein–polymer nanoconjugates at water droplet/oil interfaces, a protein-enriched microcapsule (proteinosome) was successfully prepared.24–27 Typically, the polymer poly(N-isopropyl acrylamide) (PNIPAAm) was covalently attached to the cationized surface of bovine serum albumin, and the resulting protein–polymer nanoconjugates then were used as membrane building blocks to stabilize water droplets dispersed in isooctanol to produce proteinosomes. The proteinosomes consisted of a closely packed monolayer of conjugated protein–polymer building blocks with an outer and inner surface of polymer and protein-rich domains, respectively. As a synthetic protocell it was shown to be capable of selective permeability, high level guest molecule encapsulation, gene-directed protein synthesis, multi-compartmentalization and spatially confined membrane-gated enzyme activity.14,24–27 Besides these rudimentary studies which have well demonstrated the design and construction of various artificial cell models with bespoke structures, functions and behaviours, recently the advanced design of synthetic artificial cells and the investigation of the communication behaviour within artificial cell communities have been attracting more and more attention from academic communities.Within this context, recent studies by Mann and coworkers successfully demonstrated a predation-based communication behaviour in an interacting community of protease-containing coacervate micro-droplets and proteinosomes,28 as well as a rudimentary form of artificial phagocytosis based on controlled engulfment by taking micro-compartmentalized colloidal objects as a model of a synthetic protocell community.29 Raghavan et al. demonstrated a microscale polymer capsule that was able to show an enzyme-triggered degradation of a certain type of polymeric microbead in their immediate vicinity.30 Chemical communication between populations of synthetic vesicles and bacterial cells via carbohydrate,31 biological autoinducer32,33 or chemical signals was also developed.34–37 Although these studies well indicated the possibility of engineering protocell models as microreactor networks capable of chemical interactions with living organisms, the advanced design of the protocells as a multiplex consortia towards programmable interaction with living organisms, which could lead to a deeper understanding of information-processing among multicellular communities and then allow for the development of life-inspired materials, still needs our great effort.
In this study, we constructed a positively charged and thermo-sensitive proteinosome with loaded acid-sensitive hydrogel composed of aldehyde-functionalized dextran (Dex-CHO), cationized bovine serum albumin (BSA-NH2) and L-arginine grafted chitosan oligosaccharides (COS-Arg) cross-linked by the formation of Schiff base bonds. For the first time we showed a programmed recycled interaction between the proteinosome-based artificial cell and living organism E. coli upon receiving different external stimuli. At 37 °C and in deionized water solution, the living organism E. coli could be highly adsorbed onto the surface of the constructed proteinosomes with the coverage of ca. 93 E. coli per proteinosome. With the amount of attached E. coli increasing, the E. coli colonization on the surface of the proteinosomes would result in an acidic local environment due to the metabolism of the E.coli, and thus the loaded COS-Arg inside the proteinosomes would be triggered to release out, which leads to the death of the E. coli. Interestingly, the dead E. coli could be desorbed from the surface of the proteinosome by cooling down the temperature to 25 °C associated with the addition of 50 mM phosphate buffered saline (PBS) pH 7.4, which then allowed renewal of the designed proteinosomes for the next round of predation towards E. coli.
Results and discussion
To fabricate a positively charged and thermo-sensitive proteinosome, a quaternary-ammonium monomer methacryloxyethyl dimethylethane ammonium bromide (MEDAB) was specially synthesized. It was then employed to copolymerize with the temperature sensitive monomer N-isopropyl acrylamide (NIPAAm) by reversible addition–fragmentation chain-transfer (RAFT) polymerization, thus giving an α-end-capped mercaptothiazoline-activated polymer (PNIPAAm-co-PMEDAB) by using a designed trithiol RAFT agent (Mn 8040 gmol−1, PDI 1.21, monomer repeat units of NIPAAm and MEDAB 52 and 8, respectively, Supplementary methods and Fig. S1–S3, ESI†). The coupling of mercaptothiazoline-activated PNIPAAm-co-PMEDAB polymer chains with primary amine groups of cationized BSA-NH2 was proceeded in 50 mM PBS pH 8.0 to produce the protein–polymer nanoconjugates (BSA-NH2/PNIPAAm-co-PMEDAB). Based on the ultraviolet-vis spectroscopic measurements (Fig. S4 and Table S1, ESI†), it showed that there were ca. ten PNIPAAm-co-PMEDAB chains per BSA molecule which was in agreement with the measured high molecular weight band in SDS-PAGE gel compared with that of the free BSA-NH2 (Fig. S5, ESI†). Next by mixing an aqueous solution of BSA-NH2/PNIPAAm-co-PMEDAB with isooctanol at a water/oil volume fraction (φw) of 0.06, a good dispersion of proteinosomes was obtained with the modulated size ranging from 20 to 50 μm by controlling the concentration of the protein–polymer nanoconjugates from 8.0 to 2.0 mg mL−1 (Fig. S6, ESI†). After cross-linking, the proteinosomes could be transferred into a continuous water phase without the loss of structural integrity (Fig. 1a, b and Fig. S7, ESI†). Also by increasing the temperature to above the LCST of PNIPAAm (ca. 33 °C), the hydrophobicity transition of PNIPAAm on the surface of the proteinosome would allow 23.4% shrinkage in size of a single proteinosome, accompanied by aggregation behaviour due to the hydrophobic interaction (Fig. S8, ESI†).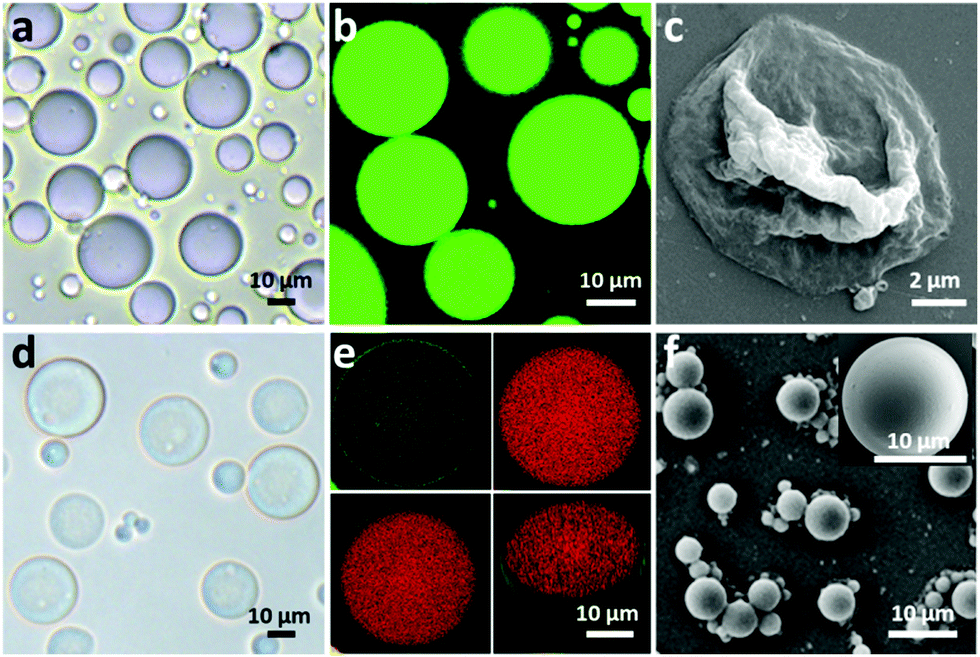 | ||
| Fig. 1 Structural and morphological characterization of proteinosomes. (a) Optical microscopy images of bare proteinosomes dispersed in oil. (b) Fluorescence microscopy images of bare proteinosomes dispersed in water with encapsulated FITC-Dextran. (c) Scanning electron microscopic images (SEM) of bare proteinosomes. (d) Optical microscopy images of hydrogel (BSA-NH2/Dex-CHO) encapsulated proteinosomes dispersed in water. (e) Confocal fluorescence microscopy images (CLSM) of hydrogel-loaded (red) proteinosomes (green) dispersed in oil and the corresponding 3D structure. (f) SEM of hydrogel-loaded proteinosomes. | ||
The membrane permeability of the constructed proteinosomes was evaluated based on the reverse osmosis method to measure the difference in the fluorescence intensity inside and outside the proteinosomes by selectively using different molecular weights of FITC-dextran (from 4 kDa to 150 kDa). In general, the results indicated that when the molecular weight of FITC-dextran was above 70 kDa, they could not pass through the membrane and for those FITC-dextrans with molecular weight below 4 kDa, they pass through the membrane freely (Fig. S9, ESI†). Based on the above observations, in this study, to further improve the spherical structural stability of the empty proteinosomes, a hydrogel-based matrix was designed to form inside the proteinosomes where the building block of a high molecular weight dextran (70 kDa) was selectively used to avoid any possible leaking. In detail, we loaded formyl benzoic acid-functionalized dextran with the molecular weight of ca. 70 kDa (Dex-CHO; 8 μL, 200 mg mL−1, ≈46 aldehyde groups/dextran, Fig. S10 and S11, ESI†) and cationized BSA (BSA-NH2; 68 kDa, 8 μL, 200 mg mL−1, ca. 78 surface primary amine groups per molecule, Fig. S12 and Table S2, ESI†) into the proteinosomes. Then, by forming the acid-sensitive Schiff base linkages at pH 7.4, the protein-polysaccharide hydrogel was generated inside the proteinosomes where the proteinosome membrane was labelled by fluorescein isothiocyanate (FITC) and Dex-CHO of the hydrogel was labelled by rhodamin B isothiocyanate (RBITC), so that the morphology of a single proteinosome with loaded hydrogel was clearly observed by confocal fluorescence microscopy (CLSM), as seen from the green fluorescent membrane with the loading of red fluorescence throughout (Fig. 1e). As expected, with the formation of the hydrogel matrix within the proteinosome, both structural stability against drying and the interfacial contrast when viewed in water by optical microscopy were improved obviously (Fig. 1d and Fig. S7b, ESI†). As seen in Fig. 1f, the spherical structure of the proteinosome-encapsulated hydrogel could be well-maintained with a uniform surface texture even after vacuum drying.In contrast, water-filled proteinosomes prepared without encapsulated hydrogel showed very low interfacial contrast when viewed in water by optical microscopy (Fig. S7b, ESI†) and displayed a completely collapsed morphology in the dry state (Fig. 1c). Also the presence of the quaternary ammonium monomers enabled the constructed proteinosomes to exhibit a higher positively charged surface in DI water (ζ = 14.5 ± 0.5 mV, Fig. S13, ESI†), which provided the possibility to study the interactions between the proteinosomes and negatively charged living organisms.
Based on the above observations, given the positively charged surface and the tunable hydrophobic moiety of the PNIPAAm in the constructed proteinosomes, negatively charged E. coli (DH5α) was employed as an example to study the interaction with the designed proteinosomes. We mixed E. coli (5 × 108 cells mL−1, 0.2 mL) with the constructed hydrogel-loaded proteinosomes (7.27 × 105 units mL−1, 1 mL) in DI water solution at 37 °C, and after different incubation intervals both the free E. coli and proteinosomes with attached E. coli were collected from the supernatant and sediment via low-speed centrifugation (100 rpm, 60 s), respectively. The absorption of the E. coli onto the surface of the proteinosomes was confirmed by the optical microscopic investigation as seen from the increase in the roughness of the proteinosomes in comparison with the smooth surface of the bare proteinosomes (Fig. 2a–c). After drying the samples under vacuum, the attached E. coli on the surface of the proteinosomes were also confirmed by SEM where the E. coli could be well discerned on the surface of the proteinosomes (Fig. 2d). Furthermore, attaching kinetics of E. coli onto the surface of the proteinosomes was studied (Fig. 2e), where one can observe that at 37 °C the whole adsorbing procedure took 8 hours to reach the plateau. There were ca. 93 E. coli per proteinosome after reaching adsorbance saturation (Fig. 2f), and if assuming all the E. coli were horizontally arrayed on the surface of the proteinosome, the surface coverage of the proteinosome by E. coli was about 49.5%.
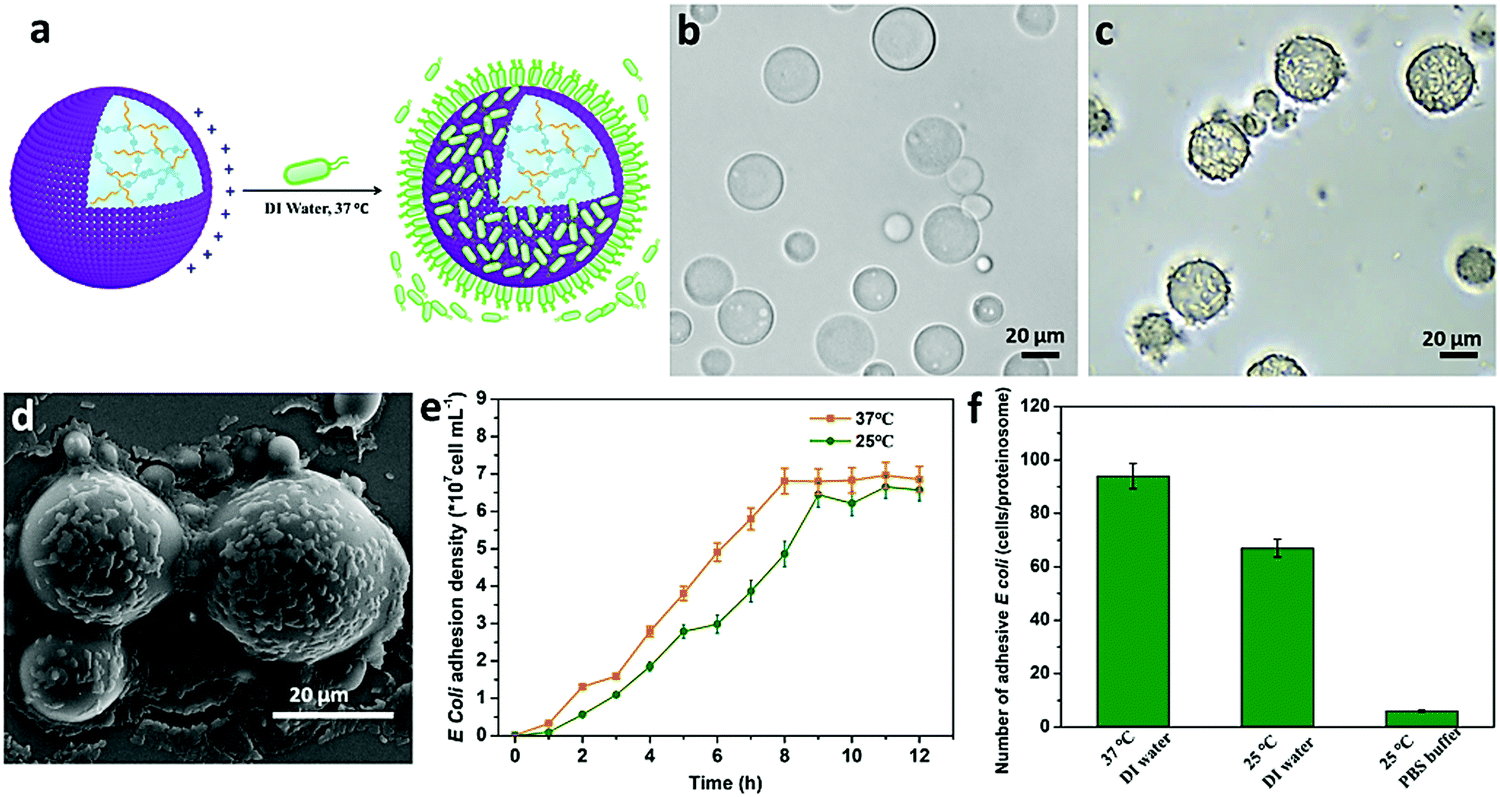 | ||
| Fig. 2 The interaction between the hydrogel-loaded proteinosomes and E. coli. (a) Schematic illustration showing the capturing of E. coli by a higher positively charged surface of hydrogel-loaded proteinosomes in DI water at 37 °C. (b) Optical microscopy images of the bare hydrogel-loaded proteinosomes dispersed in DI water. (c) Optical microscopy images of hydrogel-loaded proteinosomes with attached E. coli at 37 °C in DI water, and (d) the corresponding SEM images. (e) The attachment kinetics of E. coli onto the hydrogel-loaded proteinosomes in DI water, determined at 25 °C and 37 °C, respectively. (f) The average number of attached E. coli per hydrogel-loaded proteinosome under different conditions. Error bars indicate the standard deviation of three replicating measurements. | ||
To further study the interaction behaviour between E. coli and proteinosomes, a fluorescence activated cell sorter (FACS) was used where E. coli was stained by DAPI and hydrogel-loaded proteinosomes were labelled using the red dye RBITC. Control experiments using a single population of the E. coli or proteinosomes gave two-dimensional (2D) pseudo-color plots of Pacific Blue™ dye area (PB450-A) versus phycoerythrin area (PE-A) that were readily distinguishable (Fig. 3a and b). As a consequence, immediately after mixing, two distinct populations were clearly resolved in the 2D pseudo-color plots obtained after mixing dispersions of the E. coli and proteinosomes (t = 0; volume ratio 0.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; number ratio 4:1, respectively) (Fig. 3c), and after 8 hours incubation, the disappearance of the E. coli population well suggested the successful attachment of E. coli onto the surface of the proteinosomes (Fig. 3d). By plotting the corresponding histograms of PE-A against count for the samples, significant changes in the frequency distribution of E. coli after mixing with the proteinosomes for 8 hours were also obviously observed (Fig. 3e and f). As summarized in Fig. 3g, after 8 hours incubation, the proteinosome population remained essentially constant whereas the E. coli counts reduced linearly to approximately a quarter of their initial value. In contrast, after 8 hours incubation, the single populations of the E. coli or proteinosomes un-mixed were observed in control experiments where the pseudo-color plots were basically the same as before (Fig. S14, ESI†). Taken together, these statistical observations suggest that the interaction between the proteinosomes and E. coli could happen widely in the solution, which was also consistent with the attachment kinetics study (Fig. 2e). In addition, it should be mentioned that these interactions exhibited a certain degree of universality, where using another type of E. coli (ATCC 25922) and the Gram-positive bacteria (Staphylococcus aureus, ATCC6538) showed similar adsorbing ability (Fig. S15, ESI†).
:1; number ratio 4:1, respectively) (Fig. 3c), and after 8 hours incubation, the disappearance of the E. coli population well suggested the successful attachment of E. coli onto the surface of the proteinosomes (Fig. 3d). By plotting the corresponding histograms of PE-A against count for the samples, significant changes in the frequency distribution of E. coli after mixing with the proteinosomes for 8 hours were also obviously observed (Fig. 3e and f). As summarized in Fig. 3g, after 8 hours incubation, the proteinosome population remained essentially constant whereas the E. coli counts reduced linearly to approximately a quarter of their initial value. In contrast, after 8 hours incubation, the single populations of the E. coli or proteinosomes un-mixed were observed in control experiments where the pseudo-color plots were basically the same as before (Fig. S14, ESI†). Taken together, these statistical observations suggest that the interaction between the proteinosomes and E. coli could happen widely in the solution, which was also consistent with the attachment kinetics study (Fig. 2e). In addition, it should be mentioned that these interactions exhibited a certain degree of universality, where using another type of E. coli (ATCC 25922) and the Gram-positive bacteria (Staphylococcus aureus, ATCC6538) showed similar adsorbing ability (Fig. S15, ESI†).
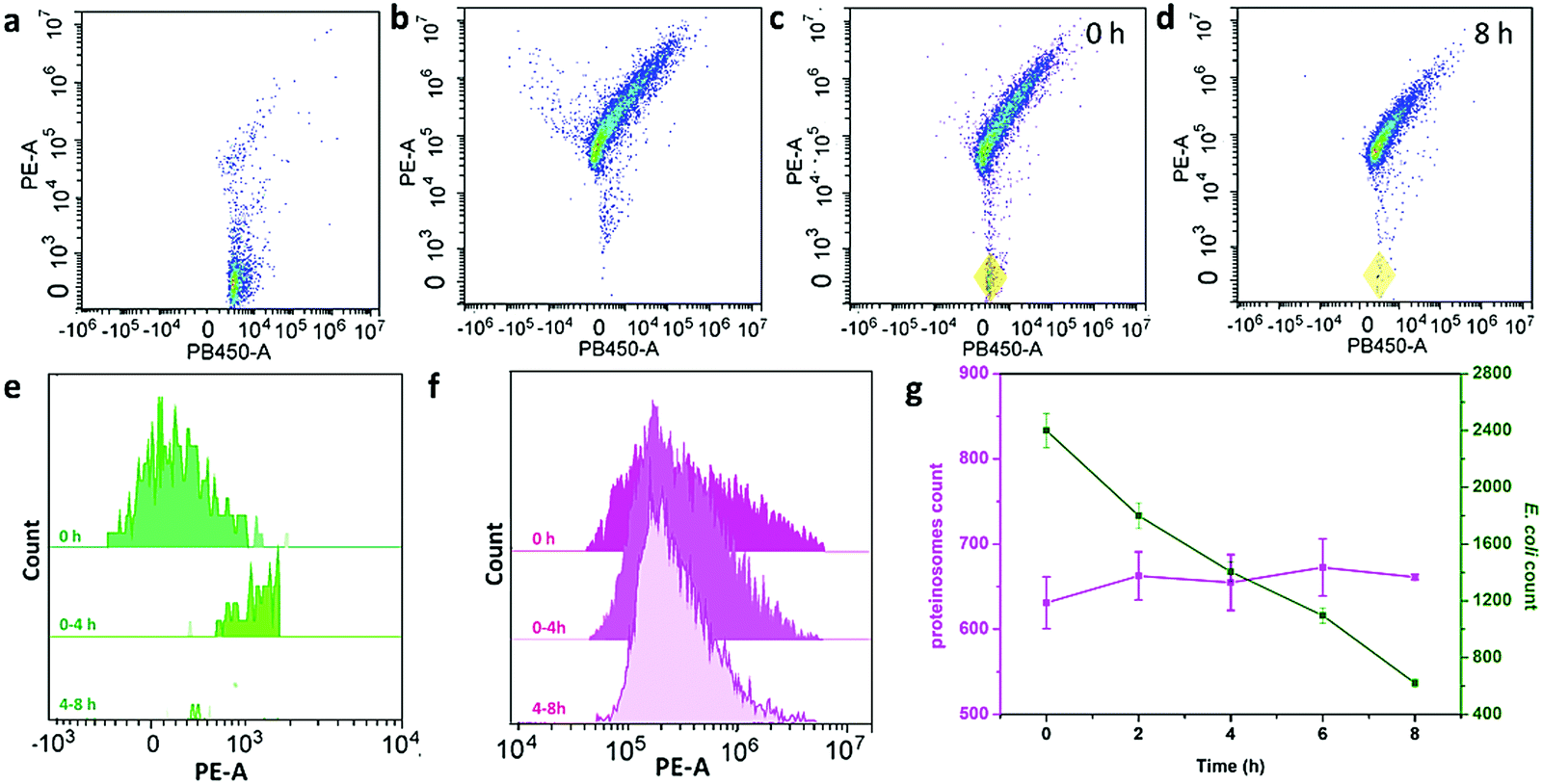 | ||
| Fig. 3 The FACS study of the interaction behaviour of E. coli and proteinosomes. 2D pseudo-color plots for (a) E. coli stained by DAPI, (b) hydrogel-loaded proteinosomes labeled with RBITC, (c) binary population of E. coli and proteinosomes after immediately mixing (t = 0 h, total particles = 10000, number ratio of E. coli:proteinosomes = 4:1) where the coexistence of E. coli was marked by yellow domain, and (d) binary population after mixing for 8 h. (e and f) Time-dependent FACS-derived histograms for a binary population showing the decrease of PE-A for E. coli (e) and essentially unchanged fluorescence for proteinosomes (f) after mixing for up to 8 h. (g) Plot of time-dependent changes in counts over an initial period of 8 h for E. coli (green) and proteinosomes (purple). Error bars indicate the standard deviation of three replicating measurements. | ||
Interestingly, such interaction could be controlled by the programmable modulation of the electrostatic attraction and hydrophobic interaction by increasing the ionic strength or cooling down the temperature (Fig. 4a). To demonstrate the interaction, the E. coli attachment efficiency onto the surface of the proteinosomes under different conditions was determined. In DI water at 25 °C, with the fading of the hydrophobic moieties in the membrane of the proteinosome due to the hydrophilic transition of the PNIPAAm, it showed a 30% decrease in the E. coli attachment efficiency compared with that at 37 °C under the same conditions after 8 hours incubation (Fig. 4b). Alternatively, upon increasing the ionic strength of the mixed solution by adding 50 mM PBS at pH 7.4, which would result in the charge screening, no obvious interaction between E. coli and the proteinosome was observed (Fig. 4b). Based on these observations, on the contrary, we were wondering whether the attached E. coli could be desorbed from the surface of the proteinosomes by controlling the interactions. To do so, we added 50 mM PBS at pH 7.4 into the E. coli attached proteinosome solution and cooled down the temperature to 25 °C. As expected, after 4 h incubation, as seen in Fig. 4c and d, the rough surface of the proteinosomes became relatively smooth and more than 81% of the attached E. coli was desorbed from the proteinosomes (Fig. 4e). Interestingly, thereafter, by centrifuging to remove the salt from the solution, such adsorption and desorption of E. coli on the surface of the proteinosomes could be continuously cycled by controlling the ionic strength and temperature (Fig. 4e). However, after three cycle tests, due to the deformed morphology of the hydrogel-loaded proteinosomes, the number of attached E. coli on the surface of the proteinosome decreases obviously (Fig. S16, ESI†).
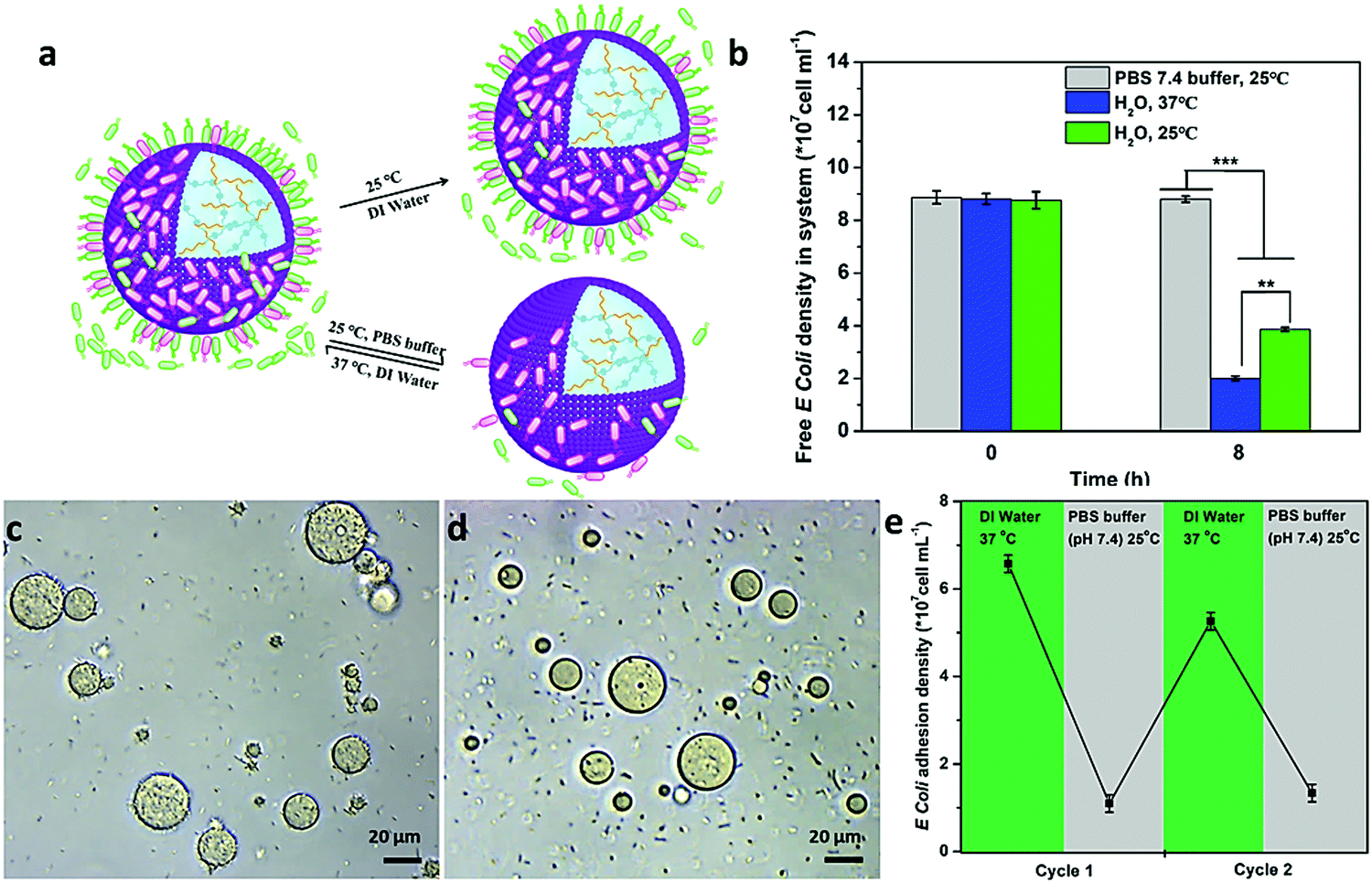 | ||
| Fig. 4 Programmable modulation of the interaction behaviour between the hydrogel-loaded proteinosomes and E. coli. (a) Schematic illustration showing that the interaction between hydrogel-loaded proteinosomes and E. coli could be controlled by increasing the ionic strength (PBS buffer) or cooling down the temperature (25 °C) to modulate the electrostatic attraction and hydrophobic interaction. (b) The average number of free E. coli was determined after 8 h under different conditions. (c) Optical microscopy images of the attached E. coli on the surface of the proteinosomes at 25 °C in water. (d) Optical microscopy images of the attached E. coli on the surface of the proteinosomes at 25 °C in PBS. (e) Reversible binding and release of attachment and de-attachment of E. coli from the surface of the proteinosomes in DI water at 37 °C or PBS buffer (pH 7.4) at 25 °C, respectively. The number of bacteria in solution and detached from the surfaces was quantified using the plate counting method. **p < 0.01. Error bars indicate the standard deviation of three replicating measurements. | ||
Upon attaching of E. coli onto the surface of the proteinosomes, the quaternary ammonium salt in the membrane of the proteinosome could then disrupt the cell membrane and lead to the lysis of the E. coli,38 as observed from Fig. 2d where the morphology of the attached E. coli was deformed. This was also confirmed by staining E. coli with both propidium iodide (PI) and fluorescein diacetate (FDA), and as shown in Fig. 5b, after 2 hours incubation, one can see that most attached E. coli was dead from the appearance of the red fluorescence. However, such adsorbing and then killing interaction only happened on the surface of the proteinosome, and most of the free suspended E. coli in the solution was still alive. To further extend such interaction to a collective interaction behaviour among the hybrid community solution, inspired by the quorum sensing in the living organism population, we wondered whether we could advance the design by constructing an aggregation-induced release of chemical signals behaviour and then result in the annihilation of all the free E. coli in the solution. To do so, an L-arginine modified chitosan oligosaccharide was synthesized as a chemical signal molecule (antimicrobial) (Fig. S17, ESI†), which was then incorporated into the hydrogel matrix inside the proteinosomes by forming the Schiff base with the aldehyde groups (Fig. S18, ESI†). Given that the bacterial colonization can form when the number of attached E. coli is increased, which then makes the local environment become acidic due to the production of acids resulting from the bacterial metabolism,39 this would trigger the release of the bactericide (COS-Arg) from the hydrogel due to the cleavage of the Schiff base bonds. As a control experiment, the release of the COS-Arg from the proteinosomes at different time intervals was confirmed by immersing the samples in 50 mM PBS at pH 5.5 (according to the acidic environment of bacteria) and PBS buffer of pH 7.4, respectively. From the UV-vis spectroscopy, the release of the COS-Arg was recorded at 348 nm by forming a chromogenic derivative with 2,4,6-trinitrobenzene sulfonic acid (TNBSA) (Fig. S19, ESI†). As expected, it showed a much faster release rate of the COS-Arg at pH 5.5 than that of the release at pH 7.4 (Fig. 5c). Then, we mixed the hydrogel-loaded proteinosomes containing COS-Arg with E. coli (1 × 108 cells mL−1) incubated in DI water at 37 °C, by employing litmus as a pH indicator, and with more E. coli absorbed onto the surface of the proteinosomes, it could be observed that their metabolized acidic products dropped the pH of the solution to ca. 5.5 within 1 h (Fig. S20, ESI†). As a consequence, the release of the COS-Arg would be triggered which then killed the free suspended E. coli in the solution associated with the appearance of the red fluorescence (Fig. 5d–g). The killing procedure was also observed in situ from the fluorescence microscopy image (Fig. 5h), and within 8 h, about 97% of the E. coli was killed in this system compared with 44% of E. coli being killed by the hydrogel-loaded proteinosomes in the absence of COS-Arg (Fig. 5i). Moreover, from the study of the COS-Arg release kinetics (Fig. 5c), there was only ca. 11% of the loaded COS-Arg released out within 8 h. Therefore, by adding 50 mM PBS at pH 7.4 into the solution and cooling down the temperature to 25 °C, with the desorption of the attached dead E. coli, the proteinosomes could be recovered and then could be used for the second round of the interaction study with E. coli from the adsorption, aggregation-induced self-suicide to desorption. It should be mentioned that such interaction cycle was strictly modulated in sequence by the ionic strength and temperature, where the whole cycle could be triggered at 37 °C in deionized water. In contrast, when mixing the hydrogel-loaded proteinosomes containing COS-Arg with E. coli in the presence of 50 mM PBS pH 7.4 at 25 °C, neither the absorption nor the aggregation-induced self-suicide like behaviour could be observed obviously.
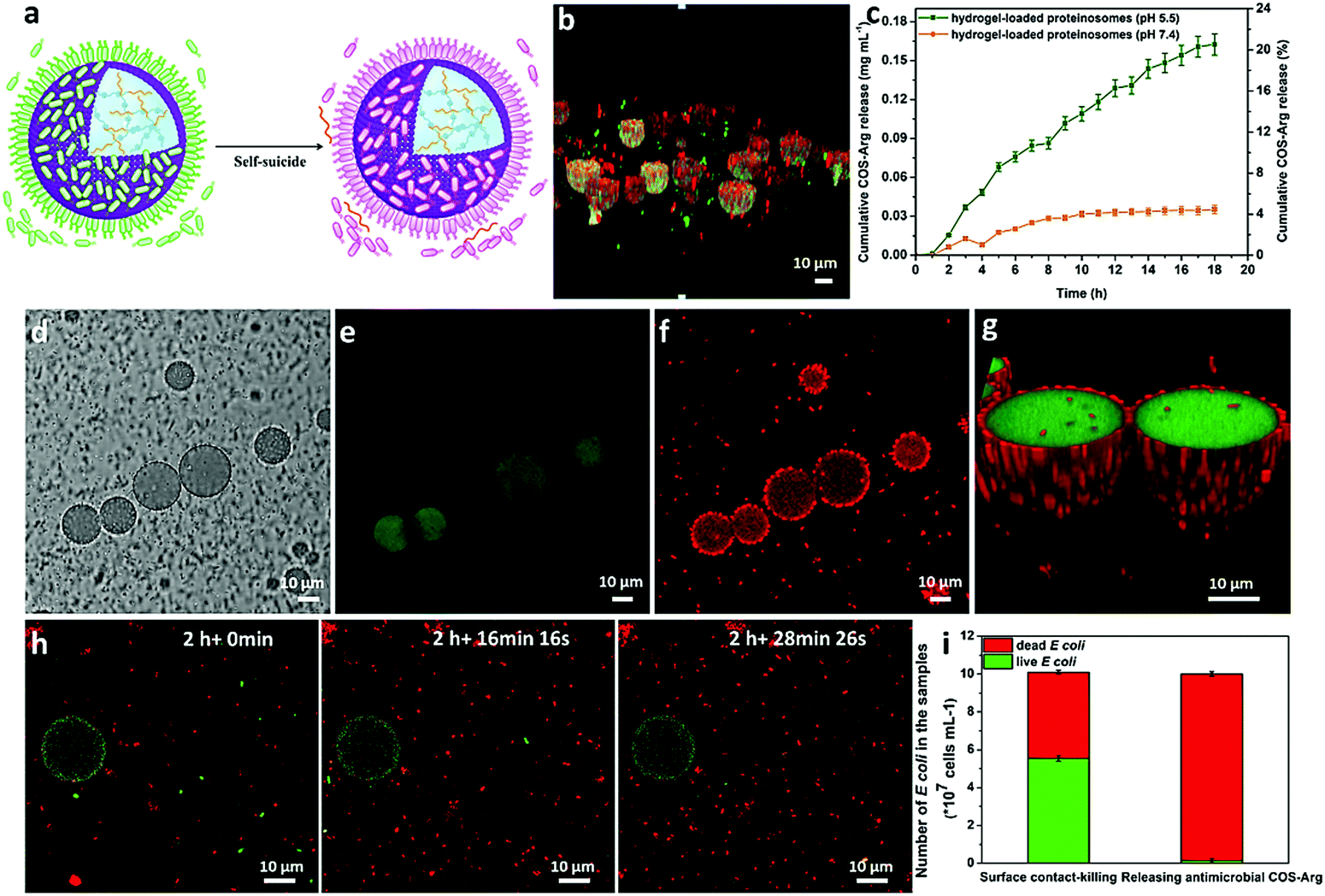 | ||
| Fig. 5 Bactericidal activity of the hydrogel-loaded proteinosomes. (a) Schematic illustration showing the aggregation-induced self-suicide like behaviour of E. coli by the quaternary ammonium salt contact-killing on the surface of proteinosomes and the releasing of the bactericide (COS-Arg) from the proteinosomes due to the decrease in pH. (b) CLSM 3D merge images of the quaternary ammonium salt contact-killing on the proteinosome surface (green staining indicates live bacteria, red staining indicates dead bacteria). (c) Release profiles of COS-Arg from hydrogel-loaded proteinosomes at 37 °C under different pH (7.4 and 5.5) and corresponding percentage of total release in proteinosomes. (d–g) CLSM of the hydrogel-loaded proteinosomes containing COS-Arg with E. coli in DI water, (d) bright field, (e) green fluorescence channel showing no live E. coli in the system via FDA staining, (f) red fluorescence channel showing that the dead E. coli were both around the proteinosomes and in the solution via PI staining, and (g) the 3D merge images. (h) CLSM images showing the death of the free suspended E. coli in the solution in situ at different incubation times. (i) Quantitative analysis of the viability of E. coli in the solution based on the surface contact-killing and releasing of the COS-Arg (1 × 108 cells mL−1). Error bars indicate the standard deviation of three replicating measurements. | ||
Conclusions
In this study, by incorporating a quaternary ammonium-containing monomer into BSA–PNIPAAm nanoconjugates, a type of thermo-sensitive proteinosome with a highly positively charged surface was synthesized. After forming an acid-sensitive hydrogel matrix inside the proteinosomes, by controlling the ionic strength and temperature, the interaction between the designed proteinosome and E. coli was successfully demonstrated including adsorption, aggregation-induced self-suicide, desorption and renewal. Significantly, by the incorporation of L-arginine grafted chitosan oligosaccharides into the hydrogel matrix, the E. coli aggregation-induced self-suicide behaviours allowed the designed proteinosomes to show a highly efficient bactericidal behaviour and more than 97% of E. coli could be annihilated in the system within 8 hours. Interestingly, the activity of the proteinosomes could be recovered by adding salt and cooling down the temperature and then could be used for the second round study. In general, considering the proteinosome as an artificial cell, our results should illustrate a way to design synthetic protocells capable of recycling the capture-killing-release of living organisms, which would contribute to the development of advanced mutual interaction behaviours in protocell and living organism hybrid microsystems, and the proposed concept such as capture-killing-release could also play an important role in bridging the gap design between biology and materials science. In particular, given the low cytotoxicity and good biocompatibility shown by the constructed proteinosomes (Supplementary methods, ESI†), it is anticipated that the studied interaction behaviours between the constructed proteinosomes and E. coli should offer a potential possibility for the development of proteinosome-based smart antibacterial agents towards the challenge of bacterial contamination and the associated risk of infection in a variety of areas including public health, medical equipment and water purification etc.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknoeledge NSFC (51873050, 21871069 and 51703043) and the China Postdoctoral Science Foundation (2015M571401) and the Thousand Young Talent Program for financial support.References
- S. Mann, Acc. Chem. Res., 2012, 45, 2131 CrossRef CAS.
- W. K. Spoelstra, S. Deshpande and C. Dekker, Curr. Opin. Biotechnol., 2018, 51, 47 CrossRef CAS.
- F. Caschera and V. Noireaux, Curr. Opin. Chem. Biol., 2014, 22, 85 CrossRef CAS PubMed.
- N. N. Deng, M. Yelleswarapu, L. Zheng and W. T. Huck, J. Am. Chem. Soc., 2017, 139, 587 CrossRef CAS PubMed.
- S. Seiffert, J. Thiele, A. R. Abate and D. A. Weitz, J. Am. Chem. Soc., 2010, 132, 6606 CrossRef CAS PubMed.
- B. C. Buddingh and J. C. M. van Hest, Acc. Chem. Res., 2017, 50, 769 CrossRef CAS PubMed.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572 RSC.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377 RSC.
- X. Liu, P. Formanek, B. Voit and D. Appelhans, Angew. Chem., Int. Ed., 2017, 56, 16233 CrossRef CAS.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053 CrossRef CAS PubMed.
- Y. Tu, F. Peng, A. Adawy, Y. Men, L. K. Abdelmohsen and D. A. Wilson, Chem. Rev., 2016, 116, 2023 CrossRef CAS PubMed.
- H. Che and J. van Hest, ChemNanoMat, 2019, 5, 1 CrossRef.
- J. Gaitzsch, D. Appelhans, L. Wang, G. Battaglia and B. Voit, Angew. Chem., Int. Ed., 2012, 51, 4448 CrossRef CAS.
- H. Che, B. C. Buddingh and J. C. M. van Hest, Angew. Chem., Int. Ed., 2017, 56, 12581 CrossRef CAS.
- W. Jiang, Y. Zhou and D. Yan, Chem. Soc. Rev., 2015, 44, 3874 RSC.
- S. S. Yadavalli, Q. Xiao, S. E. Sherman, W. D. Hasley, M. L. Klein, M. Goulian and V. Percec, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 744 CrossRef CAS.
- M. Li, R. L. Harbron, J. V. Weaver, B. P. Binks and S. Mann, Nat. Chem., 2013, 5, 529 CrossRef CAS.
- L. Hosta-Rigau, O. Shimoni, B. Stadler and F. Caruso, Small, 2013, 9, 3573 CrossRef CAS.
- S. D. Hann, K. J. Stebe and D. Lee, Langmuir, 2017, 33, 10107 CrossRef CAS.
- P. Torre, C. D. Keating and S. S. Mansy, Langmuir, 2014, 30, 5695 CrossRef CAS.
- L. Wang, Y. Lin, Y. Zhou, H. Xie, J. Song, M. Li, Y. Huang, X. Huang and S. Mann, Angew. Chem., Int. Ed., 2019, 131, 1079 CrossRef.
- S. Koga, D. S. Williams, A. W. Perriman and S. Mann, Nat. Chem., 2011, 3, 720 CrossRef CAS PubMed.
- A. F. Mason, B. C. Buddingh, D. S. Williams and J. C. M. van Hest, J. Am. Chem. Soc., 2017, 139, 17309 CrossRef CAS.
- X. Huang, M. Li, D. C. Green, D. S. Williams, A. J. Patil and S. Mann, Nat. Commun., 2013, 4, 2239 CrossRef.
- X. Huang, A. J. Patil, M. Li and S. Mann, J. Am. Chem. Soc., 2014, 136, 9225 CrossRef CAS.
- X. Liu, P. Zhou, Y. Huang, M. Li, X. Huang and S. Mann, Angew. Chem., Int. Ed., 2016, 55, 7095 CrossRef CAS.
- M. Li, X. Huang, T.-Y. D. Tang and S. Mann, Curr. Opin. Chem. Biol., 2014, 22, 1 CrossRef CAS.
- Y. Qiao, M. Li, R. Booth and S. Mann, Nat. Chem., 2017, 9, 110 CrossRef CAS.
- P. Gobbo, A. J. Patil, M. Li, R. Harniman, W. H. Briscoe and S. Mann, Nat. Mater., 2018, 17, 1145 CrossRef CAS.
- C. Arya, H. Oh and S. R. Raghavan, ACS Appl. Mater. Interfaces, 2016, 8, 29688 CrossRef CAS PubMed.
- P. M. Gardner, K. Winzer and B. G. Davis, Nat. Chem., 2009, 1, 377 CrossRef CAS.
- A. Zargar, D. N. Quan, N. Abutaleb, E. Choi, G. F. Payne, J. L. Terrell and W. E. Bentley, Biotechnol. Bioeng., 2017, 114, 407 CrossRef CAS PubMed.
- A. Gupta, J. L. Terrell, R. Fernandes, M. B. Dowling, G. F. Payne, S. R. Raghavan and W. E. Bentley, Biotechnol. Bioeng., 2013, 110, 552 CrossRef CAS PubMed.
- R. Lentini, S. P. Santero, F. Chizzolini, D. Cecchi, J. Fontana, M. Marchioretto, C. Del Bianco, J. L. Terrell, A. C. Spencer, L. Martini, M. Forlin, M. Assfalg, M. D. Serra, W. E. Bentley and S. S. Mansy, Nat. Commun., 2014, 5, 4012 CrossRef CAS PubMed.
- M. Schwarz-Schilling, L. Aufinger, A. Muckl and F. C. Simmel, Integr. Biol., 2016, 8, 564 CrossRef CAS PubMed.
- X. Qin, C. Engwer, S. Desai, C. Vila-Sanjurjo and F. M. Goycoolea, Colloids Surf., B, 2017, 149, 358 CrossRef CAS PubMed.
- H. Kang, H. J. Jung, D. S. H. Wong, S. K. Kim, S. Lin, K. F. Chan, L. Zhang, G. Li, V. P. Dravid and L. Bian, J. Am. Chem. Soc., 2018, 140, 5909 CrossRef CAS PubMed.
- S. J. Lam, N. M. O'Brien-Simpson, N. Pantarat, A. Sulistio, E. H. Wong, Y. Y. Chen, J. C. Lenzo, J. A. Holden, A. Blencowe, E. C. Reynolds and G. G. Qiao, Nat. Microbiol., 2016, 1, 16162 CrossRef CAS PubMed.
- Y. Lu, Y. Wu, J. Liang, M. R. Libera and S. A. Sukhishvili, Biomaterials, 2015, 45, 64 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mh00976k |
| This journal is © The Royal Society of Chemistry 2020 |