
Novel chiral aggregation induced emission molecules: self-assembly, circularly polarized luminescence and copper(II) ion detection†
Guangxi
Huang
 ab,
Rongsen
Wen
a,
Zhiming
Wang
b,
Bing Shi
Li
*a and
Ben Zhong
Tang
*bc
ab,
Rongsen
Wen
a,
Zhiming
Wang
b,
Bing Shi
Li
*a and
Ben Zhong
Tang
*bc
aKey Laboratory of New Lithium-Ion Battery and Mesoporous Material, Department of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen 518060, China. E-mail: phbingsl@szu.edu.cn; Fax: +86-755-26536141; Tel: +86-755-26558094
bHong Kong University of Science & Technology (HKUST) – Shenzhen Research Institute, No. 9 Yuexing 1st RD, South Area, Hi-tech Park, Nanshan, Shenzhen 518057, China
cDepartment of Chemistry, Institute for Advanced Study, Institute of Molecular Functional Materials, and State Key laboratory of Molecular Neuroscience, The Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
First published on 14th August 2018
Abstract
This work provides a feasible strategy for future design of CPL materials and ion sensors. By introducing chiral alanine to hydrazone derivatives, two novel chiral molecules 1 and 2 with aggregation-induced emission enhanced (AIEE) characteristics were designed and synthesized. Molecule 1 had the two phenyl groups connecting to hydrazone covalently bonded, so its fluorophore is more conjugated than that of 2. This subtle structural variation between 1 and 2 had a considerable influence on their photophysical properties, chiral optical properties and self-assembly behaviors. Both kinds of molecules could self-assemble into long helical nanofibers with lengths up to several microns upon aggregation. They both possessed AIEE properties, but 1 emitted red fluorescence and 2 had yellow fluorescence upon aggregation. They also had aggregation induced circular dichroism (CD) and circularly polarized luminescence (CPL) properties, but 1 had better performance because its fluorophore was more conjugated than that of 2. However, only 2 had a specific interaction with Cu2+ and it showed a highly selective and sensitive “turn-off” fluorescence response to Cu2+.
Introduction
Supramolecular self-assembly based on non-covalent interactions is a simple and efficient strategy to fabricate various ordered nanostructures with well-defined shapes and dimensions. Helices, twists or springs with chirality, a unique category of nanostructures, have drawn considerable attention due to their fascinating features and potential applications in optoelectronic devices, biosensors and catalysis.1 Natural molecules, such as proteins, self-assemble into helices due to the chiral codes embedded in their amino acid components. This strategy was also applicable to synthetic molecules. When naturally occurring chiral building blocks, such as saccharides, amino acids, cholesterol or chiral alkyl chains, are hybridized with synthetic scaffolds, their chiral offspring is also capable of self-assembling into helical architectures. The parent molecules can provide judicious patterns of non-covalent interactions, such as hydrogen bonds, van der Waals interactions or hydrophobic interactions for the self-assembly and subtle changes of the chemical structures may lead to the formation of diverse assemblies. Meanwhile, the novel functions of the parent molecules might also be inherited by their offspring. When the parent molecules carry fluorophores, the helical architectures might show differential emission of left- versus right-circularly polarized light, namely circularly polarized luminescence (CPL) properties. Materials with CPL properties have attracted increasing interest due to their potential application as display devices, optical storage devices, probes and sensors.2In 2001, Tang's group reported the intense fluorescence emission of silole molecules upon aggregation, which is the well-known aggregation-induced emission (AIE) phenomenon.3 Since most common applications of luminescent materials are in the solid state, AIE fluorophores are of great importance to design CPL materials as they do not have to suffer from aggregation caused quenching (ACQ). By melding chirality with AIE fluorophores, a group of novel materials with self-assembly ability and impressive CPL properties have been obtained in recent years.4 For example, Liu et al. synthesized a silole derivative with two mannose-containing side-chains, which showed extraordinarily high gem-factors ranging from −0.08 to −0.32 depending on the experimental conditions.5 Our group successfully utilized amino acid pendants to guide the AIE fluorophore siloles, tetraphenylethylene (TPE) and phenanthro[9,10]imidazole, to self-assemble into blue helical luminescent fibers with excellent CPL properties.6 In spite of the progress in the exploration of chiral AIE molecules, the AIE fluorophores and their luminescent color remain to be explored. The influence of chemical structures on the optical structures and self-assembling behaviors also needs to be further explored. The underlying mechanism that governs the optical properties and self-assembling process remains to be revealed with more systematic work.
In this work, we choose a Schiff base group as the luminophore and modify it with the chiral component alanine, and synthesize two chiral molecules 1 and 2 (Scheme 1). Such modification aims to endow the molecules with the properties of AIE, CD, CPL and helical self-assembly. Meanwhile, salicylaldehyde Schiff base compounds have the ability to act as fluorescent probes to selectively recognize metal ions and our product molecules are also expected to have specific interactions with target metal ions and can serve as fluorescence sensors for metal ions. Molecule 1 differs from molecule 2 in that its two phenyl groups of hydrazone are locked with a single bond, so that molecule 1 has a more planar conformation and enhanced conjugation than molecule 2. Though the difference in their chemical structure is subtle, it causes a dramatic difference in their photophysical properties, chiral optical properties and self-assembly behaviors. We have carried out a systematic work to explore the influence of the chemical structures on their optical properties and self-assembling behavior. Both molecules have AIEE characteristics, but molecule 1 emits red fluorescence, which is in sharp contrast to the yellow fluorescence emitted by molecule 2. Both molecules have the ability to self-assemble into nanohelical fibers, but molecule 1 has much better CD and CPL performance than molecule 2. Thus molecule 1 is an ideal candidate for making display devices. Because of the subtle difference in their chemical structure, the two molecules have diverse responses to metal ions: molecule 1 is silent to all metal ions, but molecule 2 has a specific fluorescence response to Cu2+. The SEM image further suggests that Cu2+ interacts with molecule 2 and causes dramatic changes in its self-assemblies. The self-assembly of molecule 2 was transformed from helical fibers into irregular polyhedral particles in the presence of Cu2+, corresponding to a switching off of the fluorescence. This property makes molecule 2 an ideal candidate for fabricating a highly selective and sensitive “turn-off” fluorescent chemosensor for Cu2+ detection. Detection of excessive Cu2+ is of great importance as it is biologically toxic and closely related to some severe diseases such as Wilson's and Alzheimer's disease.7 Fast, convenient and low cost fluorescent chemosensors with high sensitivity and selectivity are always in high demand.
 | ||
| Scheme 1 Molecular structures and synthetic routes of compounds 1–3. | ||
Our systematic explorations help in revealing how a subtle change in the chemical structure of molecules can lead to their diverse optical properties and self-assembly behaviors. Hopefully our work can help enrich our understanding of the influence of chemical structures on the properties of molecules and improve the design of molecular structures with desirable properties.
Results and discussion
Synthesis and characterization
The molecular structures and synthetic routes of compounds 1 and 2 are shown in Scheme 1. Compound 3 was prepared by a Sonogashira reaction between 5-ethynylsalicylaldehyde and (R)-methyl 2-(4-bromobenzamido)propanoate. 1 and 2 were obtained by the condensation reaction between hydrazone and compound 3 at r.t. After filtration, 1 and 2 were of high purity, and did not need further purification. Their structures were characterized and confirmed by NMR, high resolution mass spectroscopy (HRMS) and elemental analysis (Experimental section and Fig. S8–S16, ESI†).UV absorption and CD spectra
The absorption spectra of compounds 1 and 2 in THF solution are shown in Fig. 1. Molecule 1 exhibited two characteristic absorption peaks at 327 nm and 395 nm (Fig. 1a), corresponding to the absorption of the phenyl group connected to the Ala attachment and the conjugated hydrazone plane, respectively. For molecule 2, which has two phenyl groups of the hydrazone plane in the free state, the absorption peaks blue shifted to 313 nm and 380 nm (Fig. 1b) due to a decrease in conjugation compared with that of molecule 1. After the evaporation of solvent, the absorption spectra of both 1 and 2 had a red shift in the film state, suggesting that aggregates were formed by the molecules. CD spectra of 1 and 2 in solution and in the film state were then measured. Both the compounds were CD silent in THF solution because they were well dissolved and randomly orientated in the good solvent, and CD absorptions of molecules likely counterpart each other and generated no signal (Fig. 1a and b). When they were cast into films, bisignate Cotton effects (CEs) were observed, showing typical aggregation induced circular dichroism (AICD).6 Molecule 1 showed negative CEs at 288 nm and strong positive CEs at 342 nm, while 2 showed negative CEs at 272 nm and 422 nm and positive CEs at 343 nm. The intensity of the CD spectrum of 2 was weaker than that of 1. Because an alanine unit is CD-silent at wavelengths longer than 270 nm,8 so the new peaks must stem from the absorption of the fluorophores, implying the successful formation of the self-assembly helical nanofibers in the aggregate state.6 | ||
| Fig. 1 UV and CD spectra of 1 (a) and 2 (b) in THF solution (10 μM) and in the film state. Inset: HOMO and LUMO energy levels of 1 (a) and 2 (b). | ||
To better understand the absorption spectra of 1 and 2, density functional theory calculations were performed. The HOMO and LUMO energy levels of 1 and 2 are inserted in Fig. 1. The HOMO energy level of 1 is dominated by the orbitals from the alkyne group and the adjacent phenyl rings, while the orbitals from the corresponding hydrazone moiety and adjacent phenyl ring contribute mainly to the LUMO level (Fig. 1a). For 2, the HOMO and LUMO energy levels are similar to that of 1 (Fig. 1b). The energy band gap (ΔE) was determined to be 3.13 eV and 3.38 eV for 1 and 2, respectively. ΔE of 1 is smaller than that of 2, revealing that 1 has more enhanced conjugation than that of 2 because of the joining of the two phenyl groups. Hence, the absorption of 1 occurred at a longer wavelength than that of 2, which is in agreement with the aforementioned absorption spectra. These results reveal that the minor structural change of the fluorophore has a considerable influence on the absorption spectra of the molecules.
Fluorescence and CPL spectra
The fluorescence spectra of 1 and 2 were measured to check if they still possessed AIE properties after the modification with amino acid attachments. As shown in Fig. 2, both 1 and 2 had weak emission in THF and the emission became slightly decreased with the addition of water. For 1, when the water content reached the turning point (fw = 60%), the emission intensity began to become gradually boosted at a wavelength of 635 nm (Fig. 2a and b). At a fw of 99%, it reached the highest intensity which is four times higher than that in THF solution and the solution showed red color emission under UV light (Fig. 2b). For 2, the turning point lagged behind that of molecule 1 (fw = 80%) and after the turning point the fluorescence intensity started to become abruptly enhanced at a wavelength of 586 nm. When the fw increased to 99%, the fluorescence intensity was four times that in pure THF and the solution emitted a bright yellow color under UV light (Fig. 2c and d). In THF/water mixtures, water served as the poor solvent for the molecules to induce their aggregation and increase the emission intensity as the nonradiative intramolecular decay process, including C–N and N–N single bond rotation, was restricted. Thus both molecules exhibited AIEE properties at higher water content.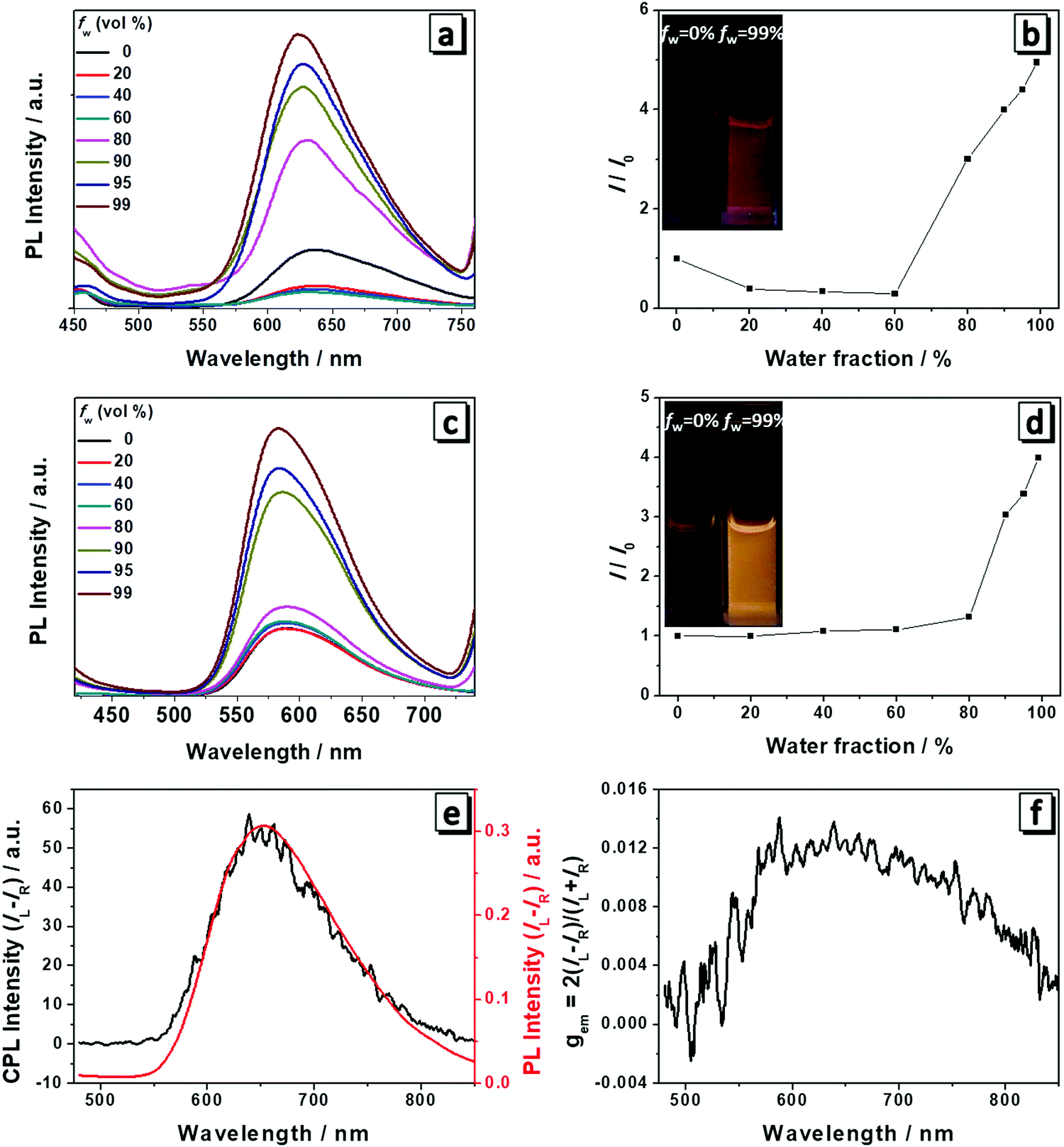 | ||
| Fig. 2 Fluorescence spectra of 1 (a) and 2 (c) in THF/water mixtures with different water fractions fw. Concentration: 10 μM. Plots of I/I0versus water fractions of 1 (b) and 2 (d), where I0 is the emission intensity at 625 nm and 586 nm in THF, respectively. Inset: Fluorescence images at fw = 0% and 99%. Plots of CPL and PL (e) and CPL dissymmetry factor gem (f) of the cast film of 1 formed by evaporation of its THF solution. | ||
Because the cast films of 1 and 2 have obvious fluorescence emission and CD absorption as well, it is natural to expect that both molecules are also CPL-active. A Jasco CPL-300 was employed to investigate the CPL and the gem value was directly obtained from the equipment. gem is the CPL dissymmetry factor, which is used to evaluate the magnitude of CPL. It is defined as gem = 2(IL − IR)/(IL + IR), where IL and IR denote the emission intensities of left- and right-handed circularly polarized light, respectively. We then prepared the cast films of both molecules and they emitted red and yellow fluorescence under a fluorescence microscope (Fig. S1, ESI†). Plots of the CPL, PL and gem of 1 in the film state are presented in Fig. 2e and f. Molecule 1 displayed an intense CPL signal and the maximum emission peak was at a wavelength of 650 nm, which corresponded well with its fluorescence spectrum, implying that the fluorescent aggregates had dominant chirality originated from the self-assembly helical nanofibers.6 The calculated value of gem was 0.013 for 1 at a wavelength of 650 nm and this value is quite impressive considering that the typical gem values for most CPL materials based on chiral organic fluorescence molecules are in the range from 10−5 to 10−2. Hence, 1 displayed excellent CPL performance. In contrast, 2 only showed a very weak CPL signal and low gem due to the subtle difference in the chemical structure (Fig. S2, ESI†).
Chiral self-assembly
Molecules 1 and 2 exhibited different CD and CPL properties in the aggregation state, so their self-assembling structures were crucial to their optical properties. We then investigated their self-assembling structures formed upon the evaporation of its good solvent and addition of a poor solvent, respectively. Scanning electron microscopy (SEM) images of 1 in THF (100 μM) upon solvent evaporation showed the formation of helical fibers with a width of about 50–70 nm and a helical pitch of 150–200 nm (Fig. 3a). Both molecules are small, but they can form helical fibers with lengths longer than 2 μm, revealing that the helical fibers were formed by hierarchical self-assembly. The helical fibers were left-handed and thinner fibers further interlaced with each other to form thicker ones, implying that the association of helical fibers was hierarchical, as indicated with an arrow in Fig. 3a. With the increase of water content, a large number of left-handed helical fibers were observed with lengths up to several micrometers (Fig. 3b–d), demonstrating that the chirality of the molecules has been amplified in the supramolecular self-assembly. When the content of water increased to 90%, the helical fibers got much thicker and twisted more easily with each other. From the labeled area, left-handed rotation of the helical fibers can still be discerned (Fig. 3d).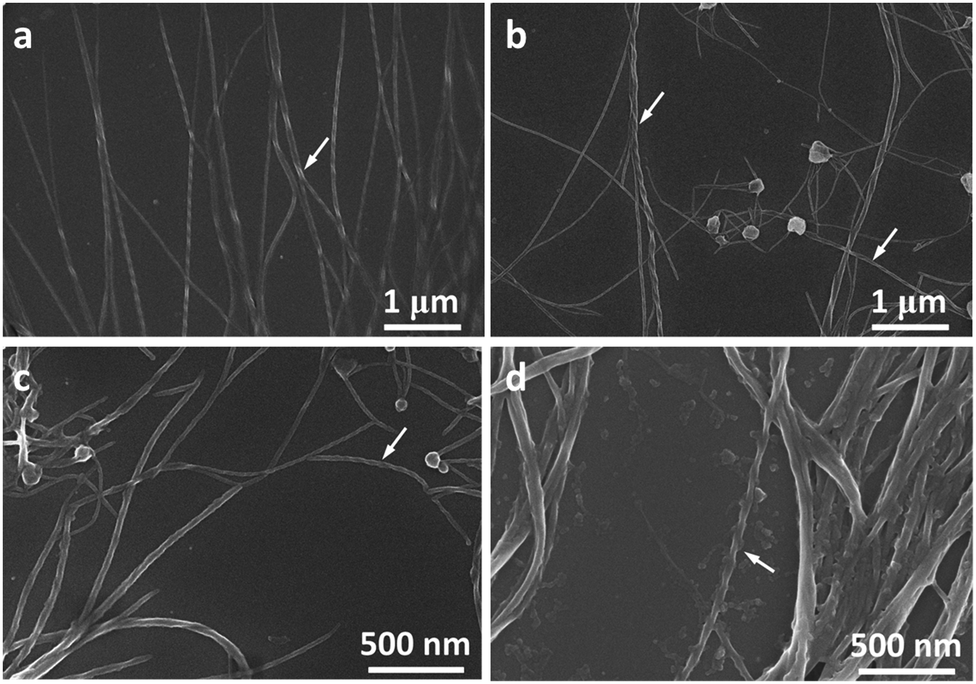 | ||
| Fig. 3 SEM images of 1 at high magnification obtained from the evaporation of (a) THF, (b) THF/H2O 5/5, (c) THF/H2O 2/8 and (d) THF/H2O 1/9. Solution concentration: 100 μM. Arrows in the images point to the areas where helices are most obviously discerned. | ||
For molecule 2, the self-assembly of molecules was investigated upon the evaporation of THF mixture at a concentration of 10 μM (Fig. 4a–d) and 100 μM (Fig. 4e and f), respectively. Molecule 2 self-assembled into helical fibers upon the evaporation of THF, as shown in Fig. 4a. The helical fibers have obvious left-handed rotation with width and helical pitch of ∼50 and 100 nm, respectively. When the water fraction was 50%, helical fibers got thicker and helical pitch became longer (Fig. 4b). With a further increase of water content, the helical fibers became much thicker and the edge of the helical fibers also became obscure, but the left-handed rotation could still be discerned (Fig. 4c). When the water content increased to 90%, some of the helical fibers were twisted into small knots which were decorated along the helical fibers, like pearl necklaces (Fig. 4d). When the concentration of the solution was increased to 100 μM, the morphologies of the self-assembly also changed. After the evaporation of THF solution of 2, some fibers showed distinguishable left-handedness of helical segments (labeled with arrows in Fig. 4e) and some had the appearance of ribbons without obvious helicity. Accordingly, the helical pitches varied from several tens of nanometers to more than 200 nm and they were not as uniform as that formed at lower concentration. At a water fraction of 50%, many cumulated helical fibers with left-handedness were observed (Fig. 4f). When the water content was increased to 80% and 90%, most of the assemblies had the appearance more like helical ribbons (Fig. 4g and h). The helical turns of the pitches became even more loosely stacked than those formed at lower water content.
 | ||
| Fig. 4 SEM images of 2 obtained from the evaporation of different THF/H2O mixtures of concentration 10 μM (a–d) and 100 μM (e–h), respectively. Arrows in the images point to the areas where helices are most obviously discerned. | ||
Optical response toward Cu2+
Both molecules have a salicylaldehyde Schiff base scaffold and it is well-known that Schiff base compounds can be used as a fluorescent probe to selectively bind to metal ions. Among various metal ions, Cu2+ has a strong thermodynamic affinity to ligands possessing N and O atoms. Once Cu2+ is bound to the ligand, it can strongly quench the fluorescence of the ligand through photo-induced metal-to-fluorophore, electron or energy transfer mechanism.9 We then investigate whether the two molecules have specific interactions with metal ions. Firstly, we measured the spectral change of 1 and 2 towards 1 equiv. of various metal ions (Li+, Mg2+, Ca2+, Co2+,Cd2+, Mn2+, Cu2+, Zn2+, Ni2+, Al3+, Cr3+, Fe2+, Fe3+, Ag+, Pb2+) in THF/H2O (1/9, v/v). Each type of metal ion was added to the solution and then the absorption and fluorescence spectra were recorded. Though molecules 1 and 2 have quite similar chemical structures, molecule 1 had no obvious changes in the absorption and fluorescence response with the addition of metal ions (Fig. S3, ESI†). Only molecule 2 has responses to the metals (Fig. 5a and b). As depicted in Fig. 5b, Cu2+ induced a dramatic change in the absorption spectrum in that the solution changed from colorless to yellow, which was easily distinguished even with the naked eye. In the presence of Al3+, Fe2+ or Fe3+, there was a slight increase in the absorption intensity around 380 nm and other metal ions made no changes to the absorption of molecule 2. Therefore, 2 showed a rapid, naked eye recognizable specific detection ability in the case of Cu2+. The fluorescence response of 2 towards various metal ions was also studied. It was found that the fluorescence emission of 2 at 586 nm was quenched by 1 equiv. of Cu2+ (Fig. 5b). However, no obvious changes occurred in the presence of other metal ions except that Al3+, Fe2+ or Fe3+ caused a slight fluorescence enhancement, which was in accordance with their absorption spectrum change. The fluorescence images showed that only Cu2+ induced dramatic fluorescence quenching of molecule 2, demonstrating its high specificity to copper ions. The selectivity of 2 toward Cu2+ in the presence of competing metal ions was also measured. As shown in Fig. 5b, the solutions of 2 in the presence of different metal ions (100 equiv. of Li+, Mg2+ or Ca2+, 10 equiv. of Co2+, Cd2+, Mn2+, Cu2+, Zn2+, Ni2+, Cr3+, Ag+ or Pb2+, or 1.0 equiv. of Al3+, Fe2+ or Fe3+) all exhibited an efficient quenching of emission after addition of 1 equiv. of Cu2+, which demonstrated the high anti-interference of 2 toward Cu2+.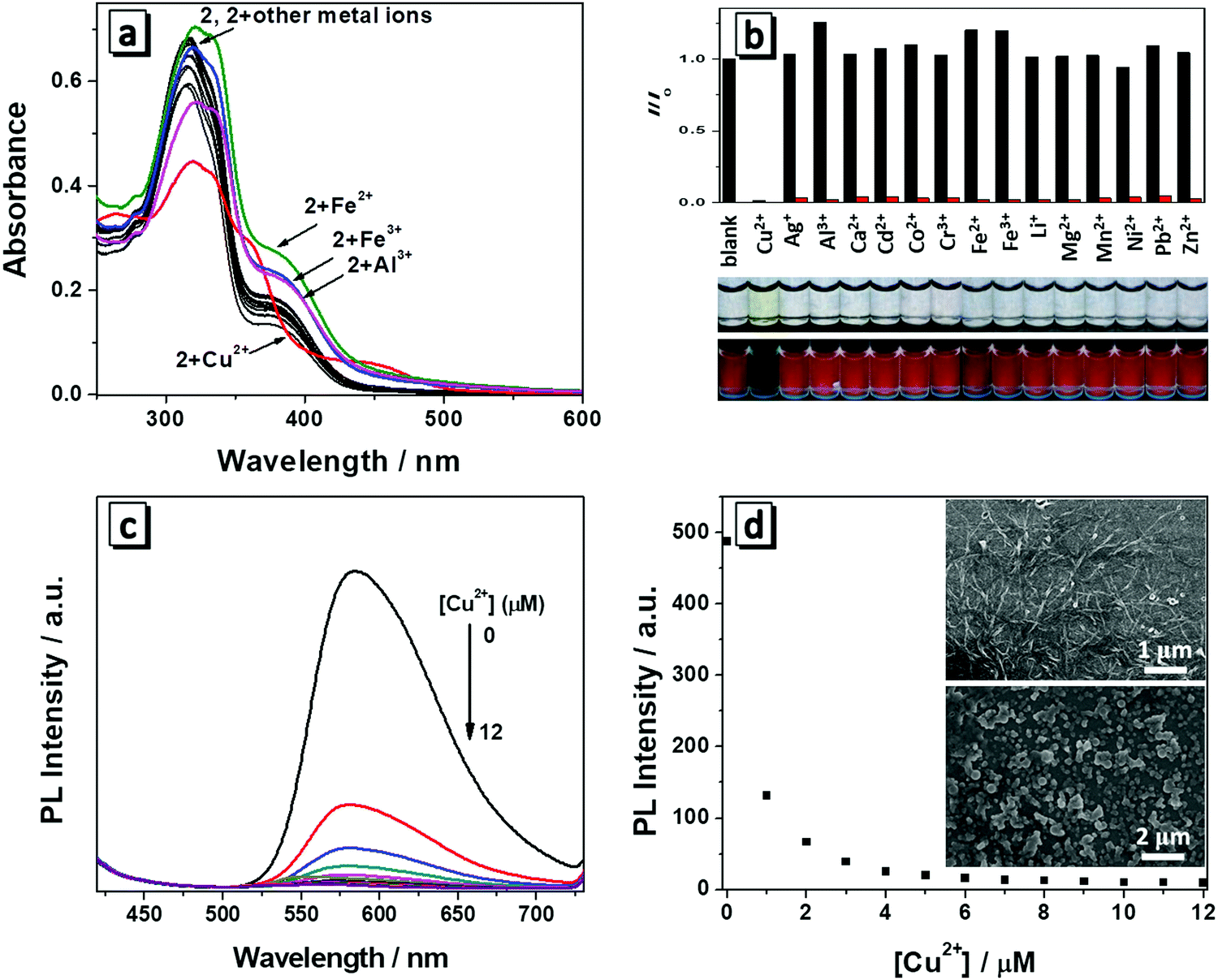 | ||
| Fig. 5 (a) UV spectra of 2 (10 μM) upon the addition of various metal cations (10 μM) in THF/H2O (1/9, v/v); (b) variation of the fluorescence intensity at 586 nm (λex = 380 nm) of 2 (10 μM) after addition of various metal cations. Black bars represent the addition of different metal ions (10 μM) to the solution of 2, whereas the red bars represent the subsequent addition of Cu2+ (10 μM) to the solution containing a specific metal ion (the concentration of Li+, Mg2+ and Ca2+ was 1 mM, Al3+, Fe2+ and Fe3+ was 10 μM, other transition metal ions was 100 μM); the photographs show 2 (10 μM) upon addition of 1 equiv. of various metal ions in THF/H2O (1/9, v/v) taken under natural light (upper) and UV light (lower). (c) Fluorescence titration spectra of 2 (10 μM) with 0–12 μM Cu2+; (d) the plot of fluorescence intensity at 586 nm versus Cu2+ concentrations; insets: SEM images of 2 (10 μM) in the absence (upper) and presence (lower) of 1 equiv. of Cu2+ in THF/H2O (1/9, v/v). | ||
To further investigate the interaction between 2 and Cu2+, a titration process with increasing concentration of Cu2+ (0–1.2 equiv.) was performed and the absorption and fluorescent spectra were monitored. It can be seen that the absorption maxima at 380 nm disappeared and two new absorption bands centred around 365 nm and 445 nm appeared with gradual addition of Cu2+. In the titration process, multiple isosbestic points around 278, 345, 380 and 410 nm were observed, which suggested that a new species was generated due to the interaction between 2 and Cu2+ (Fig. S4, ESI†). Correspondingly, the fluorescence emission intensity of 2 had a significant decrease with gradual addition of Cu2+ (Fig. 5c), indicating the new complex formation and confirming their potential application for Cu2+ detection in aqueous solution. When the concentration of Cu2+ was increased to 10 μM, the fluorescence of 2 was almost totally quenched (Fig. 5d). The emission intensity of 2 was linearly proportional to the concentration of Cu2+ ranging from 0 to 0.6 μM (Fig. S5, ESI†). The fitting curve of the relationship between the fluorescence emission intensity of 2 and Cu2+ concentration was y = −528x + 436.1. The detection limit was calculated to be 0.067 μM on the basis of equation DL = 3σ/m, where σ is the standard deviation of a blank signal, and m is the slope of the above fitting curve. The detection limit is much lower than the maximum allowable level of Cu2+ in drinking water according to the U.S. Environmental Protection Agency (20 μM).
In order to understand the interaction and binding nature between 2 and Cu2+, Job's plot and mass spectrum were recorded. The absorbance plots at 380 nm against the molar fraction of 2 have a maximum absorbance value at [Cu2+]/[Cu2+ + 1] = 0.5, confirming that the stoichiometric ratio for the complexation of Cu2+ and 2 is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S6, ESI†). This result indicated the formation of a 1:1 complex between 2 and Cu2+. As shown in the ESI-mass spectrum of 2 and Cu2+ in Fig. S7 (ESI†), in the presence of Cu2+, the ion peak m/z found at 591.1211 corresponded to [2-H+ + Cu2+]+ (calculated: 591.1214). So the mass spectrum also confirmed that 2 had bound to Cu2+ with the stoichiometric ratio of 1:1. Based on the previous papers,10 Cu2+ likely binds to the nitrogen of the imine group and oxygen of the phenolic hydroxyl group (deprotonated form) of 2.
:1 (Fig. S6, ESI†). This result indicated the formation of a 1:1 complex between 2 and Cu2+. As shown in the ESI-mass spectrum of 2 and Cu2+ in Fig. S7 (ESI†), in the presence of Cu2+, the ion peak m/z found at 591.1211 corresponded to [2-H+ + Cu2+]+ (calculated: 591.1214). So the mass spectrum also confirmed that 2 had bound to Cu2+ with the stoichiometric ratio of 1:1. Based on the previous papers,10 Cu2+ likely binds to the nitrogen of the imine group and oxygen of the phenolic hydroxyl group (deprotonated form) of 2.
To further understand the interaction process of 2 with Cu2+, SEM was employed to examine the morphological transition of 2 (10 μM) in THF/H2O (1/9, v/v) after treatment with 1 equiv. of Cu2+. As shown in the inset in Fig. 5d, the SEM images revealed that 2 self-assembled into helical fibers prior to the addition of Cu2+. After the addition of Cu2+, only irregular polyhedral particles with size ranging from tens of nanometers to several microns were observed. Apparently, the addition of Cu2+ destructed the formation of the ordered helical self-assembly of 2. It further suggests that hydrogen bonding between molecules plays an important role in stabilizing the helical assemblies, especially the hydroxyl groups connected to imine groups make a major contribution. When they coordinates with Cu2+, hydrogen bonding between molecules will be destroyed, thus helical assemblies are disassociated.
Thus, by deliberately designing molecules with both AIE characteristics and chirality, we have revealed how a subtle change in the chemical structure of molecules can lead to their diverse optical properties and self-assembly behaviors. The structure–property relationship can be explained as follows (Scheme 2): molecule 2 has two freely rotating phenyl rings whereas these two phenyl rings are locked in molecule 1, therefore 1 has better planarity than 2. Therefore large steric hindrance exists in 2 in the process of self-assembly and leads to relatively loose and disordered packing. Consequently, molecule 2 displayed worse CD and CPL properties than 1. On the other hand, the nitrogen of the imine group and oxygen of the phenolic hydroxyl group (deprotonated form) of 2 can easily bind to Cu2+. In contrast, molecule 1 had the two phenyl groups connecting to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N locked and it did not facilitate coordination of copper ions with N and O atoms due to the steric hindrance of the planar conjugated fluorescence scaffold. Thus, molecule 2 can serve as a fluorescence sensor towards copper ions, whereas 1 cannot.
N locked and it did not facilitate coordination of copper ions with N and O atoms due to the steric hindrance of the planar conjugated fluorescence scaffold. Thus, molecule 2 can serve as a fluorescence sensor towards copper ions, whereas 1 cannot.
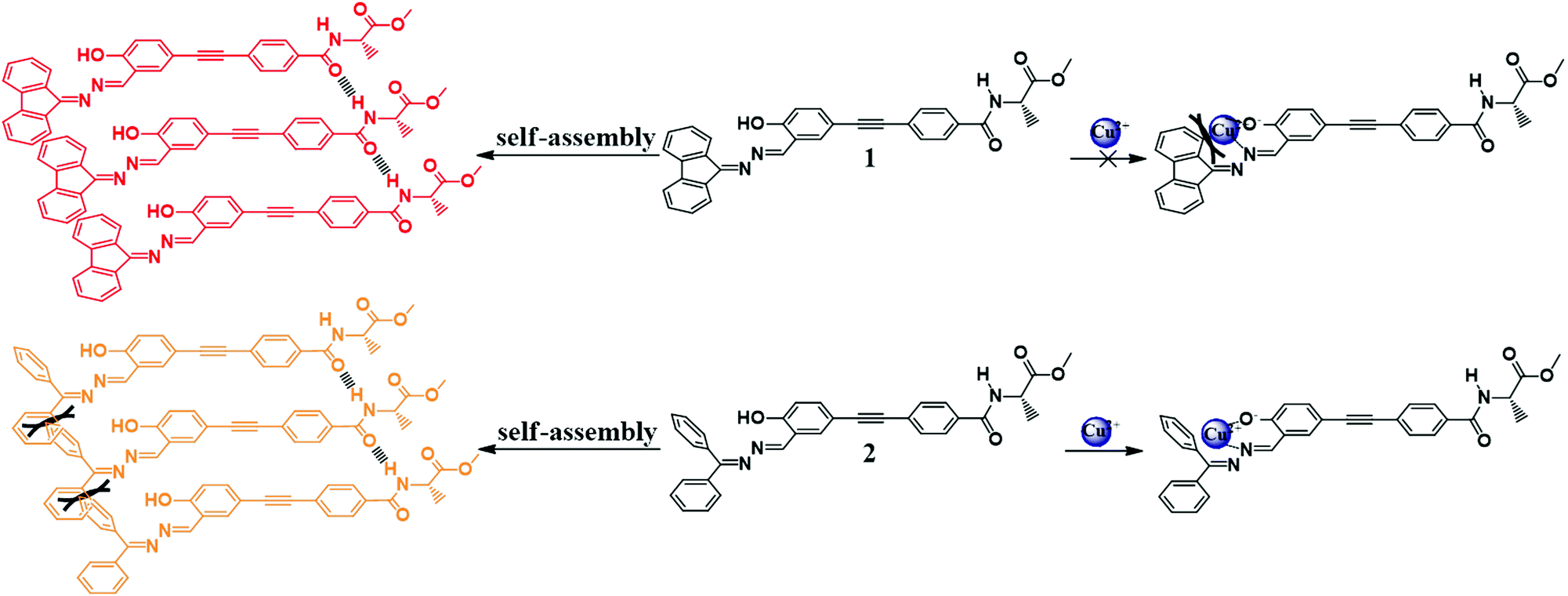 | ||
| Scheme 2 Proposed self-assembly and binding mode towards Cu2+ of 1 and 2. | ||
Conclusions
In conclusion, we have designed two novel chiral molecules 1 and 2 by introducing a chiral alanine attachment to a Schiff base AIE scaffold. Both molecules have CD, AIE, and CPL optical properties and can self-assemble into long helical fibers or ribbons. Molecule 1 displayed much better CD and CPL activity than 2 and it is likely due to the much more enhanced conjugation of the fluorophore of 1; the conjugated structure of 1 on the other hand was too bulky to allow its complexation with Cu2+; while the less conjugated molecule 2 has a specific interaction with Cu2+ and it is an ideal sensor for Cu2+ with a detection limit of 0.067 μM. Thus, this work provides important insights into the design of chiral AIE molecules with a subtle chemical structure variation to achieve multiple functions and applications in the optical field.Experimental section
Sample preparation for SEM
A solution of 1 or 2 with certain concentration was first prepared. 20 μL of the as prepared solution was then dropped onto the surface of a silicon wafer (5 × 5 mm). After evaporation of the solvent under ambient conditions, the samples were characterized by SEM.Sample preparation for CD and CPL measurements
A THF solution of 1 or 2 (100 μM) was first prepared. 1 mL of the as prepared solution was deposited dropwise onto quartz plates with a diameter of 2 cm. After evaporation of the solvent under ambient conditions, the samples were characterized by CPL.Synthesis
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21574085, 21490570 and 21490574), the National Natural Science Foundation of Guangdong Province (2016A030312002 and 2017A030313067), Fundamental Foundation of Shenzhen (JCYJ20170302143846672) and the Science and Technology Plan of Shenzhen (JCYJ20160229205601482).Notes and references
- (a) J. Jiang, Y. Meng, L. Zhang and M. Liu, J. Am. Chem. Soc., 2016, 138, 15629–15635 CrossRef PubMed; (b) L. Zhang, T. Wang, Z. Shen and M. Liu, Adv. Mater., 2016, 28, 1044–1059 CrossRef PubMed; (c) W. Zou, Y. Yan, J. Fang, Y. Yang, J. Liang, K. Deng, J. Yao and Z. Wei, J. Am. Chem. Soc., 2014, 136, 578–581 CrossRef PubMed; (d) Y. Yan, R. Wang, X. Qiu and Z. Wei, J. Am. Chem. Soc., 2010, 132, 12006–12012 CrossRef PubMed; (e) Y. Zhang, P. Chen, L. Jiang, W. Hu and M. Liu, J. Am. Chem. Soc., 2009, 131, 2756–2757 CrossRef PubMed; (f) T. Tu, W. Fang, X. Bao, X. Li and K. H. Dötz, Angew. Chem., Int. Ed., 2011, 50, 6601–6605 CrossRef PubMed.
- (a) M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 CrossRef PubMed; (b) M. Li, S. H. Li, D. Zhang, M. Cai, L. Duan, M. K. Fung and C. F. Chen, Angew. Chem., Int. Ed., 2018, 57, 2889–2893 CrossRef PubMed; (c) D. Zhao, H. He, X. Gu, L. Guo, K. S. Wong, J. W. Y. Lam and B. Z. Tang, Adv. Opt. Mater., 2016, 4, 534–539 CrossRef; (d) R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC; (e) S. Huo, P. Duan, T. Jiao, Q. Peng and M. Liu, Angew. Chem., Int. Ed., 2017, 56, 12174–12178 CrossRef PubMed; (f) J. Han, P. Duan, X. Li and M. Liu, J. Am. Chem. Soc., 2017, 139, 9783–9786 CrossRef PubMed.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef PubMed.
- R. Jesse, T. B. Zhong and W. K. Sing, Small, 2016, 12, 6495–6512 CrossRef PubMed.
- J. Liu, H. Su, L. Meng, Y. Zhao, C. Deng, J. C. Y. Ng, P. Lu, M. Faisal, J. W. Y. Lam, X. Huang, H. Wu, K. S. Wong and B. Z. Tang, Chem. Sci., 2012, 3, 2737–2747 RSC.
- (a) B. S. Li, R. Wen, S. Xue, L. Shi, Z. Tang, Z. Wang and B. Z. Tang, Mater. Chem. Front., 2017, 1, 646–653 RSC; (b) H. Li, J. Cheng, H. Deng, E. Zhao, B. Shen, J. W. Y. Lam, K. S. Wong, H. Wu, B. S. Li and B. Z. Tang, J. Mater. Chem. C, 2015, 3, 2399–2404 RSC; (c) H. Li, J. Cheng, Y. Zhao, J. W. Y. Lam, K. S. Wong, H. Wu, B. S. Li and B. Z. Tang, Mater. Horiz., 2014, 1, 518–521 RSC; (d) H. Li, S. Xue, H. Su, B. Shen, Z. Cheng, J. W. Y. Lam, K. S. Wong, H. Wu, B. S. Li and B. Z. Tang, Small, 2016, 12, 6593–6601 CrossRef PubMed; (e) H. Li, X. Zheng, H. Su, J. W. Y. Lam, K. Sing Wong, S. Xue, X. Huang, X. Huang, B. S. Li and B. Z. Tang, Sci. Rep., 2016, 6, 19277 CrossRef PubMed; (f) J. C. Y. Ng, H. Li, Q. Yuan, J. Liu, C. Liu, X. Fan, B. S. Li and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 4615–4621 RSC.
- (a) C. Vulpe, B. Levinson, S. Whitney, S. Packman and J. Gitschier, Nat. Genet., 1993, 3, 7 CrossRef PubMed; (b) D. J. Waggoner, T. B. Bartnikas and J. D. Gitlin, Neurobiol. Dis., 1999, 6, 221–230 CrossRef PubMed; (c) T. Francesco, M. Cristina, P. Marina, P. Maura and S. Carlo, Med. Res. Rev., 2010, 30, 708–749 Search PubMed.
- E. Molteni, G. Onida and G. Tiana, J. Phys. Chem. B, 2015, 119, 4803–4811 CrossRef PubMed.
- (a) Q. Wu and E. V. Anslyn, J. Am. Chem. Soc., 2004, 126, 14682–14683 CrossRef PubMed; (b) H.-H. Zeng, R. B. Thompson, B. P. Maliwal, G. R. Fones, J. W. Moffett and C. A. Fierke, Anal. Chem., 2003, 75, 6807–6812 CrossRef PubMed.
- (a) H. Wang, B. Fang, L. Zhou, D. Li, L. Kong, K. Uvdal and Z. Hu, Org. Biomol. Chem., 2018, 16, 2264–2268 RSC; (b) L. Gao, J. Q. Wang, L. Huang, X. X. Fan, J. H. Zhu, Y. Wang and Z. G. Zou, Inorg. Chem., 2007, 46, 10287–10293 CrossRef PubMed; (c) W. Tang, Y. Xiang and A. Tong, J. Org. Chem., 2009, 74, 2163–2166 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, instruments and methods; fluorescence images of 1 and 2; absorption spectra and fluorescence intensity of 1 upon the addition of various metal cations; absorption and fluorescence titration spectra, Job's plot, mass spectrum of 2 with Cu2+; NMR and mass spectra of 1–3. See DOI: 10.1039/c8qm00294k |
| This journal is © the Partner Organisations 2018 |