
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fast and simple B–C bond formation in metallacarboranes avoiding halometallacarboranes and transition metal catalysts†
Ana B.
Buades
 ,
Zsolt
Kelemen
,
Víctor S.
Arderiu
,
Adnana
Zaulet
,
Clara
Viñas
and
Francesc
Teixidor
*
,
Zsolt
Kelemen
,
Víctor S.
Arderiu
,
Adnana
Zaulet
,
Clara
Viñas
and
Francesc
Teixidor
*
Institut de Ciència de Materials de Barcelona – CSIC, Campus UAB, 08193, Bellaterra, Barcelona, Spain. E-mail: clara@icmab.es
First published on 11th February 2020
Abstract
An electrophilic substitution on metallacarboranes by using a stabilized carbocation that can be made in situ is reported for the first time. This new synthetic methodology provides a new perspective on easy metallacarborane derivatization with organic fragments, which enhances the properties of both fragments and widens their possible applications.
Introduction
Metallabisdicarbollides are sandwich complexes in which a central metal ion (commonly Co or Fe) is η5 coordinated by two dicarbollide anions [C2B9H11]2−. Metallacarboranes have high thermal and chemical stability, show reversible redox behaviour1 and can be stored for years in open shelves without taking much care. They can be derivatized with C/B, and the number of possible derivatizations is higher than that with most similar molecular weight organic compounds. Commonly, derivatization of metallacarboranes with organic moieties leads to preservation of their mentioned characteristics and the incorporation of properties associated with the new fragment. As a consequence, much effort has been made over the last decades to develop new synthetic routes for the derivatization of metallacarboranes with organic fragments. The formation of B–C bonds is the most relevant reaction, although the formation of CCarborane–C, B–N, B–S, and B–P bonds has been widely studied.1a,b,2While the formation of B–C bonds is one of the most studied reactions in icosahedral boron cluster derivatization, this has been much less studied in metallacarboranes.2a,3 It is not surprising that most of the procedures used to produce B–C bonds are modifications of methods applied in classical C–C bond formation. Commonly what works in Y–C organic coupling reactions works differently in metallacarborane or borane chemistry.4 It has been previously reported that Friedel–Crafts type, palladium-catalyzed cross coupling and thermal decomposition reactions are successful tools for B–C cluster bond formation.5 Palladium-catalyzed cross coupling reactions require, in addition to a high loading of the Pd catalyst, to start with a metallacarborane having a preformed B–I bond before reaction; conversely, the Friedel–Crafts type reaction with AlCl3 requires cobaltabisdicarbollide [Co(C2B9H11)2]− ([1]−) as a starting reagent. Electrophile-induced nucleophilic substitution works well for halogens, OH, OR and other nucleophiles6 but is less efficient in producing B–C bonds. For this, Pd catalyzed reactions have been successful, particularly the Kumada and Heck5d,7 reactions. This procedure requires to begin with a B–I iodo-borane, iodo-carborane or iodo-metallacarborane. It could be concluded that to produce a B–C bond in a boron cluster, it is required to start from an iodinated borane and to have an active Pd or other suitable transition metal catalysts or a stretched iodonium.8 Herein we give the primary results of the formation of a B–C bond in the absence of any metal catalysts and not necessitating any B–I containing boron clusters.
Results and discussion
Before turning to the relevant matter of this work it is important to note that the characterization of the compounds reported herein and the conclusions drawn derive primarily from MALDI-TOF-MS and secondly from NMR, particularly 1H-NMR and 11B-NMR. MALDI-TOF of [Co(C2B9H11)2]− derivatives is key as it provides the molecular mass, and from the height we can derive the molecular ratio of the distinct derivatives of [1]− in the crude of the reaction, as we had proved earlier.9 We have found it very relevant to be able to analyse the products directly from the reaction crude with valuable information from MALDI-TOF-MS such as the molecular mass and the molecular ratio. The errors in the interpretation of these reactions are minimized. Important information on the number of isomers can be obtained from the metallacarborane C–H peak positions and their areas in the 1H-NMR spectra. Information on the nature of the isomers generated can be obtained by careful analysis of the aromatic region, but this has proved to be more challenging due to the difficulty in isolating the component isomers. It is to be noted that we have used for simplicity the terminology MALDI-TOF to describe the matrix-assisted laser desorption/ionization (MALDI), time-of-flight (TOF) mass spectrum (MS) technique, although we do not use any matrix for derivatives of [1]−. Further evidence of the measurable properties of MS, in this case, is included at the end of the paper under the title MALDI-TOF spectral analysis.We discovered the formation of a B–C bond in the absence of any metal catalysts and any B–I containing boron clusters by accident when we sought for a second B–C bond on a metallacarborane that already had a preformed B–Csp2 bond. The sought, but unsuccessful, reaction is depicted in Fig. 1 (top). The triphenylmethyl cation (Tr+) is commonly used for three purposes, as a starter for cationic polymerization, as a weak cation or as a hydride scavenger.10 In this work we hypothesized that it would act as a hydride scavenger. Although crystal structures have shown that aryl metallacarboranes tend to exhibit trans disposition,5b,d it is also true that most of the pristine ortho-metallacarboranes exhibit cis-disposition.11 Therefore it was reasonable that the formation of a B–Csp2 bond could occur due to the relative movement of the clusters in solution following hydride removal by Tr+ and B+ formation. First experiments were done to study the compatibility of Cs[Co(C2B9H11)(8-C10H7-C2B9H10)]5d (Cs[2]) and TrBF4, and the compatibility of the latter with tetrahydrofuran (THF). Upon mixing a solution of Cs[2] in THF with TrBF4 under nitrogen and stirring overnight, a highly viscous mixture was obtained. The MALDI-TOF-MS of the crude of the reaction displayed only one peak at 450 m/z corresponding to the starting cobaltabisdicarbollide substituted with naphthalene. The increase in viscosity came from the polymerization of THF under the reaction conditions.12 Then, dimethoxyethane, DME, was chosen as the alternative solvent because it does not polymerize as THF and because the reagents are also soluble in it.
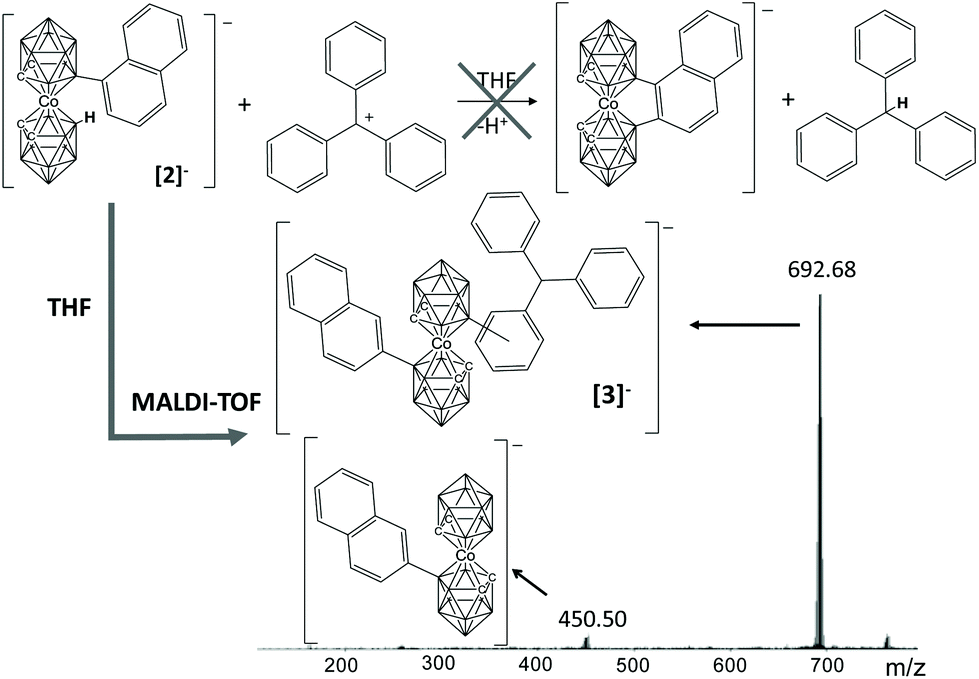 | ||
| Fig. 1 Reaction initially proposed and MALDI-TOF spectra of the crude of the reaction, leading to the formation of a B–Csp2 bond. | ||
Following the reaction in DME, under the same conditions as for THF, the crude of the reaction was analysed by MALDI-TOF-MS (Fig. 1). The target product was not obtained; conversely, as previously observed for the [CB11H12]− boron cluster13 upon reaction with TrBr, an electrophilic substitution occurred, leading to [3]−. The mass of the base peak on MALDI-TOF-MS is the sum of the molecular peak of the initial metallacarborane (450 m/z) plus a triphenylmethyl moiety (243 m/z) resulting in a new and unexpected molecule with 692.68 m/z ([3]−). We say unexpected based on the results given by Reed and co-workers earlier where they did not notice any B–C formation in a very similar reaction.10e To confirm the formation of the B–C bond, we run the metallacarborane/trityl reaction in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in DME this time using [1]−. The result was again the formation of the B–C bond with the base peak in MALDI-TOF-MS at 566.59 m/z corresponding to the triphenyl derivative ([4]−) (see ESI†).
:1 ratio in DME this time using [1]−. The result was again the formation of the B–C bond with the base peak in MALDI-TOF-MS at 566.59 m/z corresponding to the triphenyl derivative ([4]−) (see ESI†).
The reaction was also performed with 2 equivalents of TrBF4. The MALDI-TOF-MS of the crude of the reaction displayed the base peak at 808.74 m/z which corresponds to a cobaltabisdicarbollide disubstituted with two triphenylmethyl moieties ([5]−). Thus, depending on the stoichiometry of the reaction, one or two units of the triphenylmethyl group can be bonded to cobaltabisdicarbollide. Furthermore, a reaction with 3 equivalents of TrBF4 was done leading again to [5]−, implying that the next B–H entities to be replaced are not nucleophilic enough to initiate the substitution. This led clearly to 8 or 8,8′ substitution. See MALDI-TOF-MS of the purified TLC in the ESI.†
The NMR spectra (see the ESI†) are fundamental to demonstrate the formation of the B–C bond between the cobaltabisdicarbollide B(8) site and one aromatic C of triphenylmethane. The NMR spectra were run after preparative TLC on silica, by which a layer incorporating a mixture of the trityl derivatives of [1]− was analysed. The B–Csp2 resonance appears at 12.3 ppm in the 11B NMR spectra4c,5d,11b consistent with the resonance at 5.5 ppm in the 1H NMR spectra corresponding to the H–Csp3 of triphenylmethane, and therefore the B–C bond shall be between an aromatic carbon and a boron. Moreover, the 4 signals corresponding to H–Ccluster, 2 + 2 reveal the presence of two isomeric products in a 2:3 ratio, ortho-[4]− and meta-[4]−, respectively (see full characterization in the ESI†). Lavallo also found isomeric non-separable mixtures of meta- and para- upon the reaction of TrBr with [CB11H12]− in FC6H5.13
Reactions via carbocation generation in situ
Just to explore if this method could have a wider application without the need to use preformed stable electrophiles, we have attempted to produce carbocations in situ. One way of producing carbocations is by mixing halogenated reagents RX with soluble silver salts, resulting in the precipitation of the AgX salt and the formation of the carbocation.14 In our case, we have taken advantage of the anionic nature of the metallacarborane and we have prepared its silver salt (see ESI†), with the expectation that Ag+ will precipitate with the halogen and the in situ formed carbocation will react directly with [1]−. The silver salt of cobaltabisdicarbollide is soluble in DME or THF. To achieve this target, we have utilized bromomethane derivatives with three, two (shown in Scheme 1) or one phenyl groups. It was expected that the higher the number of benzene rings, the higher the purity of the resulting “combined isomers”, in agreement with the stability of the carbocations. So we have used bromotriphenylmethane (TrBr), benzhydryl bromide (BzhBr) and benzyl bromide (BnBr). These were added in air to Ag[1] in THF in a molar ratio of 1:1. Immediately, due to the high speed of the reaction, a white precipitate of AgBr appears, while the colour of the solution remains orange, the typical colour of cobaltabisdicarbollide. The solution was immediately filtered to avoid oxidation by Ag+ and aqueous sodium chloride was added to the reaction mixture to produce two phases. The organic phase was dried and analyzed by MALDI-TOF-MS. As expected the reactions of TrBr and BzhBr with Ag[1] proved the formation of mono and disubstituted products, consequently leaving behind unreacted [1]−. Both mixtures were purified by chromatography obtaining an isolated yield of 75% and 76% for the isomeric mixtures [o-, m-4]− and [o-, m-6]−, respectively. The isomers in these mixtures had similar physical properties and would have required many sequential TLC steps. As our intention was to demonstrate the feasibility of the B–C bond formation, which had been accomplished, we decided to work with the isomeric mixture. 1D and 2D 1H-NMR spectroscopy provided sufficient clues to permit a consistent but not unambiguous assignation (see full characterization in the ESI†). The 1H NMR spectra of the mixture of NMe4[4] show a singlet at 7.13 ppm, indicating the presence of the meta-isomer, and the two triplets at 7.05 and 7.16 ppm can only be explained by the existence of the ortho-isomer in the mixture. The 1H-NMR interpretation of compound Cs[6] is equal with the singlet and triplets in the same region (7.19, 7.04 and 7.15 ppm respectively). In addition, around 8% of the product in each reaction corresponds to the disubstituted compounds [5]− and [7]−, along with 8% of unreacted [1]−, respectively. Moreover, it is possible to obtain the ortho:meta (2:3) and (1:1) ratio from the integration of some aromatic protons in the 1H-NMR spectrum of [4]− and [6]−, respectively (see Fig. S8 and S28 of ESI†). The high velocity of the reaction and perhaps the enhanced nucleophilic character of the B(8)′–H moiety after the first B–Tr formation could explain the tendency to form the disubstituted species. If the reactions are carried out with 2 equivalents of TrBr or BzhBr the main products are [5]− and [7]− with yields of 83 and 87%, respectively (Scheme 1). In addition, the 1H NMR spectra of both compounds show three peaks at around 3.8 ppm (due to H–Ccluster) which may be explained by the existence of three isomers in the mixture. Based on the 1H-NMR interpretation of the monosubstituted species which indicates both ortho- and meta-isomers, we suggest for the disubstituted species the isomers [o,m-], [o-,o-] and [m,m-] of compounds [5]− and [7]−. This is purely tentative and, obviously, it is not possible to determine the isomeric ratio.
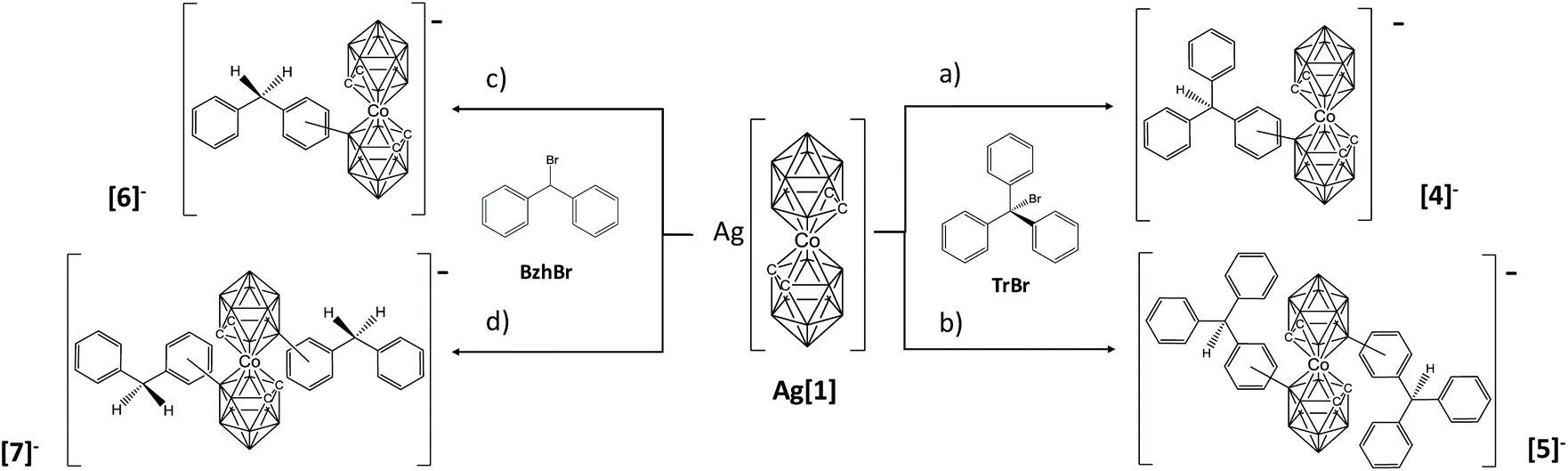 | ||
| Scheme 1 Reaction conditions for the synthesis of compounds [4]−, [5]−, [6]− and [7]−. Reagents: (a) 1 equiv. TrBr; (b) 2 equiv. TrBr; (c) 1 equiv. BzhBr; (c) 2 equiv. BzhBr. Conditions: (a–d) the reaction was performed in THF or DME as solvent, at room temperature and the products were obtained instantaneously. In all cases, the products are isomeric mixtures between the isomers ortho and meta. | ||
The reaction of BnBr with Ag[1] was not as successful as the others in THF. The mass spectra of the crude from BnBr display 4 major peaks: one corresponding to unreacted cobaltabisdicarbollide at 324 m/z, which is the more intense (see Fig. S42, on the left†), and the other three corresponding to [Co(C2B9H11)(8-OH-C2B9H10)]− at 340 m/z, [Co(C2B9H11)(8-Br-C2B9H10)]− at 403 m/z and [Co(C2B9H11)(8-CH3C6H4-C2B9H10)]− at 414 m/z, indicating the formation of the target compound. We suggest substitution at the 8 position, as it is the most plausible and the one shown by the computational study in Fig. 2, but we have not done any exhaustive experimental study that provides unambiguous information on where the substitution occurs.
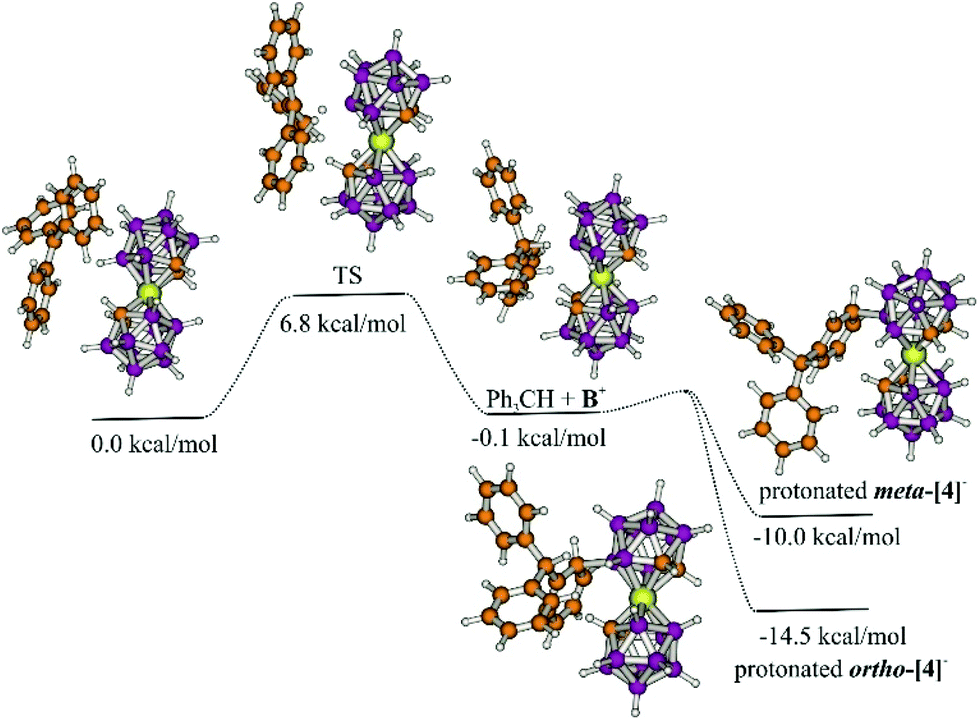 | ||
| Fig. 2 Calculated reaction mechanism of the hydride abstraction and the nucleophilic attack of Ph3CH. | ||
For the reactions with TrBr and BzhBr, THF as solvent is not influential for the desired outcome of the reaction. However, in the reaction between Ag[1] and BnBr the outcome is compromised by the polymerization of the solvent as shown by the MALDI-TOF spectra. To avoid this drawback, the reaction was performed in DME. The MALDI-TOF-MS (Fig. S42, on the right†) spectra show five peaks separated by 90 m/z corresponding to the sequential addition of up to four benzyl group units, the base peak being observed for [1]−. The reaction presumably is so fast that the multiple substitutions occur. To determine the major product the experiment was done under different conditions by modifying the ratios of benzyl bromide (to two, three and ten), decreasing the reaction temperature to −20 °C and replacing benzyl bromide by chloride to slow down the reaction speed. However, the results show that the major component in the mixture is always [1]−.
MALDI-TOF spectral analysis
In the Introduction we indicated how important is the information obtained from MALDI-TOF-MS concerning [1]− and its derivatives. Noticeable is that these studies have permitted us to demonstrate another example of the quantitative ability of MS. Commonly, MS has been used as a tool for qualitative chemical analysis, although much progress is being done to convert it also into a quantitative tool. We noticed in 2009 that the MS-height of related cobaltabisdicarbollide derivatives was proportional to the concentration.9,15 This represented a great advance in cobaltabisdicarbollide research. However, we had not found a distinct type of experiment to the one described to test the validity of the approach. As seen in Fig. S42† on the right for an equimolar ratio of Ag[1] and BnBr five peaks are formed, the more intense being unreacted [1]−. This was observed for the crude of the reaction, and so all reagents were present. The addition of the heights of the five peaks corresponds to the total amount of [1]−, [A], whereas the total amount of benzyl, [B], corresponds to the addition of the height of the peaks times the number of benzyl units in each peak. If the reactant ratio was 1:1 and the reaction went to completion, the two numbers A and B should be identical, indicating that MS is quantitative. A discrepancy of less than 1.3% has been found. These results are supported by the NMR data (see the ESI†).
Reaction mechanism
To get more insight into the reaction mechanism, DFT calculations at the ωB97XD/6-31+G* level of theory were performed. The initial step (which is the rate limiting step with 6–8 kcal mol−1 barrier) is the hydride transfer which results in the formation of the adduct of the formal B+ intermediate (B+) and triphenylmethane (Fig. 2). As seen in Fig. 2, the transition state (TS) shows a boronium ion and a carbocation sharing a hydride. This step is followed by the nucleophilic attack of triphenylmethane on the boronium, which seems to be almost barrierless. Unfortunately, all attempts to localise this transition state failed, but scan calculations (decreasing stepwise the B–C distance by 0.1 Å) showed that the energy of the system decreases continuously, indicating no significant barrier for this process. In agreement with the proposed very small barrier, the geometry optimization of the slightly modified geometry of the complex of Ph3CH and B+ gives the protonated ortho-[4]−, meta-[4]− and para-[4]− which are more stable by 14.5, 10.0 and 11.9 kcal mol−1, respectively. The last step is proton abstraction (see ESI† for ortho- and meta-) which results in the final products. The small barriers were in agreement with the observed fast reaction; the initially formed triphenylmethane immediately attacks the electrophilic center. Indeed Lavallo proposed an ion pair complex,16,19e.g. indicated as 3[CPh3]+, although structurally not informative, which is not so far from the transition state in our proposed mechanism, in which the H is shared by the [CPh3]+ and the boronium in [Co(C2B9H11)(C2B9H10)]0. The difference is that in the mechanism proposed by Lavallo the anionic boron cluster acts as a nucleophile on the Ph on Tr+, whereas in our computationally described mechanism there is an electrophilic attack on the Ph on Tr+. This suggested reaction mechanism was also supported by the experimental reaction of Tr+ and [1]− in the presence of 10 eq. of benzene, to have a possible competing reaction between the two arenes PhH and Ph3CH. However, the derivatives of Ph3CH according to MALDI-TOF-MS were dominant compared to those of PhH in a ratio of 11:2 which suggests a very fast reaction, as Ph3CH is the one closest to the boronium produced (see more details in the ESI†).
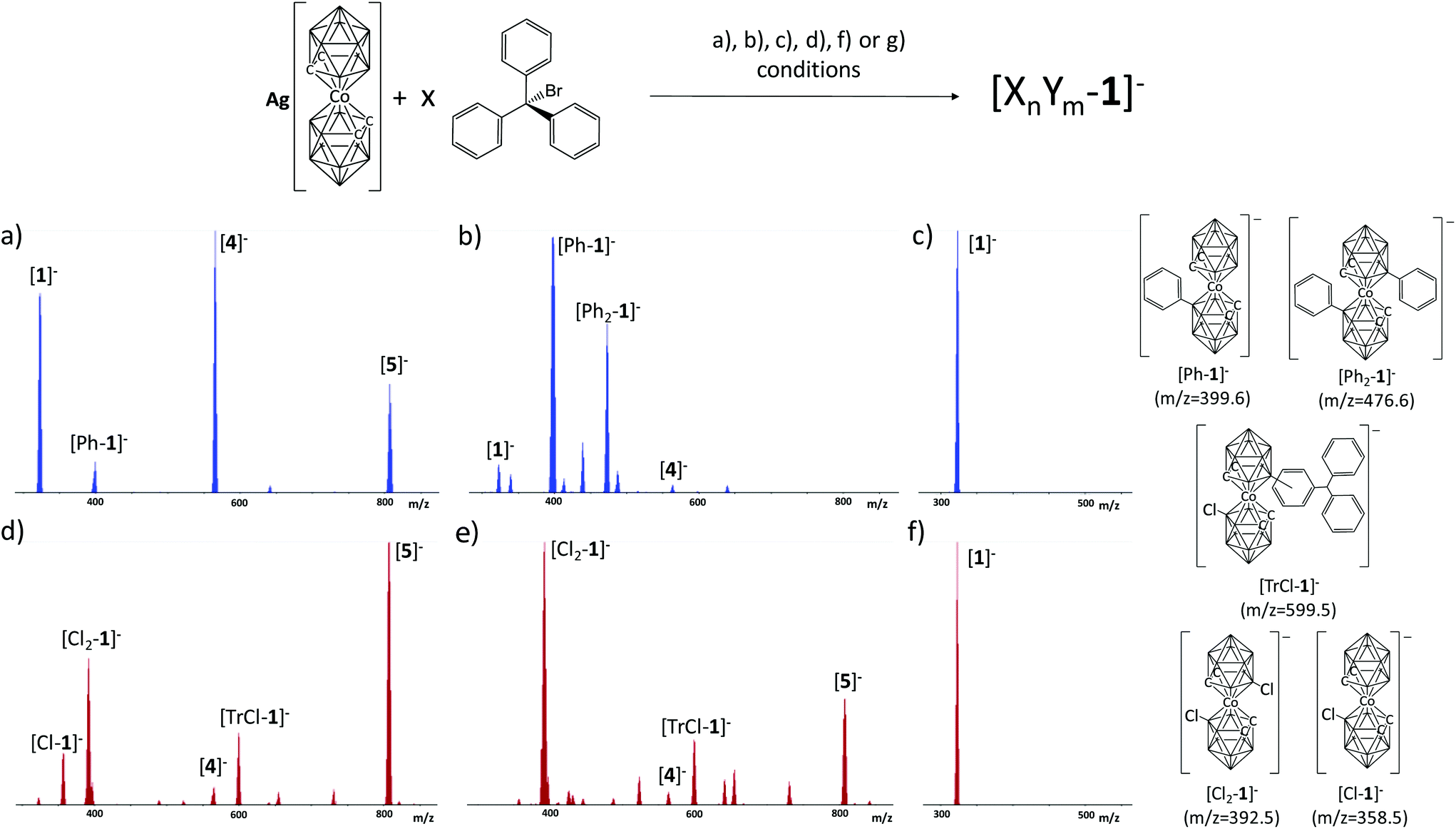 | ||
| Fig. 3 MALDI-TOF-MS spectra of the synthesis conditions leading to [4]− but in different solvents and at different temperatures: (a) in benzene at room temperature; (b) in benzene at reflux temperature; (c) in benzene at reflux temperature in the absence of TrBr; (d) in DCM at room temperature; (e) in DCM at reflux temperature; (f) in DCM at reflux temperature in the absence of TrBr. All compounds formed are represented by the formula XnYm, where X = Tr, Ph or Cl, Y = Tr, Ph or Cl, n and m are the numbers 1 or 0. | ||
The outcome of the reaction at room temperature with benzene and toluene is the same as in THF, but is different for DCM. The main product is always [4]−, but for DCM the MALDI-TOF-MS spectra show some minor peaks corresponding to mono ([Cl1-1]−), dichloro ([Cl2-1]−), chlorotrityl cobaltabisdicarbollide ([TrCl-1]−) and [5]−, which is the dominant product. Conversely, the results are different under refluxing conditions. In refluxing toluene the reaction does not take place, but very small peaks appeared in the MALDI-TOF-MS spectra in addition to that of [1]−. As could be deduced from the reaction of Tr+ and [1]− in the presence of 10 eq. of benzene, in neat benzene as a solvent, only benzene derivatives of cobaltabisdicarbollide are produced, which suggests that Tr+ indeed behaves as a hydride scavenger. The MALDI-TOF-MS spectrum displays two major peaks at 399 and 476 m/z corresponding to [Ph-1]− and [Ph2-1]−, respectively (Fig. 3b). The reaction in refluxing DCM shows the same three major peaks in the MALDI-TOF spectra (Fig. 3f) as at room temperature, but now the major peak is due to [Cl2-1]−. In (c) and (f), the same conditions as for (a) and (b), on the one side, and (d) and (e) on the other are applied, but for the absence of TrBr. In (c) and (f), the MALDI-TOF-MS spectrum is the same at room temperature and under refluxing conditions. As seen, no reaction occurred as only the peak due to [1]− is found. These results are quite conclusive to understand the role of TrBr in these B–C coupling processes. The reaction in PhH explains that TrBr indeed acts as a hydride scavenger, and given that the electrophilic attack by the boronium is very fast it attacks one of the aryl groups in Ph3C–H. However, under refluxing conditions the molecules move much quicker than at room T and under these conditions the attack occurs on the more available arenes, which in this case is the benzene solvent.
Conclusions
Herein we report for the first time a fast procedure to form B–C bonds in metallacarboranes without the use of any catalysts (Lewis acid or Pd) and iodometallacarboranes. This has been proved with benzyl stabilized carbocations; three or two aryl groups are preferable to one. In these cases, the reaction is successful with high to very high yields although a mixture of isomers (ortho- and meta-) has been detected. Noticeable is that they are ortho- and meta- and not ortho- and para- as in the classical electrophilic aromatic substitution due to the very high velocity of the reaction. On the other hand, less stable carbocations such as allyl or alkyl that do not have a “built in” nucleophile do not lead to the target B–C bond formation. The high velocity of the reaction permits us to obtain distinct products depending on the solvent utilized, the temperature applied and the existence of one arene reagent in the reaction medium. This B–C bond coupling reaction opens the way to rich positive derivatization of the cobaltabisdicarbollide and probably other sandwich metallacarboranes in a simple and fast reaction, which needs to be further developed. It is a step ahead of the Kumada and Heck reactions, although they can complementarily skip the need for [Pd] catalysts, Grignard reagents and metallacarborane's iodo derivatives. The fact that it works with just generated carbocations is a great advantage, as the possibilities of available reagents are very high.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Spanish Ministerio de Economía y Competitividad (CTQ2016-75150-R), the Generalitat de Catalunya (2017SGR1720) and European Union's Horizon 2020 Marie Skłodowska-Curie grant agreement MSCA-IF-2016-751587. A. B. Buades is enrolled in the PhD program of UAB.Notes and references
- (a) R. N. Grimes, Carboranes, US, 3rd edn, 2016 Search PubMed; (b) M. Corsini, F. F. de Biani and P. Zanello, Coord. Chem. Rev., 2006, 250, 1351–1372 CrossRef CAS; (c) R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed.
- (a) D. Olid, R. Nuñez, C. Viñas and F. Teixidor, Chem. Soc. Rev., 2013, 42, 3318–3336 RSC; (b) B. Grüner, V. Šícha, D. Hnyk, M. G. S. Londesborough and I. Císařová, Inorg. Chem., 2015, 54, 3148–3158 CrossRef; (c) A. B. Buades, V. S. Arderiu, D. Olid-Britos, C. Viñas, R. Sillanpää, M. Haukka, X. Fontrodona, M. Paradinas, C. Ocal and F. Teixidor, J. Am. Chem. Soc., 2018, 140, 2957–2970 CrossRef CAS; (d) I. B. Sivaev, M. Y. Stogniy, S. A. Anufriev, M. V. Zakharova and V. I. Bregadze, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 192–196 CrossRef CAS; (e) S. A. Anufriev, S. A. Erokhina, K. Y. Suponitsky, I. A. Godovikov, O. A. Filippov, F. Fabrizi de Biani, M. Corsini, A. O. Chizhov and I. B. Sivaev, Eur. J. Inorg. Chem., 2017, 4444–4451 CrossRef CAS; (f) A. V. Shmal'ko, S. A. Anufriev, A. A. Anisimov, M. Y. Stogniy, I. B. Sivaev and V. I. Bregadze, Russ. Chem. Bull., 2019, 68, 1239–1247 CrossRef.
- (a) X. Zhang and H. Yan, Coord. Chem. Rev., 2019, 378, 466–482 CrossRef CAS; (b) X. Wu, J. Guo, W. Jia, J. Zhao, D. Jia and H. Shan, Dyes Pigm., 2019, 162, 855–862 CrossRef CAS; (c) C. Tang, J. Zhang, J. Zhang and Z. Xie, J. Am. Chem. Soc., 2018, 140, 16423–16427 CrossRef CAS; (d) K. Z. Kabytaev, A. V. Safronov, S. S. Jalisatgi and M. F. Hawthorne, J. Organomet. Chem., 2014, 749, 106–108 CrossRef CAS; (e) I. D. Kosenko, N. V. Dudarova, I. V. Ananyev, V. I. Bregadze and A. A. Semioshkin, Russ. Chem. Bull., 2018, 67, 500–503 CrossRef CAS.
- (a) I. Rojo, F. Teixidor, R. Kivekäs, R. Sillanpää and C. Viñas, J. Am. Chem. Soc., 2003, 125, 14720–14721 CrossRef CAS PubMed; (b) P. Farràs, D. Olid-Britos, C. Viñas and F. Teixidor, Eur. J. Inorg. Chem., 2011, 2525–2532 CrossRef; (c) V. S. Arderiu, C. Viñas and F. Teixidor, J. Organomet. Chem., 2015, 798, 160–164 CrossRef CAS; (d) D. Olid, C. Viñas and F. Teixidor, Chem. – Eur. J., 2012, 18, 12936–12940 CrossRef CAS PubMed.
- (a) J. Plesek and S. Hermanek, Collect. Czech. Chem. Commun., 1978, 43, 1325 CrossRef CAS; (b) A. Franken, J. Plesek and C. Nachtigal, Collect. Czech. Chem. Commun., 1997, 62, 746 CrossRef CAS; (c) M. D. Mortimer, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1996, 35, 5750–5751 CrossRef CAS; (d) I. Rojo, F. Teixidor, C. Viñas, R. Kivekäs and R. Sillanpää, Chem. – Eur. J., 2003, 9, 4311–4323 CrossRef CAS; (e) I. P. Beletskaya, V. I. Bregadze, V. A. Ivushkin, P. V. Petrovskii, I. B. Sivaev, S. Sjöberg and G. G. Zhigareva, J. Organomet. Chem., 2004, 689, 2920–2929 CrossRef CAS; (f) J. Francis, C. Jones and M. Hawthorne, J. Am. Chem. Soc., 1972, 94, 4878–4881 CrossRef CAS.
- (a) I. B. Sivaev, A. A. Semioshkin, B. Brellochs, S. Sjöberg and V. I. Bregadze, Polyhedron, 2000, 19, 627–632 CrossRef CAS; (b) T. Peymann, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 2000, 39, 1163–1170 CrossRef CAS; (c) M. F. Hawthorne, D. C. Young, T. D. Andrews, D. V. Howe, R. L. Pilling, A. D. Pitts, M. Reintjes, L. F. Warren Jr. and P. A. Wegner, J. Am. Chem. Soc., 1968, 90, 879–896 CrossRef CAS; (d) B. Grüner, J. Plešek, J. Báča, I. Císařová, J. F. Dozol, H. Rouquette, C. Viñas, P. Selucký and J. Rais, New J. Chem., 2002, 26, 1519–1527 RSC; (e) I. Rojo, F. Teixidor, R. Kivekäs, R. Sillanpää and C. Viñas, Organometallics, 2003, 22, 4642–4646 CrossRef CAS.
- (a) M. Lupu, A. Zaulet, F. Teixidor, E. Ruiz and C. Viñas, Chem. – Eur. J., 2015, 21, 6888–6897 CrossRef CAS PubMed; (b) X. Zhang, H. Zheng, J. Li, F. Xu, J. Zhao and H. Yan, J. Am. Chem. Soc., 2017, 139, 14511–14517 CrossRef CAS PubMed.
- (a) V. I. Bregadze, I. D. Kosenko, I. A. Lobanova, Z. A. Starikova, I. A. Godovikov and I. B. Sivaev, Organometallics, 2010, 29, 5366–5372 CrossRef CAS; (b) I. D. Kosenko, I. A. Lobanova, I. A. Godovikov, Z. A. Starikova, I. B. Sivaev and V. I. Bregadze, J. Organomet. Chem., 2012, 721, 70–77 CrossRef.
- P. González-Cardoso, A.-I. Stoica, P. Farràs, A. Pepiol, C. Viñas and F. Teixidor, Chem. – Eur. J., 2010, 16, 6660–6665 CrossRef PubMed.
- (a) C. Schade and H. Mayr, Makromol. Chem., Rapid Commun., 1988, 9, 477–482 CrossRef CAS; (b) Y. N. Smirnov, V. Volkov, E. Oleinik, B. Komarov, B. Rozenberg and N. Yenikolopyan, Polym. Sci. U.S.S.R., 1974, 16, 846–854 CrossRef; (c) Y. Xiong, T. Szilvási, S. Yao, G. Tan and M. Driess, J. Am. Chem. Soc., 2014, 136, 11300–11303 CrossRef CAS PubMed; (d) N. J. Patmore, M. J. Ingleson, M. F. Mahon and A. S. Weller, Dalton Trans., 2003, 14, 2894–2904 RSC; (e) Z. Xie, T. Jelinek, R. Bau and C. A. Reed, J. Am. Chem. Soc., 1994, 116, 1907–1913 CrossRef CAS.
- (a) E. J. Juárez-Pérez, R. Núñez, C. Viñas, R. Sillanpää and F. Teixidor, Eur. J. Inorg. Chem., 2010, 2385–2392 CrossRef; (b) P. Farràs, A. D. Musteti, I. Rojo, C. Viñas, F. Teixidor and M. E. Light, Inorg. Chem., 2014, 53, 5803–5809 CrossRef.
- V. M. Retivov, E. Y. Matveev, M. V. Lisovskiy, G. A. Razgonyaeva, L. I. Ochertyanova, K. Y. Zhizhin and N. T. Kuznetsov, Russ. Chem. Bull., 2010, 59, 550–555 CrossRef CAS.
- J. F. Kleinsasser, S. P. Fisher, F. S. Tham and V. Lavallo, Eur. J. Inorg. Chem., 2017, 4417–4419 CrossRef CAS.
- D. Sharp and N. Sheppard, J. Chem. Soc., 1957, 674–682 RSC.
- P. L. Urban, Philos. Trans. R. Soc., A, 2016, 374, 20150382 CrossRef PubMed.
- S. P. Fisher, S. G. McArthur, V. Tej, S. E. Lee, A. L. Chan, I. Banda, A. Gregory, K. Berkley, C. Tsay, A. L. Rheingold, G. Guisado-Barrios and V. Lavallo, J. Am. Chem. Soc., 2020, 142, 251–256 CrossRef CAS PubMed.
- (a) V. I. Bregadze, S. V. Timofeev, I. B. Sivaev and I. A. Lobanova, Russ. Chem. Rev., 2004, 73, 433 CrossRef CAS; (b) D. Astruc, Organometallic Chemistry and Catalysis, Springer, Berlin, 2007 Search PubMed; (c) H. Mayr, J. Ammer, M. Baidya, B. Maji, T. A. Nigst, A. R. Ofial and T. Singer, J. Am. Chem. Soc., 2015, 137, 2580–2599 CrossRef CAS PubMed; (d) L. Cabrera, G. C. Welch, J. D. Masuda, P. Wei and D. W. Stephan, Inorg. Chim. Acta, 2006, 359, 3066–3071 CrossRef CAS; (e) V. Šícha, J. Plešek, M. Kvíčalová, I. Císařová and B. Grüner, Dalton Trans., 2009, 5, 851–860 RSC.
- G. Olah and S. Kobayashi, J. Am. Chem. Soc., 1971, 93, 6964–6967 CrossRef CAS.
- One of the reviewers is convinced that the observed reaction follows the recently reported electrophilic arylation pathway.16.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C9DT04695J |
| This journal is © The Royal Society of Chemistry 2020 |