
Nitrogen-doped graphene oxide for effectively removing boron ions from seawater†
Fuming
Chen
,
Lu
Guo
,
Xiaoming
Zhang
,
Zhi Yi
Leong
,
Shengyuan
Yang
and
Hui Ying
Yang
*
Pillar of Engineering Product Development, Singapore University of Technology and Design, Singapore 487372, Singapore. E-mail: yanghuiying@sutd.edu.sg
First published on 22nd November 2016
Abstract
Elemental boron exists in the form of boric acid or borate salts in aqueous solution. The human body is very sensitive to the amount of boron, and boron contamination in drinking water affects our health adversely. However, boron is not easily removed due to its small ionic size and is a problem to water treatment systems. Herein, we report a new method to remove boron using nitrogen-doped graphene oxide (N-GO). The maximum adsorption capacity we have obtained is 58.7 mg g−1 and this makes N-GO one of the best materials to adsorb boron. Real seawater with 5 mg L−1 as boron is used as a feed for testing and the absorption capacity is shown to be up to 2.42 mg g−1. This high adsorption capacity is mainly attributed to the large amount of hydroxyl groups distributed across the high surface area of graphene oxide and the enhanced adsorption that results from nitrogen-doped sites. Once N-GO is saturated with boron ions, it can be easily regenerated via acid treatment. Our proposed technique has high commercial value and we believe that it is very valuable to the water treatment industry.
1. Introduction
Boron is a naturally occurring element in the environment and in aqueous form, it exists as boric acid or borate salts. At high enough concentrations, it poses an environmental risk and when present in drinking water, it is a health hazard. In the semiconductor industry, large amounts of pure water are used and the boron content in the water used needs to be in the ppb range since high concentrations of boron may cause defects in chip manufacturing.1 There are other areas where the boron concentration has to be within a specific range. For example, the boron concentration in the water used in irrigation cannot be so low as to cause stunted growth, yet it cannot be so high (>1 ppm) as to cause death to plants.2–6 For potable water, the World Health Organization (WHO) recommends a maximum allowable concentration of 2.4 mg L−1.7 Any higher concentration will cause problems to the cardiovascular, coronary, nervous and reproductive systems. Hence, it is crucial to remove boron in water treatment processes and one major process is desalination.Common desalination strategies include reverse osmosis, capacitive desalination, and electro-dialysis.8–13 However, none of these technologies are able to efficiently remove boron from seawater due to its small size and uncharged state. Hence, additional post-treatment processes are required to remove boron. These post-treatment processes include electrocoagulation, chemical precipitation, ion exchange processes, and liquid–liquid extraction.8–35 Electrocoagulation has been proven to be highly effective for solutions with a high boron concentration.14,15,17,24 However, it has also been shown that these two methods are inefficient for solutions with low boron concentrations.14,15,17,24 On the other hand, the use of reverse osmosis membranes and electro-dialysis methods is very effective for ions such as chloride, sodium, sulfate, magnesium, calcium, potassium and bicarbonate,10,13,19,23,31,32,35 but fails to reject boron as boric acid can easily diffuse through the membranes at neutral pH levels. In order to achieve high boron rejection, adjustment of pH is required. Presently, removal of boron is most effective through ion exchange resins using an anionic exchanger and is seen as the most promising method in the industry.11,13,16,20,29 This resin based adsorption method utilizes chelating resins with ligands containing –OH hydroxyl groups, which are highly selective towards boron via the formation of borate complexes.
In this paper, we report the synthesis of a novel boron removal medium, namely nitrogen-doped graphene oxide (N-GO), via a simple two-step process. Boron can be efficiently removed through the formation of complexes between boron species and N-GO. The enhanced boron adsorption capacity is attributed to the enhanced surface charge that promotes electrostatic attraction of boron ions to the N-GO surface. At low boron concentrations (20 ppm), we can obtain an adsorption capacity of 6.55 mg g−1 and the adsorption capacity is as high as 58.70 mg g−1. In real seawater with 5 mg L−1 as boron, the adsorption capacity is up to 2.42 mg g−1 for N-GO. N-GO is reusable and the adsorptive properties can be regenerated using an acid treatment followed by a DI water rinse. Other environmental factors such as pH levels were also investigated and the results are reported. Significantly, N-GO can be directly used in seawater without any treatment, which is not common for most of the other current technologies. This facile method of producing effective materials for removing boron ions will be of great significance to the water industry.
2.Materials and method
2.1. Materials
Graphite powder, aqueous ammonia (NH3·H2O) (28 wt%), hydrogen peroxide (H2O2) (30 wt%), sodium nitrate (NaNO3), K2S2O8, P2O5 and potassium permanganate (KMnO4) were purchased from Sigma-Aldrich. Nitric acid (HNO3) (65%) and sulfuric acid (H2SO4) (98 wt%) were obtained from Merck. All reagents were used as received without further purification.2.2. Preparing graphene oxide
Preparation of graphene oxide (GO): graphene oxide (GO) was synthesized using natural graphite powders by a modified Hummers’ method.36–38 Briefly, 2 g of graphite powder was added into a mixture of 12 mL of 98% H2SO4, 2.5 g K2S2O8, and 2.5 g of P2O5. The solution was kept under stirring at 80 °C for 4.5 h followed by thorough washing with DI water (through filtration) and oven drying at 60 °C. Subsequently, the as-treated graphite was put into a beaker, and 100 mL of H2SO4 were added while keeping the beaker in an ice bath. Afterwards, 13 g of KMnO4 was then added slowly. After 5 min, the ice bath was removed, and the solution was heated up and kept at 35 °C under vigorous stirring for 4 h, followed by the slow addition of 200 mL of water. Finally, 16 mL H2O2 was added, followed by centrifugation and washing with 1 L dilute HCl (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DI water 1:10). The exfoliation of GO was achieved by sonication.
:DI water 1:10). The exfoliation of GO was achieved by sonication.
2.3. Preparing N-doped graphene oxide as a boron removal medium
Some amount of aqueous ammonia was added into the above dispersion of GO (37.5 mL, 4 mg mL−1) under magnetic stirring. Then the mixture was transferred to a Teflon-lined autoclave at room temperature and heated at 40 °C–140 °C for 5 h without stirring before natural cooling. The products of nitrogen doped graphene oxide were rinsed with DI water to remove the excess or physisorbed ammonia, and the powder form of products was collected as a boron removal medium.2.4. Boron removal and measurements
In the batch system, 25 ml of a boron feed solution with 20 ppm boron was mixed with 40 mg of nitrogen-doped graphene oxide, and kept under stirring for 48 hours followed by centrifugation or filtration with 220 nm nylon filter paper; the filtered solution was collected for boron analysis. The following equation was used to calculate the boron adsorption capacity at equilibrium: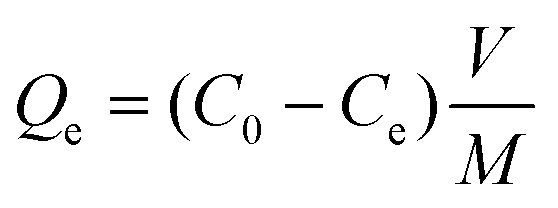 | (1) |
2.5. Regeneration process of boron removal media
For a typical regeneration process, 3–5 bed volumes of an aqueous solution of 5% HCl or H2SO4 are applied and the regenerate contact time was 60 minutes. Thereafter, the medium is rinsed with 8–10 bed volumes of DI water. At the conversion step, 3–5 bed volumes of 2.5% NaOH were used followed by the DI water rinse.3. Results and discussion
3.1. N-GO synthesis process and analysis
Fig. 1 describes the synthesis of N-GO in two simple steps. During the first step, graphite is oxidized and exfoliated into graphene oxide (GO) by a modified Hummers’ method.36 Following this, a hydrothermal step involving GO and aqueous ammonia is applied to obtain N-GO.39 After the hydrothermal treatment, the color of N-GO solutions changed from brown to dark brown, as shown in Fig. S1,† suggesting partial removal of oxygen-containing functional groups.39 XPS spectra of GO and N-GO-x are shown in Fig. 2. The samples are denoted as N-GO-x, where x represents the reaction temperature (in °C) during hydrothermal treatment. An increase in the intensities of the N 1s peak at ∼400 eV with increasing reaction temperatures indicates that nitrogen doping has occurred for the GO. The doping of GO occurs via the formation of covalently bonded nitrogen and simultaneous de-oxygenation of several oxygenated functional groups on the surface of GO, and the atomic ratio of N/C and O/C is shown in Fig. 2(b) and Table S1† based on the XPS results. The extent of nitrogen doping is directly proportional to the reaction time and hydrothermal temperature39,40 where longer reaction times and higher temperatures produce a greater amount of nitrogen doped sites. | ||
| Fig. 1 The two step process to synthesize nitrogen-doped graphene oxide. Graphite was oxidized into graphene oxide, followed by nitrogen doping through hydrothermal treatment using ammonia. | ||
 | ||
| Fig. 2 (a) XPS spectra of GO and N-GO-x; (b) atomic ratio of N/C and O/C of the samples calculated from XPS results. | ||
3.2. Boron absorption properties of N-GO-x and characterization
In a typical boron removal experiment, 25 mL of boron feed solution at a concentration of 20 ppm boron is mixed with 40 mg of N-doped GO and stirred for 48 h followed by centrifugation or filtration in a batch study. The filtered solution is collected for boron analysis and the adsorption capacity is calculated. Fig. 3(a) shows the boron adsorption capacity of GO and N-GO-x (where x = room temperature, 40 °C, 60 °C, 80 °C, 100 °C and 140 °C). The optimal hydrothermal temperature is around 60 °C, where the boron adsorption capacity is up to 6.55 mg g−1 for N-GO. A summary of our experiments is shown in Table S2.† At an increased temperature above 80 °C, the boron adsorption capacity decreases exponentially with increasing reaction temperatures. This can be attributed to a significant reduction of –OH functional groups. As a result, the boron complexes are unable to form. The nitrogen-doping amount at room temperature and 40 °C is less than that at 60 °C as shown in Fig. 3d, causing a lower boron absorption capacity. The ammonia concentration used during the hydrothermal step is another factor which can affect the adsorption capacity. As shown in Fig. S2,† the optimal ammonia concentration is between 2% and 3% for 150 mg GO. At low ammonia levels, there is negligible nitrogen doping, which results in the low boron adsorption capacities. At high ammonia concentrations, the increase in nitrogen doping and the reduction effect from the excess ammonia may result in the excessive loss of the hydroxyl group in GO.39 The detailed data are shown in Table S3.† | ||
| Fig. 3 (a) The boron absorption capacity of N-GO at various temperatures; (b) XRD patterns of GO, N-GO-60 and N-GO-60 + B; (c) XPS spectra of GO + B, N-GO-x + B, and boric acid; (d) N-doped atomic ratio of N/C, and the ratios of B/C of the samples calculated from XPS results; (e) high resolution N 1s spectra of GO and N-GO-60 and (f) high resolution B 1s spectra of boric acid and N-GO-60 + B. | ||
The effects of de-oxygenation and boron-adsorption were analyzed using X-ray diffraction, as shown in Fig. 3(b). The diffraction peak of GO is located at 9.22°, which corresponds to an interlayer distance of 1.09 nm. The hydrothermal treatment with ammonia at 60 °C reduces the interlayer distance to 0.91 nm due to the de-oxygenation effect. However, the interlayer distance increases to 0.98 nm when the boron species were absorbed on the surface of N-GO-60.
X-ray photon spectroscopy (XPS) was used to characterize the extent of nitrogen doping and the amount of boron adsorbed by quantitatively analyzing the elemental composition of the samples from the peak area (C 1s, N 1s and B 1s). Fig. 3(c) shows the XPS profiles of N-GO-x after boron absorption. The appearance in the peak of the B 1s at ∼192 eV indicates that boron absorption has occurred for the N-GO. The nitrogen-doping causes N-GO to have an overall positive charge with respect to undoped GO. The increased surface charge induces an electrostatic attraction between the nitrogen-doped sites on the GO surface and the negatively charged B(OH)4− species. As the B(OH)4− species are brought close to the N-GO surface, hydrogen-bonding donor–acceptor interactions occur between the –OH from the boric acid/borate ion and the –OH/–NH– from N-GO and this results in the formation of a stable boron complex.
Fig. 3(d) further shows that the nitrogen content (N/C) of the samples has increased from 0.06 to 0.08 as the reaction temperatures increased from room temperature, 40 °C to 140 °C, shown in Table S4.† This implies that there is an increase of nitrogen content in the GO with increasing hydrothermal temperature. On the other hand, it can be observed that the B/C content is inversely proportional to the reaction temperature where the highest B/C ratio is 0.19 for the N-GO-60 sample. Hence, this suggests that 60 °C is the optimal temperature for the synthesis of N-GO with the highest boron adsorption capacity (Fig. 3(a)). This can be explained by considering the unique roles of both nitrogen and –OH functional groups where the former facilitates the localized positive attraction of boric acid to GO surfaces and the latter allows the formation of a stable complex between boric acid and N-GO. However, as nitrogen doping results in the simultaneous deoxygenation of GO, an optimal condition is necessary. The high-resolution XPS N 1s spectrum is illustrated in Fig. 3(e) for the GO and the optimal sample N-GO-60. The main peak of the N 1s spectrum present at 397.75 eV corresponds to the pyridinic N while the weaker peak comes from pyrrolic N (399.75 eV), and the minor peak is assigned to graphitic N (402 eV).41
The high resolution XPS of B 1s is shown in Fig. 3(f). The B 1s peak significantly blue shifts to higher binding energy after a boron complex is formed. This might be due to a reduction effect from the N-GO. In the B 1s peak for the N-GO-60 + B, there are two peaks overlapping each other.41–43 The peak at 191.75 eV can be likely attributed to the edge effects of the sample and the other minor peak can be assigned to the configuration shown at the center.
3.3. The boron kinetics absorption of N-GO-60 and the effect of pH value
In order to explore the kinetic adsorption of boron, the batch experiments were conducted at room temperature using 5 ppm, 20 ppm, and 100 ppm boron feed. The pH value was kept constant at the original value of the solutions (∼8.5). The results obtained are shown in Fig. 4(a). In general, the boron adsorption capacity increases with increasing contact time until adsorption saturation. Based on the curve, equilibrium is reached after twenty hours for these three solutions. The saturated adsorption capacity is 6.55 mg g−1 for a solution with an initial concentration of 20 ppm, which corresponds to 52.40% boron rejection. The detailed data are shown in Tables S5–S7.† However, the feed concentration of 5 ppm delivers the highest efficient boron rejection, which can be up to 91.12%. When the amount of N-GO is increased to 110 mg from 40 mg, 0.0630 ppm boron product can be obtained in the product solution prepared with deionized water, and the boron rejection rate is calculated to be up to 98.74%. The detailed data are presented in Table S8.†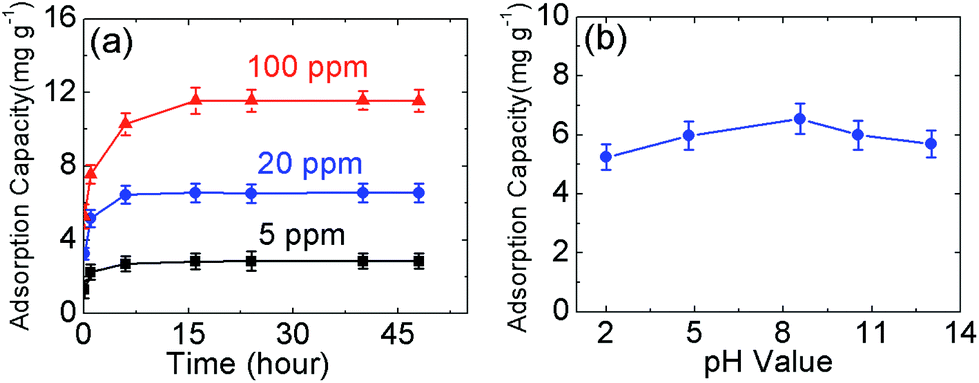 | ||
| Fig. 4 (a) The kinetics absorption of boron (25 ml boron feed solution with 20 ppm boron was mixed with 40 mg N-GO-60); (b) the effect of pH value of N-GO-60. | ||
As shown in previous reports,9,17,18,21,25 the pH value plays an important role in the boron adsorption process. Therefore, we investigated the pH effect and the results are shown in Fig. 4(b). The experiments were carried out with an initial concentration of 20 ppm B with 25 ml feed. 40 mg of N-GO-60 medium was added during the batch testing. The pH value was adjusted with HCl or NaOH. The mixture was stirred for 48 hours followed by centrifugation or filtration with 220 nm nylon filter paper; the filtered solution was collected for boron analysis. As displayed in Fig. 4(b), N-GO-60 shows a high selectivity for boron at pH 8.6. The lowest adsorption capacity was achieved at pH ∼ 2.0. The detailed data are shown in Table S9.† The pH value in the solution controls the distribution of boric acid molecules and borate ions,9 which can affect the efficiency of boron removal. At low pH levels, boric acid predominates in the aqueous solution. This uncharged species will suppress the complex formation and result in low boron removal efficiency. At high pH (>9) levels, the borate ion is the primary species in solution. However, as the boron complex formation at basic pH is carried out by hydroxyl groups on GO surfaces, this implies that the surface of N-GO-60 is more negatively charged in the basic medium due to the high amount of OH ions present in the solution, which results in low boron loading. The favorable pH range is shown to be between 6 and 10. Despite this, the influence of pH in the medium only resulted in a small fluctuation of boron adsorption capacity (maximum 20% over the pH range 2–13). It is noted that seawater has a pH value of 8.4, which is within the highest adsorption range for our materials. This indicates that N-GO-60 can be highly useful for seawater boron ion removal.
3.4. Boron adsorption equilibrium isotherm, the adsorption performance in real seawater, and the regeneration of N-GO-60
A Langmuir isotherm model was chosen to analyze the boron adsorption equilibrium isotherm based on the adsorption data. The equation of the Langmuir adsorption isotherm is presented as follows: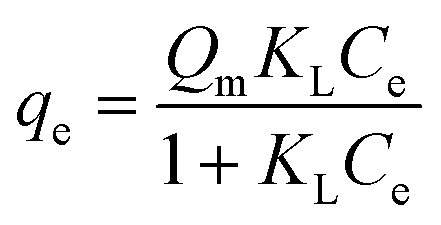 | (2) |
 | ||
| Fig. 5 (a) Langmuir isotherms at the temperature of 25 °C, pH 8.5. Insert: the adsorption performance in DI water, synthetic seawater and real seawater (25 ml boron feed solution with 5 ppm boron was mixed with 40 mg N-GO medium for 48 hours); (b) the regeneration of N-GO-60 using acid (25 ml boron feed solution with 20 ppm boron was mixed with 40 mg N-GO medium for 48 hours). | ||
| Equation: qe = Qm × KL × Ce/(1 + KL × Ce) | ||
|---|---|---|
| R 2 | 0.9853 | |
| Value | Standard error | |
| Q m | 58.69875 | 2.63124 |
| K L | 0.00409 | 8.81507 × 10−4 |
| Boron absorption media | Maximum absorption capacity (mg g−1) | Ref. |
|---|---|---|
| N-GO | 58.70 | This work |
| Dowex 2 × 8 anion exchange resin | 16.98 | 45 |
| Polystyrene-based resin grafted with glycidol | 1.3 | 46 |
| Polymer supported iminodipropylene glycol functions | 32 | 47 |
| Sorbitol-modified poly (N-glycidyl styrene sulfonamide) | 13.18 | 48 |
| Iminobis-(propylene glycol) modified chitosan beads | 29.19 | 49 |
| Unmodified rice husk | 4.23 | 50 |
| Calcine alunite | 3.39 | 51 |
| Neutralized red mud | 5.99 | 52 |
| Fly ash | 20.9 | 53 |
| Cotton | 11.3 | 54 |
| Curcumin-impregnated activated carbon | 5.00 | 55 |
| Magnetic carbon nanotube improved with tartaric acid | 1.53 | 56 |
| Purolite S108 resin | 12.87 | 57 |
| Crosslinking of poly(vinylbenzyl chloride) with a cyclic diamine piperazine | 28 | 58 |
| Aquatic booster | 1.42 | 59 |
| Hydrotalcite-like compounds | 14.0 | 60 |
| Acetyl-meglumine resin | 28.1 | 61 |
In order to evaluate the interference from other ions or molecules, a comparison study was conducted as shown in the insert of Fig. 5(a). In a feed solution prepared with deionized water, the adsorption capacity is 2.84 mg g−1 for N-GO-60. Real seawater with 2.67 ppm boron was collected from the ocean of East Coast Park, Singapore. Pre-filtration was conducted to remove the particles. Additional boric acid was added to reach the average 5 ppm boron as feed. The adsorption capacity drops a little to 2.42 mg g−1 for N-GO-60, which suggests that as high as 85% adsorption capacity can be maintained even in real seawater. The detailed data are shown in Table S11.† This implies that the nitrogen doped graphene oxide is unreactive to other ions or molecules and is highly selective towards boron.
As shown in Fig. 5(b), this boron removal medium can be regenerated with acid, which means that the trapped boron can be removed from the exhausted N-GO through the addition of an acid solution (HCl or H2SO4) into the medium. Strong acids were required to break down the formed association of the borate–N-GO complex, where boron was released to elute. 5% HCl was chosen for regeneration. 3–5 bed volumes (BV) of aqueous HCl solution were very effective with a 1 hour contact time for the removal of boron from N-GO. After that, 3–5 BV of 2% NaOH rinse was used as a neutralization step to neutralize the added acid, followed by an 8–10 BV DI water rinse. After regeneration, the N-GO medium may be reused for the next loading cycle. The regenerated result is shown in Fig. 5(b), four cycles were carried out during the test. After regeneration, the capacity can still be maintained at ∼86% and ∼81% for the 2nd and 3rd cycles. The detailed data are shown in Table S12.† Based on the image of inserted SEM in Fig. 5(b), after four cycles, the morphology of N-GO remains the same, which indicates that it can continue to recycle.
3.5 Boron removal mechanism
Boron atoms exist in the form of negatively charged B(OH)4− ions and boric acid. It is known that some compounds with adjacent hydroxyl groups have a good affinity to boron acid or borate ions. The possible reaction mechanism for the removal of boron appears to be the formation of complexes, and a schematic diagram of the reaction pathway is shown in Fig. 6. The removal of boron ions occurs in two steps: physisorption of boron ions and chemical bonding of boron ions to specific sites.11,20,29 Since GO is intrinsically negatively charged, boron ions are electrostatically repelled from the surface of GO and this results in a low adsorption capacity. Nitrogen doped GO has two advantageous effects: (1) increased electrostatic attraction of B(OH)4− ions to the N-GO surface and (2) enhancement in binding sites. Firstly, the presence of nitrogen atoms induces a positive charge density in the carbon atoms adjacent to them and this will result in an enhanced adsorption of boron ions to the surface of N-GO.44 Secondly, –OH groups attached to carbon atoms adjacent to nitrogen doped sites experience an increased affinity to boron ions and are able to form complexes more effectively. In order to show the function of N doping for GO modification, we have also measured the zeta potential for both GO and N-GO in Fig. S3† and the result shows an improvement in surface charge for N-GO over GO. The overall zeta potential for N-GO is still negative because induced positive charges are only localized at nitrogen doping sites. The enhancement in binding sites is further verified through Density Functional Theory (DFT) calculations. The calculation process and results are shown in Fig. S4.† The bonding energies for N-GO and GO are calculated to be −2.67 eV and −1.87 eV, respectively, which indicates higher possibility to promote the chemical reaction for the N-GO.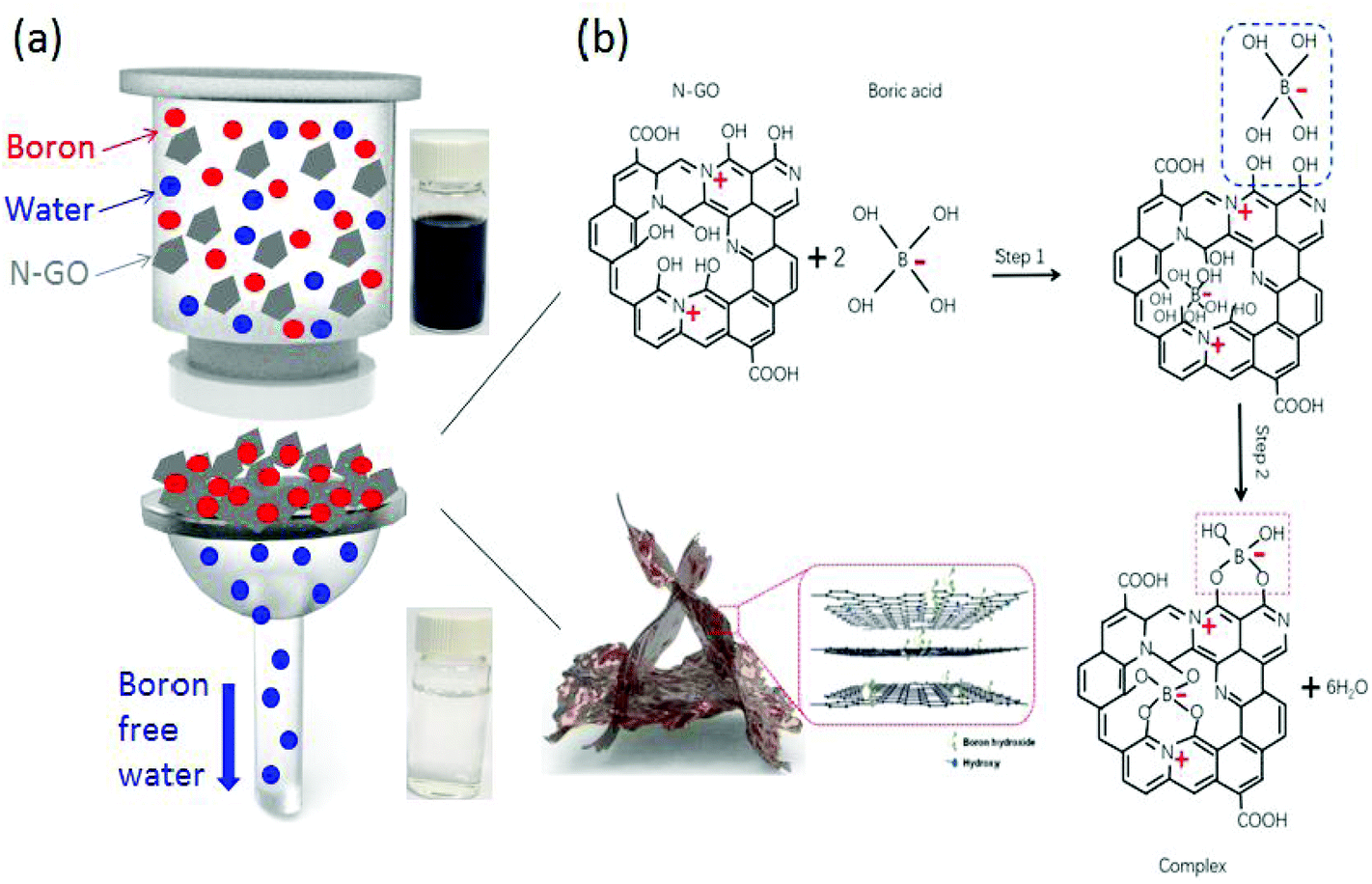 | ||
| Fig. 6 (a) Schematic representation of the boron removal by the filtration process; (b) schematic diagrams for removing boron by N-GO. | ||
4. Conclusion
In summary, a novel carbon-based boron removal medium was synthesized by a two-step process: (1) oxidation of carbon material to form the –OH group, (2) nitrogen-doping by the hydrothermal method to incorporate nitrogen into graphene oxide in order to enhance the boron adsorption ability. N-GO exhibits an adsorption capacity of 6.55 mg g−1 medium at 25 ml of feed with 20 ppm boron for 40 mg medium, and the corresponding rejection is 52.40%. In 5 ppm boron feed, this medium can achieve a boron rejection of up to 91.12%. In real seawater with 5 ppm of boron, N-GO also shows an up to 2.42 mg g−1 capacity, which corresponds to 77.44% rejection. The maximum adsorption capacity is 58.7 mg g−1 based on the Langmuir adsorption isotherm, which is the highest among all the sorbent products at present. The high adsorption capability of N-GO was shown to be attributed to the high amount of hydroxyl group and the surface area of graphene oxide, as well as the enhanced adsorption from the nitrogen-doped effect. The influences from pH, other ions, and kinetics adsorption were investigated. It was found that the N-GO medium is extremely effective in a wide range of pH conditions as well as under real conditions of seawater. Moreover, this boron removal medium can be easily regenerated for further use with a simple acid treatment. Therefore, the promising result reported herein has a strong potential to greatly impact the field of boron removal.Acknowledgements
The research project was supported by the Singapore National Research Foundation under its Environmental & Water Technologies Strategic Research Programme and administered by the Environment & Water Industry Programme Office (EWI) of the PUB.Notes and references
- R. Wickham and R. Godec, Semiconductor Pure Water and Chemicals Conference, Proceedings, 2001, pp. 15–33.
- J. A. Rajaratnam, J. B. Lowry, P. N. Avadhani and R. H. V. Corley, Science, 1971, 172, 1142–1143 CAS.
- W.-W. Choi and K. Y. Chen, Environ. Sci. Technol., 1979, 13, 189–196 CrossRef CAS.
- J. W. Mitchell, W. M. Dugger and H. G. Gauch, Science, 1953, 118, 354–355 CAS.
- W. J. McIlrath, J. W. Mitchell, I. R. Schneider and H. G. Gauch, Science, 1960, 132, 898–899 CAS.
- S. Lee and S. Aronoff, Science, 1967, 158, 798–799 CAS.
- WHO-Guidelines, Background document for development of WHO Guidelines for Drinking-water Quality, 2009, WHO/HSE/WSH/09.01/2 Search PubMed.
- N. Hilal, G. J. Kim and C. Somerfield, Desalination, 2011, 273, 23–35 CrossRef CAS.
- E. H. Ezechi, M. H. Isa and S. R. B. M. Kutty, J. Appl. Sci., 2012, 12, 402–415 CrossRef CAS.
- N. Kabay, M. Bryjak, S. Schlosser, M. Kitis, S. Avlonitis, Z. Matejka, I. Al-Mutaz and M. Yuksel, Desalination, 2008, 223, 38–48 CrossRef CAS.
- J. Wolska and M. Bryjak, Desalination, 2013, 310, 18–24 CrossRef CAS.
- R. Bernstein, S. Belfer and V. Freger, Environ. Sci. Technol., 2011, 45, 3613–3620 CrossRef CAS PubMed.
- M. V. S. Elena Borokhov Akerman and V. Gitis, Desalin. Water Treat., 2012, 46, 285–294 CrossRef.
- A. E. Yilmaz, R. Boncukcuoğlu, M. M. Kocakerim, M. T. Yilmaz and C. Paluluoğlu, J. Hazard. Mater., 2008, 153, 146–151 CrossRef CAS PubMed.
- E. H. Ezechi, M. H. Isa, S. R. M. Kutty and A. Yaqub, Process Saf. Environ. Prot., 2014, 92, 509–514 CrossRef CAS.
- M. Parsaei, M. S. Goodarzi and M. M. Nasef, 2011 2nd International Conference on Environmental Science and Technology, 2011, 6, V1-398–V391-402.
- K. Missaoui, W. Bouguerra, C. Hannachi and B. Hamrouni, J. Water Resour. Prot., 2013, 5, 867–875 CrossRef CAS.
- M. del Mar de la, F. García-Soto and E. M. Camacho, Sep. Purif. Technol., 2006, 48, 36–44 CrossRef.
- N. Öztürk, D. Kavak and T. E. Köse, Desalination, 2008, 223, 1–9 CrossRef.
- M.-O. Simonnot, C. Castel, M. NicolaÏ, C. Rosin, M. Sardin and H. Jauffret, Water Res., 2000, 34, 109–116 CrossRef CAS.
- N. Geffen, R. Semiat, M. S. Eisen, Y. Balazs, I. Katz and C. G. Dosoretz, J. Membr. Sci., 2006, 286, 45–51 CrossRef CAS.
- A. Iizuka, M. Takahashi, T. Nakamura and A. Yamasaki, Ind. Eng. Chem. Res., 2014, 53, 4046–4051 CrossRef CAS.
- X. Zhai, J. Meng, R. Li, L. Ni and Y. Zhang, Desalination, 2011, 274, 136–143 CrossRef CAS.
- J.-Q. Jiang, Y. Xu, K. Quill, J. Simon and K. Shettle, Ind. Eng. Chem. Res., 2007, 46, 4577–4583 CrossRef CAS.
- H. Polat, A. Vengosh, I. Pankratov and M. Polat, Desalination, 2004, 164, 173–188 CrossRef CAS.
- F. M. Nima Zohdi, L. C. Abdullah and T. S.Y Choong, J. Environ. Health Sci. Eng., 2014, 12, 1–12 CrossRef PubMed.
- Ç. D. H. Ö. Ö. N. B. L. Yılmaza, Sep. Sci. Technol., 2002, 37, 1257–1271 CrossRef.
- M. Badruk and N. Kabay, International Geothermal Conference, Reykjavík, Sept. 2003, 2003, Session #14, pp. 8–13.
- C. Yan, W. Yi, P. Ma, X. Deng and F. Li, J. Hazard. Mater., 2008, 154, 564–571 CrossRef CAS PubMed.
- K. Ikeda, D. Umeno, K. Saito, F. Koide, E. Miyata and T. Sugo, Ind. Eng. Chem. Res., 2011, 50, 5727–5732 CrossRef CAS.
- L. J. Banasiak and A. I. Schäfer, J. Membr. Sci., 2009, 334, 101–109 CrossRef CAS.
- R. Wen, S. Deng and Y. Zhang, Desalination, 2005, 181, 153–159 CrossRef CAS.
- N. H. T. Azhar Abdul Halim, N. Awang and M. T. Latif, Am. J. Environ. Sci., 2012, 8, 322–327 CrossRef.
- Y. Xu and J.-Q. Jiang, Ind. Eng. Chem. Res., 2008, 47, 16–24 CrossRef CAS.
- H. Nagasawa, A. Iizuka, A. Yamasaki and Y. Yanagisawa, Ind. Eng. Chem. Res., 2011, 50, 6325–6330 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- F. Chen, S. Liu, J. Shen, L. Wei, A. Liu, M. B. Chan-Park and Y. Chen, Langmuir, 2011, 27, 9174–9181 CrossRef CAS PubMed.
- L. Wei, F. Chen, H. Wang, T. H. Zeng, Q. Wang and Y. Chen, Chemistry – Asian J., 2013, 8, 437–443 CrossRef CAS PubMed.
- G. Wang, L.-T. Jia, Y. Zhu, B. Hou, D.-B. Li and Y.-H. Sun, RSC Adv., 2012, 2, 11249–11252 RSC.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- P. Ahmad, M. U. Khandaker, Y. M. Amin, N. Muhammad, A. R. Usman and M. Amin, New J. Chem., 2015, 39, 7912–7915 RSC.
- A. Pakdel, X. Wang, C. Zhi, Y. Bando, K. Watanabe, T. Sekiguchi, T. Nakayama and D. Golberg, J. Mater. Chem., 2012, 22, 4818–4824 RSC.
- T. He, X. Guo, K. Zhang, Y. Feng and X. Wang, RSC Adv., 2014, 4, 5880–5886 RSC.
- Y. Shao, S. Zhang, M. H. Engelhard, G. Li, G. Shao, Y. Wang, J. Liu, I. A. Aksay and Y. Lin, J. Mater. Chem., 2010, 20, 7491–7496 RSC.
- N. Öztürk and T. E. Köse, Desalination, 2008, 227, 233–240 CrossRef.
- J. Kluczka, T. Korolewicz, M. Zołotajkin and J. Adamek, Water Resour. Ind., 2015, 11, 46–57 CrossRef.
- B. F. Senkal and N. Bicak, React. Funct. Polym., 2003, 55, 27–33 CrossRef CAS.
- N. Biçak and B. F. Şenkal, J. Appl. Polym. Sci., 1998, 68, 2113–2119 CrossRef.
- M. Gazi and S. Shahmohammadi, React. Funct. Polym., 2012, 72, 680–686 CrossRef CAS.
- H. Che Man, W. H. Chin, M. R. Zadeh and M. R. M. Yusof, BioResources, 2012, 7(3), 3810–3822 Search PubMed.
- D. Kavak, J. Hazard. Mater., 2009, 163, 308–314 CrossRef CAS PubMed.
- Y. Cengeloglu, A. Tor, G. Arslan, M. Ersoz and S. Gezgin, J. Hazard. Mater., 2007, 142, 412–417 CrossRef CAS PubMed.
- N. Öztürk and D. Kavak, Fresenius Environ. Bull., 2003, 12, 1450–1456 Search PubMed.
- R. Liu, W. Ma, C.-y. Jia, L. Wang and H.-Y. Li, Desalination, 2007, 207, 257–267 CrossRef CAS.
- T. T. Kurkumin, A. A. Halim, N. A. Roslan, N. S. Yaacub and M. T. Latif, Sains Malays., 2013, 42, 1293–1300 Search PubMed.
- N. Zohdi, F. Mahdavi, L. C. Abdullah and T. S. Choong, J. Environ. Health Sci. Eng., 2014, 12, 1–12 CrossRef PubMed.
- F. B. A. Korkmaz M., C. Özmetin and Y. Yaşar, Bulg. Chem. Commun., 2014, 46, 594–601 Search PubMed.
- N. Thakur, S. A. Kumar, R. N. Shinde, A. K. Pandey, S. D. Kumar and A. V. R. Reddy, J. Hazard. Mater., 2013, 260, 1023–1031 CrossRef CAS PubMed.
- X. T. Z. M. H. S. Ismail and M. F. M. Lazim, Pol. J. Environ. Stud., 2013, 22, 403–408 Search PubMed.
- O. P. Ferreira, S. G. de Moraes, N. Durán, L. Cornejo and O. L. Alves, Chemosphere, 2006, 62, 80–88 CrossRef CAS PubMed.
- N. Wang, F.-T. Cao and X.-N. Liu, Chem. Res. Chin. Univ., 2012, 33, 2795–2800 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr07448k |
| This journal is © The Royal Society of Chemistry 2017 |