DOI:
10.1039/C6NR06851K
(Paper)
Nanoscale, 2017,
9, 334-340
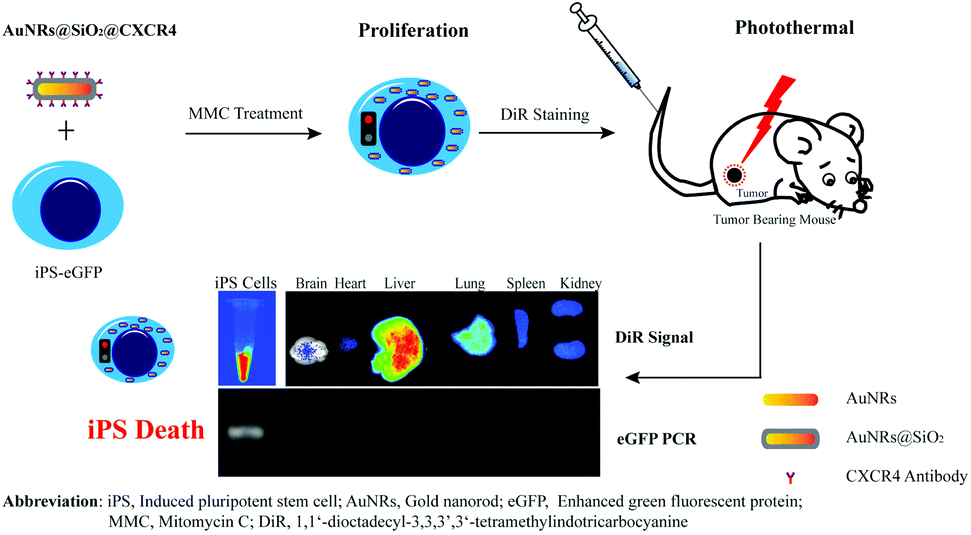
Mitomycin C-treated human-induced pluripotent stem cells as a safe delivery system of gold nanorods for targeted photothermal therapy of gastric cancer†
Received
30th August 2016
, Accepted 13th November 2016
First published on 16th November 2016
Abstract
Human-induced pluripotent stem cells (iPS) possess an intrinsic tumor tropism ability. However, iPS cells are impeded in clinical applications of tumor therapy due to the formation of teratomas and their survival in normal organs such as the liver, lungs, spleen and kidneys. Mitomycin C (MMC) can overcome this limitation by suppressing iPS proliferation. Herein, we fabricated a safe delivery system of iPS cells treated with MMC loading with gold nanorods (AuNRs) for the targeted photothermal treatment of gastric cancer. Our results showed that the tumor cells were efficiently killed by the heat generated from the gold nanorods, and the iPS cells ultimately died due to the action of MMC seven days after the photothermal treatment. This suggested that pre-treated iPS cells with MMC can be used as a novel and safe approach for targeted tumor therapy. This paves the road for clinical translation in the future.
Introduction
Stem cell-based cancer therapy is emerging as a promising strategy in recent years.1,2 Multiple types of stem cells display intrinsic tropism towards tumors and the SDF1/CXCR4 axis plays an important role in the process.2–4 Briefly, tumor cells express SDF1, and the SDF1 receptor CXCR4 is expressed on stem cells. When stem cells are engineered to deliver therapeutic agents, these cellular vehicles can effectively migrate to the tumor site following the SDF1 concentration gradient. Due to the high internal pressure in tumors, it is very difficult for anticancer agents, such as nanoparticles, to gain entry into the tumor. However, stem cells with anticancer agents can disseminate in whole solid tumors and achieve more effective therapeutic results.5,6
Researchers have used mesenchymal stem cells (MSCs)7–11 and neural stem cells (NSCs)5,12–14 as micro-vehicle anticancer agents. However, it is a challenge to obtain adult stem cells from human bodies. Induced pluripotent stem (iPS) cells appear more attractive for clinical applications since these cells can be relatively easily generated through reprogramming of differentiated somatic cells with transcription factors.15 iPS cells may be good candidates for stem cell-based cancer therapy. The feasibility of using iPS6 or iPS-derived stem cells16 as vehicles for antitumor agents have been tested. Albeit iPS cells can effectively target a tumor site and will be killed by the photothermal energy of the released gold nanoparticles, as seen in our previous research.6 Similar to what has been observed in other studies, we have also observed that iPS cells can get distributed in organs such as liver, lungs, spleen, and kidney.16 As we know, iPS cells possess proliferation and differentiation ability.15 The residual stem cells in the abovementioned organs may also induce an unintended side effect. Therefore, ideally, when stem cells are used as a vehicle for anticancer agents, stem cell death should occur in the entire body after the tumor therapy results are achieved. To achieve these goals, Zhu et al. used engineered NSCs derived from iPS. In their experiments, there was no toxicity to normal non-targeted organs, but therapeutic effects were observed in tumors in a pH-dependent manner.17
In this study, we tried to use iPS cells pre-treated with mitomycin C (MMC) as an alternative source for the cellular vehicle. MMC is a DNA alkylating and crosslinking agent and has been used to arrest cell proliferation.18,19 In culturing of embryonic stem cell and iPS cells, MMC has been used to treat fibroblasts to restrict fibroblast proliferation; however, the cells have also expressed some proteins to support stem cell growth.20 Cells have been observed to stop proliferation and die several days after MMC treatment. Nanoparticles have been widely studied in tumor therapy. Particularly, gold nanoparticles have the ability to convert laser light to thermal energy which can be used for tumor photothermal therapy.5,6,21–25 In our previous study, we found that silica-coated gold nanorods (AuNRs@SiO2) conjugated with a CXCR4 antibody (AuNRs@SiO2@CXCR4) can improve the loading efficiency of nanoparticles into iPS.6 In the present study, we used iPS cells expressing enhanced green fluorescent protein (iPS-eGFP) as micro-vehicle cells for AuNRs@SiO2@CXCR4. In a gastric cancer model, we found that the iPS-eGFP cells in normal organs died after tumor photothermal therapy (Scheme 1). Our findings demonstrate that using pre-treated iPS cells with MMC is a safe strategy for nanoparticle vehicle.
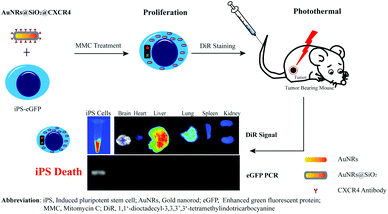 |
| | Scheme 1 The schematic of Mitomycin C-treated human-induced pluripotent stem cells as a safe micro-vehicle in a gastric cancer model. | |
Experimental
Materials
Gold (III) chloride trihydrate (HAuCl4·3H2O, 99%), cetyltrimethyl ammonium bromide (CTAB), sodium borohydride (NaBH4), poly(vinylpyrrolidone) (PVP), tetraethylorthsilicate (TEOS), tetrahydrofuran, 3-aminopropyltrimethoxysilane (APTS) and succinic anhydride were obtained from Sinopharm® Chemical Reagent Co., Ltd (Shanghai, China). 1-Ethyl-3-(3-dimethyl aminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) were obtained from Aladdin Reagent Co., Ltd (Shanghai, China). Mitomycin C was purchased from Zhejiang Hisun Pharmaceutical Co., Ltd (Zhejiang, China). Accumax cell detachment solution was obtained from Chemicon®. 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine (DiR) was purchased from Invitrogen. Anti-mouse CXCR4 antibody, anti-mouse GAPDH antibody and goat anti-mouse HRP second antibody were purchased from Proteintech® Group, Inc. (Chicago, USA). A whole RNA extract kit was purchased from Shanghai Promega biological products Ltd (Shanghai, China). A first strand cDNA synthesis kit ReverTra Ace-α- was purchased from Toyobo Co., Ltd (Kita-ku Osaka, Japan). A PCR proliferation kit and HE staining solution were obtained from Beyotime Biotechnology (Jiangsu, China). Deionized water with a resistivity of 18.2 MΩ was used in all of the experiments (Millipore® Co., USA).
AuNRs@SiO2@CXCR4 synthesis
AuNRs were synthesized according to the seed-mediated template-assisted protocol. The synthesis of a mesoporous silica coating on AuNRs was carried out according to a previous protocol,26 with some modifications. A 40 mL aliquot of an AuNRs solution was centrifuged twice at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to remove excess reagents and was redispersed in 20 mL deionized water, followed by the addition of 4 mL of a 25 mg mL−1 PVP aqueous solution before gentle stirring for 15 h. The mixture was centrifuged at 8500 rpm for 6 min and dispersed in 10 mL of deionized water. Then, 100 μL 0.1 M NaOH solution was added under stirring. Next, 15 μL of 20% TEOS in methanol was injected three times under gentle stirring at 30 minute intervals. The mixed solution was then gently stirred for 3 days at 28 °C. After this step, the synthesized AuNRs@SiO2 were collected by centrifugation at 11000 rpm for 6 min, washed twice with deionized water and ethanol, and then gently suspended into 5 mL of N,N-dimethylformamide for further reaction. A 400 μL aliquot of APTS and 168.89 μg of succinic anhydride were added in 20 mL of N,N-dimethylformamide under vigorous stirring at 65 °C for 3 h. Subsequently, 5 mL of AuNRs@SiO2 solution was added and vigorously stirred at 65 °C for 5 h. Finally, the solution was washed with deionized water, and dried at −50 °C for 12 h to obtain AuNRs@SiO2–COOH. Covalent binding of the CXCR4 antibody to AuNRs@SiO2–COOH was carried out according to the standard EDC–NHS reaction method. AuNRs@SiO2–COOH was activated by an EDC/NHS solution for 30 min. Next, 1.5 μL of CXCR4 antibody was added to form a mixed solution and allowed to react under gentle stirring at room temperature overnight. It was then washed three times with deionized water and centrifuged at 11000 rpm each time to remove the unreacted chemical. Finally, the purified AuNRs@SiO2@CXCR4 was redispersed into 5 mL of PBS for further characterization and application. The morphology of the nanoparticles was examined using transmission electron microscopy and UV-Vis spectra.
000 rpm to remove excess reagents and was redispersed in 20 mL deionized water, followed by the addition of 4 mL of a 25 mg mL−1 PVP aqueous solution before gentle stirring for 15 h. The mixture was centrifuged at 8500 rpm for 6 min and dispersed in 10 mL of deionized water. Then, 100 μL 0.1 M NaOH solution was added under stirring. Next, 15 μL of 20% TEOS in methanol was injected three times under gentle stirring at 30 minute intervals. The mixed solution was then gently stirred for 3 days at 28 °C. After this step, the synthesized AuNRs@SiO2 were collected by centrifugation at 11000 rpm for 6 min, washed twice with deionized water and ethanol, and then gently suspended into 5 mL of N,N-dimethylformamide for further reaction. A 400 μL aliquot of APTS and 168.89 μg of succinic anhydride were added in 20 mL of N,N-dimethylformamide under vigorous stirring at 65 °C for 3 h. Subsequently, 5 mL of AuNRs@SiO2 solution was added and vigorously stirred at 65 °C for 5 h. Finally, the solution was washed with deionized water, and dried at −50 °C for 12 h to obtain AuNRs@SiO2–COOH. Covalent binding of the CXCR4 antibody to AuNRs@SiO2–COOH was carried out according to the standard EDC–NHS reaction method. AuNRs@SiO2–COOH was activated by an EDC/NHS solution for 30 min. Next, 1.5 μL of CXCR4 antibody was added to form a mixed solution and allowed to react under gentle stirring at room temperature overnight. It was then washed three times with deionized water and centrifuged at 11000 rpm each time to remove the unreacted chemical. Finally, the purified AuNRs@SiO2@CXCR4 was redispersed into 5 mL of PBS for further characterization and application. The morphology of the nanoparticles was examined using transmission electron microscopy and UV-Vis spectra.
Cell culture
The human gastric cancer cell line MGC803 was cultured in Dulbecco's DMEM supplemented with 10% FBS. The iPS-eGFP cells were cultured in an iPS medium. Both cell lines were maintained at 37 °C in a humidified incubator containing 5% CO2.
Animal and tumor model
Female BALB/c athymic nude mice of 6–8 weeks of age were purchased from Shanghai LAC Laboratory Animal Co., Ltd and housed in an SPF grade animal center. All animals received care in compliance with the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University. The mice were anesthetized and 2 × 106 MGC803 cells suspended in 50 μL of PBS were subcutaneously transplanted into the mice.
iPS-eGFP co-culture with AuNRs@SiO2@CXCR4
According to our previous results,6 40 μg mL−1 of AuNRs was an optimal concentration. In this study also, we used 40 μg mL−1 of AuNRs as a cultured concentration, and the iPS cells were co-cultured with AuNRs@SiO2@CXCR4 for 24 h.
MMC-treated iPS-eGFP cells
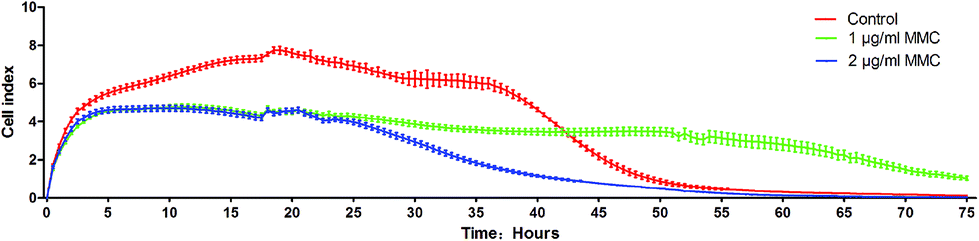
The iPS cells loaded with AuNRs@SiO2@CXCR4 were co-cultured with 1 μg mL−1 and 2 μg mL−1 MMC or with the same concentration of PBS as a control for 2 hours. Then, the iPS cells were dispersed into single cells by the Accumax cell detachment solution. Each group has 5 E-Plate-16 wells and 3000 cells were added in one well. The three groups of cells were observed by the xCELLigence DP system. The culture medium was changed every 24 h.
Two-photon microscopy and quantification of uptake amount
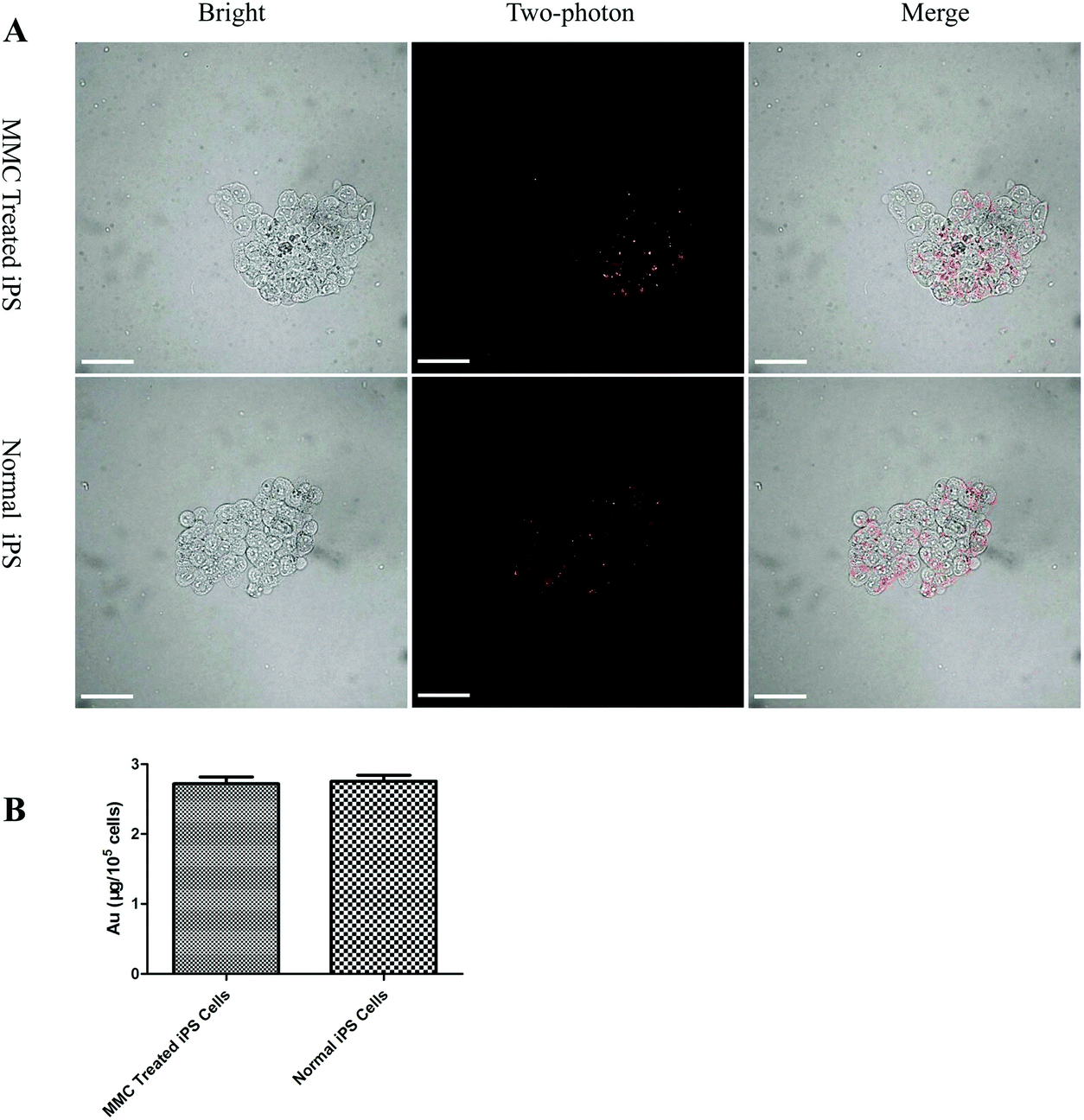
The iPS cells were incubated with AuNRs@SiO2@CXCR4 for 24 h, washed three times with PBS to remove unloaded nanoparticles, and treated with 1 μg mL−1 of MMC for 2 h. After 24 h, two-photon fluorescent images of AuNRs were obtained using a two-photon fluorescent microscope with excitation at 780 nm and emission at 601–657 nm. The AuNRs@SiO2@CXCR4-loaded MMC-treated iPS cells were diluted into 1 mL of PBS with 2% HNO3. The amount of gold was detected using inductively coupled plasma-mass spectrometry (ICP-MS).
DiR label iPS-eGFP cells
MMC-treated iPS-eGFP cells were labeled with 5 μg mL−1 of DiR overnight. Then, the cultured iPS-eGFP was dispersed into single cells for the following experiment.
In vitro transwell boyden migration assay
As previously described,6 500 μL of target media (either DMEM or MGC803 culture supernatant medium) per well was added in a 24-well microplate. MMC-treated iPS cells in DMEM were placed in the upper transwell chamber and incubated at 37 °C overnight. After incubation, the upper cells were removed using a cotton swab. The transwell chambers were placed in a new 24-well cell culture plate containing DAPI, incubated for 5 min, and washed twice with PBS. Then, the cells were observed under a fluorescence microscope.
In vivo image of iPS-eGPF in tumor bearing mice
1.8 × 106 DiR-labeled iPS-eGFP cells were injected into the nude mice bearing gastric tumors. After 3 days, an in vivo imaging system (excitation: 750 nm; emission: 780 nm) was used to determine the whole-animal distribution of the iPS-eGFP cells. After 7 days, ex vivo organ imaging was performed.
Tumor photothermal therapy
The nude mice bearing MGC-803 tumors were anaesthetized and tail vein injections of PBS, free AuNRs@SiO2 (50 μg) and 1.8 × 106 MMC-treated iPS cells loaded with AuNRs@SiO2@CXCR4 were performed. Three days later, the tumor region was irradiated with an 808 nm laser. The laser treatment was carried out for 3 min at a power density of 1.5 W cm−2 (30 s interval after every 1 min of exposure). An infrared thermal imaging camera was used to monitor the temperature of the tumor sites.
eGFP PCR
The total RNA of organs and iPS cells were extracted and then reverse transcribed to obtain cDNA. A pair primer (forward: AAGGGCGAGGAGCTGTTCA; reverse: TTACTTGTACAGCTCGTCCATGC) of eGFP was designed to amplify a 720 bp length of the eGFP gene.
Western blot
Total proteins were harvested from normal iPS cells and MMC-treated iPS cells. CXCR4 and GAPDH were detected by Western blot.
HE staining
The 4% paraformaldehyde-fixed tumor was cut, placed on a 5 μm slide, and then stained with hematoxylin and eosin.
Statistical analysis
Data are presented as mean ± SD, unless otherwise stated. Statistical significance was determined using a two-tailed Student's t-test (*P < 0.05).
Results and discussion
The fabrication of AuNRs@SiO2@CXCR4
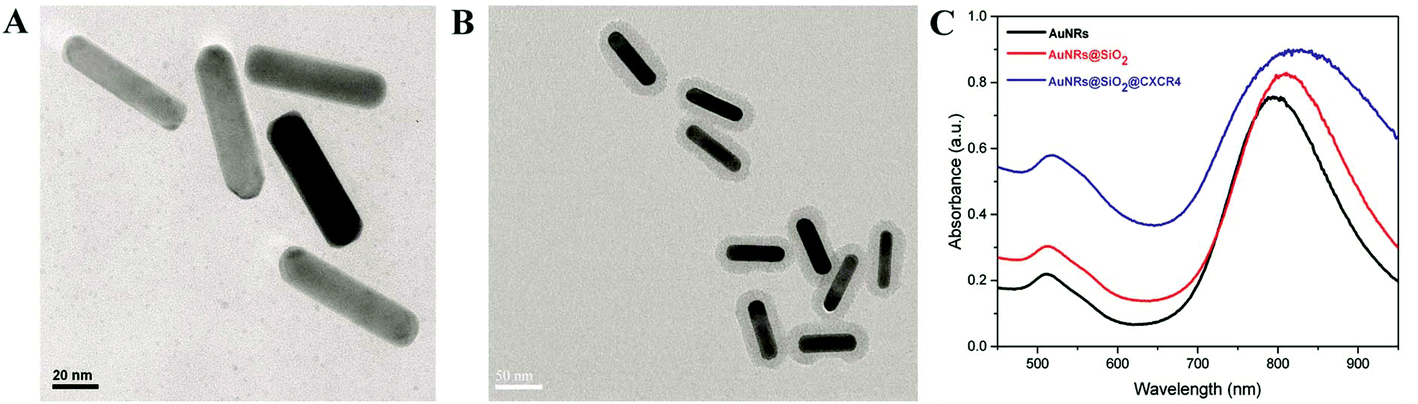
According to our previously reported method,6 CTAB-stabilized AuNRs were synthesized by the seed-mediated growth method as shown in Fig. 1A. For the conjugate CXCR4 antibody, the AuNRs were coated with SiO2 (Fig. 1B). Then, AuNRs@SiO2@CXCR4 were fabricated using APTES and succinic anhydride. As shown in Fig. 1C, the UV-Vis spectra absorbance peak slightly red-shifted after AuNRs were coated with SiO2 and conjugated with the CXCR4 antibody. The conjugated CXCR4 antibody of AuNRs will help more AuNRs enter CXCR4 expressed iPS cells and thus elevate the photothermal therapeutic efficiency.
 |
| | Fig. 1 AuNRs@SiO2@CXCR4 synthesis. TEM images of (A) AuNRs and (B) AuNRs@SiO2 (C) UV-Vis spectra of AuNRs, AuNRs@SiO2 and AuNRs@SiO2@CXCR4. | |
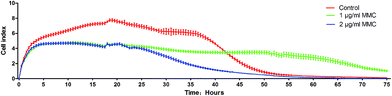 |
| | Fig. 2 The different proliferation of normal iPS cells and iPS cells treated with 1 μg mL−1 and 2 μg mL−1 MMC for 2 hours. | |
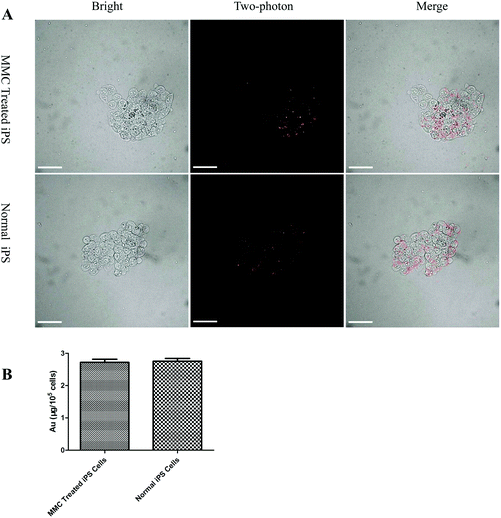 |
| | Fig. 3 Amount of AuNRs in iPS cells. (A) Cellular uptake of AuNRs@SiO2@CXCR4 in MMC-treated iPS cells (upper panel) and normal iPS cells (lower panel). All scale bars are 100 μm. (B) ICP-MS result of the amount of Au per 105 iPS cells. | |
The tumor tropism ability assay
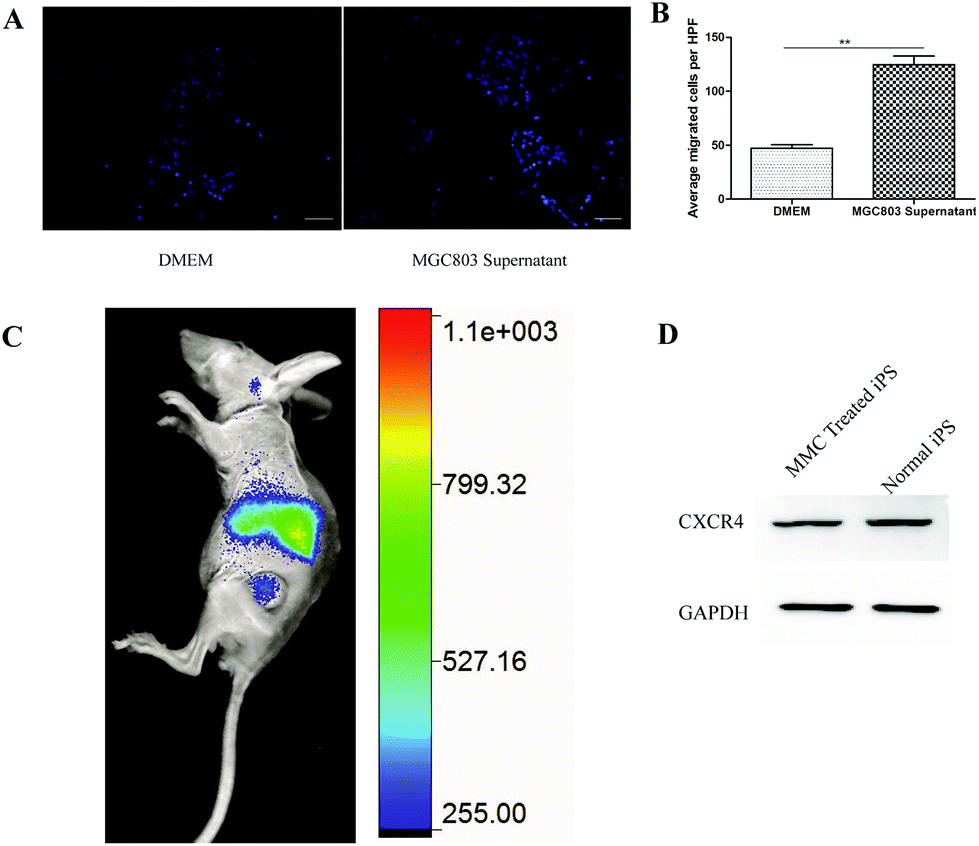
A transwell experiment was used to detect the migration of mouse-induced pluripotent stem cells into the glioma-conditioned medium.33In vitro, we used the transwell to indicate that MMC treatment of iPS cells had gastric tumor tropism (Fig. 4A). As shown in Fig. 4B, more MMC-treated iPS cells transferred from the upper chamber to the lower chamber if the lower chamber contained MGC803 culture supernatant. As is well known, several tumor cells2,4 secrete SDF1, which induce the migration of the expressed CXCR4 cells. In this transwell experiment, the secreted SDF1 by MGC803 may play an important role in inducing more iPS cells to migrate to the lower chamber. Although the iPS cells express eGFP, the eGFP fluorescence (excitation: 470 nm; emission: 535 nm) signal cannot penetrate the bodies of the mice, and the auto fluorescence of organs will also disturb the eGFP fluorescent signal. DiR is an ideal in vivo imaging dye for the near infrared excitation/emission (750/780 nm), which easily penetrates mouse organs with almost no auto fluorescence at these wavelengths.4 We used DiR to label MMC-treated iPS-eGFP cells for rapid in vivo observation. After DiR staining, 1.8 × 106 iPS-eGFP cells and the same volume of PBS and 50 μg of AuNRs@SiO2 (control) were injected into tumor bearing mice through the tail vein. After 3 days, the in vivo imaging results demonstrated that the MMC-treated iPS-eGFP also displayed tumor tropism as the DiR signal was mainly located in liver and tumor (Fig. 4C). Since the SDF1/CXCR4 axis plays an important role in the tumor tropism process,2 we detected the CXCR4 expression of MMC-treated iPS-eGFP after 3 days and found that the expression of CXCR4 protein did not change compared with that in the normal iPS cells (Fig. 4D). This result can explain the unaffected tumor tropism ability after MMC treatment in some way.
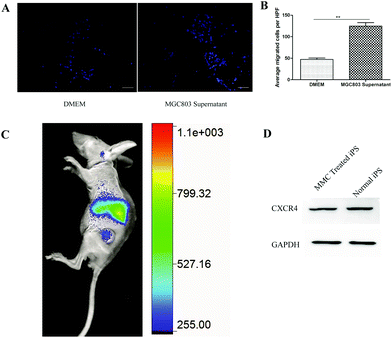 |
| | Fig. 4 The ability of MMC-treated iPS cells target to tumor. (A) The target media of the lower chamber of the transwell are DMEM (left panel) and MGC803 (right panel) culture supernatant. (B) The quantitative result for the iPS cells. (C) MMC treated iPS cells can target to tumor site. (D) MMC treated iPS and normal iPS have same CXCR4 expression, GAPDH as control. | |
Photothermal therapy of tumor
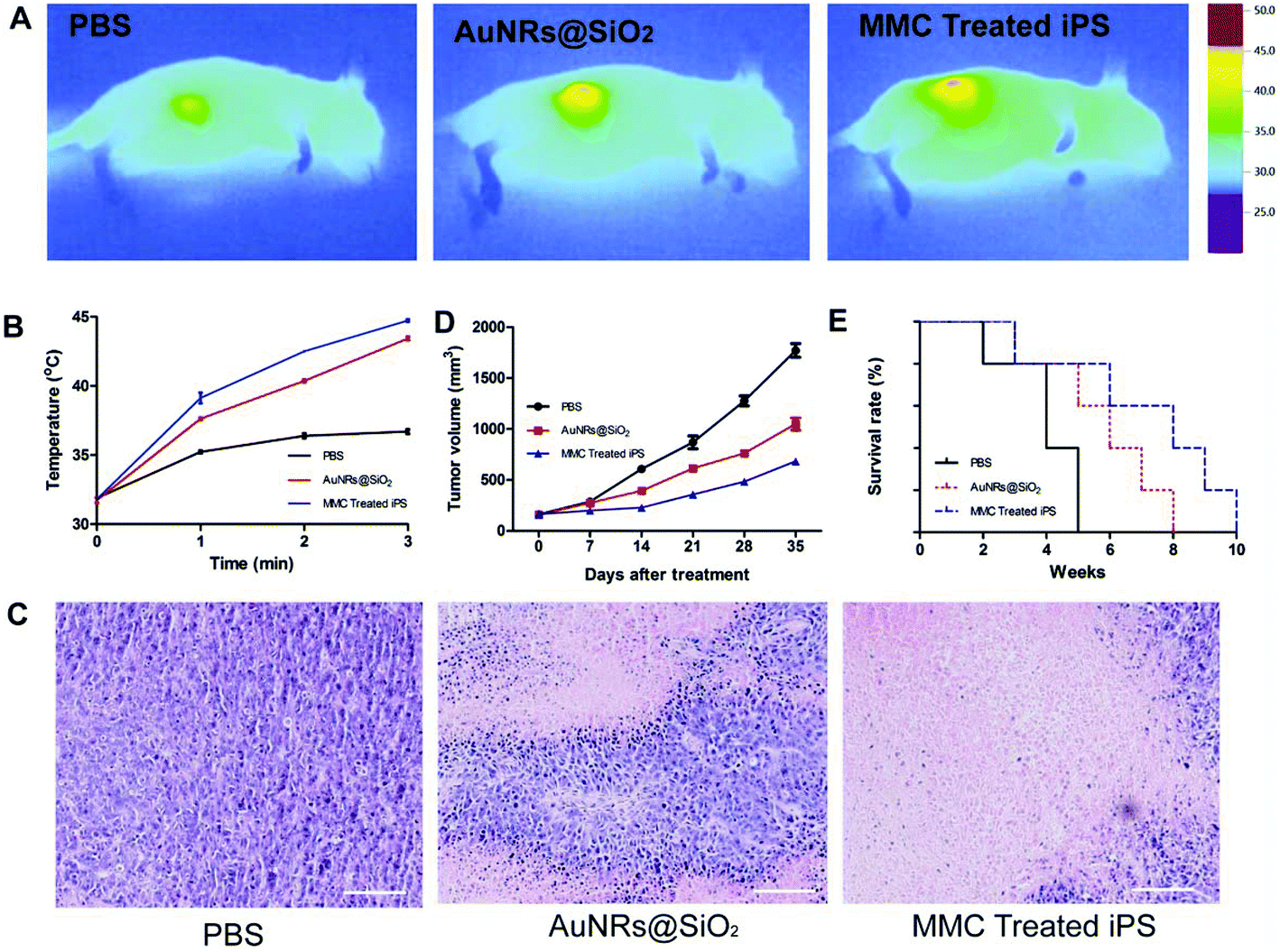
Three days after iPS cell injection, an 808 nm laser was used for photothermal therapy of the tumor. Song et al.34 have demonstrated that rodent tumors can be destroyed by heating at 42–43 °C with minimal damage to normal tissue vasculature, and higher temperatures likely cause vascular damage resulting in tumors becoming hypoxic. In order to control overheating from photothermal treatment, we applied the strategy6 that was carried out for 3 min (30 s interval after every 1 min of exposure) at a power density of 1.5 W cm−2. Stem cells loaded with anticancer agents can disseminate in whole solid tumors, and then achieve a more effective therapy result.5,6 MMC- treated iPS cell group achieve better treatment results. As shown in Fig. 5A and B, compared to the PBS and AuNRs@SiO2 group, the tumor temperature increase was higher` in the MMC-treated iPS cell group. Seven days after photothermal treatment, some photothermal-treated mice were sacrificed to remove the tumor and organs (brain, heart, liver, lung, spleen and kidney) and the others were used to observe the survival time. The tumor was fixed in 4% paraformaldehyde for HE staining. Organs were used to extract RNA, which was then reverse transcribed to cDNA after ex vivo imaging. As shown in Fig. 5C, more damaged cells were presented in the MMC-treated iPS cell group than in the AuNRs@SiO2 and PBS groups. The tumor size reduced (Fig. 5D), and survival time was also prolonged in the MMC-treated iPS cell group (Fig. 5E).
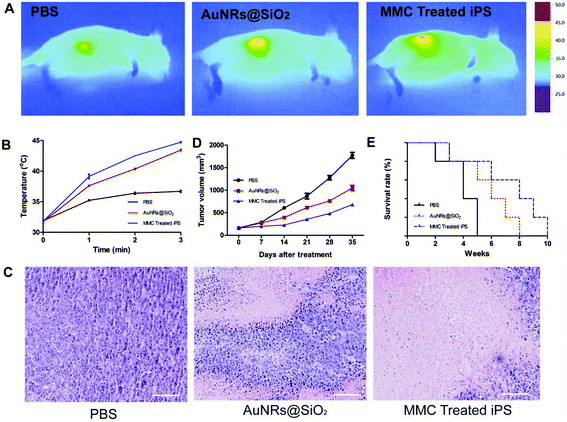 |
| | Fig. 5 Tumor photothermal therapeutic results. (A) Infrared microscopic imaging: NIR laser irradiation of the tumor bearing mice after injections of PBS, AuNRs@SiO2 and MMC-treated iPS cells after 3 days. (B) The corresponding temperature rise profiles at the tumor site (C) HE staining after phototherapy treatment. All scale bars are 100 μm. (D, E) Tumor size and survival time of mice injected with PBS, AuNRs@SiO2 and MMC-treated iPS cells. | |
The state of iPS distributed in normal organs
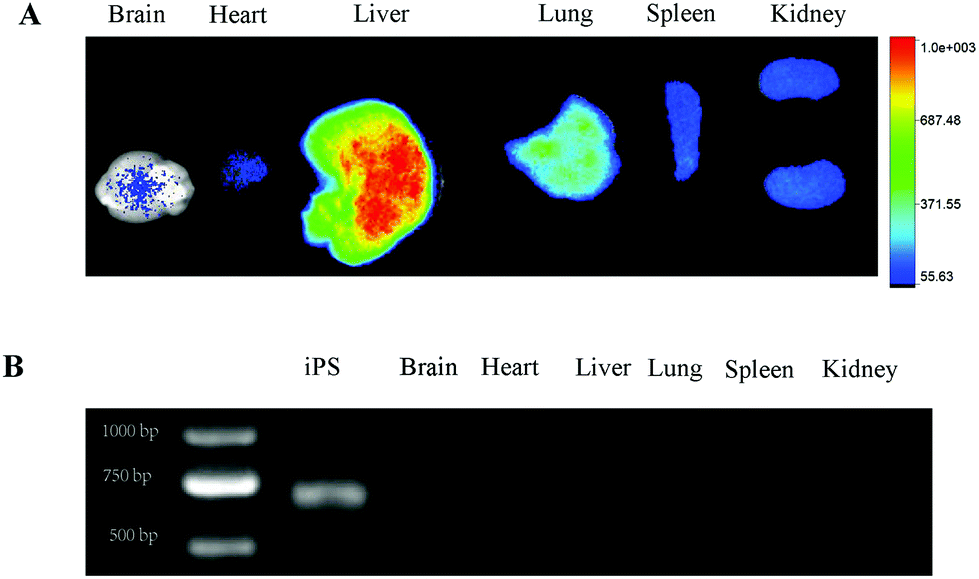
To determine the distribution of iPS-eGFP in the body, ex vivo imaging of organs was performed. Fig. 6A showed that in the iPS-eGFP group, the DiR signal demonstrated that the iPS cells were still retained in normal organs. Our preliminary experiment has indicated that the DiR signal can be detected even after iPS cell death one month in vitro. Therefore, in order to verify the state of iPS-eGFP cells in the organs in vivo, observing the fluorescent signal was not the correct way as the fluorescent signal exists even after cell death. As the iPS cells have eGFP gene, eGFP gene can be detected if iPS cells survive and vice versa. It is feasible to demonstrate the state of iPS cell survival by detecting the eGFP gene expression. As shown in Fig. 6B, except in the iPS cells (positive control), there was no eGFP expression in organs. The results demonstrate that the MMC-treated iPS cells died in the organs. MMC treated iPS cells cannot form teratomas. When applying iPS cells as a cargo carrier, the clinical immune response of the patients should be considered. Fortunately, the body lacks an immune response to iPS cells as iPS cells are derived from the body.35
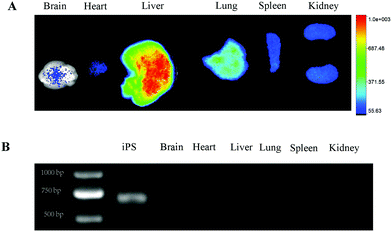 |
| | Fig. 6 The MMC-treated iPS cell death in organs. (A) The DiR signal of brain, heart, liver, lung, spleen and kidney. (B) eGFP PCR results of iPS cells, brain, heart, liver, lung, spleen and kidney. | |
Conclusions
In this study, we attempted to confirm the feasibility of using MMC-treated iPS cells as an alternative gold nanoparticle micro vehicle for gastric cancer therapy. We found that both MMC-treated iPS cells and normal iPS cells have tumor tropism ability both in vitro and in vivo and also show CXCR4 expression. The MMC-treated iPS cells showed good tumor photothermal therapy results by loading AuNRs. The most important is the MMC-treated iPS cells death in normal organs and cannot form teratoma. In conclusion, our findings solves the hurdle of iPS cell-based cancer therapy and paves the road for clinical translation in the future.
Acknowledgements
This study was supported by the National Key Basic Research Program (973 Project) (Project No. 2010CB933901), National Natural Scientific Foundation of China (Grant No. 81225010, 81028009, and 31170961), 863 project of China (Project No. 2014AA020700), and Shanghai Science and Technology Fund (13NM1401500).
Notes and references
- M. Roger, A. Clavreul, M. C. Venier-Julienne, C. Passirani, L. Sindji, P. Schiller, C. Montero-Menei and P. Menei, Biomaterials, 2010, 31, 8393–8401 CrossRef CAS PubMed.
- D. W. Stuckey and K. Shah, Nat. Rev. Cancer, 2014, 1–9 Search PubMed.
- M. Kucia, R. Reca, K. Miekus, J. Wanzeck, W. Wojakowski, A. Janowska-Wieczorek, J. Ratajczak and M. Z. Ratajczak, Stem cells, 2005, 23, 879–894 CrossRef CAS PubMed.
- J. Ruan, H. Song, C. Li, C. Bao, H. Fu, K. Wang, J. Ni and D. Cui, Theranostics, 2012, 2, 618–628 CrossRef CAS PubMed.
- R. Mooney, L. Roma, D. Zhao, D. Van Haute, E. Garcia, S. U. Kim, A. J. Annala, K. S. Aboody and J. M. Berlin, ACS Nano, 2014, 12450–12460 CrossRef CAS PubMed.
- Y. Liu, M. Yang, J. Zhang, X. Zhi, C. Li, C. Zhang, F. Pan, K. Wang, Y. Yang, J. Martinez De La Fuentea and D. Cui, ACS Nano, 2016, 10, 2375–2385 CrossRef CAS PubMed.
- G. Xu, X.-D. Jiang, Y. Xu, J. Zhang, F.-H. Huang, Z.-Z. Chen, D.-X. Zhou, J.-H. Shang, Y.-X. Zou, Y.-Q. Cai, S.-B. Kou, Y.-Z. Chen, R.-X. Xu and Y.-J. Zeng, Cell Biol. Int., 2009, 33, 466–474 CrossRef CAS PubMed.
- P. Gao, Q. Ding, Z. Wu, H. Jiang and Z. Fang, Cancer Lett., 2010, 290, 157–166 CrossRef CAS PubMed.
- L. Li, Y. Guan, H. Liu, N. Hao, T. Liu, X. Meng, C. Fu, Y. Li, Q. Qu, Y. Zhang, S. Ji, L. Chen, D. Chen and F. Tang, ACS Nano, 2011, 5, 7462–7470 CrossRef CAS PubMed.
- E. K. Mader, G. Butler, S. C. Dowdy, A. Mariani, K. L. Knutson, M. J. Federspiel, S. J. Russell, E. Galanis, A. B. Dietz and K.-W. Peng, J. Transl. Med., 2013, 11, 20 CrossRef CAS PubMed.
- C. Altaner, V. Altanerova, M. Cihova, K. Ondicova, B. Rychly, L. Baciak and B. Mravec, Int. J. Cancer, 2014, 134, 1458–1465 CrossRef CAS PubMed.
- K. Aboody, A. Brown, N. Rainov, K. Bower, S. X. Liu, W. Yang, J. Small, U. Herrlinger, V. Ourednik and P. Black,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2000, 23, 12846–12851 CrossRef PubMed.
- A. B. Brown, W. Yang, N. O. Schmidt, R. Carroll, K. K. Leishear, N. G. Rainov, P. M. Black, X. O. Breakefield and K. S. Aboody, Hum. Gene Ther., 2003, 14, 1777–1785 CrossRef CAS PubMed.
- K. S. Aboody, J. Najbauer, M. Z. Metz, M. D'Apuzzo, M. Gutova, A. J. Annala, T. W. Synold, L. A. Couture, S. Blanchard, R. A. Moats, E. Garcia, S. Aramburo, V. V. Valenzuela, R. T. Frank, M. E. Barish, C. E. Brown, S. U. Kim, B. Badie and J. Portnow, Sci. Transl. Med., 2013, 5, 184ra59–184ra59 Search PubMed.
- K. Takahashi and S. Yamanaka, Cell, 2006, 126, 663–676 CrossRef CAS PubMed.
- J. Yang, D. Lam, S. Goh, E. Lee and Y. Zhao, Stem cells, 2012, 30, 1021–1029 CrossRef CAS PubMed.
- D. Zhu, D. H. Lam, Y. I. Purwanti, S. L. Goh, C. Wu, J. Zeng, W. Fan and S. Wang, Mol. Ther., 2013, 21, 1621–1630 CrossRef CAS PubMed.
- D. Werner, A. Atmaca, C. Pauligk, A. Pustowka, E. Jäger and S.-E. Al-Batran, Cancer Med., 2013, 2, 325–333 CrossRef CAS PubMed.
- C. Li, S. Liang, C. Zhang, Y. Liu, M. Yang, J. Zhang, X. Zhi, F. Pan and D. Cui, Biomaterials, 2015, 54, 177–187 CrossRef CAS PubMed.
- A. Nieto, C. M. Cabrera, P. Catalina, F. Cobo, A. Barnie, J. L. Cortés, A. Barroso del Jesus, R. Montes and A. Concha, Cell Biol. Int., 2007, 31, 269–278 CrossRef CAS PubMed.
- C. C. Bao, J. Conde, F. Pan, C. Li, C. L. Zhang, F. R. Tian, S. J. Liang, J. M. de la Fuente and D. Cui, Nano Res., 2016, 9, 1043–1056 CrossRef CAS.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Lasers Med. Sci., 2008, 23, 217–228 CrossRef PubMed.
- T. Stuchinskaya, M. Moreno, M. J. Cook, D. R. Edwards and D. A. Russell, Photochem. Photobiol. Sci., 2011, 10, 822–831 CAS.
- J. Han, J. Zhang, M. Yang, D. Cui and J. M. de la Fuente, Nanoscale, 2015, 8, 492–499 RSC.
- W. Hou, X. Zhao, X. Qian, F. Pan, C. Zhang, Y. Yang, J. M. de la Fuente and D. Cui, Nanoscale, 2016, 8, 104–116 RSC.
- I. Gorelikov and N. Matsuura, Nano Lett., 2008, 8, 369–373 CrossRef CAS PubMed.
- C. Bird and S. Kirstein, Nat. Methods, 2009, 6, 5–6 Search PubMed.
- A. Ozdemir and M. Ark, Niche J., 2014, 2, 15–17 CrossRef.
- K. Moodley, C. E. Angel, M. Glass and E. S. Graham, J. Neurosci. Methods, 2011, 200, 173–180 CrossRef CAS PubMed.
- N. J. Durr, T. Larson, D. K. Smith, B. A. Korgel, K. Sokolov and A. Ben-Yakar, Nano Lett., 2007, 7, 941–945 CrossRef CAS PubMed.
- H. F. Wang, T. B. Huff, D. A. Zweifel, W. He, P. S. Low, A. Wei and J. X. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15752–15756 CrossRef CAS PubMed.
- W. Zhang, Y. Ji, X. Wu and H. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 9856–9865 CAS.
- S. Koizumi, C. Gu, S. Amano, S. Yamamoto, H. Ihara, T. Tokuyama and H. Namba, Oncol. Lett., 2011, 2, 283–288 CAS.
- C. W. Song, H. J. Park, C. K. Lee and R. Griffin, Int. J. Hyperthermia, 2005, 21, 761–767 CrossRef CAS PubMed.
- P. Guha, J. W. Morgan, G. Mostoslavsky and N. P. Rodrigues, Cell Stem Cell, 2013, 12, 407–412 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr06851k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.