
Extraction of gallium from simulated Bayer process liquor by Kelex 100 dissolved in ionic liquids†
Stijn
Raiguel
 ,
Wim
Dehaen
and
Koen
Binnemans
*
,
Wim
Dehaen
and
Koen
Binnemans
*
KU Leuven, Department of Chemistry, Celestijnenlaan 200F, P.O. box 2404, B-3001 Leuven, Belgium. E-mail: Koen.Binnemans@kuleuven.be
First published on 21st February 2020
Abstract
Two 1,2,3-triazolium ionic liquid diluents were evaluated for the extraction of gallium from spent Bayer process liquor by Kelex® 100. The first of these is a hydrophobic ionic liquid with a low water content, which allows the extraction of gallium directly from untreated Bayer process liquor. The second is based on a hydrophilic ionic liquid and which forms an aqueous biphasic system with concentrated salt solutions. The aqueous biphasic system homogenizes upon cooling, which allows homogeneous liquid–liquid extraction of gallium from carbonated Bayer process liquors. The influence of the ionic liquids on the extraction kinetics, mechanism, thermodynamics and selectivity is investigated. Stripping was examined from both systems and their potential for industrialization is discussed. Ionic liquids are shown to beneficially influence the extraction process from thermodynamic and kinetic perspectives. They are therefore promising to solve the issues which have thus far prevented the industrial application of resource-efficient solvent extraction techniques for the recovery of gallium from spent Bayer process liquor.
Introduction
While its production totaled only 410 tons in 2018, gallium is an economically important material, considered to be a critical raw material by the European Commission.1,2 Moreover, the demand for gallium has been increasing steadily over the past several years, and is expected to increase to over 5000 tons by 2030.3 The main applications driving this demand are semiconductor materials consisting of gallium compounded with nitrogen (GaN) or arsenic (GaAs).3 One important usage of these is as power amplifiers in mobile phones, particularly smartphones. The increase in popularity of these devices in Asian markets have therefore led to a steady rise in gallium demand.1 Other important applications of gallium-based semiconductors are photovoltaics, light-emitting diodes, integrated circuits and laser diodes.1,3Gallium does not readily form minerals in which it is a major constituent. Consequently, it is only obtained as a by-product from the processing of other raw materials.3,4 Three major, exploitable sources of gallium are generally recognized: (1) bauxite residues, (2) zinc ores and (3) coal deposits. Coal deposits comprise the largest gallium reserves, but gallium can only be recovered from fly ash after combustion due to the very low concentration of gallium in most coal deposits.3,5 Gallium-enriched zinc refinery residues contain residual zinc in the form of zinc ferrite, in which gallium may be present by substitution.4 After leaching of the residue with sulfuric acid, solvent extraction is generally used to recover gallium from the leachate.6 This source only contributes to about 10% of worldwide gallium production. Most gallium is instead obtained from bauxite.3
Bauxite is the major ore for aluminum production, and mainly consists of aluminum hydroxides and oxide-hydroxides, iron oxides and oxide-hydroxides, clay minerals, silicon dioxide and titanium dioxide, often in various polymorphic mineral phases.7,8 The concentration of gallium in bauxite ranges from below 10 ppm to over 800 ppm, with an average concentration of 57 ppm.9 Actual production of aluminum is preceded by the production of alumina in the Bayer process. Herein, bauxite is digested by a concentrated sodium hydroxide solution at elevated temperatures. The resulting slurry is then separated from the residual gangue solids, cooled and seeded with aluminum hydroxide crystals to induce precipitation of the dissolved sodium aluminate as aluminum hydroxide.10 The caustic liquor is recycled after precipitation, but over time impurities tend to accumulate, including organics, inorganic anions, silica, vanadium and gallium.10–12 This leads to a reduction in precipitation yield, contamination of the product and a reduction of the quality of the final product's morphology and crystallinity.12 Final gallium concentrations in spent Bayer liquor are between 0.1–0.3 g L−1, but residual aluminum concentrations are around 50 g L−1, making the selective recovery of gallium highly challenging.13
Gallium has been recovered on an industrial scale from such spent, impurity-rich Bayer process liquor by a number of different methods, including fractional precipitation, electrolysis, cementation and ion exchange with amidoxime-functionalized resins.14 Of these methods, ion exchange is currently considered the most effective, resulting in it being the most widely applied.14 These resins are highly selective for gallium over aluminum and exhibit high affinities and loading capacities.15 As a result, gallium recovery yields are high and little aluminum is lost from the Bayer process.14 However, stripping of the resins takes place in acidic media and the gallium-binding amidoxime groups are not stable with respect to hydrolysis under acidic conditions, with carboxylic acids and hydroxamic acids as possible decomposition products.14,16 Consequently, the resins slowly degrade under stripping and regeneration conditions, resulting in a loss of gallium binding capacity.15,16 The resins also bind tightly to vanadium, poisoning the resin due to the progressive occupation of binding sites.15,17 Adsorbed vanadium can be removed by scrubbing with highly concentrated sodium hydroxide (9 mol L−1), but this process is slow and not all vanadium can be scrubbed.15 Moreover, the production of aqueous waste by adsorption processes in column setup is substantial.15,18
From the late 1970s to the early 1990s, considerable interest had arisen in an alternative technique for sequestering gallium from spent Bayer process liquor: solvent extraction.13,19–22 Gallium was found to be preferentially extracted over aluminum by 7-(4-ethyl-1-methyloctyl)-8-hydroxyquinoline (better known under the commercial name Kelex® 100, Fig. 1), usually dissolved in kerosene and modified with 10 vol% 1-decanol. Solvent extraction seemed to offer the prospect of a simple process, the consumption of relatively small quantities of reagents and a very limited impact on the Bayer process itself, as the Bayer process liquor is not decomposed by the extraction process itself.14 In practice, however, the extraction process turned out to be impractically slow, and as a consequence it was never employed on an industrial scale.13,14,19 The use of surfactant-stabilized microemulsions or ultrasonic agitation were proposed in order to enhance extraction kinetics.21,23–26 Puvvada observed a decrease in the required equilibration time by factor of 10 upon addition of Versatic Acid 10 to the organic phase, but the system exhibited poor reusability.25 Systems with Kelex® 100 dissolved in n-butanol as organic phase form stable microemulsions and exhibit very strong, relatively rapid extraction of gallium.23,26 Unfortunately, these systems exhibit poor selectivity of gallium over aluminum.26 Pesic and Zhou observed a 10- to 20-fold increase of the extraction rate constant by applying ultrasonic agitation during extraction, but this technique has not been applied industrially.21
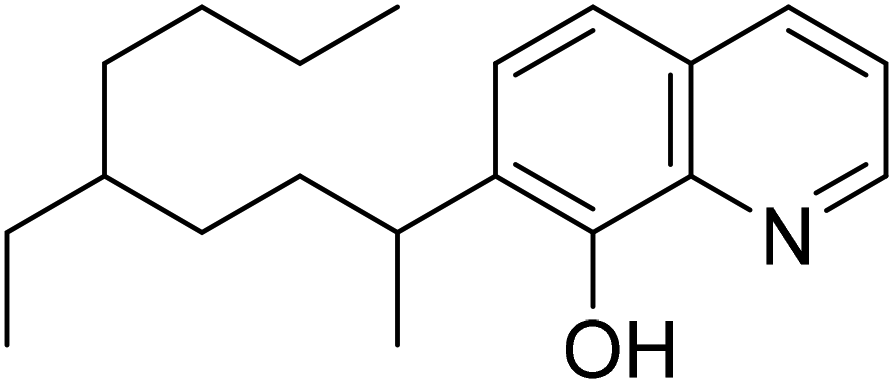 | ||
| Fig. 1 Structure of 7-(4-ethyl-1-methyloctyl)-8-hydroxyquinoline (Kelex® 100). | ||
The extraction mechanism of gallium by Kelex® 100 and the cause of these poor kinetics have been the subject of previous studies. By carefully studying the parameters affecting the extraction equilibrium, Sato and Oishi established the net extraction mechanism to be that shown in eqn (1):20
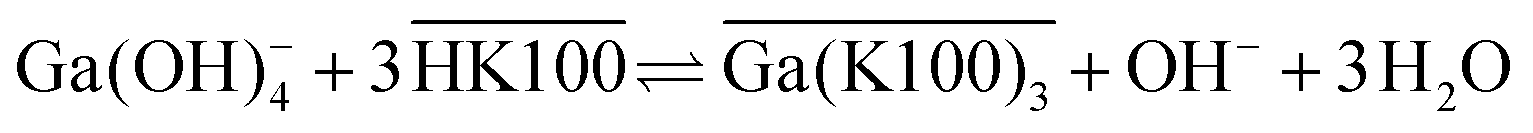 | (1) |
Overbars denote organic phase species and HK100 denotes the acidic (uncharged) form of Kelex® 100. Sato and Oishi hereby affirmed earlier hypotheses concerning the extraction mechanism of gallium.13 In this earlier work it was also hypothesized that aluminum is extracted analogously and sodium is extracted via a neutralization reaction (eqn (2)):
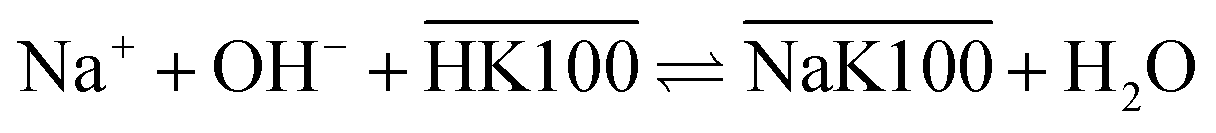 | (2) |
Sato and Oishi further observed that, at high hydroxide concentrations pertinent in spent Bayer process liquors, the extraction rate constant is of inverse first order with respect to the hydroxide concentration, implying that the extraction rate is limited by the loss of a hydroxide ligand to form an intermediate trihydroxo complex.20 Indeed, the species Ga(OH)3 never predominates in aqueous solutions, and only small equilibrium concentrations are present.27 This ligand loss may be necessary due to the impossibility of stabilizing the charged Ga(OH)4− species in the apolar organic phase. A viable method of improving extraction kinetics could thus consist of designing an extraction system capable of delivering Ga(OH)4− to the extractant molecules dissolved in the organic phase, without a prerequisite, unfavorable change in inner-sphere coordination.
In this work, we present two ionic liquid-based solvent extraction systems for the selective extraction of gallium(III) from spent Bayer liquor. Two strategies are employed. The first strategy involves the use of a hydrophobic ionic liquid, in which Kelex® 100 has been dissolved, for the direct extraction of gallium(III) from untreated Bayer liquor analogous to conventional extraction using a hydrocarbon diluent. The second strategy makes use of an ionic liquid with temperature-dependent miscibility with the aqueous phase, allowing a homogeneous phase to form at upon cooling and allowing Ga(OH)4− to diffuse to the extractant molecules directly, without traversing an interfacial barrier. This type of extraction, known as homogeneous liquid–liquid extraction (HLLE), has been used to extract metal ions in several previous studies.28–34 The ionic liquids employed as diluents for these systems are both 1,2,3-triazolium ionic liquids, which were previously reported by our group to be very stable in alkaline media.35,36 From a perspective of sustainability, a solvent extraction-based process offers the advantage of a relatively low production of aqueous waste compared to adsorption-based processes in column setup.15,18 In contrast to many other techniques, solvent extraction has a fairly limited impact on the composition of the spent Bayer liquor, facilitating its recycling after gallium recovery.14 The process flowsheet is comparatively simple and little processing is required after solvent extraction.14 Moreover, the use of ionic liquids is particularly advantageous in industrial-scale solvent extraction, as this mitigates the production of large quantities of fumes related to the use of volatile organic solvents. In addition, the low volatility and intrinsic conductivity of ionic liquids both work to prevent fires related to sparking after the buildup of static electricity, which commonly occurs in conventional solvent extraction due to the intensive handling of non-conducting liquids.37
Experimental
Chemicals
2-Ethylhexylamine (99%), triethylamine (99%), 1-hexanol (98%), 1-octanol (99%), 1-decanol (98%), petroleum ether (boiling range 180–280 °C), chloroform-d (99.8 atom% D), sodium aluminate (technical grade) and sodium azide (99%) were purchased from Acros Organics (Geel, Belgium). Sodium dihydrogen phosphate (anhydrous, 98%), heptane (mixture of isomers, 99%), 4-aminobenzoic acid (99%), nitric acid (65%) gallium standard solution (1000 ppm), aluminum standard solution (1000 ppm) and scandium standard solution (1000 ppm) were purchased from Chem-Lab (Zedelgem, Belgium). 4-Heptanone (99%), 2-ethylhexanol (99%) and gallium sulfate hydrate (99.999% metals basis) were purchased from Alfa Aesar (Karlsruhe, Germany). Ammonia (25 wt% in water), sodium carbonate (analytical reagent grade), ethyl acetate (technical grade), toluene (reagent grade), magnesium sulfate (dried), sodium sulfate (analytical reagent grade) and sodium hydroxide (analytical grade) were purchased from VWR (Oud-Heverlee, Belgium). Methanesulfonyl chloride (98%), was purchased from Sigma-Aldrich (Overijse, Belgium). Sodium hydrogen sulfate (93%) was purchased from Karl Roth GmbH (Karlsruhe, Germany). Sulfuric acid (98%) and hydrochloric acid (37%) were purchased from Fischer Scientific (Hampton, New York, USA). 7-(4-Ethyl-1-methyloctyl)-8-hydroxyquinoline (Kelex® 100) was purchased from Boc Scientific (Shirley, New York, USA). Hydrogen bistriflimide (80 wt% in water) and lithium bistriflimide were purchased from Iolitec (Heilbronn, Germany). All reagents were used as received, without further purification. 5-Ethyl-1,3-dihexyl-4-methyl-1,2,3-triazolium bis(trifluoromethylsulfonyl)imide and 1,3-bis(2-ethylhexyl)-4-methyl-5-propyl-1,2,3-triazolium sulfate were synthesized according to previously published procedures.35,36 Their purity was assessed using 1H NMR (spectra shown in ESI, Fig. S1 and S2†).Instrumentation
pH measurements were performed on a Mettler−Toledo SevenCompact pH meter in combination with a Mettler−Toledo InLab Micro glass electrode.1H NMR spectra were recorded on a Bruker Avance III HD 400 MHz spectrometer operating at 400 MHz. Samples of individual compounds were diluted in chloroform-d and referenced internally to tetramethylsilane (TMS). Mixed samples used to probe intermolecular interactions were not diluted, but a glass insert filled with D2O was used as external reference. Analyses of metal concentrations in the aqueous phase were performed on a PerkinElmer Avio 500 inductively coupled plasma-optical emission spectrometer, equipped with a GemCone High Solids Nebulizer, baffled cyclonic spray chamber, alumina injector and demountable quartz glass torch. Samples were prepared by collecting an appropriate aliquot of the aqueous solution and diluting to 5 mL with 2 wt% HNO3 solution. Scandium (60 ppm) was used as internal standard. The calibration curve was constructed by measuring an external standard series spanning the expected concentration range, prepared by diluting gallium or aluminum standard solution to the desired concentration with 2 wt% HNO3 solution and standardized internally with 60 ppm scandium. The lines at 417, 396 and 361 nm were measured in axial viewing mode for gallium, aluminum and scandium, respectively. Analysis of metal concentrations in water-soluble ionic liquids was performed analogously. Samples were diluted using 2 wt% H2SO4 (as opposed to 2 wt% HNO3) to prevent the ionic liquid from separating out of the aqueous solution. Additionally, a reagent blank was measured and subtracted from the sample spectra. This solution contained the ionic liquid in the same concentration as that present in the samples and 60 ppm scandium as internal standard, and was diluted in 2 wt% H2SO4.The analysis of samples with known concentrations, prepared by making appropriate dilutions of standard solutions of gallium and aluminum using the procedures discussed above, allowed us to make an estimate of the combined error on sample preparation and analysis. The median relative error was found to be about 4%. Propagation of this error leads to an estimated error of 7% on the percentage extraction and the distribution ratio for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 phase ratio.
:1 phase ratio.
Solvent extraction
Two-phase solvent extraction experiments were carried out in 4 mL glass screwcap vials and the organic phase volume was kept at 500 μL (unless stated otherwise), the aqueous phase volume was adjusted as necessary. Unless otherwise stated, the aqueous phase contained 2.000 mol L−1 NaOH, with gallium and aluminum concentrations of 200 ppm each. For the biphasic ionic liquid extraction system, the organic phase consisted of 5 vol% Kelex 100 and 10 vol% 1-decanol in bistriflimide ionic liquid. For the benchmark (kerosene) system, the organic phase consisted of 5 vol% Kelex 100 and 10 vol% 1-decanol in kerosene. The organic phase was presaturated with 2 mol L−1 NaOH before extraction. Unless stated otherwise, samples were equilibrated by magnetic stirring at 1200 rpm using an 8 mm by 3 mm stirring bar. Phase separation was aided by centrifugation in a Heraeus Labofuge 200 (2 min, 5000 rpm). The metal content in the aqueous phase was analyzed measured before and after extraction. Using concentration data for the aqueous phase before and after extraction, the percentage extraction was calculated using eqn (3):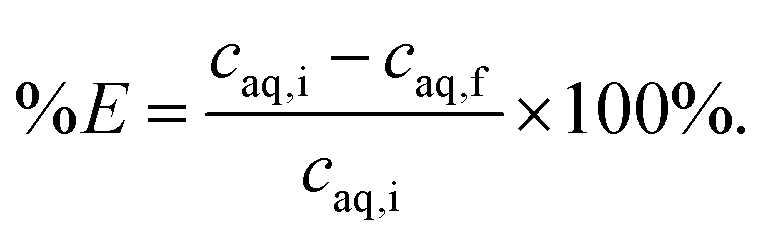 | (3) |
The distribution ratio was calculated using eqn (4):
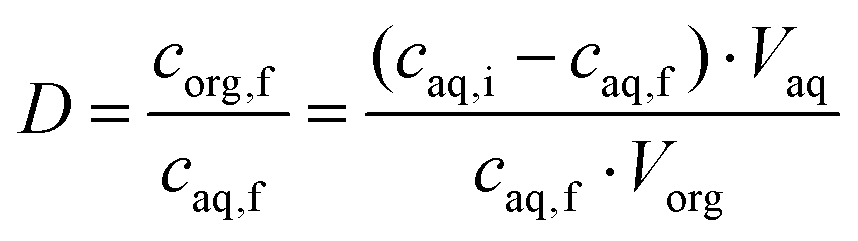 | (4) |
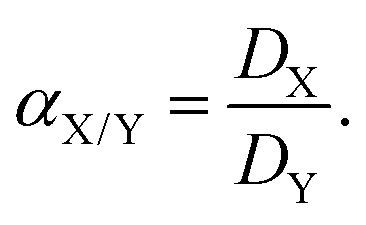 | (5) |
In eqn (3), DX and DY represent the distribution ratios for elements X and Y, respectively. Elements X and Y are defined such that DX > DY and hence the separation factor is greater than 1 by definition.
All homogeneous liquid–liquid extraction (HLLE) experiments were preceded by presaturation of the organic phase, in order to prevent the transfer of water from the aqueous to the organic phase during extraction. This was done by dissolving 2.00 mL of ionic liquid and 100 μL of Kelex® 100 in 10.00 mL of sodium carbonate solution (1.20 mol L−1) and heating this mixture to a temperature arbitrarily set at 80 °C, sufficiently high above the cloud point temperature to obtain a stable final phase ratio. The final aqueous salt concentration could be estimated at about 1.7 mol L−1 from the reduction in aqueous phase volume (determined using a graduated cylinder, with an error of approximately 1%). The organic phase resulting from this procedure is thus at equilibrium with a 1.7 mol L−1 solution of sodium carbonate at 80 °C, and has a Kelex® 100 concentration of 5 vol% with respect to the total organic component. Therefore, the aqueous sodium carbonate concentration was kept at 1.7 mol L−1 for all HLLE experiments. The resulting extraction systems have cloud point temperatures close to room temperature. The concentration of gallium and aluminum in the aqueous phase was 200 ppm each. In mechanistic studies, only the metal being investigated was present in the aqueous phase.
HLLE experiments were performed in 4 mL glass vials equipped with magnetic stirring bars. The samples were homogenized for 10 minutes (unless stated otherwise) using an ice bath under magnetic stirring at 500 rpm. Phases were separated by reheating to 80 °C, followed by centrifuging at 5000 rpm for 2 min using a Heraeus Labufuge 200 centrifuge. When no precipitation occurred during solvent extraction, the percentage extraction and distribution ratio were determined by measuring the aqueous phase before and after extraction using eqn (3) and (4) as discussed above. When a precipitate had formed during extraction, both phases were measured after extraction in order to determine the percentage extraction and the distribution ratio using eqn (6) and (4), respectively.
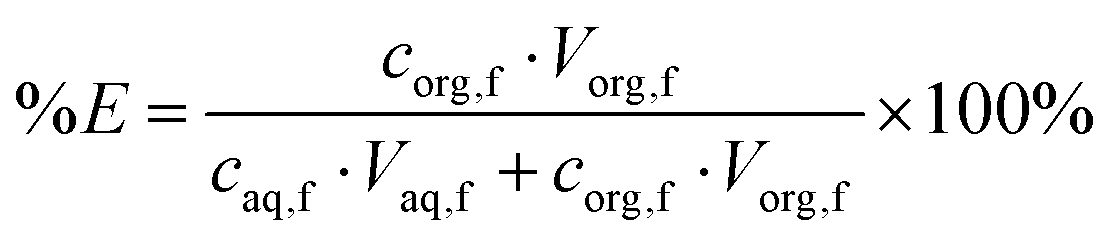 | (6) |
Results and discussion
Preliminary extraction trials showed that the gallium(III) could be extracted from 2 mol L−1 NaOH solutions by Kelex® 100 dissolved in bis(trifluoromethylsulfonyl)imide (bistriflimide) ionic liquids. Bistriflimide ionic liquids are generally very hydrophobic and are hence suitable as diluents in solvent extraction with minimal losses to the aqueous phase.38 Therefore, a bistriflimide ionic liquid previously reported by our group was selected for use in the two-phase (non-HLLE) solvent extraction system.35 This ionic liquid, 4-ethyl-5-methyl-1,3-dihexyl-1,2,3-triazolium bis(trifluoromethylsulfonyl)imide or [HHT12][Tf2N], is highly base-stable and can therefore safely be contacted with hot caustic solution without the risk of decomposition (Fig. 2).35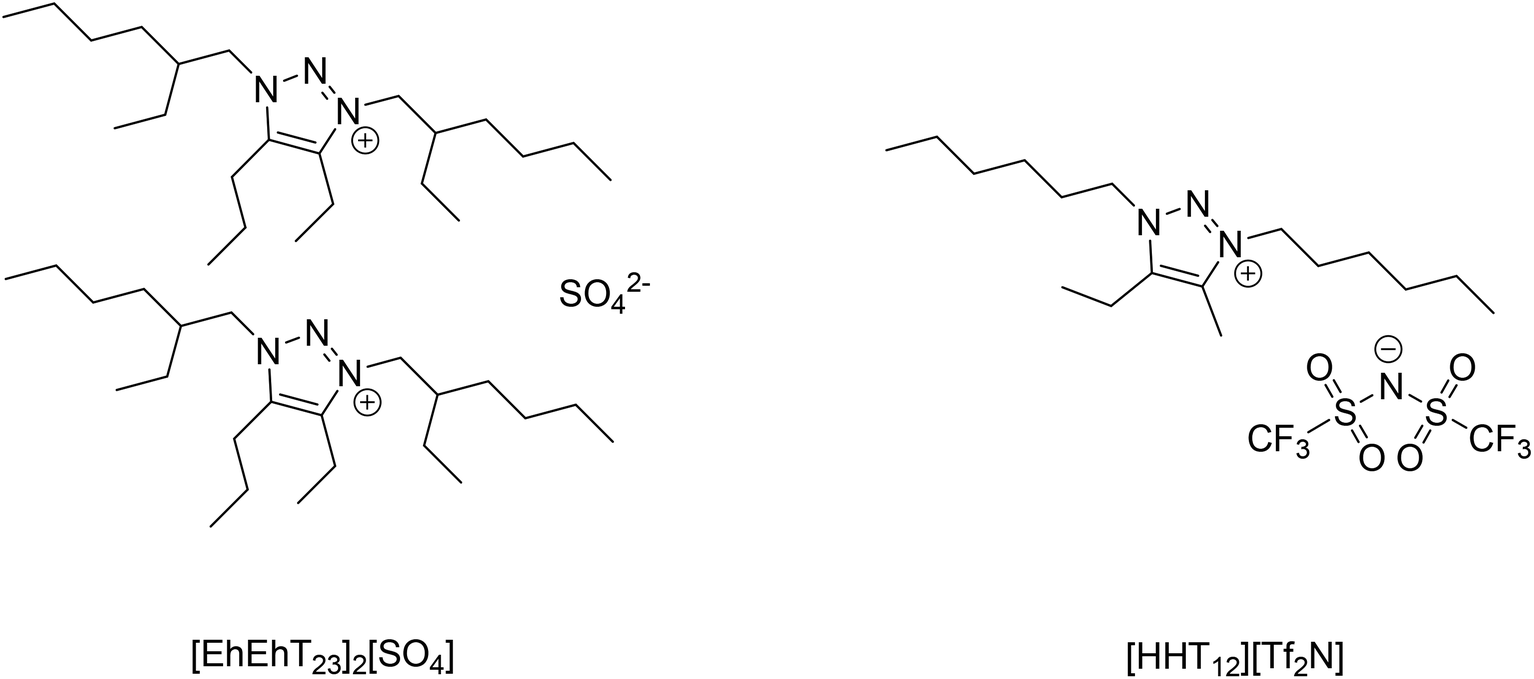 | ||
| Fig. 2 Structures of the ionic liquids used in this study: 4-ethyl-1,3-bis(2-ethylhexyl)-5-propyl-1,2,3-triazolium sulfate and 4-ethyl-5-methyl-1,3-dihexyl-1,2,3-triazolium bis(trifluoromethylsulfonyl)imide. | ||
A more hydrophilic ionic liquid was required for the HLLE system. A sulfate ionic liquid with a similar cation was chosen: 4-ethyl-1,3-bis(2-ethylhexyl)-5-propyl-1,2,3-triazolium sulfate, or [EhEhT23]2[SO4]. This ionic liquid was also previously reported by our group and was found to be miscible with pure water, but immiscible with concentrated sodium hydroxide solutions.36 At intermediate electrolyte concentrations, this ionic liquid is observed to exhibit a lower critical solution temperature (LCST behavior). Dupont et al. suggested that this type of behavior can be expected from ionic liquids with large, hydrophobic cations and highly hydrated anions, as the hydration of the anion entails a favorable enthalpy term but an unfavorable entropy term.38 Therefore, demixing of aqueous solutions of such salts is entropy-driven and occurs upon heating of the mixture. When the system is in its homogeneous state, metal ions and extractant molecules diffuse freely and interfacial phenomena no longer control extraction kinetics. This forms the basis of the HLLE technique. After homogeneous extraction, the solution is reheated for phase separation.
Some preliminary solvent extraction studies were carried out from sodium hydroxide solutions in order to evaluate the feasibility of extracting gallium from untreated spent Bayer process liquor using the HLLE technique. However, extraction of gallium was very poor from these solutions.1H NMR showed that saponification of the Kelex® 100 extractant occurred at high sodium hydroxide concentrations (spectra shown in ESI, Fig. S3†). This prevents the extractant from functioning properly, as protonation of the hydroxo ligands bound to gallium is an integral part of the extraction mechanism (eqn (1)).20 HLLE from sodium hydroxide solutions was thus not investigated any further. In contrast, gallium was extracted very efficiently from carbonate solutions by Kelex® 100 diluted in [EhEhT23]2[SO4]. Therefore, [EhEhT23]2[SO4] is proposed as a diluent for the extraction of gallium(III) by Kelex® 100 from carbonated Bayer process liquor, rather than untreated Bayer process liquor. Carbonation is a process in which CO2 is injected in the Bayer liquor in order to reduce the pH and partly precipitate the aluminum present in the solution.14 This process converts most of the sodium hydroxide in solution to sodium carbonate. As a result of the carbonation, the Ga/Al ratio in the liquor increases and the solvent extraction of gallium(III) becomes possible using [EhEhT23]2[SO4] in a HLLE process. As mentioned above, carbonation is not required if the more hydrophobic [HHT12][Tf2N] is used as diluent, as the extractant does not saponify in this ionic liquid even under highly alkaline conditions. The decrease of the pKa of the extractant in [EhEhT23]2[SO4] can be attributed to the high water content of the ionic-liquid rich phase, even after demixing, facilitating the stabilization of charged species.
Phase behavior of [EhEhT23]2[SO4] with concentrated salt solutions
The chief solutes in carbonated Bayer process liquor are sodium carbonate and unreacted sodium hydroxide. These act as salting-out agents that induce phase separation of the organic and aqueous phases upon heating. Since the ionic liquid exhibits lower critical solution temperature (LCST) behavior with aqueous salt solutions, the binodal curve is expected to be roughly U-shaped, with the cloud point temperature reaching a minimum at the critical point. However, the critical point of the systems investigated here occurs at very low aqueous to organic phase ratios, which are of little practical relevance for solvent extraction. Therefore, only the latter part of the phase diagram is investigated, in which the cloud point temperature (CPT) rises with increasing mass fractions of aqueous salt solution. In addition to the mass fraction of aqueous salt solution (χm,aq), several other factors also influence the CPT of a certain mixture, including the Kelex® 100 mass fraction, the nature of the salting-out agent, and its concentration. The constructed phase diagrams are shown in Fig. 3 and 4.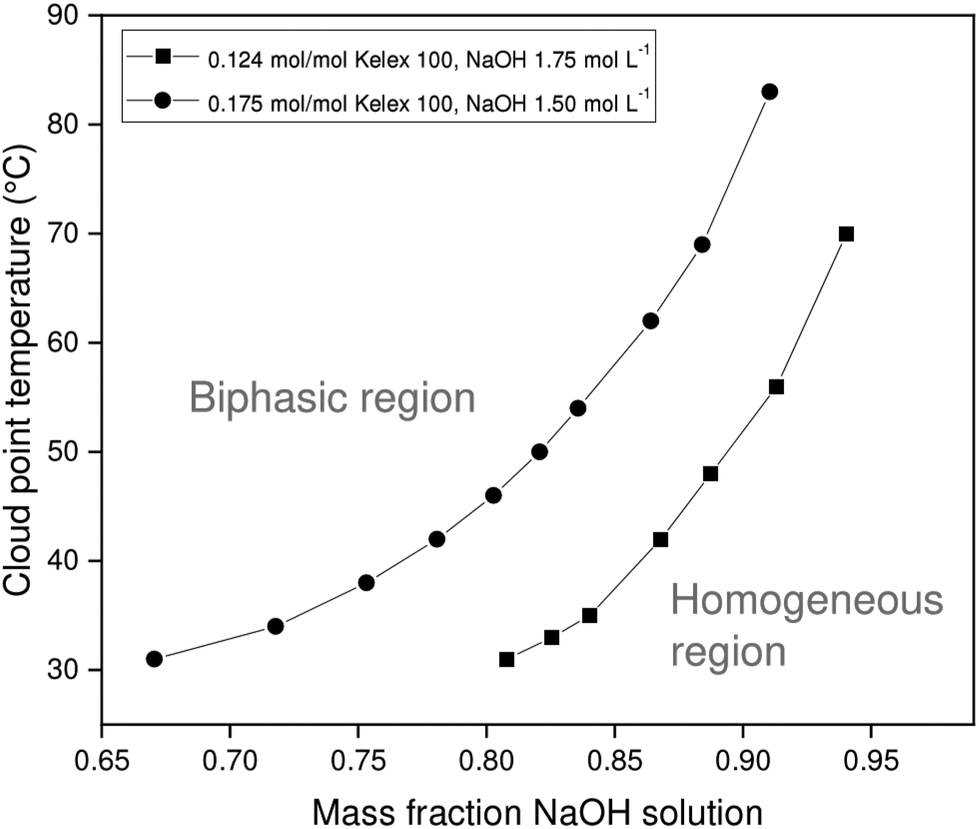 | ||
| Fig. 3 Variation of cloud point temperature of [EhEhT23]2[SO4] and sodium hydroxide solutions with the composition of the mixture. | ||
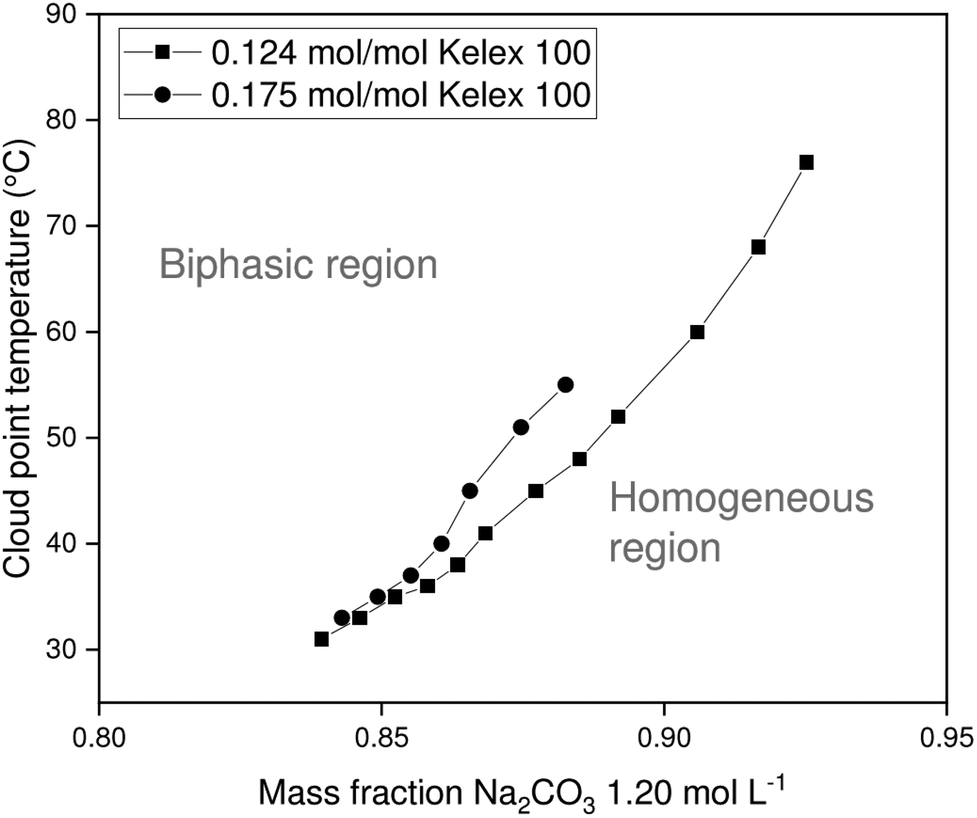 | ||
| Fig. 4 Variation of cloud point temperature of [EhEhT23]2[SO4] and sodium carbonate solutions with the composition of the mixture. | ||
Aqueous solutions of sodium hydroxide (1.500 mol L−1 or 1.750 mol L−1) and sodium carbonate (1.200 mol L−1) were used in constructing phase diagrams with ionic liquid-Kelex® 100 mixtures. Lower sodium carbonate concentrations are required to induce phase separation than those required for sodium hydroxide. This stronger salting-out effect can be attributed both to the high charge of the carbonate anion and the release of two equivalents of sodium cations by sodium carbonate upon dissolution. Two concentrations of Kelex® 100 were evaluated: one corresponding to a 200:10 volume fraction at 22 °C, and one corresponding to a 200:15 volume fraction. These are equivalent to mole fractions of 0.124 and 0.175, respectively. As Kelex® 100 is poorly soluble in pure water, one may intuitively predict that increasing its concentration in an aqueous biphasic system increases the cloud point temperature of the system. Hence, lower sodium hydroxide concentrations are required to obtain a thermomorphic system when using higher extractant concentrations. The system containing Kelex® 100 in 0.175 molar ratio with the ionic liquid is only thermomorphic with sodium carbonate (1.200 mol L−1) over a limited composition range, as Kelex® 100 becomes insoluble in the homogeneous phase at higher values of χm,aq. Transferring these data to solvent extraction systems is not straightforward, as mutual miscibility of the phases results in a transfer of a large quantity of water from the aqueous phase to the organic phase. In the Experimental section, more details are given concerning the method used to obtain a constant phase ratio throughout the extraction process.
Mechanistic studies
While the extraction mechanism of Kelex® 100 dissolved in hydrocarbon diluents is well-established, the drastically different solvent environment of ionic liquids can lead to a change in the net extraction mechanism of an otherwise identical extractant.39–41 The sulfate ionic liquid [EhEhT23]2[SO4] is the most likely of the two investigated ionic liquids to affect the extraction mechanism, due to the strongly coordinating nature of the sulfate anion and the high equilibrium concentration of water in the ionic liquid. Hence, mechanistic studies were performed on this ionic liquid. Acidic extractants, in particular, have been observed to form anionic complexes when dissolved in ionic liquids, while they generally form neutral complexes in aliphatic diluents.39 It is unlikely for gallium to acquire a coordination number greater than 6, thus a complex of the form [Ga(K100)4]− is unlikely to exist (where K100 denotes the anionic form of Kelex® 100). However, a mechanism such as that shown in eqn (7) is possible: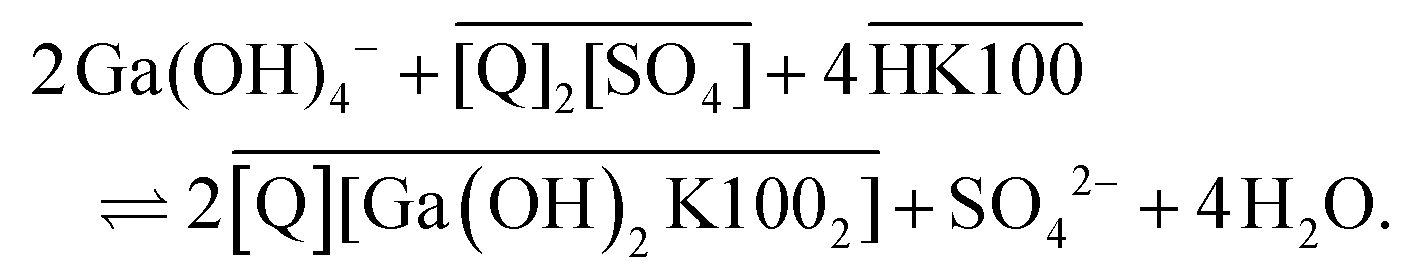 | (7) |
Q denotes the ionic liquid cation in eqn (7). The mechanism in hydrocarbon diluents, which was already mentioned above, proceeds as shown in eqn (8).
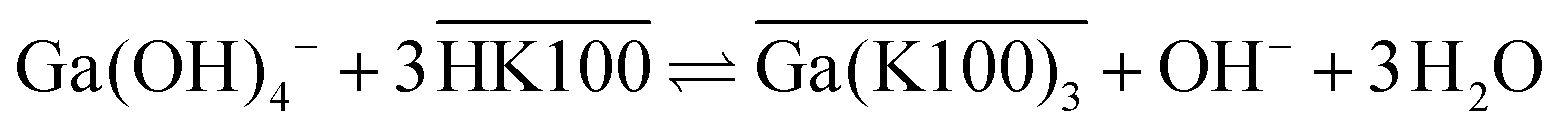 | (8) |
In both equations, overbars denote organic phase species and HK100 denotes the acidic (uncharged) form of Kelex® 100.
The usual mechanism shown in eqn (8) is characterized by the release of a hydroxide anion into the aqueous phase. This results in a pH-dependence of gallium extraction. Increasing the pH should suppress gallium extraction, rather than enhancing it as is generally the case for acidic extractants. The pH dependence of extraction was evaluated by varying the equilibrium pH of the aqueous phase. This was accomplished by the addition of various concentrations of NaOH to the feed solution, between 0 and 50 mmol L−1. Note that the potentiometrically determined pH values reflect proton activity, rather than proton concentration. A linear relationship was observed between the logarithm of the distribution ratio (log(DGa)) and the pH, with a slope of close to −1. Bearing in mind the relationship shown in eqn (9),
| pH = 14 − pOH = 14 + log[OH−]. | (9) |
It can be inferred that hydroxide anions are expelled to the aqueous phase upon gallium extraction with 1:1 stoichiometry. A plot of the obtained extraction results is shown in the ESI (Fig. S4†).
A similar strategy was used to evaluate the involvement of sulfate anions in the interfacial exchange mechanism. A series of extractions were performed from solutions containing 1.7 mol L−1 sodium carbonate using 0.124 mol mol−1 Kelex® 100 in [EhEhT23]2[SO4] in a 1:2 phase volume ratio (organic to aqueous). The composition of the aqueous phase was varied by the addition of various concentrations of Na2SO4 to the feed solution. The sulfate concentration was hereby varied between 0.001 and 200 mmol L−1. Extraction was found to be nearly independent of the sulfate concentration, strongly suggesting that sulfate anions are not involved in maintaining charge neutrality of the phases upon extraction. This result also suggests that the mechanism is unaltered by the use of ionic liquids as diluents. A plot of the obtained extraction results is shown in the ESI (Fig. S5†).
Kinetic studies
As mentioned above, the largest drawback of Kelex® 100 is the poor interfacial kinetics when dissolved in hydrocarbon diluents. Therefore, the sulfate ionic liquid-based HLLE system was compared to the benchmark system of Kelex® 100 in kerosene.13 A third system was also included in the study, namely a phosphonium ionic liquid system which does not possess the thermomorphic property of the triazolium ionic liquid system. This system consisted of 5 vol% Kelex® 100 diluted in the ionic liquid denoted as [P66614]2[SO4]. This phosphonium ionic liquid is the sulfate analog of the commercially available Cyphos® IL101 and was described by our group in a recent paper.36 Thus, all systems contained the same concentration of Kelex® 100 (5 vol%) with respect to the organic component. The composition of the aqueous and the phase volume ratio (1:1) were constant for all evaluated systems. These extractions were performed from sodium carbonate media (1.700 mol L−1). For both heterogeneous systems, the stirring speed and the temperature were identical (1200 rpm at 22 °C), while a lower stirring speed and temperature were applied to the homogeneous system (500 rpm and 0 °C). For the homogeneous system, ‘contact time’ refers to the time between homogenization and phase separation.
Intuitively, one expects the HLLE system to reach equilibrium much more quickly than the biphasic systems. Fig. 5 shows that the benchmark system takes about 2 hours to reach equilibrium for gallium extraction under the conditions evaluated here. While the triazolium HLLE system did outperform the benchmark kerosene system, both ionic liquid systems reached equilibrium for gallium extraction within 5 minutes. Thus, significant improvements in the gallium extraction kinetics can be obtained by using an ionic liquid as diluent, regardless of thermomorphic behavior. These results appear to indicate a kinetic advantage inherent to the use of ionic liquids as diluents. Note that the extraction of aluminum by the HLLE system is not shown in Fig. 5 as aluminum partially precipitates during extraction. This is discussed further below.
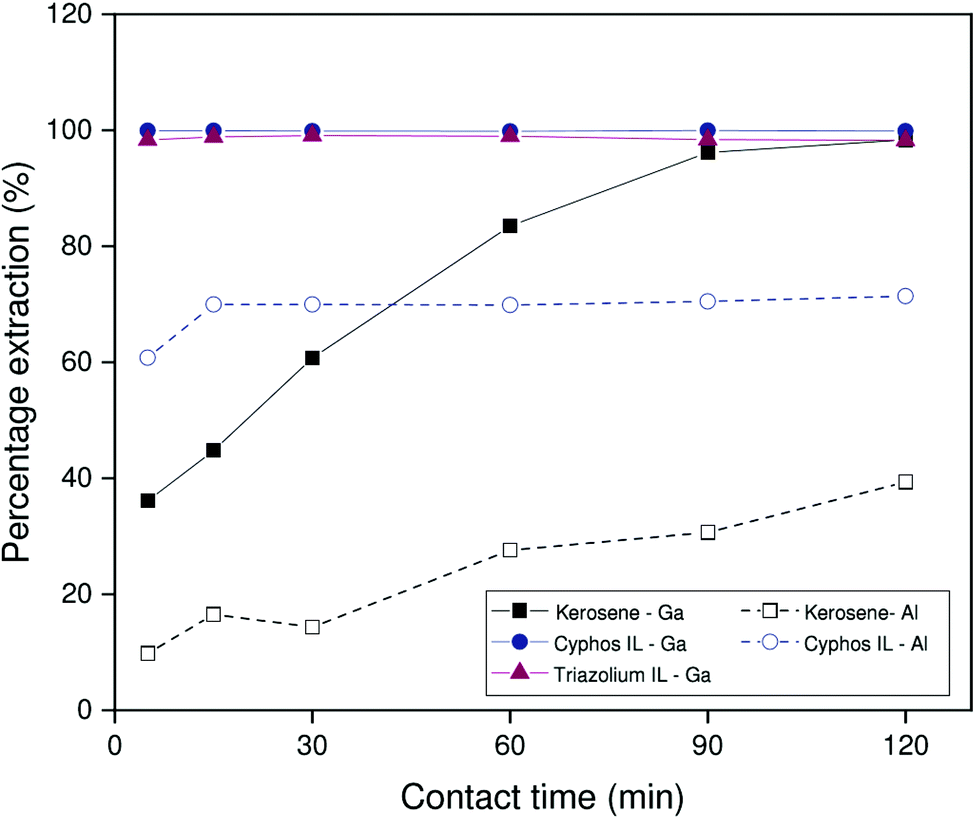 | ||
| Fig. 5 Percentage extraction as a function of time for 200 ppm solutions extracted from 1.700 mol L−1 sodium carbonate by 5 vol% Kelex® 100 in [EhEhT23]2[SO4], 5 vol% Kelex® 100 in [C101]2[SO4] and 5 vol% Kelex® 100 in kerosene modified with 10 vol% 1-decanol. Phase ratio: 1:1 organic to aqueous. The HLLE procedure was followed for [EhEhT23]2[SO4]. | ||
Similar experiments were performed on the non-HLLE ionic liquid system with [HHT12][Tf2N] as diluent. In this case, extractions are performed from sodium hydroxide medium (2.000 mol L−1). The stirring speed was 1200 rpm at a temperature of 22 °C. As is evident from Fig. 6, the benchmark system suffers from poor reproducibility under these conditions, which may be related to small differences in interfacial area between individual samples. The ionic liquid system, by contrast, shows a far more consistent trend as a function of time. What is clear, however, is that the ionic liquid exhibits modestly improved kinetics over the benchmark system: a percentage extraction of 75% is reached after about 15 minutes, while the benchmark system only reaches this point after about 60 minutes. Since the extraction rates of Kelex® 100 in the benchmark system are similar for both carbonate and hydroxide feed solutions, it is thus reasonable to deduce the following general trend in extraction rates by Kelex® 100: sulfate ionic liquids > bistriflimide ionic liquids > kerosene.
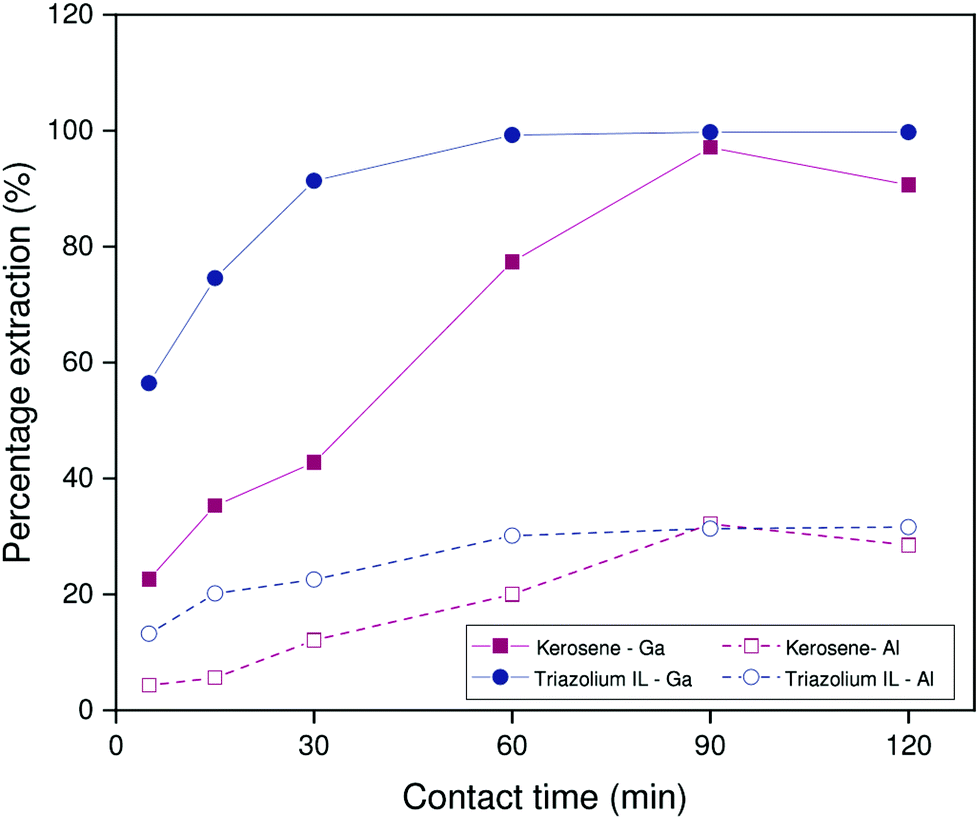 | ||
| Fig. 6 Percentage extraction as a function of time for 200 ppm solutions extracted from 1.700 mol L−1 sodium carbonate by 5 vol% Kelex® 100 in [HHT12][Tf2N] modified with 10 vol% 1-decanol, and 5 vol% Kelex® 100 in kerosene modified with 10 vol% 1-decanol. Phase ratio: 1:1 organic to aqueous. Temperature: 22 °C. | ||
An elegant hypothesis to explain these findings invokes anion exchange, a process possible only in ionic liquids and which becomes more favorable with increasing hydration enthalpy of the ionic liquid anion.38 In conventional systems, gallium must lose a hydroxo ligand before traversing the interface (eqn (8)), as anions are poorly stabilized in alkanes, even with the extractant present.20 This ligand loss is very unfavorable, causing the concentration of the mobile intermediate to be very low. Thus, the rate of interfacial mass transfer is limited. The contrasting, rapid interfacial kinetics of Kelex® 100 in ionic liquid systems would, according to this hypothesis, result from the occurrence of the reaction described by eqn (10) as rate determining step.
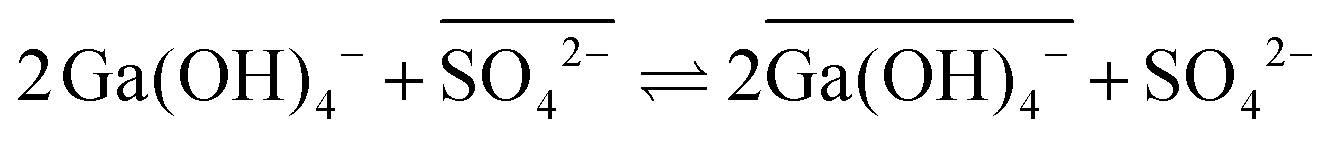 | (10) |
Overbars in eqn (10) represent organic phase species. While this equilibrium is unfavorable (sulfate ionic liquids without Kelex® 100 do not extract gallium from alkaline solutions), it does provide a mechanism by which gallium can transverse the interface without any changes in inner-sphere coordination. The bistriflimide anion has a lower hydration enthalpy than the sulfate anion, and as a result, the analogous process is less favorable for ionic liquids containing this anion, which would then explain the intermediate extraction rates of these ionic liquids.
If this hypothesis is indeed valid, the addition of a catalytic amount (5 vol%) of [EhEhT23]2[SO4] to the organic phase of the bistriflimide ionic liquid system should induce rapid kinetics, similar to pure sulfate ionic liquids. However, this addition was found to have no positive influence on the extraction kinetics. Hence, the cause of the rapid kinetics of the sulfate ionic liquid systems should be sought elsewhere. We propose that the high water content of sulfate ionic liquids allows the Na+–Ga(OH)4− ion pairs to diffuse into the organic phase. Earlier authors have concluded that water tends to form polar microphases in the bulk structure of ionic liquids, through which charged solutes may rapidly diffuse and in which they can be solvated.42,43 The water content of the bistriflimide ionic liquid is lower (0.51% at saturation35), reducing the solubility of ion pairs in the organic phase and reducing their mobility.
In the homogeneous extaction system, the ionic liquid, extractant and gallate(III) anions diffuse freely in the same phase, and an interfacial exchange mechanism is no longer needed. However, since extraction is very rapid to Kelex® 100 in sulfate ionic liquids, the added energy expenditure to perform the HLLE procedure may not be justified. A more energy-efficient solution to using Kelex® 100 dissolved in [EhEhT23]2[SO4] may thus be to select a sufficiently high extractant or salt concentration such that the system is biphasic under all operating conditions.
Effect of alkalinity on extraction and selectivity
The co-extraction of aluminum in the HLLE (sulfate) system was investigated over a range of pH values. The feed solution was spiked with various concentrations of sodium hydroxide (0–50 mM) to vary the equilibrium pH value. A phase volume ratio of 1:1 was applied. The equilibrium pH of the HLLE system is buffered and varies between about 11 and 12 (see Fig. S6 in the ESI† for an initial vs. equilibrium pH plot). Note that the potentiometrically determined pH values reflect proton activity, rather than proton concentration. Under these conditions, the pH has a limited influence on the ionic strength of the solution. As is evident from Fig. S7 (ESI†), the logarithm of the distribution ratio of aluminum(III) varies linearly with the equilibrium pH under working conditions, with a slope of −0.64. This is indicative that aluminum, as is the case in aliphatic diluents, is extracted via the same mechanism as gallium(III) (eqn (8)).13 Consequently, the alkalinity has little influence on the nominal selectivity factor of gallium(III) over aluminum(III). However, the solubility of aluminum is limited by the precipitation of Al(OH)3, and thus the organic phase concentration of aluminum will be lower than predicted purely on the basis of the distribution ratios and the initial aluminum concentration. This precipitation results from the reduction of the pH of the aqueous phase upon extraction.27 The nominal selectivity factor αGa/Al varies from about 20.70 to 17.04 between a pH of 11.29 to a pH of 11.67. The aqueous aluminum concentrations varied from 50.10 to 67.65 ppm over this range. A precipitate was present which was found to contain aluminum but no gallium (analysis performed with ICP-OES). While it cannot be stated with certainty that equilibrium aluminum solubility is reached, it does appear true that the maximal organic phase concentration of aluminum(III) is limited by the solubility of aluminum in the aqueous phase.
The non-HLLE (bistriflimide) system is compatible with the high alkalinities of untreated spent Bayer liquor. The pH is not buffered and all processes are subject to the strong variations in ionic strength resulting from the highly variable NaOH concentrations. Hence, strong variations on the selectivity of gallium over aluminum are to be expected depending on the alkalinity of the feed solution. To examine the extraction of both elements as a function of the alkalinity, extractions were performed from solutions with sodium hydroxide concentrations ranging from 1.000 to 5.000 mol L−1. A phase volume ratio of 1:2 (organic to aqueous) was used at 25 °C. Fig. 7 shows that large variations occur both in the distribution ratio of gallium and the selectivity factor αGa/Al. The latter varies by two orders of magnitude, although the range of hydroxide concentrations corresponds to less than one formal pH unit. This variation is large even accounting for variations in ionic strength. It is thus likely that saponification of the extractant contributes to the loss of affinity for gallium.
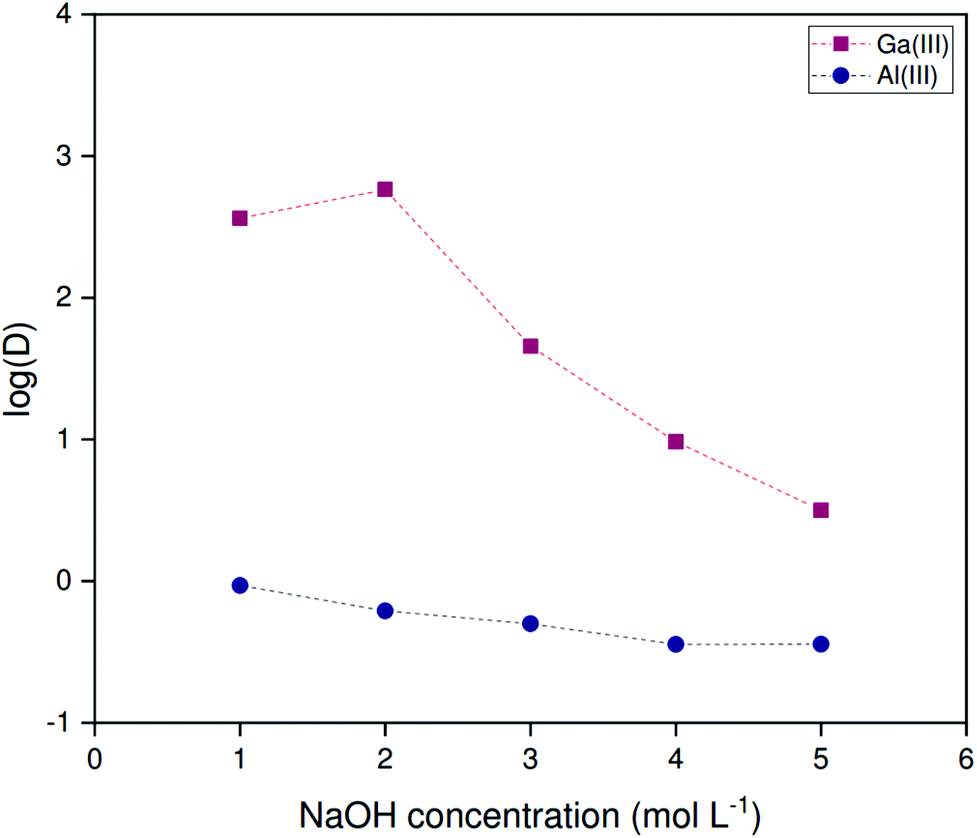 | ||
| Fig. 7 Variation of the logarithmic distribution ratios of gallium and aluminum with the sodium hydroxide concentration of the feed for 200 ppm solutions extracted by 5 vol% Kelex® 100 in [HHT12][Tf2N] modified with 10 vol% 1-decanol. Phase ratio: 1:2 (organic to aqueous). Temperature: 25 °C. | ||
Thermodynamics
The molar enthalpy and entropy of extraction (respectively ΔexH and ΔexS) were determined using van ‘t Hoff's method for the non-HLLE system.44 This method is inaccurate for the HLLE system as the phase ratio and composition is temperature-dependent. Data for the kerosene system (identical NaOH, extractant and modifier concentrations) were found in the literature.20 A 2:1 aqueous-to organic phase volume ratio was used for experimental data collection. Data plots are shown in the ESI (Fig. S8†).
The positive slope of the natural logarithm of the equilibrium constant as a function of the reciprocal temperature indicates a suppression of the extraction performance with increasing temperature. This means that the extraction is enthalpy-driven and that the entropy of extraction is unfavorable. From the value of the slope and the intercept of linear regressions of the data, numerical values of ΔexH and ΔexS can be found for both the non-HLLE ionic liquid system and the kerosene system. For the ionic liquid system, ΔexH = (−77.4 ± 3.3) kJ mol−1 and ΔexS = (−164 ± 10) J mol−1 K−1. For the kerosene system, ΔexH = (−65.8 ± 1.2) kJ mol−1 and ΔexS = (−171 ± 4) J mol−1 K−1. The given standard errors are those on the linear regression. The enthalpic contribution is significantly more favorable in the ionic liquid system. The entropic contribution is slightly more favorable, although this is within the experimental error.
The value of ΔexH is 11.6 kJ mol−1 more favorable in the non-HLLE ionic liquid system than the kerosene system. This is somewhat surprising, as one may expect the more polar ionic liquid to diminish the coordinating ability of the Kelex® 100 ligand by solvating the electron-donating sites. However, Kelex® 100, like many acidic extractants, contains both hydrogen bond donor and acceptor sites in its structure and is thus subject to dimerization in the organic phase.45 The necessity to separate these dimers may account for the less favorable enthalpy of extraction in the kerosene system. The value of 11.6 kJ mol−1 corresponds well with the strength of 3 weak hydrogen bonds (one per extractant molecule). Moreover, this hypothesis is supported by 1H NMR measurements. The aromatic spectral region of Kelex® 100 in the kerosene system strongly resembles that of Kelex® 100 in chloroform-d. The corresponding region of the spectrum of Kelex® 100 in the ionic liquid system, on the other hand, is characterized by an upfield shift of the ortho-proton on the pyridinic ring (position 2) by about 0.45 ppm relative to the para-proton (position 4), indicative of an increased electron density near the ring nitrogen atom. As the phenolic ring is substituted in the ortho-position, no such comparison can be made for the ortho-proton. However, the para-proton of the phenolic ring (position 5) is shifted upfield by about 0.2 ppm relative to the reference proton on the pydridinic ring, indicating that the Kelex® 100 ligand is also subject to interactions other than hydrogen bonding in the ionic liquid. For instance, π–π interactions between the electron-rich phenolic ring and the electron-poor triazolium cation may give rise to the upfield shift of the para-proton on the phenolic ring while also explaining why the observed difference in ΔexH is at the weak end of the scale for hydrogen bonds. A summary of the NMR results are shown in Table 1, while the full spectra can be viewed in the ESI (Fig. S9†). Note that the signals in the ionic liquid system are broadened due to the viscosity of the ionic liquid, which reduces the mobility of solutes.
Stripping
Both scrubbing of aluminum and stripping of gallium can be conveniently achieved from the non-HLLE (bistriflimide) system using HCl solutions, as originally described by Leveque and Helgorsky.13 Due to differences in hydration enthalpy, the bistriflimide ion is stable with respect to anion exchange by the chloride anion.38 The bistriflimide ionic liquid can thus be contacted with chloride solutions without anion exchange occurring. Distribution ratios for gallium and aluminum were determined at various HCl concentrations by contacting loaded organic phase in 1:1 volume ratio. Samples were stirred at 1200 rpm for 2 h at a temperature of 22 °C. The organic phase was loaded from 200 ppm metal solutions using a 1:2 organic to aqueous phase volume ratio. Leveque and Helgorsky reported that these equilibria are established quite rapidly under acidic conditions and hence shorter contact times probably suffice.13Fig. 8 shows that aluminum is efficiently removed from the organic phase over the entire range of evaluated HCl concentrations. Gallium, on the other hand, stripped only at low HCl concentrations, while virtually no stripping of gallium occurs at a HCl concentration over 3 mol L−1. These findings can be rationalized by considering the amphiprotic nature of Kelex® 100, which behaves as both an acidic and basic extractant depending on the conditions of the aqueous phase. At high HCl concentrations, gallium(III) is extracted by Kelex® 100 as [HK100][GaCl4].13 Aluminum(III) cannot be extracted this way as it does not form stable complexes with chloride in the presence of water. Hence, aluminum can first be scrubbed by contacting the organic phase with 3 mol L−1 HCl, after which gallium can be recovered by contacting with 1 mol L−1 HCl.
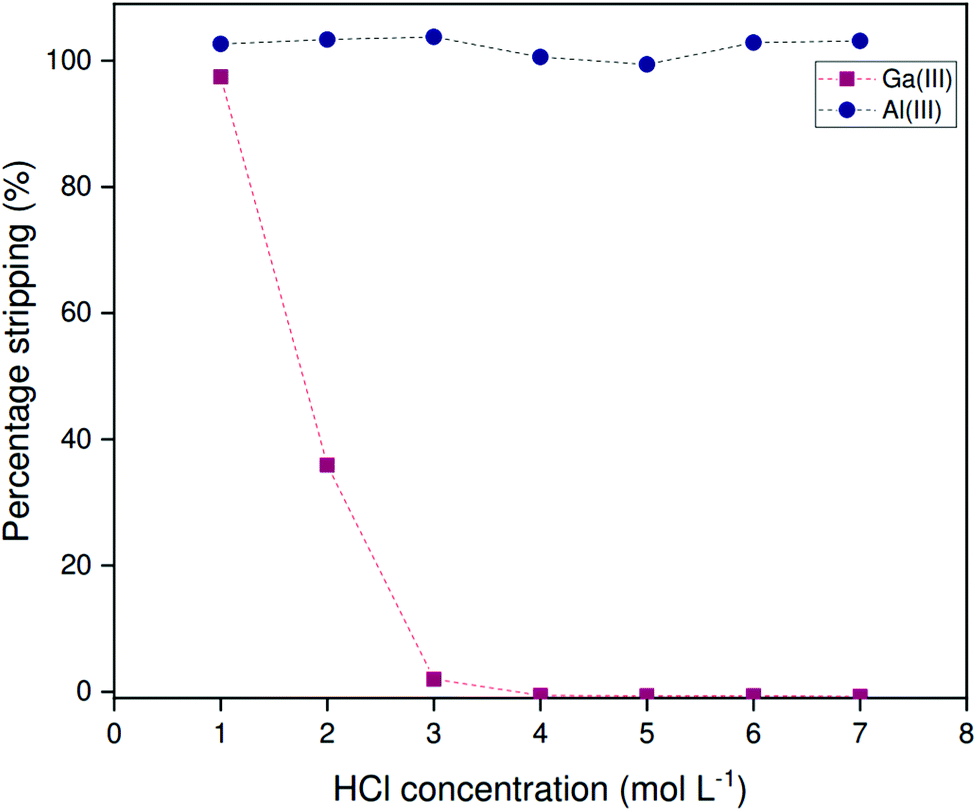 | ||
| Fig. 8 Percentage stripping of gallium and aluminum as a function of the HCl concentration in the stripping liquor for loaded 5 vol% Kelex® 100 in [HHT12][Tf2N] modified with 10 vol% 1-decanol, extracted from 200 ppm solutions containing 2.000 mol L−1 sodium hydroxide in 1:1 phase ratio (organic to aqueous). Temperature: 22 °C. | ||
Unfortunately, this scrubbing and stripping procedure is incompatible with the HLLE (sulfate) system. Firstly, the sulfate and hydrogen sulfate anions are readily displaced by chloride anions from the aqueous phase.38,46 Secondly, acidic media cause the protonation of sulfate anions to hydrogen sulfate anions. An alternative stripping method was devised, based on the controlled saponification of the extractant. Herein the loaded organic phase is contacted with a sodium hydroxide solution. Extracted metal ions are released by driving the equilibrium described by eqn (8) to the left as a result of the high hydroxide concentration and the saponification of the acidic form of Kelex® 100 (HK100). The organic phase was first loaded from 200 ppm metal solutions and subsequently contacted with solutions containing various sodium hydroxide concentrations at 0 °C. The phases were separated by heating to 80 °C. Note that volume ratios changed during the course of the extraction, and therefore only distribution ratios are reported. Sodium hydroxide concentrations below 2.25 mol L−1 did not yield biphasic systems at 80 °C. Fig. 9 shows that sodium hydroxide is effective at reducing distribution ratios of both gallium and aluminum. With increasing sodium hydroxide concentration, the distribution ratios are further lowered. The method is not selective and both gallium and aluminum are stripped together. Furthermore, several contacts will be necessary to remove all gallium from the organic phase. A fraction of both gallium and aluminum was found to precipitate during stripping, corresponding to about 8% of the gallium in the initial feed solution and 30% of the initial aluminum. As a result, some gallium is not recovered, but the remainder is purified further, to a final purity of 88–90 wt% in the aqueous phase. To regenerate the extractant after stripping, we propose the use of ammonium sulfate solutions, which are sufficiently acidic to protonate the extractant, but not to protonate or exchange the sulfate anion in the organic phase.
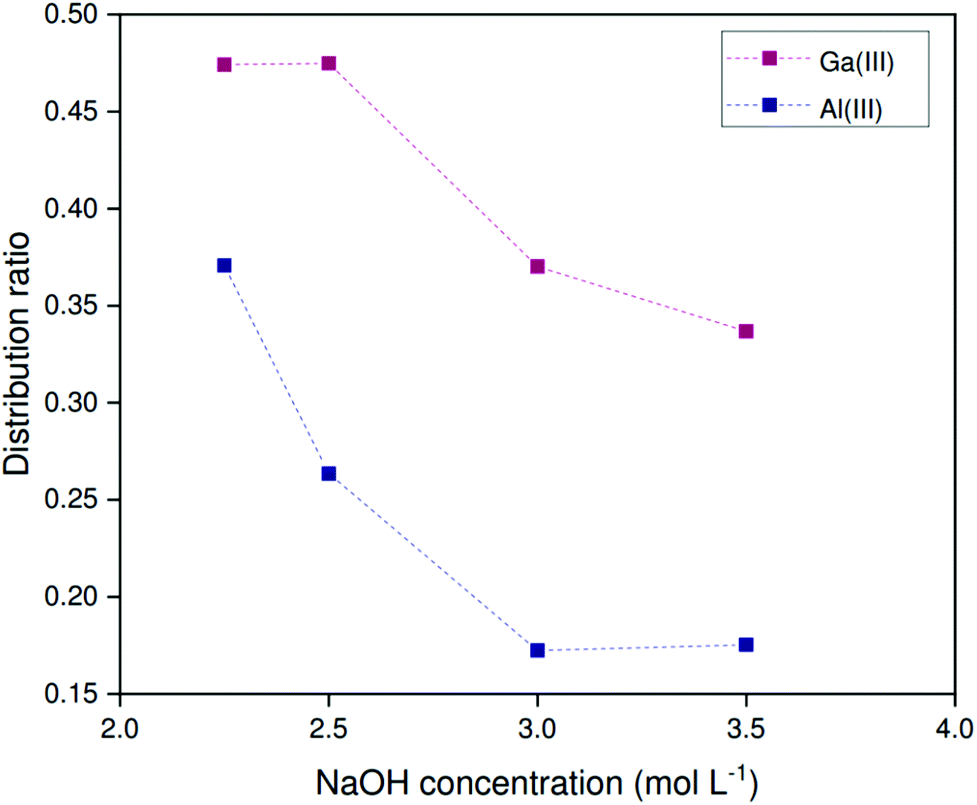 | ||
| Fig. 9 Distribution ratio of gallium as a function of the NaOH concentration in the stripping liquor for loaded 0.124 mol mol−1 Kelex® 100 in [EhEhT23]4[SO4], extracted from 200 ppm solutions containing 1.700 mol L−1 sodium carbonate in 1:1 phase ratio (organic to aqueous). Temperature: 22 °C. | ||
High aluminum concentrations
Extraction performance was investigated at high Al/Ga ratios mimicking those present in Bayer process liquors. While carbonated Bayer process liquors are limited in the concentration of gallium they can sustain in solution, untreated liquors contain concentrations of aluminum far higher than those of gallium. Leveque and Helgorsky report the concentration of sodium aluminate to be 1.569 mol L−1 at a free sodium hydroxide concentration of 3.594 mol L−1.13 Aluminum is thus superstoichiometric with respect to Kelex® 100, which means that the free extractant concentration in the organic phase will be very low at equilibrium, suppressing the extraction of gallium. Hence, aluminum extraction become competitive with gallium in binding to the limited amount of Kelex® 100 in the organic phase. A high selectivity of gallium over aluminum is therefore necessary to guarantee sufficient extraction of gallium even at high aluminum concentrations. The data presented in Fig. 7 demonstrate that the selectivity factor αGa/Al is maximal at 2 mol L−1 sodium hydroxide, reaching a value of approximately 1.0 × 103 under these conditions. Experiments were thus performed on artificial solutions, mimicking Bayer liquor diluted to 2 mol L−1 sodium hydroxide, which contained 873 mmol L−1 Al(III) as sodium aluminate and 150 ppm (about 2 mmol L−1) gallium(III) as gallium(III) sulfate. These concentrations were based on the report of Leveque and Helgorsky.13Both the kinetics of extraction and the performance over successive extraction/stripping cycles were considered. The latter was investigated by subjecting a 2.5 mL sample of [HHT12][Tf2N] containing 5 vol% Kelex® 100 and modified with 10 vol% 1-decanol to alternating cycles of extraction from simulated Bayer liquor (2.5 mL) and stripping using 1 mol L−1 hydrochloric acid (2.5 mL). Experiments were carried out at 22 °C in 10 mL glass screwcap vials under magnetic stirring at 1200 rpm using a 15 × 4.5 mm stirring bar. Equilibration times of 6 hours were used for extraction and 30 minutes for stripping.
The distribution ratio for gallium obtained after 6 hours of contact on the first extraction cycle is fairly low at only 1.40 (Fig. 10). In subsequent extraction cycles, distribution ratio is over an order of magnitude greater, at values up to 16.92. The percentage extraction remains stable over at least three successive cycles (93–94%). Stripping thus induces a chemical change in the organic phase which either results in more rapid or more complete extraction of gallium. This change is most likely to be the protonation of Kelex® 100 by hydrochloric acid, converting it to its hydrochloride form, a process which also occurs during selective scrubbing (vide supra).
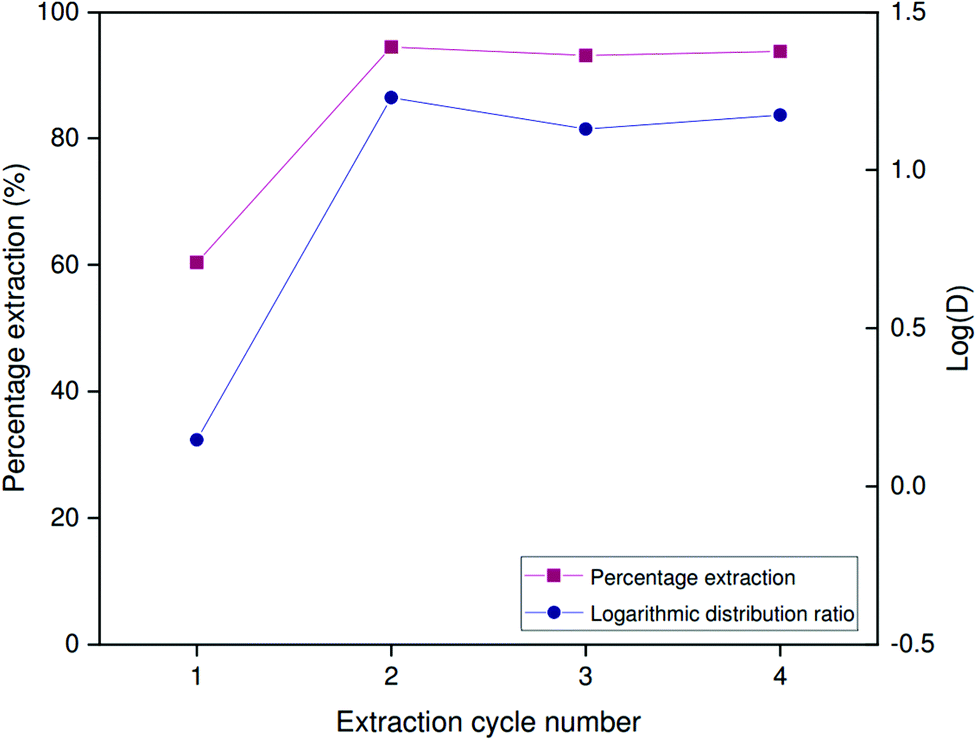 | ||
| Fig. 10 Percentage extraction and logarithmic distribution ratio of gallium over several extraction/stripping cycles. Extraction was performed in 1:1 phase ratio by 5 vol% Kelex 100 in [HHT12][Tf2N] (modified with 10 vol% 1-decanol) from 2 mol L−1 NaOH containing 873 mmol L−1 NaAl(OH)4 and 150 ppm Ga. Stripping was performed with 1 mol L−1 HCl. Temperature: 22 °C. | ||
In an identical setup, the extraction kinetics of both the kerosene and [HHT12][Tf2N] systems were evaluated after presaturation with either 2 mol L−1 sodium hydroxide or 1 mol L−1 HCl. In the samples equilibrated with sodium hydroxide, kinetics were slow both for both the ionic liquid system and in kerosene system (Fig. 11). In contrast to the results obtained for low aluminum concentrations (vide supra), the kerosene system is more selective than the ionic liquid system at high aluminum/gallium ratios. This difference in sensitivity to the aluminum concentration may result from differences in the extraction pathway, dependent on the nature of the diluent. As was evident from the results obtained from Sato and Oishi, mass transfer in the kerosene system is limited by the equilibrium concentrations of the neutral species Ga(OH)3 and Al(OH)3, which remain low even at high alumina concentrations.20 In the ionic liquid system, anionic hydroxometalate complexes can enter the organic phase without a prerequisite ligand loss. Due to the great excess of aluminate ions with respect to gallate(III) ions, the extraction of aluminum will initially be much more rapid than that of gallium. Saturation of the extractant with aluminum thus implies that gallium can only be extracted by metal exchange, which appears to be a slower process. In light of this reasoning, the lower selectivity observed for the ionic liquid system may in fact be a kinetic effect resulting from a progressively decreasing concentration of free extractant molecules, which slows gallium extraction and prevents the system from reaching thermodynamic equilibrium.
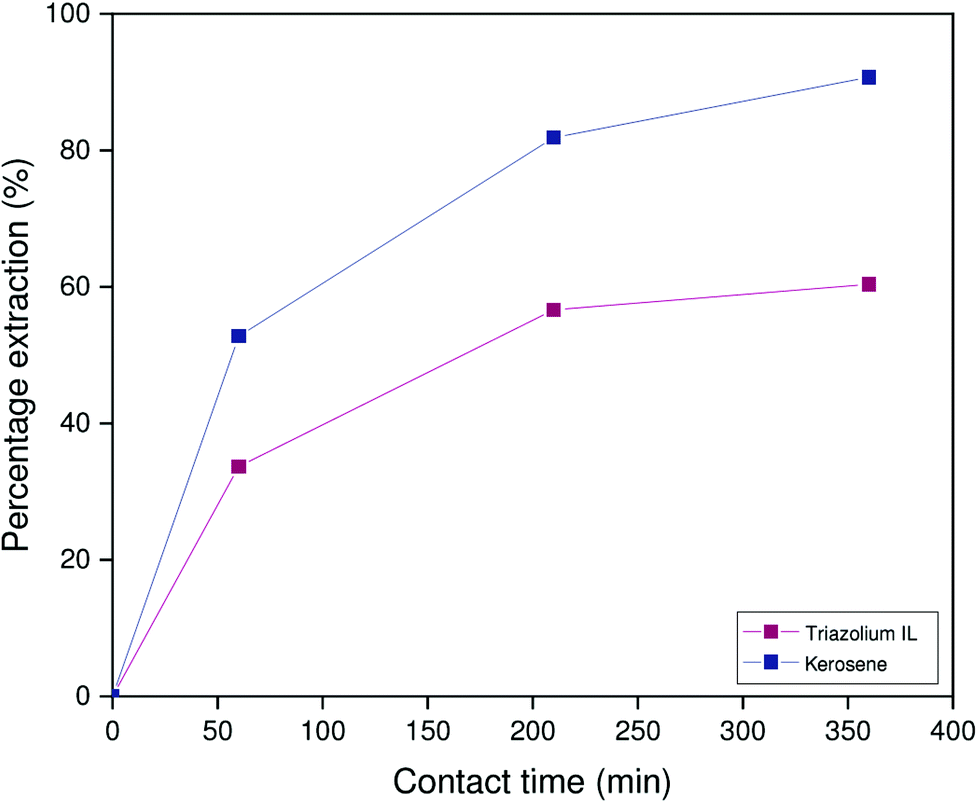 | ||
| Fig. 11 Percentage extraction as a function of time for extractions performed in 1:1 phase ratio by 5 vol% Kelex 100 in [HHT12][Tf2N] or kerosene (modified with 10 vol% 1-decanol) from 2 mol L−1 NaOH containing 873 mmol L−1 NaAl(OH)4 and 150 ppm Ga. Temperature: 22 °C. | ||
This reasoning is corroborated by the kinetic results of the HCl-saturated systems, shown in Fig. 12. Under these conditions, the ionic liquid system reaches a percentage extraction of 88% after 60 min. Extraction of gallium is thus not only faster, but more complete, which corresponds to an apparent higher selectivity over aluminum. However, at full thermodynamic equilibrium, the HCl extracted to the organic phase will be neutralized by the much higher sodium hydroxide concentration in the aqueous phase, which results in similar equilibrium conditions to those of the system presaturated with sodium hydroxide solution (taking into account the concentration of the extractant in the organic phase, the aqueous sodium hydroxide concentration will be reduced to approximately 1.85 mol L−1, and a NaCl concentration of 0.15 mol L−1 will be present). Hence, the equilibrium distribution of gallium should be similar after either sodium hydroxide or hydrochloric acid presaturation. The presence of HCl thus likely has a catalytic effect on the extraction process. Unfortunately, full kinetic results of the kerosene system presaturated with hydrochloric acid could not be obtained, due to poor phase separation resulting from the surface activity of Kelex® 100 hydrochloride. No such problems occurred for the ionic liquid system, which is an advantage for the application of the ionic liquid systems in industrial setting. However, the data obtained after a contact time of 5 minutes do suggest that the extraction kinetics of the ionic liquid system are significantly faster after equilibration with HCl than those of the kerosene system.
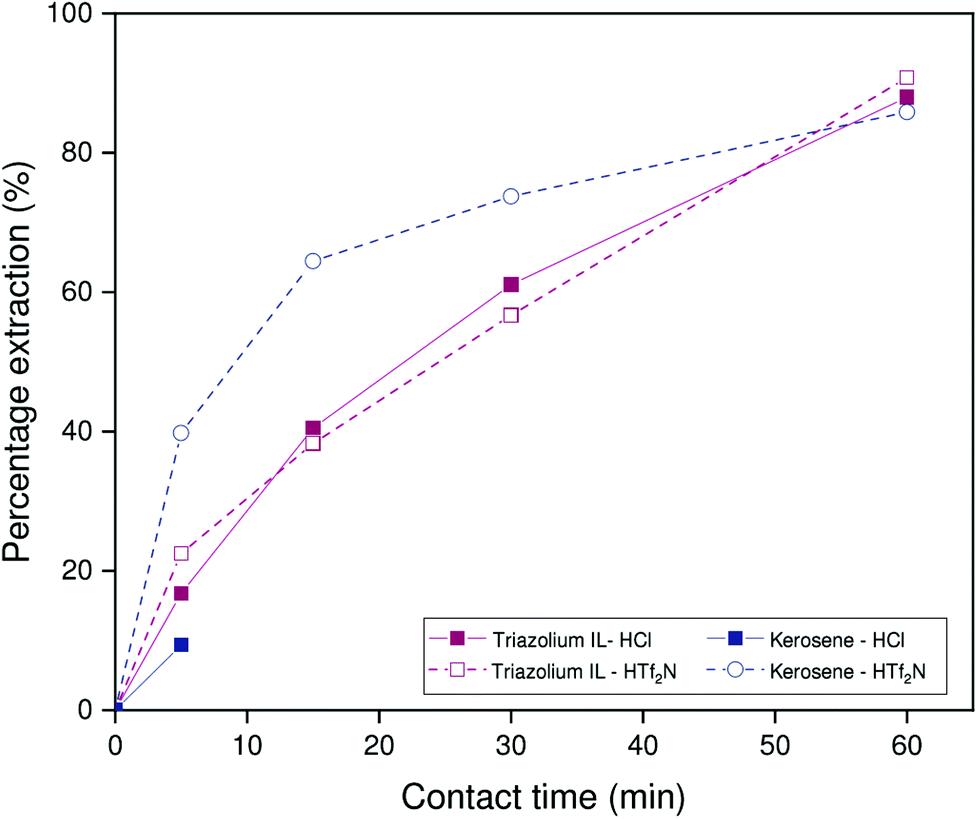 | ||
| Fig. 12 Percentage extraction as a function of time for extractions performed in 1:1 phase ratio by organic phases saturated with 1 mol L−1 HCl or hydrogen bistriflimide. The composition of the organic phase was 5 vol% Kelex 100 in [HHT12][Tf2N] or kerosene (modified with 10 vol% 1-decanol) and the aqueous phase was 2 mol L−1 NaOH containing 873 mmol L−1 NaAl(OH)4 and 150 ppm Ga. Temperature: 22 °C. | ||
The nature of the catalytic effect of extracted HCl was further investigated by performing a similar kinetic assay after presaturation of the organic phase with 1 mol L−1 hydrogen bistriflimide (HTf2N), the conjugate acid of the bistriflimide anion. This anion is poorly hydrated and thus exhibits little propensity to anion exchange.38 Other experimental parameters were kept unchanged. The results demonstrate that hydrogen bistriflimide exhibits a similar catalytic effect to HCl (Fig. 12). This eliminates any anion-related effects as potential mechanisms of catalysis. Furthermore, both the kerosene system and the ionic liquid system are affected by the catalytic effect. As the kinetics of the kerosene system are limited by the availability of kerosene-soluble metal complexes in the aqueous phase, the catalytic effect is likely either related to the accumulation of the extractant at the interface (effectively allowing aqueous phase species to interact with the extractant directly) or the influence of the aqueous phase speciation by local pH changes near the interface.
Conclusion
Two ionic liquid diluents were evaluated for the extraction of gallium from spent Bayer process liquor by Kelex® 100. The hydrophilic sulfate ionic liquid [EhEhT23]2[SO4] forms an aqueous biphasic system with concentrated salt solutions exhibiting LCST-type thermomorphic behavior, allowing it to be used for homogeneous liquid–liquid extraction. Due to the high hydrophilicity of this ionic liquid, the acidity of Kelex® 100 increases with respect to aliphatic diluents, implying that the alkalinity of Bayer liquor must first be reduced by carbonation before extraction. The pH of the solution drops further during the extraction process, causing further precipitation of aluminum. The system exhibits good selectivity for gallium (αGa/Al ≈20) and the extraction proceeds via the same mechanism as it does in conventional systems (cation exchange and loss of one hydroxo ligand). The system reaches equilibrium within 5 min. The kinetics remain fast even under non-HLLE conditions, and evidence suggests this is a result of diffusion of sodium gallate(III) ion pairs in the highly polar ionic liquid phase. Stripping can be accomplished by contacting the loaded organic phase with sodium hydroxide solutions, although this procedure is not selective and leads to the loss of some gallium by precipitation.Kelex® 100 diluted in the hydrophobic ionic liquid [HHT23][Tf2N] and modified with 1-decanol is capable of selectively extracting gallium from untreated Bayer process liquor analogously to the conventional kerosene system. The ionic liquid is advantageous over the kerosene system in that it (1) exhibits more rapid equilibration; (2) binds gallium with a higher enthalpy of extraction; (3) improves phase disengagement and (4) avoids the use of volatile and flammable organic solvents. Compared to the ion exchange system in current industrial use, the system exhibits better long-term stability and lower waste production. The system performs optimally when the aqueous sodium hydroxide concentration is 2 mol L−1, at which the system is highly selective (αGa/Al ≈1000) and a distribution ratio of nearly 1000 is reached for gallium at low aluminum concentrations. Scrubbing of aluminum can be performed using hydrochloric acid solutions of over 3 mol L−1 while solutions of under 1 mol L−1 can be used for the stripping of gallium. When extracting from feed solutions containing high concentrations of aluminum, residual acid remaining in the organic phase from the previous stripping cycle exhibits a catalytic effect on the extraction of gallium(III). Without this residual acid, extraction of gallium(III) from concentrated aluminum(III) solutions is slow. Overall, this system is the most promising for industrial application, as it does not require decomposing the Bayer liquor before extraction, yields a pure gallium product and the hydrophobicity of the diluent is high, leading to low rates of diluent loss and a stable phase ratio. Further improvements in extraction kinetics may be possible by combining the triazolium ionic liquid/Kelex® 100 system with the use of intensified solvent extraction techniques such as centrifugal contactors or by dispersion of the extracting phase on a solid support (supported ionic liquid phase).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank FWO-Flanders for funding (doctoral fellowship to SR).References
- B. Jaskula, Mineral Commodity Summaries: Gallium, U.S. Geological Survey, Reston, 2019 Search PubMed.
- Study on the review of the list of Critical Raw Materials: Executive Summary, European Commission, Brussels, 2017 Search PubMed.
- F. Lu, T. Xiao, J. Lin, Z. Ning, Q. Long, L. Xiao, F. Huang, W. Wang, Q. Xiao, X. Lan and H. Chen, Hydrometallurgy, 2017, 174, 105–115 CrossRef CAS.
- T. H. Nguyen and M. S. Lee, Miner. Process. Extr. Metall. Rev., 2018, 40, 287–291 Search PubMed.
- O. Font, X. Querol, R. Juan, R. Casado, C. R. Ruiz, Á. López-Soler, P. Coca and F. G. Peña, J. Hazard. Mater., 2007, 139, 413–423 CrossRef CAS PubMed.
- S. Nusen, T. Chairuangsri, Z. Zhu and C. Y. Cheng, Hydrometallurgy, 2016, 160, 137–146 CrossRef CAS.
- T. F. de Aquino, H. G. Riella and A. M. Bernardin, Miner. Process. Extr. Metall. Rev., 2011, 32, 137–149 CrossRef.
- H. R. Hose, in Essential Readings in Light Metals: Volume 1 Alumina and Bauxite, ed. D. Donaldson and B. E. Raahauge, Springer International Publishing, New York, 2016, pp. 21–29 Search PubMed.
- R. F. Schulte and N. K. Foley, Compilation of Gallium Resource Data for Bauxite Deposits, U.S. Geological Survey, Reston, 2014 Search PubMed.
- E. Jamieson, A. van Riessen, B. McLellan, B. Penna, C. Kealley and H. Nikraz, in Handbook of Low Carbon Concrete, ed. A. Nazari and J. G. Sanjayan, Butterworth-Heinemann, Oxford, 2017, pp. 159–193 Search PubMed.
- P. E. Jackson, J. Chromatogr. A, 1995, 693, 155–161 CrossRef CAS.
- R. Sonthalia, P. Behara, T. Kumaresan and S. Thakre, Int. J. Miner. Process., 2013, 125, 137–148 CrossRef CAS.
- A. Leveque and J. Helgorsky, in Proceedings of the International Solvent Extraction Conference, Toronto, 1977, pp. 429–431 Search PubMed.
- Z. Zhao, Y. Yang, Y. Xiao and Y. Fan, Hydrometallurgy, 2012, 125–126, 115–124 CrossRef CAS.
- P. A. Riveros, Hydrometallurgy, 1990, 25, 1–18 CrossRef CAS.
- J. M. Loureiro and A. E. Rodrigues, Sep. Sci. Technol., 1998, 33, 1585–1604 CrossRef.
- K. S. Rao, D. Sarangi, P. K. Dash and G. R. Chaudhury, J. Chem. Technol. Biotechnol., 2003, 78, 555–561 CrossRef CAS.
- J. Roosen, S. Mullens and K. Binnemans, Hydrometallurgy, 2017, 171, 275–284 CrossRef CAS.
- B. Pesic and T. Zhou, JOM, 1988, 40, 24–26 CrossRef CAS.
- T. Sato and H. Oishi, Hydrometallurgy, 1986, 16, 315–324 CrossRef CAS.
- B. Pesic and T. Zhou, JOM, 1989, 41, 42–45 CrossRef CAS.
- I. Mihaylov and P. A. Distin, Hydrometallurgy, 1992, 28, 13–27 CrossRef CAS.
- P. Fourre and D. Bauer, Solvent Extr. Ion Exch., 1983, 1, 465–483 CrossRef CAS.
- P. P. Borges and I. O. C. Masson, Miner. Eng., 1994, 7, 933–941 CrossRef CAS.
- G. V. K. Puvvada, Hydrometallurgy, 1999, 52, 9–19 CrossRef CAS.
- T. N. de Castro Dantas, M. H. de Lucena Neto and A. A. Dantas Neto, Talanta, 2002, 56, 1089–1097 CrossRef CAS.
- C. Eckberg and P. L. Brown, in Hydrolysis of Metal Ions, John Wiley & Sons, Hoboken, 2016, pp. 757–834 Search PubMed.
- K. Murata, Y. Yokoyama and S. Ikeda, Anal. Chem., 1972, 44, 805–810 CrossRef CAS PubMed.
- J. Xu, N. Rajapakse and H. L. Finston, Radiochim. Acta, 1990, 49, 135–140 CAS.
- J. D. Lamb and R. T. Peterson, Sep. Sci. Technol., 1995, 30, 3237–3244 CrossRef CAS.
- N. Alizadeh and K. Ashtari, Sep. Purif. Technol., 2005, 44, 79–84 CrossRef CAS.
- M. H. Hosseini and N. Alizadeh, Ind. Eng. Chem. Res., 2010, 49, 7068–7073 CrossRef CAS.
- T. Vander Hoogerstraete, B. Onghena and K. Binnemans, J. Phys. Chem. Lett., 2013, 4, 1659–1663 CrossRef PubMed.
- D. Depuydt, L. Liu, C. Glorieux, W. Dehaen and K. Binnemans, Chem. Commun., 2015, 51, 14183–14186 RSC.
- S. Raiguel, J. Thomas, K. Binnemans and W. Dehaen, Eur. J. Org. Chem., 2018, 4850–4485 CrossRef CAS.
- S. Raiguel, W. Dehaen and K. Binnemans, Green Chem., 2019, 21, 3948–3960 RSC.
- J. N. Chubb, P. Lagos and J. Lienlaf, J. Electrost., 2005, 63, 119–127 CrossRef CAS.
- D. Dupont, D. Depuydt and K. Binnemans, J. Phys. Chem. B, 2015, 119, 6747–6757 CrossRef CAS PubMed.
- M. P. Jensen, J. Neuefeind, J. V. Beitz, S. Skanthakumar and L. Soderholm, J. Am. Chem. Soc., 2003, 125, 15466–15473 CrossRef CAS PubMed.
- A. Rout and K. Binnemans, Dalton Trans., 2015, 44, 1379–1387 RSC.
- C. H. C. Janssen, A. Sánchez, G.-J. Witkamp and M. N. Kobrak, ChemPhysChem, 2013, 14, 3806–3813 CrossRef CAS PubMed.
- U. Schröder, J. D. Wadhawan, R. G. Compton, F. Marken, P. A. Z. Suarez, C. S. Consorti, R. F. de Souza and J. Dupont, New J. Chem., 2000, 24, 1009–1015 RSC.
- A.-L. Rollet, P. Porion, M. Vaultier, I. Billard, M. Deschamps, C. Bessada and L. Jouvensal, J. Phys. Chem. B, 2007, 111, 11888–11891 CrossRef CAS PubMed.
- J. H. van't Hoff, Etudes de dynamique chimique, Frederik Muller, Amsterdam, 1884 Search PubMed.
- X. Wang, W. Li, S. Meng and D. Li, J. Chem. Technol. Biotechnol., 2006, 81, 761–766 CrossRef CAS.
- B. Onghena, S. Valgaeren, T. Vander Hoogerstraete and K. Binnemans, RSC Adv., 2017, 7, 35992–35999 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, pH changes upon extraction from carbonate solutions, graphs for slope analysis. See DOI: 10.1039/c9dt04623b |
| This journal is © The Royal Society of Chemistry 2020 |