
Stimuli-responsive anisotropic actuation of melem-formaldehyde polymer†
Yang
Wang‡
a,
Niannian
Wu‡
a,
Congyan
Liu
a,
Mohamed K.
Albolkany
a,
Min
Wang
a,
Yan
Wang
a,
Syeda
Arooj
a,
Wenhua
Zhang
b and
Bo
Liu
 *a
*a
aHefei National Laboratory for Physics Sciences at Microscale, Fujian Innovation Institute of Chinese Academy of Sciences, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: liuchem@ustc.edu.cn
bNational Synchrotron Radiation Laboratory, Anhui Provincial Engineering Laboratory of Advanced Functional Polymer Film, University of Science and Technology of China, Hefei, Anhui 230029, China
First published on 8th August 2019
Abstract
In nature, organisms control their deformation and locomotion through multi-functional synergy in response to environmental changes. Nowadays, most synthetic biomaterial systems are capable of realizing single-mode actuation movement under external triggers through biomimetic simulation, while complicated and continuous locomotion in artificial materials with multi-dimensional responsiveness remains to be explored. Here we report an anisotropic melem-formaldehyde polymer that is self-assembled from a one-dimensional (1D) chain into a two-dimensional (2D) film. The stimuli-responsive polymer film shows anisotropic responses in both radial and axial directions on contacting with different solvents (liquid or vapor) in a single material. Complex multi-dimensional motion patterns can be actuated and reversibly controlled by virtue of different interactions within the 1D line and 2D film as well as the gradients along the thickness direction. The developed architecture with tunable and programmable anisotropy is potentially applicable in soft material actuators that can operate in various environments.
New conceptsArtificial soft and smart organic polymers (SOPs) barely achieve anisotropic and multiple actuation owing to their isotropic nature. The absence of anisotropy in SOP materials results from the intrinsically disordered structural feature or average and offset of local anisotropy as a whole. Here we create macroscopic anisotropy on melem-formaldehyde based SOP by assembling a 1D less polar threadlike polymer into a 2D film via cross-linking of the polar component, thus the film with gradients along the thickness direction exhibits anisotropic locomotion in both the radial and axial directions. Freestanding melem-formaldehyde based film (MFF) with a regular and identifiable texture enables anisotropic and flexible responses to various solvents with varied polarity. The reversible and controllable multi-dimensional locomotion of MFF is achieved upon interaction with different solvents in both liquid and vapor phases. The mechanical energy generated from the locomotion can be transferred to a gearing device. Anisotropic SOPs holds great promise for the development of soft actuators, smart robotics and intelligent sensors and beyond. |
Introduction
Through tailorable assemblies at the molecular level, researchers have been able to mimic biological functions using artificially synthesized materials with rapid and continuous responsiveness to external stimuli.1–3 In this regard, enormous efforts have been devoted towards a variety of bio-prototypes, accompanied by emerging progress on soft robotics,4–6 drug delivery,7,8 artificial muscles,9–11 microfluidic systems12,13 and intelligent sensing.14–16 In particular, soft and smart organic polymers (SOPs) adaptive to environmental changes, including electric and/or magnetic field,17–20 temperature,21–23 light,23–25 humidity and pH value,26–28 have drawn increasing interest. Among these, vapor-responsive SOPs are well developed to convert external triggers into mechanical movements such as expansion/contraction, bending, and locomotion, upon interaction with vapor in different regions.29–32 Assembling multilayer films with different structures and functions together has been used to control the transformation of films under vapor stimuli. Langer et al. reported a flexible polymer film assembled from a rigid polypyrrole matrix and a dynamic polyol-borate network, demonstrating film expansion and contraction and thus rapid, continuous locomotion driven by water-exchange with the environment.31 Elastic polymers composed of polyvinylidene fluoride and polyvinyl alcohol were designed to yield reversible chiral coiling upon exposure to acetone vapor, which is expected to be utilized for actuating dynamic elements in soft robotics.32Artificially synthesized SOPs typically respond to external stimuli in single mode owing to their isotropic properties. Complex and predictive shape-change and/or locomotion require multi-dimensional/multiple anisotropic responses of SOPs to environmental changes. Inspired by botanical shape-morphing systems, four-dimensional printing has been developed to induce complex morphology change. For example, biomimetic hydrogel composites patterned in space and time can be encoded with localized swelling anisotropy.33 To date, intrinsic anisotropic response to external stimuli for complex and programmable shape-morphing/locomotion in a single artificially synthesized material remains a great challenge. Most SOPs are isotropic owing to their intrinsically disordered structural feature. Otherwise, the anisotropy in a small locally ordered region of SOPs is averaged and offset, thereby rending most SOP films isotropic as a whole. In principle, the macroscopic anisotropy of SOPs can be created via reasonably tuning the assembly of the microstructure. Here we engineer an easy-to-make and stimuli-responsive melem-formaldehyde-based film (denoted as MFF) that is self-assembled in solution (Fig. 1), and demonstrate its robust anisotropic responses to various stimuli in both radial and axial directions, upon which complex and reversible locomotion can be controlled and rationally designed.
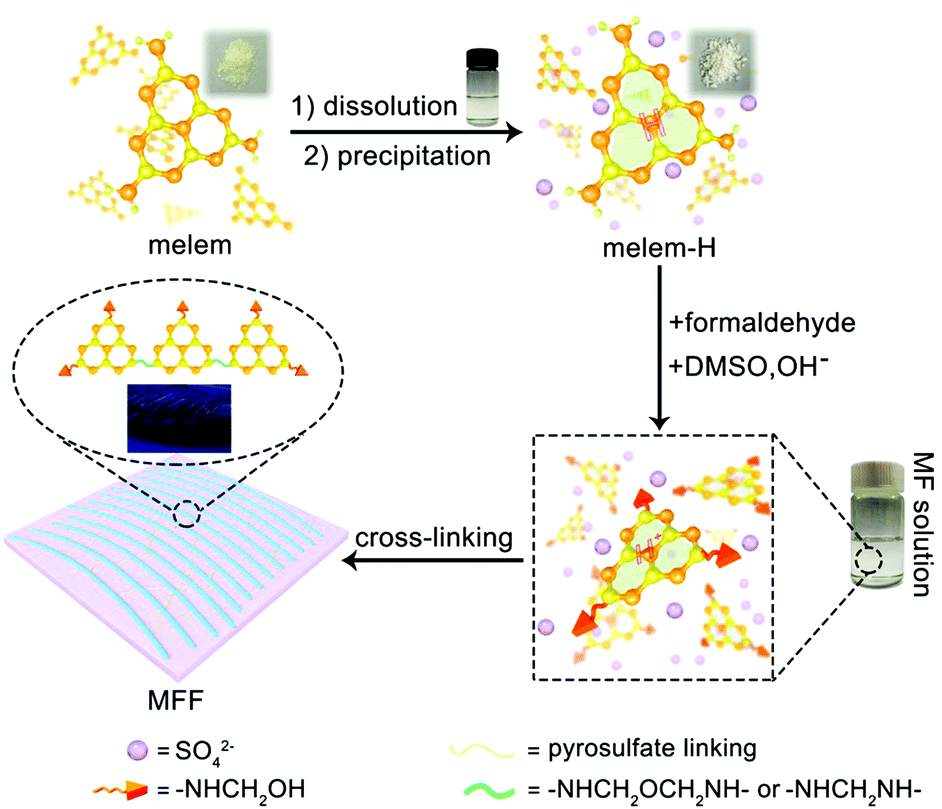 | ||
| Fig. 1 Schematic illustration of the MFF formation process. | ||
Results and discussion
General formation process of melem-formaldehyde film (MFF)
Melem (2,5,8-triamino-tri-s-triazine) is a mysterious molecule proposed by Liebig34 and considered to be the building unit of graphitic carbon nitride.35 Nevertheless, the poor solubility of melem in most solvents hinders its further use owing to the strong intermolecular hydrogen bonding. Being similar to the dissolution of graphene and single-wall carbon nanotubes in concentrated sulfuric acid (H2SO4),36,37 melem can be dissolved in H2SO4 and forms a thermodynamic solution, with a high concentration of ∼400 mg mL−1 (H2SO4, 98%) at room temperature, in which melem is structurally stable (Fig. S1, ESI†). The equivalent nitrogen atoms on the tri-s-triazine skeleton share the same ability to bind the protons of H2SO4, promoting the protonation and dissolution (Fig. 1). And hence, the positive charge of the proton is assumed to delocalize on the whole tri-s-triazine skeleton. Adding anti-solvent (methanol in this work) results in the precipitation of melem-H2SO4 composite (denoted as melem-H, see details in ESI,† Sections S1 and S2.1), which is readily soluble in dimethyl sulfoxide (DMSO). Reaction of melem-H with paraformaldehyde in DMSO at 80 °C gives rise to a clear solution (denoted as MF solution) with assistance of low concentration of sodium hydroxide (Fig. 1). Further increasing the temperature from 80 to 100 °C leads to the formation of a freestanding transparent film with regular texture in the solution, as schematically illustrated in Fig. 1 (see details in ESI,† Section S2). The blue MFF with regular texture is obtained under irradiation of a 365 nm UV lamp (Fig. 1). In sharp contrast, a yellow suspension instead of a thin film is obtained under the same conditions when melem-H is replaced with melamine or melem, although melamine and melem are structurally similar in terms of reacting with formaldehyde. Therefore, protonation of melem is a necessary step for MFF preparation.Varied techniques, including NMR, FT-IR, Raman and XPS are employed to monitor and characterize the formation process of MFF, which reveals that MFF formation undergoes a similar process with melamine-formaldehyde polymer (see details in ESI,† Section S2.2 and Fig. S2). Specifically, two evident 13C-NMR resonances shown in Fig. 2A are indicative of the corner carbon (161.4 ppm) and bay carbon (152.2 ppm) of the melem unit in melem-H, keeping good consistence with that of previously reported melem and further indicating the structural stability of melem in precipitated melem-H.38 In the powder precipitated from MF solution formed at 80 °C via adding acetone, the chemical shifts from melem-H (bay carbon (156.8 ppm) and corner carbon (163.5 ppm)), paraformaldehyde (80–90 ppm)39 and CH2-linked groups (–NH–CH2–NH– and –NH–CH2–OH, 48–54 ppm)40 are confirmed. It is noteworthy that no ether is formed as the chemical shifts are absent between 68 and 80 ppm.41,42 As MFF formed at 100 °C is incapable of dissolving in most solvents, solid-state NMR analysis is investigated. Regardless of the lower resolution compared with that of the liquid-state one, it demonstrates the existence of bay carbon (154.8 ppm) and corner carbon (163.8 ppm) assigned to melem-H, ether (68–80 ppm), –NH–CH2–NH– and –NH–CH2–OH (48–54 ppm). The 13C-NMR analyses also indicate that methylol formed from the reaction between melem-H and formaldehyde undergoes further condensation upon heating. FT-IR spectra confirm that the retained tri-s-triazine structure in MFF, accompanied by the formation of C–O vibration of ether and pyrosulfate linking (Fig. S3, ESI†). High-resolution XPS C1s spectra also help to determine the chemical environment of MFF, as shown in Fig. 2B. Specifically, the main peaks C1 and C2 centred at 288.6 eV and 287 eV stem from sp2 C atoms bonded to N (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) inside the aromatic structure of the tri-s-triazine-based framework43 and C–O ether coordination,44 respectively; another peak C3 at 284.8 eV is assigned to the signal of standard reference carbon. The structure evolution in more detail has been verified by XPS N1s, O1s, S2p and Raman spectra (see details in ESI,† Section S2.3 and Fig. S4, S5). All signal assignments keep good consistency with each other and clearly describe the MFF formation process as discussed above.
N) inside the aromatic structure of the tri-s-triazine-based framework43 and C–O ether coordination,44 respectively; another peak C3 at 284.8 eV is assigned to the signal of standard reference carbon. The structure evolution in more detail has been verified by XPS N1s, O1s, S2p and Raman spectra (see details in ESI,† Section S2.3 and Fig. S4, S5). All signal assignments keep good consistency with each other and clearly describe the MFF formation process as discussed above.
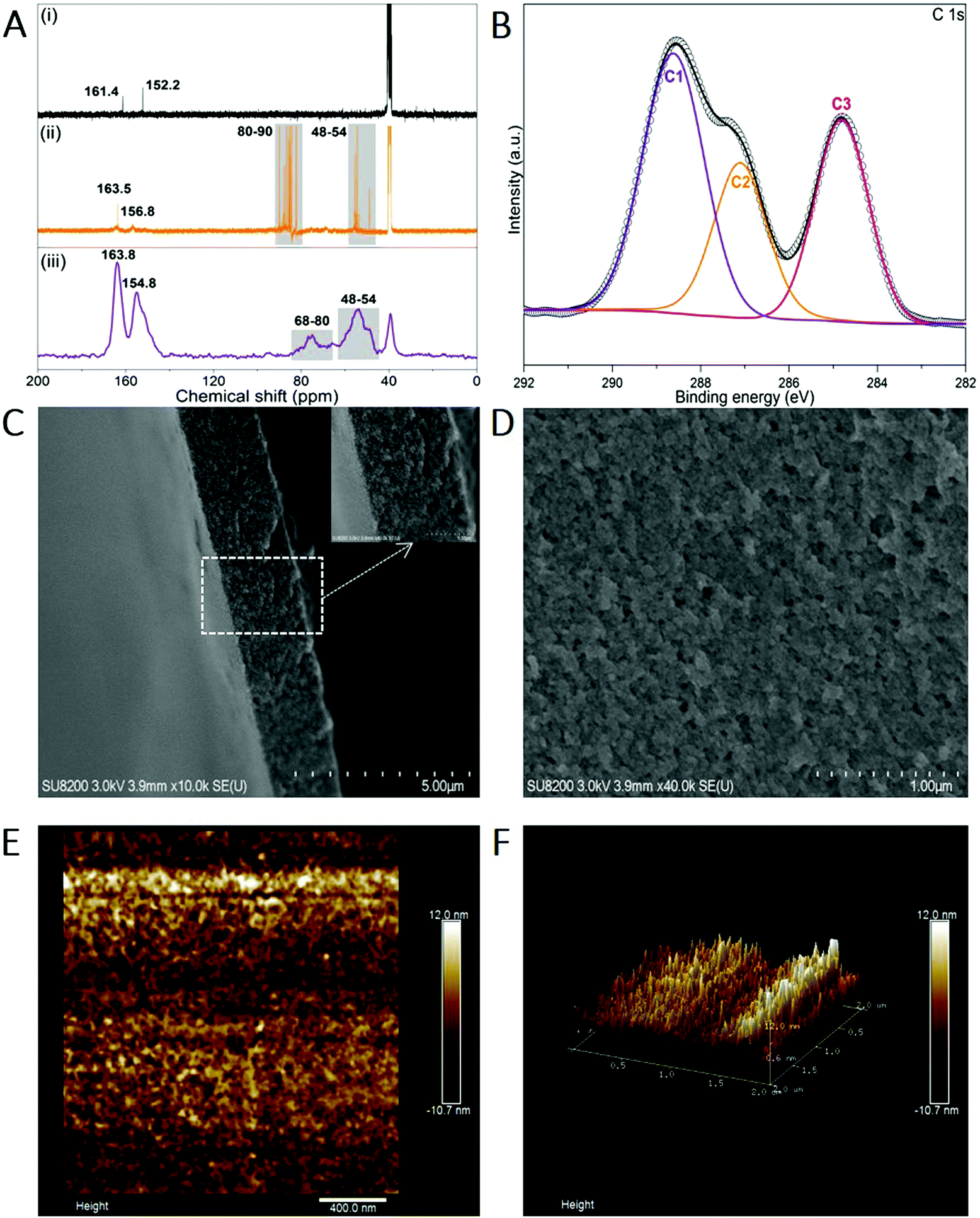 | ||
| Fig. 2 Characterizations of MFF. (A) 13C-NMR spectra of the samples at different stages of the MFF formation process. Liquid-state 13C-NMR spectra of melem-H precipitated with methanol and dissolved in d6-DMSO (i), liquid-state 13C-NMR spectra of the MF solution formed at 80 °C and then precipitated with acetone and dissolved in d6-DMSO (ii), solid-state 13C-NMR of MFF formed at 100 °C (iii). (B) High-resolution XPS C1s spectra of MFF. (C and D) Cross-sectional view (C) and top view (D) SEM images of MFF. (E and F) Two-dimensional (E) and three-dimensional (F) AFM images of MFF. | ||
Mechanism for the formation of MFF with anisotropic texture.
Along with the above analyses, we observed that the MF solution in DMSO turned muddy firstly and then a one-dimensional (1D) threadlike polymer appears, which further assembles into a two-dimensional (2D) film during the heating process at 100 °C (Video S1 and ESI,† Section S3). Remarkably, the texture of MFF is naked eye visible (inset of Fig. 1 and Video S1, ESI†). However, SEM images show that the surface of MFF (thickness: ∼2 μm) is smooth and the AFM images reveal that the surface roughness is lower than 10 nm (Fig. 2C–F), because both SEM and AFM observation only focus on a tiny area of the sample.Nevertheless, considering that the polymerized 1D MF chain is cross-linked by the pyrosulfate component (–S–O–S–), we further performed EDS analyses on the cross section of MFF (Fig. S6, ESI†) to evaluate the cross-linking and polymerization status. The results reveal that the sulfur (S) content changes obviously from the top (∼15.2 wt%) to bottom (∼24.6 wt%) surface, indicating the existence of a cross-linking gradient along the thickness direction. On the other hand, the oxygen (O) content shows an opposite variation tendency from the top (∼24.8 wt%) to bottom (∼17.0 wt%) surface. This finding may suggest the polymerization gradient of the 1D MF chain along the thickness direction, because the chain is polymerized and verified by ether formation, as shown in Fig. 1. Therefore, it is deduced that both cross-linking and polymerization gradients exist along the thickness direction, which are anticipated to regulate the response behaviors of MFF.
Both thickness and size of the 2D film can be controlled via tuning the volume of MF solution and container sizes, respectively. The threadlike polymer can be picked up using low concentration MF solution and we found that the threadlike polymer bends fast within seconds when exposed to nonpolar vapor such as n-pentane, but not to polar water vapor (Fig. 3A–C). In contrast, the 2D film turns flexural in the radial direction (along texture direction) upon exposing to n-pentane and bends in the axial direction (perpendicular to texture direction) in water vapor (Fig. 3D–I). The combined findings reveal that the hierarchical assembly from a 1D threadlike polymer into a 2D film produces macroscopic anisotropy in MFF owing to the distinct structural features inside and in-between the 1D threadlike polymer.
 | ||
| Fig. 3 Digital photos of 1D threadlike MF polymer and 2D MFF in response to n-pentane and water vapor. (A–C) Morphologies of the 1D threadlike MF polymer: pristine (A), exposed to water (B) and n-pentane (C) vapor for 2 s. (D–F) Morphologies of the 2D MFF cutting along the texture development direction: pristine (D), exposed to n-pentane (E) and water (F) vapor, respectively. (G–I) Morphologies of the 2D MFF cutting perpendicular to the texture development direction: pristine (G), exposed to n-pentane (H) and water (I) vapor, respectively. | ||
Associated with the structure of MF (Fig. 1 and details in ESI,† Section S2), we postulate that MF polymerization occurs firstly via dehydration of hydroxyl groups and decarbonylation from adjacent MF monomers at 100 °C and produces 1D threadlike polymers. We have proven this process by FTIR and NMR analyses as discussed above. Owing to stronger interaction between DMSO and sulfate ions than the polymerized chain of less polarity, the threadlike micelle forms with less polar polymerization unit inwards and polar sulfate ions outwards in DMSO. Upon further heating, sulfate ions begin to crosslink and form pyrosulfate to bundle 1D threadlike chains into a 2D film. Pyrosulfate formation (S–O–S bond) is confirmed by FTIR and Raman spectra of MFF (Fig. S3 and S4, ESI†).45,46 Accordingly, the film formation mechanism is schematically illustrated in Fig. 1. The introduction of SO42− anions not only regulates the formation of 1D melem-formaldehyde chains but also creates the macroscopic anisotropy in the 2D film, which is essentially different from melamine-formaldehyde polymer film.
Dichroism is most straightforward in materials that have uniform composition, but could prove to introduce unwanted complexities in materials that exhibit heterogeneous chemical composition and anisotropy. The polarization dependence of near-edge X-ray absorption fine structure (NEXAFS) spectra from oriented materials is a well-documented and understood phenomenon. The resulting linear dichroism spectra can be utilized to assess orientation and the degree of orientational order (anisotropy) in materials at an atomic and molecular level.47 Specifically, the linear dichroism of C and N K-edge spectra of MFF (Fig. 4) are recorded at two orthogonal azimuthal angles with the electric field vector of the incident light along and perpendicular to the texture direction of the sample, respectively. In Fig. 4A, peaks a and d around 284.4 (defects), 285.0 (CN), 287.0 (C–H) and 287.8 (C–NC) eV could be assigned to the π* resonances of the tri-s-triazine framework, while the peaks e and f around 292.8 and 294.7 eV originate from C–N δ* resonances.48–50 Additionally, two peaks a–b at 398.7 and 401.6 eV and a broad peak c centered around 406.6 eV are observed in the N-Kedge spectra of MFF (Fig. 4B). Peaks a and b are assigned to π* (C–NC) and (N–3C) resonances of the tri-s-triazine framework, while peak c is indicative of C–N δ* resonance.48,50–52 Obviously, the C-Kedge and N-Kedge spectra show pronounced linear dichroism when the electric polarization vector (E) is perpendicular to and along the texture direction of MFF; which clearly indicates a sizable in-plane anisotropy of MFF.53,54 Specifically, all π* transitions from the tri-s-triazine ring (peaks a, b and d of C-Kedge and peak a of N-Kedge) are most absorbing when the electric field vector is perpendicular to the texture aligning direction. Hence, on average the tri-s-triazine ring planes might tilt up-right with the plane parallel to the texture direction. Also, the macroscopically anisotropic structure can be clearly seen under polarized and ordinary optical microscopes (ESI,† Section S4, Fig. S7 and S8), and even with the naked eye. The anisotropic actuation of MFF upon exposing to solvents with varied polarities (described below) further supports the macroscopically anisotropic attribute.
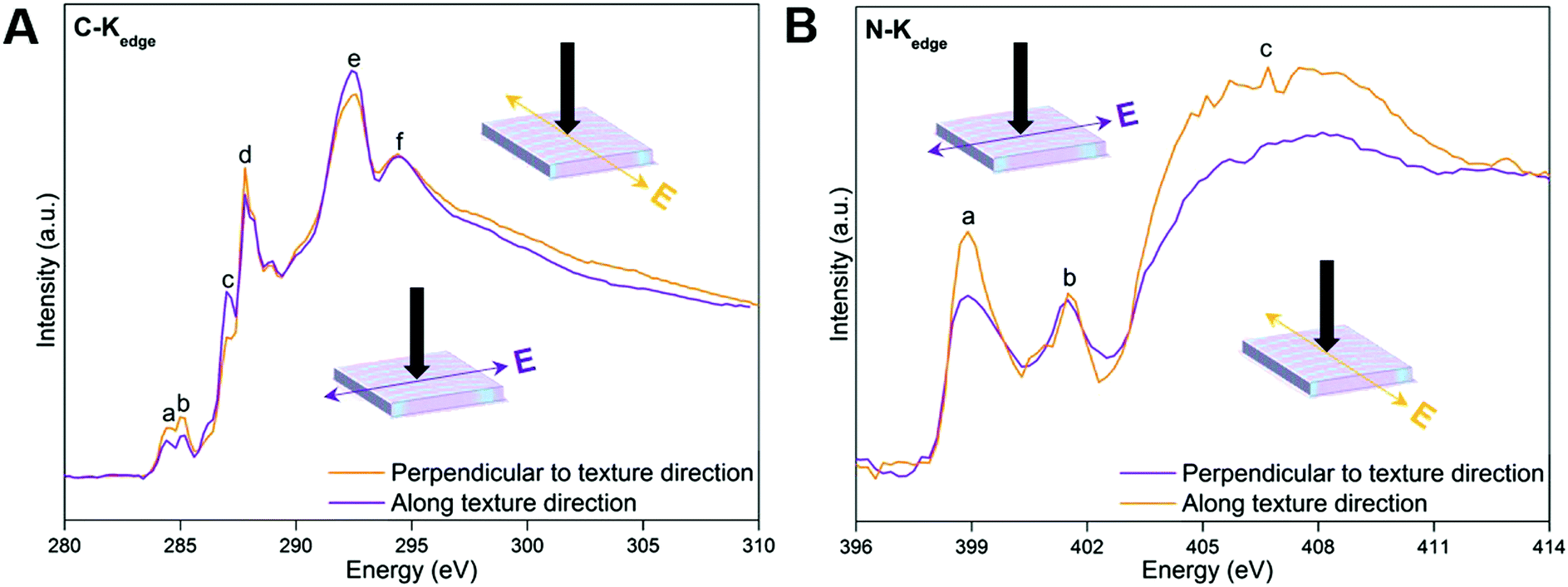 | ||
| Fig. 4 NEXAFS C K-edge (A) and N K-edge (B) spectra of MFF. The black arrows indicate the direction of incident light. | ||
Vapor-responsive mechanism for the 1D MF chain and 2D MFF
Based on the anisotropic polymer structure with gradients along the thickness direction of MFF, we can understand the stimuli-responsive mechanism of the 1D MF chain and 2D MFF upon exposing to water and n-pentane vapor, as schematically displayed in Fig. 5. Specifically, when the 1D MF chain is exposed to n-pentane vapor, the nonpolar n-pentane molecules quickly interact with the less polar melem-formaldehyde component in a van der Waals fashion. By virtue of the n-pentane vapor gradient in air, the bottom of the 1D chain which is closer to the vapor source will adsorb more n-pentane molecules and swell more compared to that of the top side, thus leading to the chain bending (Fig. 5A). However, the above-described effect is much less evident for the vapor of a polar solvent like water, which shows more sluggish interaction with the less-polar 1D chain. Instead, strong capillary action will occur and quickly balance the adsorption throughout the whole 1D chain, which finally results in negligible bending, despite the water vapor gradient (Fig. 5B). | ||
| Fig. 5 Mechanism for the bending of the 1D MF chain and 2D MFF upon contacting with different vapors. (A and B) Interaction of nonpolar n-pentane (A)/polar water (B) vapor with the 1D MF chain. (C and D) Interaction of nonpolar n-pentane (C)/polar water (D) vapor with 2D MFF. The cross-linking (pyrosulfate) and polymerization (1D chain) gradients from the top to bottom surface of MFF are also indicated in the dotted box. | ||
As for 2D MFF, in which the 1D chains are surrounded and linked by the pyrosulfate component. The interaction between the nonpolar n-pentane vapor and less-polar 1D chain (texture) with the polymerization gradient along the thickness direction contributes to the bending of the 2D MFF along the texture direction (Fig. 5C), as the polar pyrosulfate linker shows negligible interaction with nonpolar n-pentane vapor, despite the cross-linking gradient along the thickness direction. However, the polar water vapor will interact with polar pyrosulfate (–S–O–S–) via stronger hydrogen bonding, upon which the pyrosulfate linker with the cross-linking gradient along the thickness direction is more prone to bend, perpendicularly in the texture (1D chain) direction (Fig. 5D). Note that the gradients within MFF from the top to bottom surface are responsible for MFF bending, while the bending direction highly depends on the different interactions between the solvent and MFF. The combined results also highlight the important role of the gradient in controlling the response behaviors of MFF with inherent anisotropy. As such, combined with the (non)polar–(non)polar interactions between solvents and MFF comprising 1D chain with the polymerization gradient and pyrosulfate with the cross-linking gradient, one can except that MFF would bend along and perpendicularly to the texture direction when exposed to solvents with lower (such as MeOH and n-pentane) and higher (such as H2O and DMSO) polarity, respectively, as will also be shown in Fig. 6. Actually, multifunctional composite materials have been realized by introducing a diverse class of gradients, such as concentration, electrostatic complexation and cross-linking gradients during the assembly processes.55–60
 | ||
| Fig. 6 Anisotropic responsiveness of MFF upon interaction with different solvents. (A) Responses of MFF on immersing different solvents with varied polarity for 2 s. (B) Reversible anisotropic actuation of MFF alternatively immersed in DMSO and MeOH. | ||
We collect the water and n-pentane vapor adsorption–desorption curves over MFF using quartz crystal microbalance (QCM) (see details in ESI† Section S5 and Fig. S9). Using nitrogen flow as a carrier gas, the saturated adsorption of n-pentane is ∼23-fold faster than water, whereas the adsorption amount of water is ∼17-fold higher than n-pentane. The adsorption–desorption processes in both cases are reversible, indicating the weak interaction between the polar or nonpolar solvent and MFF (Fig. S10, ESI†). Furthermore, contact angle study reveals that both water and n-pentane are wettable on both sides of MFF (ESI,† Section S6.1 and Fig. S11). We also experimentally confirm that both liquids fail to permeate through MFF (ESI,† Section S6.2 and Fig. S12). It is therefore rational to conclude that the solvent adsorption in the bulk of MFF causes swelling and shrinking that is responsible for the bending behaviors. The unusual wetness behavior towards both polar and nonpolar liquids is consistent with the above structural analyses of MFF, that is, distributed hydrophobic and hydrophilic zones assembling together into a film.
Reversible anisotropic actuation and rotation of MFF
We further investigate the responsive behaviors of MFF towards the solvents in the liquid state with varied polarity. Here we select water, DMSO, methanol and n-pentane as representative solvents to cover a wide range of polarity. When MFF is immersed into the above solvents, distinct locomotion has been observed. In both water and DMSO, MFF curls perpendicularly to the texture direction, however, the film responds much faster in water than in DMSO. In the less polar solvent of MeOH, the film curls along the texture direction. In comparison, MFF immersed in n-pentane leads to much less transformation (Fig. 6A).In a single solvent, MFF curls and then stays static upon reaching the balance. However, when moving the curled film into another solvent, the exchange of the solvent causes the other mode of locomotion. For example, the film in DMSO bends perpendicularly to the texture direction into a roll firstly; the roll in MeOH restores its original shape and further bends along the texture direction into another roll (Fig. 6B). Apart from the bending along or perpendicular to the texture direction, it is of note that MFF bends inwards upon exposure to polar DMSO, resulting from the decreased cross-linking degree from the bottom to top surface. In contrast, it bends outwards in less-polar MeOH after restoration, which is ascribed to the increased polymerization degree from the bottom to top surface (Video S2, ESI†). These findings keep good consistency with the analyses of both cross-linking and polymerization gradients along the thickness direction. Furthermore, this fast responsiveness is reversible and robust without decay when the film is alternatively immersed in DMSO and MeOH (Video S2 and ESI,† Section S7.1). This phenomenon is associated with the cycles of DMSO adsorption, MeOH–DMSO exchange, MeOH adsorption and DMSO–MeOH exchange (Fig. 6B). Nevertheless, the solvent exchange effect on the film cannot be observed in solvent pairs of H2O–MeOH, MeOH–n-pentane and H2O–n-pentane, owing to the different strength of interactions between the solvents and the film and thus unavailable solvent exchange. We could conclude that the strength of the solvent interaction with MFF is in the sequence of H2O > DMSO > MeOH > n-pentane, consistent with the solvent polarity in sequence.
In addition, the negligible change of stress–strain profiles of MFF before and after being exposed to different solvents indicates that the mechanical properties after multiple cycles of anisotropic actuation would not be significantly altered (Fig. S13, Table S1 and ESI,† Section S7.2). Also, the different Young's modulus for MFF that undergoes stretching perpendicular to and along the texture direction again supports its anisotropic nature. It is therefore anticipated that nontoxic water can stimulate MFF bending and promote drug wrapping. More importantly, considering that DMSO (nontoxic within appropriate concentration) can easily permeate through the skin and facilitate the deformation, the drug can be precisely released at s fixed point by smearing DMSO onto the skin, thus showing potential in targeted drug therapy.
The continuously anisotropic actuation of MFF is indicative of the generation of mechanical energy converted from the film transformation. Therefore, we investigated and recorded the rotation behavior of MFF using the experimental facility shown in Fig. S14 (Video S3, ESI,† Section S7.3). Upon immersing into MeOH, the MFF curling drives the elastoplastic tube rotating around the glass rod clockwise. That is, the generated energy of MFF actuation in solvent can be harvested and successfully transferred to the elastoplastic tube, instead of dissipating in the ambient environment. Transferring the facility into DMSO gives rise to MFF back to the original shape and anticlockwise rotation of the elastoplastic tube upon solvent exchange, whereas the curling perpendicular to the texture direction does not occur owing to the physical hindrance of the clamping effect from the elastoplastic tube. Note that the rotation behavior is reversible when repeating the procedures described above. The demo indicates that MFF can be potentially used for driving fluid transportation via optimizing friction, resistance, gravity and other factors. Ideally, when the “bending force” of MFF is larger than the liquid resistance of solvent, the unfailing rotation could be achieved. The related work is underway.
Conclusions
In conclusion, we have fabricated a freestanding melem-formaldehyde film (MFF) with an easy and scalable solution-based self-assembly process. The inherently anisotropic attribute of MFF originates from the distinct structural features inside and in-between the 1D threadlike polymer, which contributes to the fast responsiveness upon exposing to different solvents. Also, the fabricated smart and soft MFF with gradients along the thickness direction, is capable of realizing complex and reversible anisotropic actuation movement upon interacting with different solvents with varied polarity, yielding robust and reproducible dynamic movement without performance decay after multiple cycles. The stimuli-responsive architecture that integrates macroscopic anisotropy and multi-dimensional actuation within a single material triggered by various external stimuli, holds great promise for the development of soft actuators, smart robotics, intelligent sensors and beyond.Experimental
All the procedures for experiments are given in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from Hefei National Laboratory for Physical Sciences at the Microscale, Hefei Science Center of Chinese Academy of Sciences, Fujian Institute of Innovation of Chinese Academy of Sciences, the National Natural Science Foundation of China (NSFC, 21571167, 51502282), the Fundamental Research Funds for the Central Universities (WK2060190053, WK2060190100), and Anhui Province Natural Science Foundation (1608085MB28). We thank Prof. W. H. Zhang at the BL12b beamline of National Synchrotron Radiation Laboratory (Hefei, China) for assistance with the NEXAFS measurements and data analyses. We are also grateful to the anonymous reviewers for valuable comments/suggestions that have helped to improve this work.Notes and references
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, L. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- P. Fratzl and F. G. Barth, Nature, 2009, 462, 442 CrossRef CAS PubMed.
- J. A. Burdick and W. L. Murphy, Nat. Commun., 2012, 3, 1269 CrossRef PubMed.
- D. Rus and M. T. Tolley, Nature, 2015, 521, 467 CrossRef CAS PubMed.
- L. W. Xia, R. Xie, X. J. Ju, W. Wang, Q. Chen and L. Y. Chu, Nat. Commun., 2013, 4, 2226 CrossRef.
- A. A. Stokes, R. F. Shepherd, S. A. Morin, F. Ilievski and G. M. Whitesides, Soft Robot., 2014, 1, 70 CrossRef.
- M. E. Caldorera-Moore, W. B. Liechty and N. A. Peppas, Acc. Chem. Res., 2011, 44, 1061 CrossRef CAS PubMed.
- E. R. Gillies, T. B. Jonsson and J. M. J. Frechet, J. Am. Chem. Soc., 2004, 126, 11936 CrossRef CAS PubMed.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Akahata, T. Kakuta, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270 CrossRef PubMed.
- R. H. Baughman, Science, 2005, 308, 63 CrossRef CAS PubMed.
- J. Foroughi, G. M. Spinks, G. G. Wallace, J. Oh, M. E. Kozlov, S. Fang, T. Mirfakhrai, J. D. W. Madden, M. K. Shin, S. J. Kim and R. H. Baughman, Science, 2011, 334, 494 CrossRef CAS PubMed.
- D. J. Beebe, J. S. Moore, J. M. Bauer, Q. Yu, R. H. Liu, C. Devadoss and B. H. Jo, Nature, 2000, 404, 588 CrossRef CAS PubMed.
- S. A. Morin, R. F. Shepherd, S. W. Kwok, A. A. Stokes, A. Nemiroski and G. M. Whitesides, Science, 2012, 337, 828 CrossRef CAS PubMed.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. V. Duyne, Nat. Mater., 2008, 7, 442 CrossRef CAS.
- T. P. Burg, M. Godin, S. Knudsen, W. J. Shen, G. Carlson, J. S. Foster, K. Babcock and S. R. Manalis, Nature, 2007, 446, 1066 CrossRef CAS.
- S. Singamaneni, M. C. LeMieux, H. P. Lang, C. Gerber, Y. Lam, S. Zauscher, P. G. Datskos, N. V. Lavrik, H. Jiang, R. R. Naik, T. J. Bunning and V. V. Tsukruk, Adv. Mater., 2008, 20, 653 CrossRef CAS.
- Y. Osada, H. Okuzaki and H. Hori, Nature, 1992, 355, 242 CrossRef CAS.
- E. W. H. Jager, E. Smela and O. Inganas, Science, 2000, 290, 1540 CrossRef CAS.
- F. Zhou, P. M. Biesheuvel, E. Y. Choi, W. Shu, R. Poetes, U. Steiner and T. S. Huck, Nano Lett., 2008, 8, 725 CrossRef.
- F. Hua, T. H. Cui and Y. M. Lvov, Nano Lett., 2004, 4, 823 CrossRef CAS.
- Y. Osada and A. Matsuda, Nature, 1995, 376, 219 CrossRef CAS.
- J. Kim, J. A. Hanna, M. Byun, C. D. Santangelo and R. C. Hayward, Science, 2012, 335, 1201 CrossRef CAS PubMed.
- H. J. Shepherd, I. A. Gural'skiy, C. M. Quintero, S. Tricard, L. Salmon, G. Molnar and A. Bousseksou, Nat. Commun., 2013, 4, 2607 CrossRef.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778 CrossRef CAS.
- C. L. Oosten, C. W. M. Bastiaansen and D. J. Broer, Nat. Mater., 2009, 8, 677 CrossRef.
- S. Alexander, T. Krupenkin, A. Taylor, P. Fratzl and J. Aizenberg, Science, 2007, 315, 487 CrossRef.
- T. S. Shim, S. H. Kim, C. J. Heo, H. C. Jeon and S. M. Yang, Angew. Chem., Int. Ed., 2012, 51, 1420 CrossRef CAS.
- I. Tokareva, G. L. He, C. X. Wu and J. U. Sommer, J. Am. Chem. Soc., 2004, 126, 15950 CrossRef CAS.
- Q. Zhao, J. W. C. Dunlop, X. Qiu, F. Huang, Z. Zhang, J. Heyda, J. Dzubiella, M. Antonietti and J. Yuan, Nat. Commun., 2014, 5, 4293 CrossRef.
- L. Zhang, P. Naumov, X. Du, Z. Hu and J. Wang, Adv. Mater., 2017, 29, 1702231 CrossRef.
- M. Ma, L. Guo, D. G. Anderson and R. Langer, Science, 2013, 339, 186 CrossRef CAS.
- J. Mu, G. Wang, H. Yan, H. Li, X. Wang, E. Gao, C. Hou, A. T. C. Pham, L. Wu, Q. Zhang, Y. Li, Z. Xu, Y. Guo and E. Reichmanis, Nat. Commun., 2018, 9, 590 CrossRef PubMed.
- A. S. Gladman, E. A. Matsumoto, R. G. Nuzzo, L. Mahadevan and J. A. Lewis, Nat. Mater., 2016, 15, 413 CrossRef PubMed.
- J. Liebig, Ann. Pharm., 1834, 10, 1 CrossRef.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868 CrossRef CAS PubMed.
- N. Behabtu, J. R. Lomeda, M. J. Green, A. L. Higginbotham, A. Sinitskii, D. V. Kosynkin, D. Tsentalovich, A. N. G. Parra-Vasquez, J. Schmidt, E. Kesselman, Y. Cohen, Y. Talmon, J. M. Tour and M. Pasquali, Nat. Nanotechnol., 2010, 5, 406 CrossRef CAS.
- V. A. Davis, A. N. G. Parra-Vasquez, M. J. Green, P. K. Rai, N. Behabtu, V. Prieto, R. D. Booker, J. Schmidt, E. Kesselman, W. Zhou, H. Fan, W. W. Adams, R. H. Hauge, J. E. Fischer, Y. Cohen, Y. Talmon, R. E. Smalley and M. Pasquali, Nat. Nanotechnol., 2009, 4, 830 CrossRef CAS.
- B. Jurgens, E. Irran, J. Senker, P. Kroll, H. Muller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288 CrossRef.
- Y. Zhao, N. Yan and M. W. Feng, ACS Sustainable Chem. Eng., 2013, 1, 91 CrossRef CAS.
- M. X. Tan, Y. Zhang and J. Y. Ying, ChemSusChem, 2013, 6, 1186 CrossRef CAS.
- Z. Lv, Q. Sun, X. Meng and F. S. Xiao, J. Mater. Chem. A, 2013, 1, 8630 RSC.
- S. I. Tohmura, A. Inoue and S. H. Sahari, J. Wood Sci., 2001, 47, 451 CrossRef CAS.
- X. Zhang, X. Xie, H. Wang, J. Zhang and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18 CrossRef CAS.
- G. P. Lopez, D. G. Castner and B. Ratner, Surf. Interface Anal., 1991, 17, 267 CrossRef CAS.
- R. Fehrmann, N. H. Hansen and N. J. Bjerrum, Inorg. Chem., 1983, 22, 4009 CrossRef CAS.
- M. Jariwala, J. Crawford and D. J. Lecaptain, Ind. Eng. Chem. Res., 2007, 46, 4900 CrossRef CAS.
- H. Ade and B. Hsiao, Science, 1993, 262, 1427 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. H. Zhu, L. H. Li, Y. Han, Y. Chen and A. J. Du, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- W. Che, W. Cheng, T. Yao, F. Tang, W. Liu, H. Su, Y. Y. Huang, Q. H. Liu, J. K. Liu, F. C. Hu, Z. Y. Pan, Z. H. Sun and S. Q. Wei, J. Am. Chem. Soc., 2017, 139, 3021 CrossRef CAS PubMed.
- O. Plashkevych, A. Snis, L. Yang, H. Agren and S. F. Matar, Phys. Scr., 2001, 63, 70 CrossRef CAS.
- G. P. Mane, S. N. Talapaneni, K. S. Lakhi, H. Ilbeygi, U. Ravon, K. A. Bahily, T. Mori, D. H. Park and A. Vinu, Angew. Chem., Int. Ed., 2017, 56, 8481 CrossRef CAS PubMed.
- I. Shimoyama, G. H. Wu, T. Sekiguchi and Y. J. Baba, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 0163 CrossRef.
- A. P. Smith and H. Ade, Appl. Phys. Lett., 1996, 69, 3833 CrossRef CAS.
- T. M. Jiang, K. Koshmak, A. Giglia, A. Banshchikov, N. S. Sokolov, F. Dinelli, R. Capelli and L. Pasquali, J. Phys. Chem. C, 2017, 121, 4426 CrossRef CAS.
- A. R. Studart, Chem. Soc. Rev., 2016, 45, 359 RSC.
- D. Kokkinis, M. Schaffner and A. R. Studart, Nat. Commun., 2015, 6, 8643 CrossRef PubMed.
- A. R. Studart, Adv. Funct. Mater., 2013, 23, 4423 CrossRef CAS.
- J. K. Sun, M. Antonietti and J. Yuan, Chem. Soc. Rev., 2016, 45, 6627 RSC.
- Q. Zhao, J. Heyda, J. Dzubiella, K. Täuber, J. W. C. Dunlop and J. Yuan, Adv. Mater., 2015, 27, 2913 CrossRef CAS.
- H. Lin, J. Gong, H. Miao, R. Guterman, H. Song, Q. Zhao, J. W. C. Dunlop and J. Yuan, ACS Appl. Mater. Interfaces, 2017, 9, 15148 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary discussions and 14 figures. Video S1. MFF formation process. Video S2. Reversible anisotropic actuation of MFF in DMSO and MeOH. Video S3. Rotation of MFF in MeOH and DMSO. See DOI: 10.1039/c9mh00521h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |