
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dielectric, optical and enhanced photocatalytic properties of CuCrO2 nanoparticles
Tokeer Ahmad *a,
Ruby Phula,
Parvez Alama,
Irfan H. Lonea,
Mohd. Shahazada,
Jahangeer Ahmed*b,
Tansir Ahamadb and
Saad M. Alshehribc
*a,
Ruby Phula,
Parvez Alama,
Irfan H. Lonea,
Mohd. Shahazada,
Jahangeer Ahmed*b,
Tansir Ahamadb and
Saad M. Alshehribc
aNanochemistry Laboratory, Department of Chemistry, Jamia Millia Islamia, New Delhi-110025, India. E-mail: tahmad3@jmi.ac.in
bDepartment of Chemistry, College of Science, King Saud University, Riyadh 11451, Saudi Arabia. E-mail: jahmed@ksu.edu.sa
cDepartment of Chemistry, College of Science & General Studies, Alfaisal University, Riyadh, Kingdom of Saudi Arabia
First published on 23rd May 2017
Abstract
Herein, delafossite CuCrO2 nanoparticles were synthesized through a polymeric citrate precursor method, followed by heating at 900 °C in air. The BET surface area of the CuCrO2 nanoparticles was found to be 235 m2 g−1, which was more than 500 times that of bulk CuCrO2 (0.47 m2 g−1) and 2–4 times that of the reported CuCrO2 nanoparticles (50–100 m2 g−1) (Y.-T. Nien et al., Mater. Chem. Phys., 2016, 179, 182–188 and D. Xiong et al., J. Mater. Chem., 2012, 22, 24760–24768). In the photoluminescence spectra of the CuCrO2 nanoparticles, a strong luminescent emission peak was observed at ∼460 nm in the blue light region. The resulting transmittance value of the CuCrO2 nanoparticles was found to be 77% in the visible region, and the direct band gap energy was determined to be 3.09 eV via UV-vis spectroscopy. The dielectric properties as a function of frequency and temperature and photocatalytic degradation of the organic pollutants in an aqueous solution were also investigated. CuCrO2 nanoparticles also showed remarkable enhancement in the catalytic degradation of methylene blue in H2O under sunlight irradiation. The plausible fragmentation and the structural changes in the methylene blue molecule were further demonstrated via mass spectrometry.
1. Introduction
Nanotechnology has attracted significant interest from many researchers; this is due to the fundamental scientific significance, various applications, and the peculiar and fascinating properties of the nanoparticles, superior to those of the bulk materials.3 Nanoparticles of transparent conducting oxides have shown a wide range of applications in optoelectronics,4 photovoltaics,5 gas sensors,6 batteries,7 solar cells, and electrochromic mirrors and windows.8 Generally, transparent conducting oxides (such as ZnO, SnO2, TiO2 etc.)9–11 are n-type materials, but some of them, such as NiO and ABO2, are p-type delafossite materials, which show significant electrical conductivity due to hole mobility and an optical direct band gap in the visible region.2,12–18Dye-sensitized solar cells (DSSCs) are third-generation photovoltaic devices and have recently gained attention due to their eco-friendly and economical production as compared to conventional photovoltaic devices. The DSSCs fabricated from p-type oxide materials have high surface chemical affinity, large surface area, a wide band gap, desirable valence band potential, and sufficiently high hole mobility.19 ABO2 oxides (A = Cu and B = Al, Ga, In, Sc, Y, Cr, rare earths, etc.) having delafossite structures with a band gap more than 3.1 eV have been studied as a triangular lattice antiferromagnet prototype with interesting properties, such as transparent conductivity and multiferroicity, and are mostly used in the formation of p-type DSSCs.20–22 Recently, delafossite CuCrO2 has been investigated due to its higher carrier mobility and positive flat band. In the bulk shape, the direct band gap value of CuCrO2 is found to be 2.95–3.30 eV at room temperature.23 The nanosized CuCrO2 particles have been used for constructing UV light-emitting diodes,24 transparent p–n junctions,25 and photocatalysts for hydrogen evolution,26 NO2− and NO3− removal,27,28 removal of M2+ (M = Ni, Cu, Zn, Cd, and Hg),29 combustion of methane.30 Recently, delafossite ABO2 (A = Cu and B = Al, Ga, Fe) nanoparticles were also used as catalysts in H2–O2 fuel cell applications31–33 and in the degradation of bisphenol.34 Present studies reveal that CuCrO2 nanoparticles could be used as effective photocatalysts in the degradation of organic contaminants in water. A large number of chemical and biological pollutants could be expected in groundwater. Therefore, treatment of polluted water (caused by use or disposal of waste materials in various forms including chemical, agricultural, animal products, industrial waste etc.) is the current challenge for scientists to globally control the environmental and unwanted human disease problems.
Delafossite and other nanostructured materials were also synthesized using the acid-catalyzed sol–gel method, ultrasonic process, and hydrothermal and solid state methods.2,30–33,35–38 Herein, we report the polymeric citrate precursor synthesis of CuCrO2 nanoparticles (∼40 nm) at 900 °C in 12 h in air. Various analytical techniques such as powder X-ray diffraction (XRD), BET surface area analysis, fluorescence spectroscopy, UV-vis spectrophotometry, and transmission electron microscopy (TEM) were used to characterize the CuCrO2 nanoparticles. Dielectric, optoelectronic, and photocatalytic properties of the CuCrO2 nanoparticles were also investigated. Enhanced photocatalytic efficiencies in the degradation of an organic dye (methylene blue) in an aqueous solution were examined for over 150 minutes.
2. Experimental
Ethylene glycol (Spectrochem), citric acid monohydrate (Merck; 99.0%), CuSO4·5H2O (Merck; >98%), and Cr(NO3)3·9H2O (Thomas Baker) were used as received without further purification. All the glassware was thoroughly washed with nitric acid followed by rinsing with deionized water several times before use. Deionized water was used throughout the experiments and in the preparation of stock solutions.The synthesis of nanoparticles via the polymeric citrate precursor route has already been reported in the literature.39,40 In the present protocol, ethylene glycol (1.4 mL) was mixed with 0.1 M Cr(NO3)3·9H2O (25 mL) in a beaker under constant stirring for about 10 minutes at room temperature to obtain a transparent solution. Dried citric acid (21 g) was added to the above mentioned solution in the molar ratio of ethylene glycol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :chromium ion:citric acid = 10:1:40. For the clear solution, the mixture was continuously stirred for about 2 h at room temperature followed by the addition of 0.1 M CuSO4·5H2O (25 mL) and subsequent stirring for 2 h. The temperature of the above mentioned system was maintained at 55 ± 5 °C for further 2 h to eliminate excess of water and increase the rate of the polyesterification reaction between citric acid and ethylene glycol to obtain the viscous solution. This viscous solution was kept at 135 °C for 20 h in a furnace to vaporize the solvent. A semi-solid product was recovered after polymerization, which was charred for 2 h at 300 °C followed by further heating at 500 °C for 20 h. A black-coloured precursor was obtained, which was used in the synthesis of the CuCrO2 nanoparticles. The synthesized precursor materials were heated at 900 °C for 12 h in an automatic high temperature furnace to obtain nearly green-coloured powder of the CuCrO2 nanoparticles.
:chromium ion:citric acid = 10:1:40. For the clear solution, the mixture was continuously stirred for about 2 h at room temperature followed by the addition of 0.1 M CuSO4·5H2O (25 mL) and subsequent stirring for 2 h. The temperature of the above mentioned system was maintained at 55 ± 5 °C for further 2 h to eliminate excess of water and increase the rate of the polyesterification reaction between citric acid and ethylene glycol to obtain the viscous solution. This viscous solution was kept at 135 °C for 20 h in a furnace to vaporize the solvent. A semi-solid product was recovered after polymerization, which was charred for 2 h at 300 °C followed by further heating at 500 °C for 20 h. A black-coloured precursor was obtained, which was used in the synthesis of the CuCrO2 nanoparticles. The synthesized precursor materials were heated at 900 °C for 12 h in an automatic high temperature furnace to obtain nearly green-coloured powder of the CuCrO2 nanoparticles.
The phase analysis and crystallinity of the as-synthesized nanoparticles were determined via XRD studies using a Rigaku Ultima IV diffractometer with nickel-filtered Cu-Kα radiation of wavelength 1.5416 Å, and diffraction patterns were obtained in the 2θ range from 20° to 80° with a step size of 0.05° and step time of 1 s. Fourier transform infrared spectra (FT-IR) were obtained via an IT Affinity-1, Shimadzu spectrophotometer using KBr discs as a reference. The surface area of the sample was determined via a BET (Brunauer–Emmett–Teller) surface area analyser (Quantachrome Instruments Limited, USA, Model Nova 2000e) at liquid nitrogen temperature (77 K) using the multipoint BET method. To remove the water vapour and adsorbed gases from the sample, approximately 0.06 gram of the sample was placed in the sample cell and degassed for 2 h at 200 °C in the vacuum degassing mode. The grain size and morphology of the synthesized nanoparticles were estimated using an FEI Technai G2 20 transmission electron microscope. The specimens for TEM studies were prepared by deploying a drop of sonicated nanopowder [suspension of powder sample in absolute ethanol for about 15 min using an ultrasonicator (Ultrasonic Processor Model: UP-500)] onto the TEM grid (carbon-coated copper) and drying at 45 °C in an oven for 1 h. Dielectric measurements were carried out via a Wayne Kerr high frequency LCR meter (Model: 6505P). Then, 2–3 drops of 5% PVA solution were placed on the fine powder as a fixer agent and dried at 90 °C for 1 h. Finely ground powder was transformed into pellets at a pressure of 3–4 tons, followed by heating of the pellets at 1000 °C for 12 h. Before carrying out the dielectric measurements, the pellets were coated by a silver paste (Ted Pella, Inc.) and dried at 90 °C for 1.5 h to make the surface conducting. Fluorescence studies were carried out using a Perkin-Elmer LS-55 spectrofluorophotometer after dissolving the powders in ethanol. The excitation source was a xenon lamp, and the baseline was corrected before the measurements.
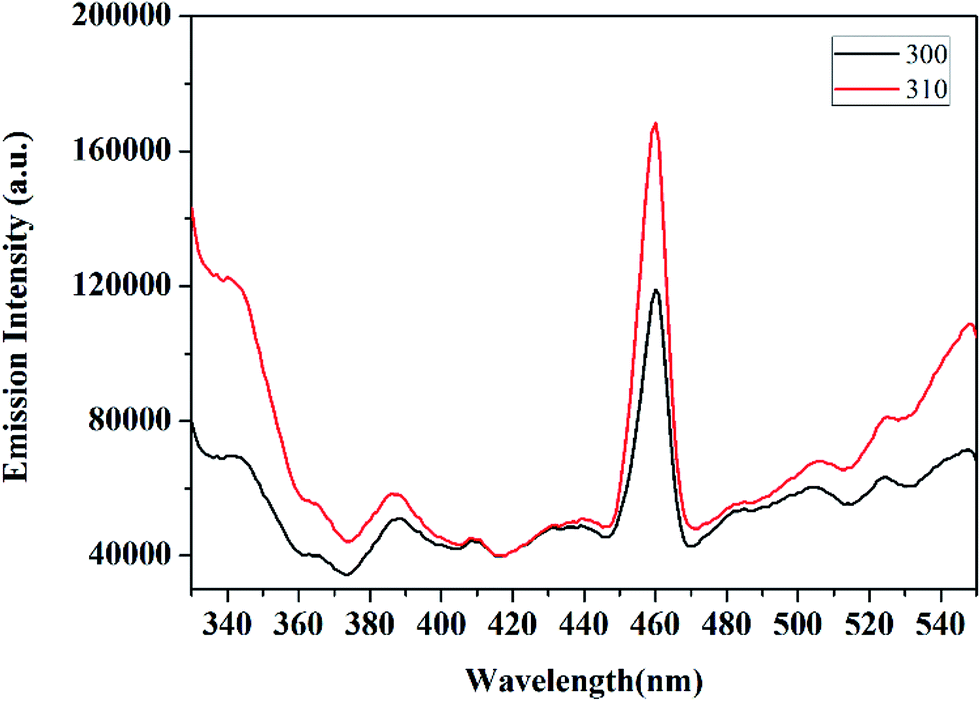
The photocatalytic efficiency of the prepared CuCrO2 nanoparticles was measured against the degradation of methylene blue (MB) under sunlight irradiation. The degradation process was investigated by UV-vis spectrophotometry (Shimadzu, UV-1650) at the maximum absorption wavelength (λmax) of 662.5 nm of MB. Herein, 1 mg of catalyst was dispersed in 50 mL MB (10 mg L−1) and placed in the dark under constant magnetic stirring to obtain the suspension with an adsorption/desorption equilibrium. The degradation process was carried out at room temperature at pH 7. Thereafter, the suspension was subjected to visible light. At 15 min intervals, the sample was obtained and centrifuged at 8000 rpm for 10 min to separate the CuCrO2 nanoparticles, and the transparent solutions were obtained to study the absorption spectra. All the experiments were repeated three times, and the results showed excellent agreement. The degradation rate constants of degraded MB, using the CuCrO2 nanoparticles were calculated using the first-order kinetic reaction as below:
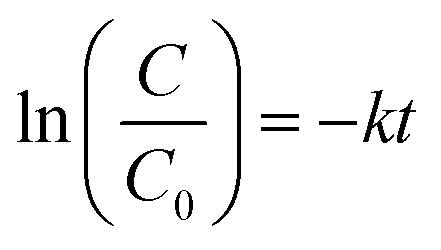
3. Results and discussion
Fig. 1a shows the powder X-ray diffraction (XRD) patterns of the black-coloured precursor and pure CuCrO2 nanoparticles. The black-coloured precursor materials contain a mixture of CuO and Cr2O3 at 500 °C (black-coloured line), whereas the pure phase of the CuCrO2 nanoparticles (red-coloured line) was obtained at 900 °C in air (Fig. 1a). The reflections appeared at the 2θ values of 31.43°, 35.22°, 36.42°, 40.90°, 47.95°, 55.90°, 62.41°, 65.60°, 71.56°, and 74.50°, assigned to (006), (101), (012), (104), (009), (018), (110), (0012), (116), and (202), in accordance with the JCPDS no. 740983 for the hexagonal crystal structure of CuCrO2. FT-IR spectrum of the CuCrO2 nanoparticles is shown in Fig. 1b. Herein, one weak (∼941 cm−1) and two strong bands (∼743 and 536 cm−1) could be assigned to the CrIII–O and M–O bond stretching frequencies of the CuCrO2 nanoparticles, respectively.2,41 The TEM image shows the formation of spherical-shaped CuCrO2 nanoparticles with the average diameter of ∼40 nm (Fig. 1c). The agglomerations of the CuCrO2 nanoparticles were also observed to a small extent, which could be due to high temperature (i.e. 900 °C) employed in the reactions of the precursors in the synthesis of the CuCrO2 nanoparticles.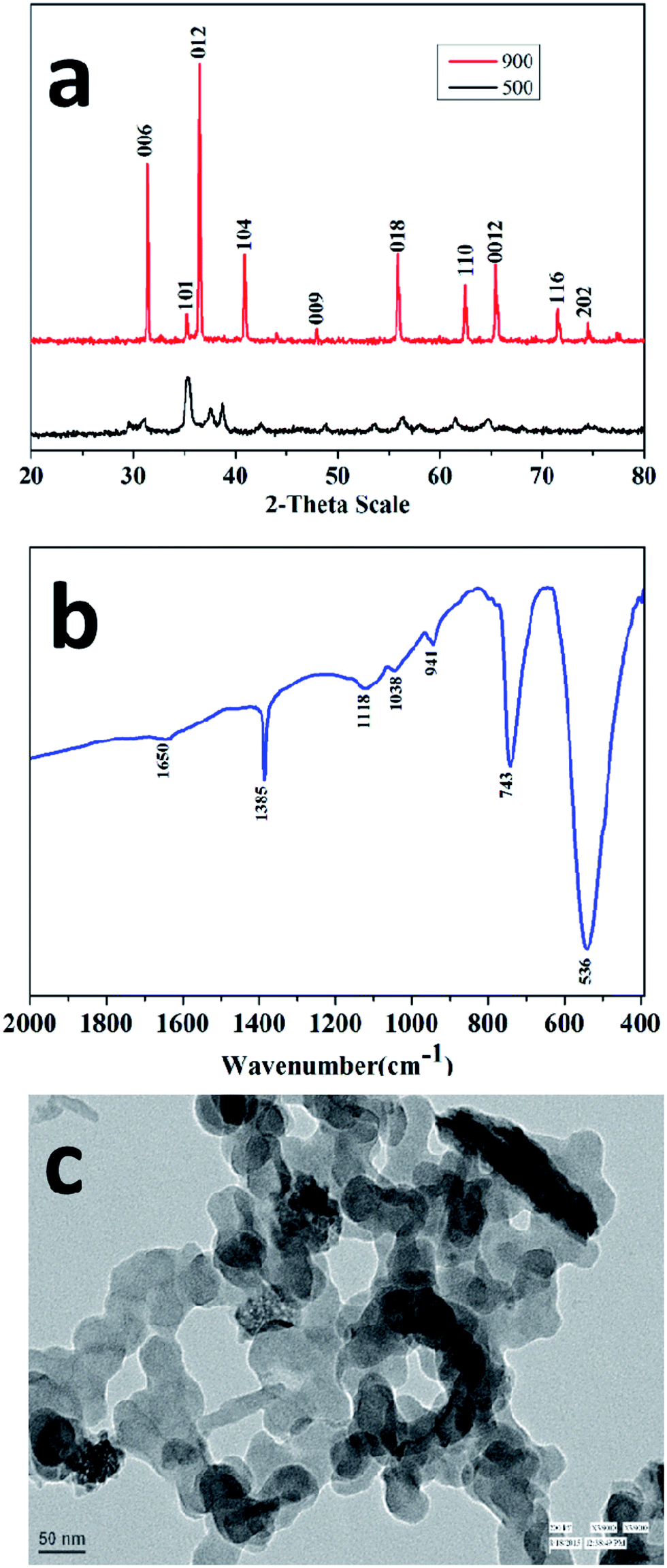 | ||
| Fig. 1 (a) XRD patterns of the precursor heated at 500 °C in air (black-coloured line) and pure phase of the CuCrO2 nanoparticles obtained at 900 °C and 12 h in air (red-coloured line). (b) FT-IR spectrum and (c) TEM image of the CuCrO2 nanoparticles obtained at 900 °C. | ||
The specific surface area of the nanocrystalline CuCrO2 particles was examined through the multipoint BET equation with the relative pressure (P/Po) in the range from 0.05 to 0.35. The surface area of the CuCrO2 nanoparticles was found to be 235 m2 g−1, as shown in Fig. 2a. Note that the present surface area of the CuCrO2 nanoparticles is >500 times the surface area of bulk CuCrO2 (0.5 m2 g−1).1 Moreover, the surface area of the as-prepared CuCrO2 nanoparticles is 2–4 times that of the previously studied CuCrO2 nanoparticles.1,2 From the DA plot (Fig. 2b), the pore radius of the CuCrO2 nanoparticles was found to be ∼15 nm, which also supported the electron microscopy images. From the TEM data, it is very clear that the particles are spherical in shape with a radius of ∼20 nm, which closely matches the particle size obtained from the Dubinin-Astakhov (DA) model.42
 | ||
| Fig. 2 (a) BET and (b) DA plots of the CuCrO2 nanoparticles. | ||
The dielectric properties of the CuCrO2 nanoparticles were determined as a function of temperature and frequency. Prior to the measurements, the thickness and diameter were calculated to measure the dielectric constant from the measured value of the capacitance. Fig. 3a shows the dependence of the dielectric constant on frequency for the CuCrO2 nanoparticles at different temperatures. The dielectric constant decreases with the increase in frequency due to the inability of the electric dipoles to align with the fast variation of the alternating applied electric field.43,44 In the low-frequency region, the dielectric constant showed dispersion and achieved a saturation limit at high frequencies. This could be due to the Maxwell–Wagner type interfacial polarization that is in excellent agreement with the Koop's model.45 Fig. 3b shows the variation of the dielectric loss as a function of frequency at different temperatures. The measurement was carried out in the frequency range from 10 kHz to 1 MHz. The dielectric loss constantly decreases as the frequency increases. Temperature-dependant studies showed that the dielectric constant was maximum at 100 °C, whereas some fluctuations in the dielectric loss were observed at this temperature. The dielectric constant and loss remained nearly stable at 200 °C. Frequency-dependant dielectric loss and dielectric constants of the CuCrO2 nanoparticles are shown in Fig. 3c, which clearly shows that the dielectric properties of the CuCrO2 nanoparticles decrease with the increase in frequency. The decrease in the dielectric constant with the increase in frequency may be associated with the fact that any species contributing to polarization was found to lag behind the applied field at high frequency and hence decreased both properties i.e. dielectric constant and dielectric loss.
 | ||
| Fig. 3 (a) Dielectric constant and (b) dielectric loss of the CuCrO2 nanoparticles at different temperatures. (c) Variation in the dielectric constant and loss of the CuCrO2 nanoparticles at room temperature as a function of frequency. | ||
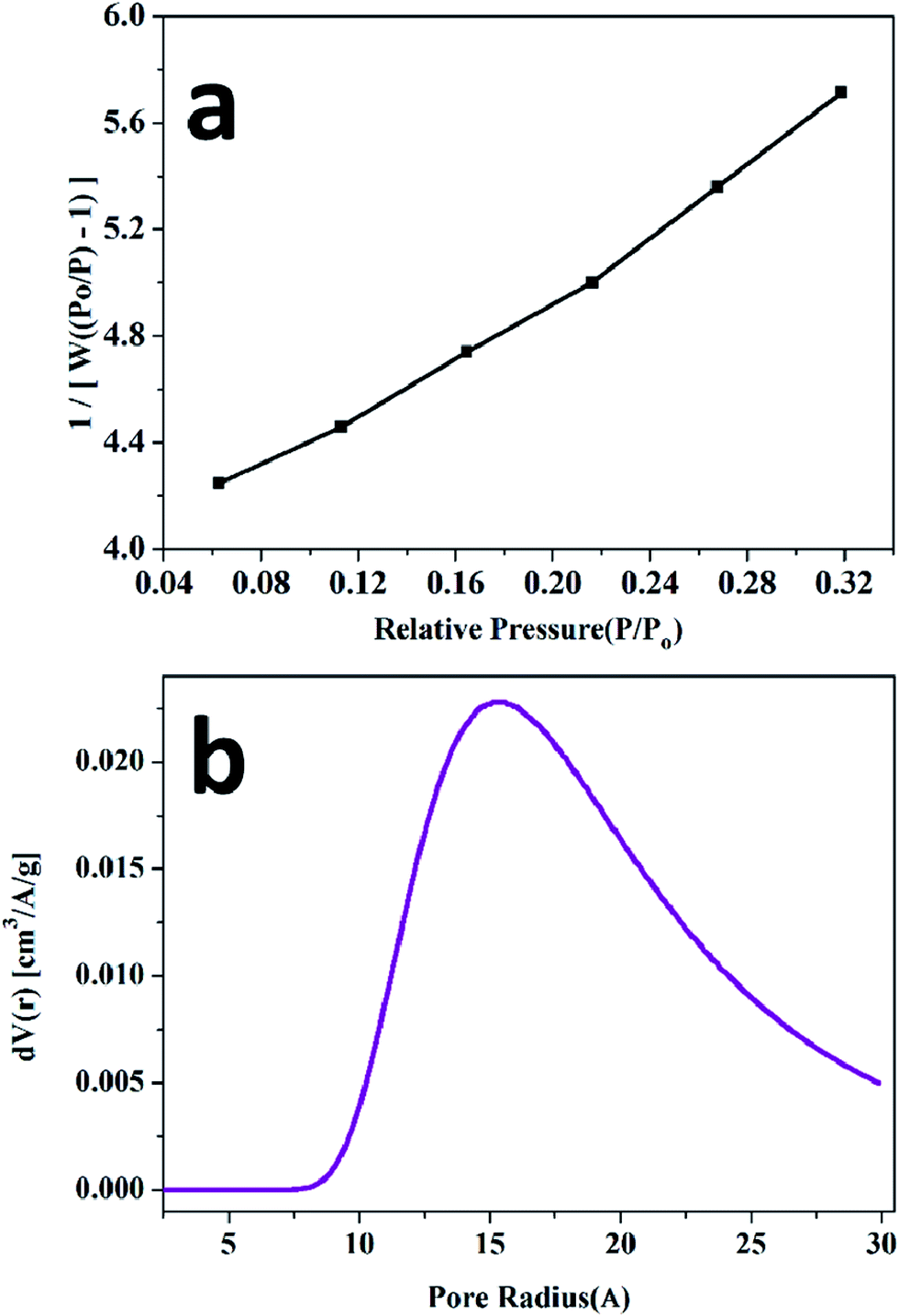
Photoluminescence (PL) is a significant technique to study the optical properties of the delafossite materials. Fig. 4 shows the photoluminescence spectra of the CuCrO2 nanoparticles. These nanoparticles show a strong luminescent emission peak at 460 nm in the blue region, and a weak emission peak at 386 nm for an excitation wavelength of 310 nm at room temperature. These transitions could be attributed to the intra-band transitions, implying 3d94s1 and 3d10 in Cu+ ions, as also reported for other Cu-based delafossite oxide insulators (such as CuYO2 and CuLaO2).46,47 On comparing CuCrO2 with CuLaO2, it was observed that the luminescence peaks of CuCrO2 shifted towards low energy. This may be a consequence of the interaction of Cu–O with the Cr atom48 since Cr ion exerts an influence on the Cu–O–Cr–O–Cu linkage owing to more favourable integration of the 3d state on Cr ion, affecting the Cu–O bond length and the Cu–Cu interaction.49
 | ||
| Fig. 4 Photoluminescence (PL) spectra of the CuCrO2 nanoparticles, excited at 300 and 310 nm. | ||
Fig. 5 shows the UV-visible spectrum of the CuCrO2 nanoparticles in the range from 200 to 800 nm. The wavelength-dependent transmittance results reveal that the light transmittance value of the CuCrO2 nanoparticles is ∼74–80% in the wavelength range from 600 to 800 nm, as shown in Fig. 5a, and the resulting values are found to be higher that the reported values for CuCrO2.50–52 The photon energy (or band gap energy) of the CuCrO2 nanoparticles was determined using the UV-visible absorption spectrum of the CuCrO2 nanoparticles. Fig. 5b shows the plot of (αhν)2 against photon energy for the CuCrO2 nanoparticles, where α, h, and ν are the absorbance (obtained from UV), Planck's constant, and frequency of the incident beam, respectively. Direct and indirect band gap energies of the solids could be calculated through the Tauc's model.53,54 Photon energy (intercept of the straight line on the x-axis) demonstrates the direct band gap of the CuCrO2 nanoparticles to be 3.10 eV, which closely matches the reported direct band gap (3.09 eV) of the CuCrO2 thin films.51 The band gap energies of the materials usually refer to the energy difference between the valence and conduction bands.
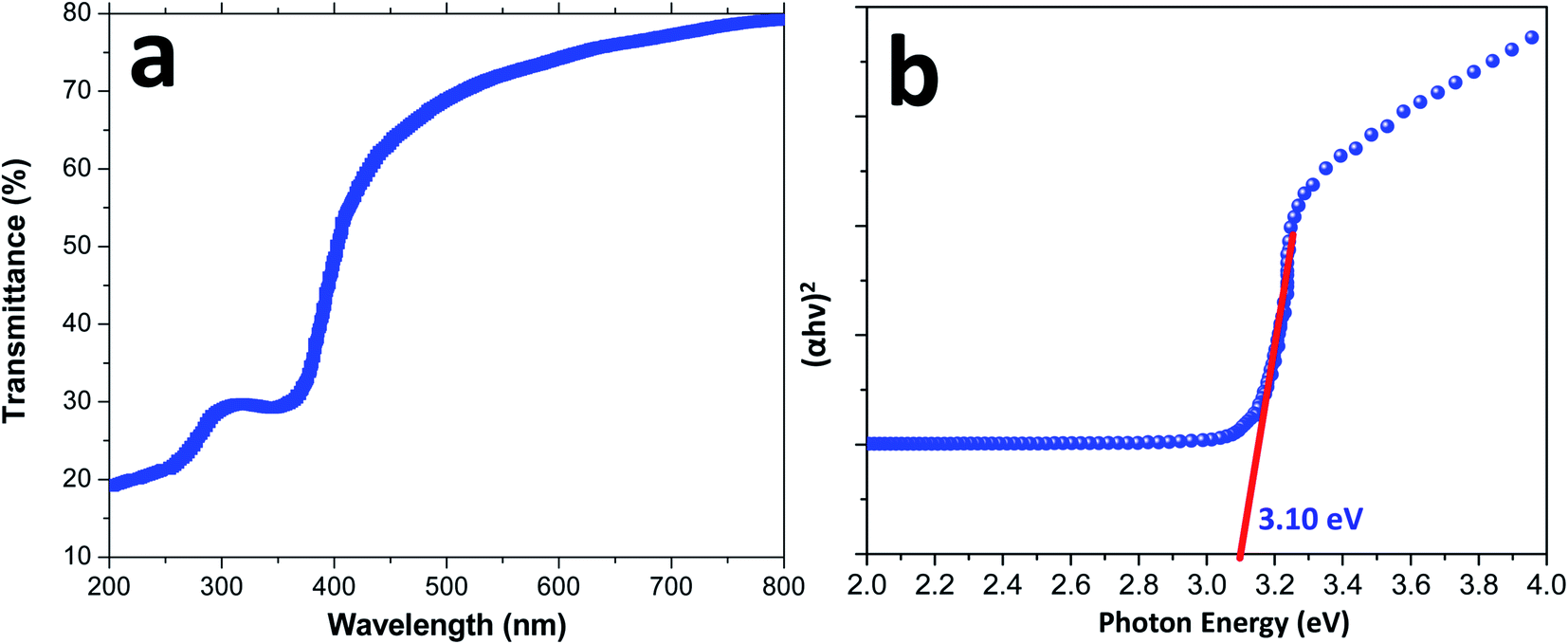 | ||
| Fig. 5 (a) UV-visible spectrum (wavelength vs. percent transmittance) and (b) plot of (αhν)2 versus photon energy (eV) for the direct band gap determination of the CuCrO2 nanoparticles. | ||
The photocatalytic activity of the CuCrO2 nanoparticles was tested in the degradation of MB under irradiation from sunlight for over 150 minutes. The degradation of MB over the CuCrO2 nanoparticles could be explained on the basis of the generation of hydroxyl free radicals (OH˙), i.e. highly oxidizing agents. When the CuCrO2 nanoparticles were irradiated in sunlight, holes (h+) were formed in the valence band of the CuCrO2 materials after the excitation of the electrons (e−) to the conduction band. This could be possible due to the larger energy of sunlight compared to the band gap energy of CuCrO2 (3.1 eV). In an aqueous medium with the presence of oxygen, the holes and electrons produced hydroxyl radicals (OH˙) and superoxide radical anions (O2−˙), respectively. The hydroxyl radicals (OH˙) are strong oxidizing agents and attack the dye molecule to provide the oxidized product, as shown in Fig. 6a and also summarized in the photochemical reactions given below:
| CuCrO2 + hν → CuCrO2 (e− + h+) |
| CuCrO2 (h+) + H2O → CuCrO2 + OH˙ + H+ |
| CuCrO2 (h+) + HO− → CuCrO2 + OH˙ |
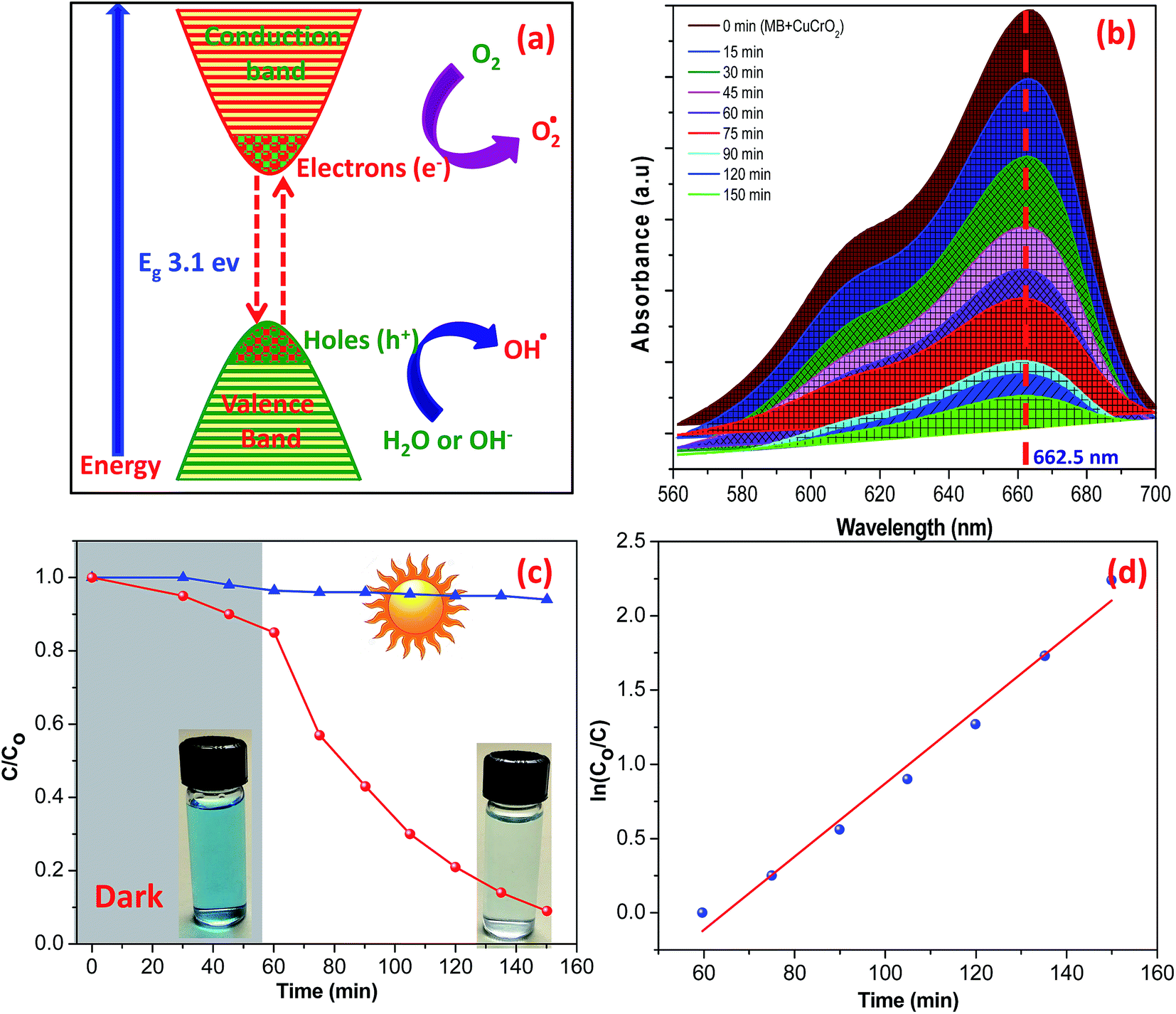 | ||
| Fig. 6 (a) Schematic of the photocatalytic degradation of MB using CuCrO2 nanoparticles. (b) UV-visible spectra and (c and d) photocatalytic degradation of aqueous MB with time (minutes) under irradiation from sunlight. | ||
In the conduction band, the electrons (e−) of CuCrO2 reduce molecular oxygen (O2) to hydroperoxyl radicals (i.e. a protonated form of superoxide):
| CuCrO2 + O2 + e− → CuCrO2 + O2˙− |
The degradation of the adsorbed compounds (i.e. MB) is likely to occur either by the direct oxidation on the surface of the photocatalysts or by hydroxyl radicals:
| OH˙ + MB → CO2 + H2O (oxidized products) |
The UV-visible spectra of MB in an aqueous solution are presented in Fig. 6b. The distinct characteristic was detected at 662.5 nm, and the absorption band intensity decreased with time under the irradiation of sunlight. The reduction in the intensity of the absorption band of aqueous MB indicates the degradation of dye by UV irradiation over the surface of the CuCrO2 nanoparticles. The degradation efficiencies of MB are shown in Fig. 6c. Note that 79% degradation of MB was observed after 1 h in sunlight, whereas ∼16.5% adsorption of MB was observed in the dark for 1 h. Photocatalytic degradation of MB over the surface of pure CuCrO2 nanoparticles (after 1 h) was found to be higher (two to three times) compared to that of the CuCrO2/TiO2 heterostructures and pure TiO2, respectively.55 In parallel, we also performed the experiments in the absence of photocatalysts and found that no degradation of MB occurred in the same time. Fig. 6d shows the linear plots of ln(C/C0) vs. irradiation time, which support the kinetic data for the degradation of MB. Previously, photocatalytic degradation efficiencies of ∼85% and 75% of MB were reported for CuCrO2/TiO2 heterostructures and pure TiO2 nanoparticles, respectively, using the comparatively very long irradiation time of 8 h.55 Note that the degradation efficiency of MB was ∼88% after just 1.5 h over the surface of the CuCrO2 nanoparticles in the presence of sunlight irradiation. This was found to be a significant enhancement in the photocatalytic efficiency of the CuCrO2 nanoparticles compared to previous reports.55,56 The kinetic plot was fitted by the pseudo-first-order rate equation using the rate constants of 0.0246 min−1, and the R2 value was found to be ∼0.9830.
Fig. 7 shows the mass spectrum for the formation of the OH radical and the subsequent oxidation of MB dye after 60 minutes of sunlight irradiation. The signals at m/z = ∼284, ∼300, and ∼316 of MB could be recognized on the basis of sequential hydroxylation of the molecule by the attack of the OH radical. However, other signals (m/z) could be due to the fragmentation of the dye molecule. The results are in good agreement with previous reports on the mineralization of the MB dye by the attack of free radicals.57,58 Table 1 compiles the resulted fragmentations of the dye molecule with the m/z values after 60 minutes of irradiation. The reaction mechanism of the projected route for the photodegradation of the MB dye molecule over the surface of nanoparticles is shown in Fig. 8.57–61 The most active bonds of the MB dye molecule are C–N and C–S, which can be easily broken by the attack of hydroxyl and superoxide free radicals to generate various fragments (oxidized organic molecules), followed by their transformation into inorganic substances such as CO2, H2O, SO42−, NO3− etc.
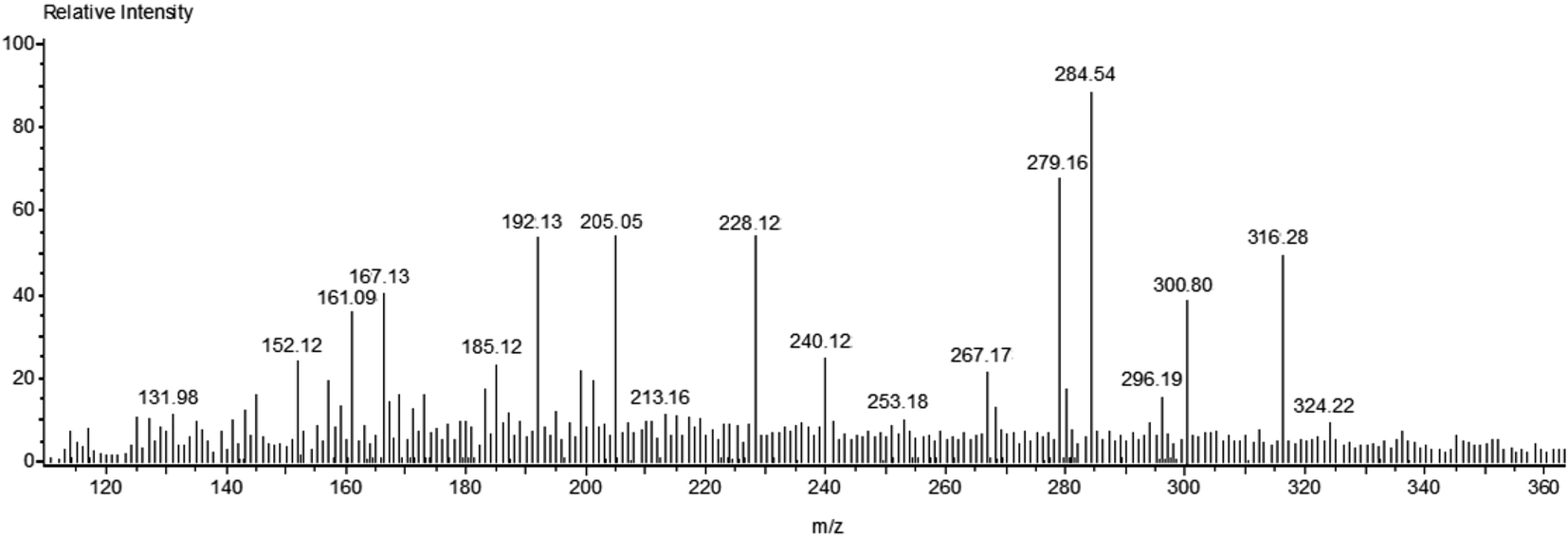 | ||
| Fig. 7 Mass spectrum of MB dye after 60 minutes of photodegradation reaction. | ||
| S. no. | Molecule | Observed m/z |
|---|---|---|
| 1 | C16H18N3S | 284.54 |
| 2 | C16H18ON3S | 300.54 |
| 3 | C16H18O2N3S | 316.28 |
| 4 | C12H10N3S | 228.12 |
| 5 | C13H15N2O3S | 279.16 |
| 6 | C7H10NSO4 | 205.05 |
| 7 | C6H8NSO4 | 192.13 |
| 8 | C8H9NO3 | 167.13 |
| 9 | C6H10NO4 | 161.09 |
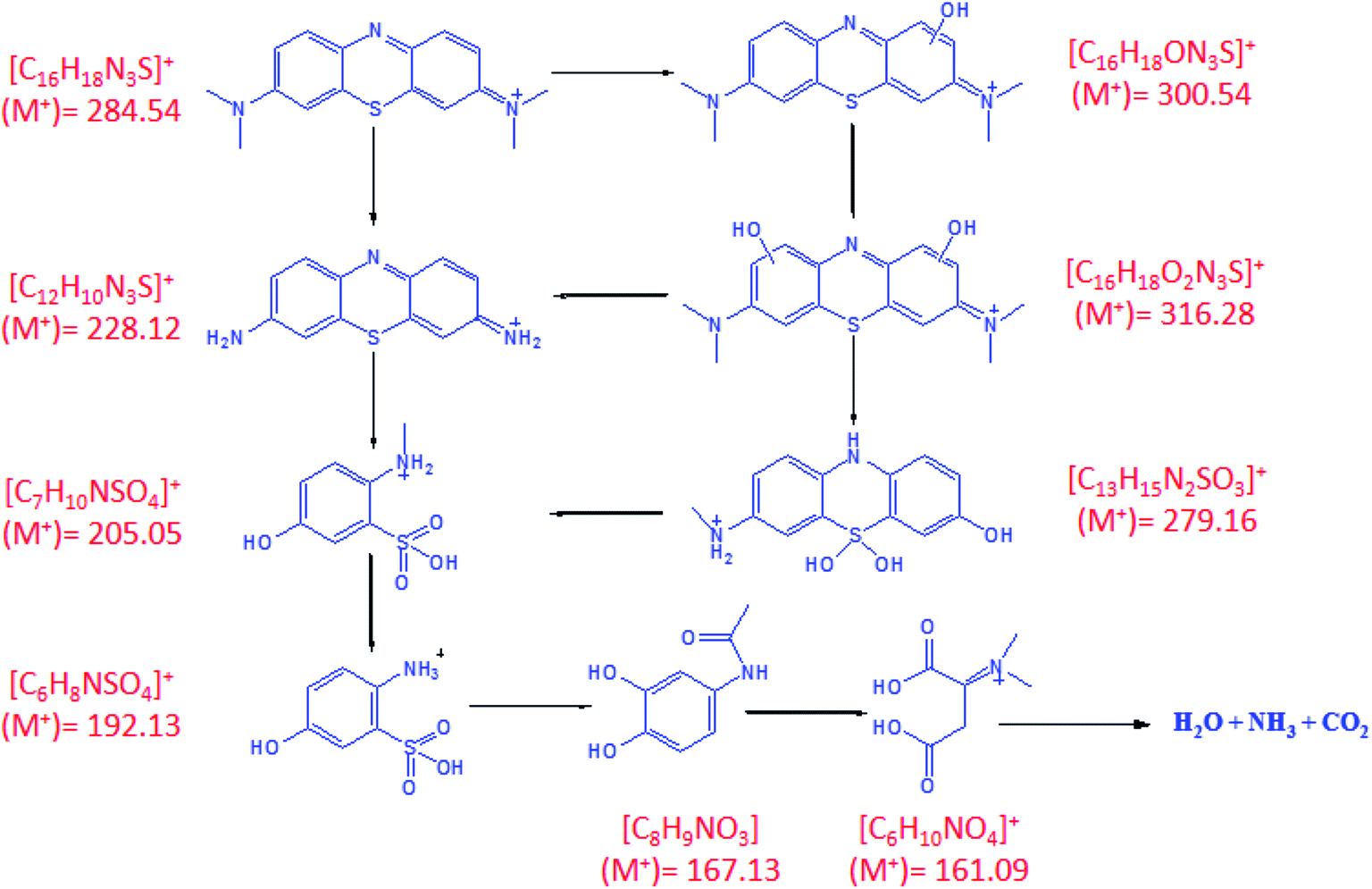 | ||
| Fig. 8 Projected route for the photodegradation of the MB dye. | ||
4. Conclusions
The wide band gap CuCrO2 nanoparticles with a delafossite structure were successfully synthesized by the polymeric citrate precursor method followed by heating at 900 °C in air. Phase purity, size, and shape of the CuCrO2 nanoparticles were investigated by XRD and electron microscopy studies. The BET surface area of the CuCrO2 nanoparticles (235 m2 g−1) was found to be more than 500 times that of bulk CuCrO2. The dielectric loss and dielectric constant of the CuCrO2 nanoparticles decreases with the increase in the frequency. CuCrO2 nanoparticles possess optical transparency and a direct band gap energy of 77% and 3.09 eV, respectively, in the visible region. The CuCrO2 nanoparticles also exhibit photoluminescence property at room temperature like other p-type TCOs, which can be utilized for practical applications such as in light-emitting diodes. A significant enhancement of the photocatalytic degradation of the organic pollutants (methylene blue) was studied over the surface of the CuCrO2 nanoparticles for over 150 minutes. The photocatalytic degradation of the MB dye using the CuCrO2 nanoparticles was further confirmed via mass spectrometry. The results suggested that the CuCrO2 nanoparticles can be considered as one of the effective photocatalysts for the degradation of organic contaminants from water.Acknowledgements
The authors thank Dr Sameer Sapra, IIT Delhi for fluorescence studies, CIF, Jamia Millia Islamia for X-ray diffraction studies, and AIIMS, New Delhi for electron microscopy studies. RP and MS thank UGC for research fellowships. The authors extend their sincere appreciation to the Deanship of Scientific Research at the King Saud University for funding this Research Group (RG-1435-007).References
- Y.-T. Nien, M.-R. Hu, T.-W. Chiu and J.-S. Chu, Mater. Chem. Phys., 2016, 179, 182–188 CrossRef CAS.
- D. Xiong, Z. Xu, X. Zeng, W. Zhang, W. Chen, X. Xu, M. Wang and Y.-B. Cheng, J. Mater. Chem., 2012, 22, 24760–24768 RSC.
- G. A. Ozin, Adv. Mater., 1992, 4, 612–649 CrossRef CAS.
- B. K. Sonawane, M. P. Bhole and D. S. Patil, Opt. Quantum Electron., 2009, 41, 17–26 CrossRef CAS.
- S. H. Lim, S. Desu and A. C. Rastogi, J. Phys. Chem. Solids, 2008, 69, 2047–2056 CrossRef CAS.
- G. Oskam and F. d. J. P. Poot, J. Sol-Gel Sci. Technol., 2006, 37, 157–160 CrossRef CAS.
- A. K. Rai, L. T. Anh, J. Gim, V. Mathew, J. Kang, B. J. Paul, J. Song and J. Kim, Electrochim. Acta, 2013, 90, 112–118 CrossRef CAS.
- B. G. Lewis and D. C. Paine, MRS Bull., 2000, 25, 22–27 CrossRef CAS.
- T. Ahmad, S. Vaidya, N. Sarkar, S. Ghosh and A. K. Ganguli, Nanotechnology, 2006, 17, 1236 CrossRef CAS.
- J. Krýsa, M. Keppert, J. r. Jirkovský, V. Štengl and J. Šubrt, Mater. Chem. Phys., 2004, 86, 333–339 CrossRef.
- W. Yu-de, M. Chun-lai, S. Xiao-dan and L. Heng-de, Nanotechnology, 2002, 13, 565 CrossRef.
- S. Mori, S. Fukuda, S. Sumikura, Y. Takeda, Y. Tamaki, E. Suzuki and T. Abe, J. Phys. Chem. C, 2008, 112, 16134–16139 CAS.
- E. A. Gibson, A. L. Smeigh, L. Le Pleux, J. Fortage, G. Boschloo, E. Blart, Y. Pellegrin, F. Odobel, A. Hagfeldt and L. Hammarström, Angew. Chem., Int. Ed., 2009, 48, 4402–4405 CrossRef CAS PubMed.
- A. Nattestad, A. J. Mozer, M. K. R. Fischer, Y. B. Cheng, A. Mishra, P. Bauerle and U. Bach, Nat. Mater., 2010, 9, 31–35 CrossRef CAS PubMed.
- M. Yu, G. Natu, Z. Ji and Y. Wu, J. Phys. Chem. Lett., 2012, 3, 1074–1078 CrossRef CAS PubMed.
- J. Ahmed, C. K. Blakely, J. Prakash, S. R. Bruno, M. Yu, Y. Wu and V. V. Poltavets, J. Alloys Compd., 2014, 591, 275–279 CrossRef CAS.
- A. Nattestad, X. Zhang, U. Bach and Y.-B. Cheng, J. Photonics Energy, 2011, 1, 011103–011109 CrossRef.
- A. Renaud, B. Chavillon, L. Le Pleux, Y. Pellegrin, E. Blart, M. Boujtita, T. Pauporte, L. Cario, S. Jobic and F. Odobel, J. Mater. Chem., 2012, 22, 14353–14356 RSC.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- H. Kawazoe, M. Yasukawa, H. Hyodo, M. Kurita, H. Yanagi and H. Hosono, Nature, 1997, 389, 939–942 CrossRef CAS.
- D. O. Scanlon and G. W. Watson, J. Mater. Chem., 2011, 21, 3655–3663 RSC.
- K. Ueda, T. Hase, H. Yanagi, H. Kawazoe, H. Hosono, H. Ohta, M. Orita and M. Hirano, J. Appl. Phys., 2001, 89, 1790–1793 CrossRef CAS.
- Y. Wang, Y. Gu, T. Wang and W. Shi, J. Alloys Compd., 2011, 509, 5897–5902 CrossRef CAS.
- J. Tate, M. K. Jayaraj, A. D. Draeseke, T. Ulbrich, A. W. Sleight, K. A. Vanaja, R. Nagarajan, J. F. Wager and R. L. Hoffman, Thin Solid Films, 2002, 411, 119–124 CrossRef CAS.
- K. Tonooka and N. Kikuchi, Thin Solid Films, 2006, 515, 2415–2418 CrossRef CAS.
- S. Saadi, A. Bouguelia and M. Trari, Sol. Energy, 2006, 80, 272–280 CrossRef CAS.
- W. Ketir, A. Bouguelia and M. Trari, Water, Air, Soil Pollut., 2009, 199, 115–122 CrossRef CAS.
- W. Ketir, A. Bouguelia and M. Trari, Desalination, 2009, 244, 144–152 CrossRef CAS.
- W. Ketir, A. Bouguelia and M. Trari, J. Hazard. Mater., 2008, 158, 257–263 CrossRef CAS PubMed.
- N. Dupont, A. Kaddouri and P. Gélin, J. Sol-Gel Sci. Technol., 2011, 58, 302–306 CrossRef CAS.
- J. Ahmed, V. V. Poltavets, J. Prakash, S. M. Alshehri and T. Ahamad, J. Alloys Compd., 2016, 688, 1157–1161 CrossRef CAS.
- J. Ahmed and Y. Mao, in Nanomaterials for Sustainable Energy, American Chemical Society, 2015, ch. 4, vol. 1213, pp. 57–72 Search PubMed.
- J. Ahmed and Y. Mao, J. Solid State Chem., 2016, 242, 77–85 CrossRef CAS.
- X. Zhang, Y. Ding, H. Tang, X. Han, L. Zhu and N. Wang, Chem. Eng. J., 2014, 236, 251–262 CrossRef CAS.
- R. Srinivasan, B. Chavillon, C. Doussier-Brochard, L. Cario, M. Paris, E. Gautron, P. Deniard, F. Odobel and S. Jobic, J. Mater. Chem., 2008, 18, 5647–5653 RSC.
- J. W. Lekse, M. K. Underwood, J. P. Lewis and C. Matranga, J. Phys. Chem. C, 2012, 116, 1865–1872 CAS.
- A. K. Ganguli, S. Vaidya and T. Ahmad, Bull. Mater. Sci., 2008, 31, 415–419 CrossRef CAS.
- A. Ganguly, T. Ahmad and A. K. Ganguli, Langmuir, 2010, 26, 14901–14908 CrossRef CAS PubMed.
- T. Ahmad and I. H. Lone, New J. Chem., 2016, 40, 3216–3224 RSC.
- T. Ahmad, I. H. Lone and M. Ubaidullah, RSC Adv., 2015, 5, 58065–58071 RSC.
- V. Pocol, L. Patron, O. Carp, M. Brezeanu, E. Segal, N. Stanica and D. Crisan, J. Therm. Anal. Calorim., 1999, 55, 143–154 CrossRef CAS.
- A. Gil, S. A. Korili and G. Y. Cherkashinin, J. Colloid Interface Sci., 2003, 262, 603–607 CrossRef CAS PubMed.
- M. Ubaidullah, I. H. Lone, O. A. Al-Hartomy, D. Kumar and T. Ahmad, Adv. Sci. Lett., 2014, 20, 1354–1359 CrossRef.
- B. K. Bammannavar, L. R. Naik and B. K. Chougule, J. Appl. Phys., 2008, 104, 064123 CrossRef.
- C. G. Koops, Phys. Rev., 1951, 83, 121–124 CrossRef CAS.
- A. Jacob, C. Parent, P. Boutinaud, G. Le Flem, J. P. Doumerc, A. Ammar, M. Elazhari and M. Elaatmani, Solid State Commun., 1997, 103, 529–532 CrossRef CAS.
- P. Boutinaud, D. Garcia, C. Parent, M. Faucher and G. Le Flem, J. Phys. Chem. Solids, 1995, 56, 1147–1154 CrossRef CAS.
- A. C. Rastogi, S. H. Lim and S. B. Desu, J. Appl. Phys., 2008, 104, 023712 CrossRef.
- R. D. Shannon, D. B. Rogers, C. T. Prewitt and J. L. Gillson, Inorg. Chem., 1971, 10, 723–727 CrossRef CAS.
- H.-Y. Chen and K.-P. Chang, ECS J. Solid State Sci. Technol., 2013, 2, P76–P80 CrossRef CAS.
- T. S. Tripathi, J.-P. Niemela and M. Karppinen, J. Mater. Chem. C, 2015, 3, 8364–8371 RSC.
- S. Götzendörfer, C. Polenzky, S. Ulrich and P. Löbmann, Thin Solid Films, 2009, 518, 1153–1156 CrossRef.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- A. Ibrahim and S. K. J. Al-Ani, Czech J. Phys., 1994, 44, 785–797 CrossRef CAS.
- D. Xiong, H. Chang, Q. Zhang, S. Tian, B. Liu and X. Zhao, Appl. Surf. Sci., 2015, 347, 747–754 CrossRef CAS.
- S. Chin, E. Park, M. Kim and J. Jurng, Powder Technol., 2010, 201, 171–176 CrossRef CAS.
- H. W. P. Carvalho, A. P. L. Batista, P. Hammer and T. C. Ramalho, J. Hazard. Mater., 2010, 184, 273–280 CrossRef CAS PubMed.
- F. Huang, L. Chen, H. Wang and Z. Yan, Chem. Eng. J., 2010, 162, 250–256 CrossRef CAS.
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard and J.-M. Herrmann, Appl. Catal., B, 2001, 31, 145–157 CrossRef CAS.
- J.-E. Lee, N. T. Khoa, S. W. Kim, E. J. Kim and S. H. Hahn, Mater. Chem. Phys., 2015, 164, 29–35 CrossRef CAS.
- H.-P. Jing, C.-C. Wang, Y.-W. Zhang, P. Wang and R. Li, RSC Adv., 2014, 4, 54454–54462 RSC.
| This journal is © The Royal Society of Chemistry 2017 |