
An electrochemical sensor for the sensitive detection of rutin based on a novel composite of activated silica gel and graphene
Jianfei Xia,
Zonghua Wang*,
Feng Cai,
Feifei Zhang,
Min Yang,
Wenjun Xiang,
Sai Bi and
Rijun Gui
College of Chemical Science and Engineering, Laboratory of Fiber Materials and Modern Textiles, The Growing Base for State Key Laboratory, Shandong Sino-Japanese Center for Collaborative Research of Carbon Nanomaterials, Collaborative Innovation Center for Marine Biomass Fiber Materials and Textiles, Qingdao University, Qingdao, Shandong 266071, China. E-mail: wangzonghua@qdu.edu.cn; 13853219173@126.com; Fax: +86-0532-85950873; Tel: +86-0532-85950873
First published on 22nd April 2015
Abstract
In this paper, a novel activated silica gel (ASiG)–graphene (G) composite was initially fabricated via a simple sonication-induced assembly and used as the substrate material to prepare an electrochemical sensor (ASiG/G/GCE) for the sensitive determination of rutin. Morphology and electrochemical properties of the composite were investigated by transmission electron microscopy (TEM), chronocoulometry, electrochemical impedance spectroscopy (EIS), cyclic voltammetry (CV) and differential pulse voltammetry (DPV). Experimental results revealed that the ASiG/G composite induced a remarkable increase of the redox currents of rutin, which could be attributed to the high surface area and excellent electric conductivity of G, as well as the strong accumulation efficiency of ASiG toward rutin. The peak current from DPV is linearly dependent on the rutin concentration in a range of 0.001 to 1.2 μmol L−1 with a detection limit of 3.3 nmol L−1. The ASiG/G/GCE also exhibited good selectivity and acceptable reproducibility. Moreover, the ASiG/G/GCE was successfully applied to the fast determination of rutin in medicine tablets and human plasma with satisfactory recoveries. Therefore, the present work offers a new way to broaden the analytical applications of functionalized graphene in pharmaceutical research.
Introduction
Since the first report in 2004,1 graphene (G) has attracted much attention due to its unique two-dimensional nanostructure, high electron conductivity,2 fast heterogeneous electron-transfer rate at the G sheet edges and basal-plane defect sites. As the thinnest carbon material until now, G has exhibited many potential applications,3–5 especially serving to modify materials for the construction of electrode interfaces. Moreover, the two-dimensional plane structure of G provides a vast platform for loading various materials, offering a new way to develop composite materials for the interface construction to fabricate electrochemical biosensors. So far, a variety of graphene-based electrodes have been applied to clinical diagnosis,6 pharmaceutical analysis7,8 and environmental monitoring.9Rutin, one of the most abundant bioactive flavonoid glycosides, exists in many plants, multivitamin preparations and drugs. In view of promising physiological activities such as anti-inflammatory,10 antiviral,11 anti-tumor12 and anticancer activities,13 rutin has been widely investigated during the past decade. Although many traditional analytical methods, such as capillary electrophoresis,14–16 chemiluminescence,17 high-performance liquid chromatography18,19 and sequential injection analysis,20 have been developed for the detection of rutin. It's still a significant and challenging work to develop a simple, sensitive and accurate analytical method to detect rutin for further study the activities and relevant functions (especially for human health) of rutin. The electrochemical method21–24 has advantages of simplicity, high sensitivity, good stability and low cost. In this case, various modified electrodes have been used for the detection of rutin.
In the development of electrochemical rutin sensing strategies, an efficient interface for rutin accumulation and permeation to the electrode surface and high-speed electronic transfer is the key factor. Graphene is an ideal candidate in constructing the interface with extraordinary electronic transport properties and high electrocatalytic activities, which facilitate the ultrasensitive detection performance for rutin. For example, β-cyclodextrin/graphene/GCE25 and PAMAM/graphene–chitosan/GCE26 have been fabricated for the detection of rutin. In addition, silica gel is another ideal candidate in constructing the interface for rutin detection. Silica gel, which is porous and insoluble in water and solvent, has excellent properties, such as strong sorption ability, high surface area, easy surface modification, etc. Particularly, the activated silica gel (ASiG) treated by hydrochloric acid showed a decreased particle size and an increased adsorption performance, which produced a higher surface area and porous structure that were favorable for analytical targets accumulation and permeation to the electrode surface27,28 to enhance the detection sensitivity. Therefore, preparation of activated silica gel (ASiG)–graphene (G) composite modified electrode is suitable for the detection of rutin. Moreover, the existence of ASiG can improve the dispersibility of graphene, which is favourable for the improvement of electrochemical determination performance. To the best of our knowledge, there is no report on the preparation of activated silica gel (ASiG)–graphene (G) composite modified electrode.
Herein, a novel ASiG/G composite was prepared and further applied to the fabrication of a chemically modified electrode for the detection of rutin. Morphology and electrochemical properties of the composite were investigated by transmission electron microscopy (TEM), chronocoulometry, electrochemical impedance spectra (EIS), cyclic voltammetry (CV) and differential pulse voltammetry (DPV). With the synergistic effects of ASiG and G, the electrochemical response to rutin was greatly enhanced, improving the sensitivity of rutin detection. And the ASiG/G/GCE was successfully applied to the fast determination of rutin in medicine tablets and human plasma with satisfactory recoveries.
Experimental
Reagents and apparatus
Rutin (C27H30O10·3H2O, Mr = 664.56), was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Silica gel (200–300 mesh) was purchased from Qingdao Ocean Chemical Co., Qingdao, China. Graphite was provided by Qingdao Fujin graphite Co., Ltd (Qingdao, China). The rutin tablets were purchased from market. Human plasma was taken from a healthy body and stored at −20 °C. The supporting electrolyte was a 0.1 M phosphate buffer saline (PBS, pH 6.8) that was prepared by NaH2PO4·2H2O and Na2HPO4. Cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and differential pulse voltammetry (DPV) experiments were performed on a CHI-660A electrochemical workstation (Shanghai Chen Hua Instrument Co., Ltd.) with a conventional three-electrode cell. Transmission electron microscope (TEM) measurements were performed on a JEOL 2100 HRTEM instrument (JEOL Ltd., Tokyo, Japan) using an accelerating voltage of 200 kV. All of the solutions were purged with pure nitrogen for at least 15 min to remove oxygen prior to experiments.Graphene preparation
Graphite-oxide nanosheets (GO) were synthesized from natural graphite powder according to our previously reported method29 and then dispersed in water to yield a yellow-brown dispersion by ultrasonication for 2 h, followed by centrifugation at 3000 rpm to remove any unexfoliated GO. Subsequently, the homogeneous GO dispersion (100.0 mL) was mixed with 0.5 mL of hydrazine hydrate and allowed to react for 24 h at 90 °C. Finally, the G was obtained by three filtration and water-washing cycles and then dried in vacuum.Preparation of ASiG and ASiG/G
Typically, the reflux treatment of silica gel (5.0 g) dispersed in hydrochloric acid (5 wt%, 300 mL) was performed under continuous stirring for 4 h. After that, the resulting products were filtered, repeatedly washed with doubly distilled water to the neutral and dried at 80 °C to obtain ASiG. The as-prepared ASiG (2 mg) and G (2 mg) were dispersed in 1.0 mL dimethylformamide (DMF), followed by ultrasonicating for 6 h to form a uniform suspension of ASiG/G. And also different mass ratios of ASiG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G (1:1, 2:1, 3:1 and 1:2) were prepared with the G concentration 2 mg mL−1.
:G (1:1, 2:1, 3:1 and 1:2) were prepared with the G concentration 2 mg mL−1.
Preparation of ASiG/G/GCE
Glassy carbon electrode (GCE) was polished successively using 0.3 μm and 0.5 μm alumina powder, then rinsed ultrasonically with doubly-distilled water and dried at room temperature. Then 5.0 μL of well-dispersed ASiG/G solution was coated onto the pretreated GCE surface and dried at room temperature to obtain the ASiG/G/GCE.Electrochemical procedure
All the cyclic voltammetric experiments were carried out at a quiescent PBS with the scan rate of 100 mV s−1. Differential pulse voltammograms (DPVs) were recorded in the potential range from 0.7 to 0.2 V with instrumental parameters as: pulse amplitude 0.005 V, pulse width 0.05 s and pulse period 0.2 s. EIS was recorded at the equilibrium potential (Eeq = −0.18 V), with an amplitude of 10 mV s−1 and in the frequency range between 10 mHz and 10 kHz.Sample preparation
Two pieces of rutin tablets were carefully ground in the agar, transferred to a10 mL calibrated tube and diluted to the scale with ethanol. Then100 μL sample solution was taken and diluted with pH 3.0 PBS in a 10 mL calibrated tube and DPVs was recorded in the potential range from 0.2 to 0.7 V.Aliquots of 2.0 mL of acetonitrile and 2.0 mL of PBS (pH of 3.0) were added to 1.0 mL of human plasma. After vortexing for 3 min, the mixture was centrifuged at 10000 rpm for 10 min, the supernatant was collected and used as the sample solution.
Results and discussion
Characterization of the modified electrodes
The well dispersed G and ASiG/G were characterized by TEM. As shown in Fig. 1A, the wrinkled G sheets display a typical crumpled and fold structure like crumpled silk veils. In comparison with the G sheets, the ASiG/G composites display a lot of particles and clusters structure (Fig. 1B), revealing that ASiG was well attached on the G sheets. Chronocoulometry was used to evaluated the electrochemically effective area of the electrodes. Fig. 1C shows the chronocoulometric curve of the bare GCE (curve a) and ASiG/G/GCE (curve b) in 1 mmol L−1 K3Fe(CN)6 containing 0.1 mol L−1 KCl. The corresponding Q–t1/2 plots are shown in the inset. According to the formula given by Anson, Q = 2nFAcD1/2t1/2/(π1/2), where A (cm2) is the area of the electrode, F is the Faraday constant, D (cm2 s−1) is the diffusion coefficient of the oxidized form, hexacyanoferrate(III), c (mol cm−3) is the bulk concentration of the oxidized form, t (s) is the time. The slope of the Q–t1/2 plots can be expressed as 2nFAcD1/2/π1/2. For 1 mmol L−1 K3Fe(CN)6, n = 1, D = 7.6 × 10−6 cm2 s−1. According to the slopes value of the straight lines, the electroactive surface areas of the bare GCE and ASiG/G/GCE were calculated to be 0.1442 cm2 and 0.4328 cm2, respectively, which confirmed the enlarged effect of ASiG/G composite on the electrode surface area. | ||
| Fig. 1 TEM images of (A) G and (B) ASiG/G. (C) Chronocoulometric curves of the 1.0 mmol L−1 K3[Fe(CN)6] containing 0.1 mol L−1 KCl at the bare GCE (a) and ASiG/G/GCE (b). Inset: the corresponding Q–t1/2 plots (D) EIS results of GCE, ASiG/GCE, G/GCE and ASiG/G/GCE with 5 mmol L−1 [Fe(CN)6]3−/4− in 0.1 mol L−1 of KCl at the frequencies swept from104 to 10−2 Hz. | ||
Electrochemical impedance spectrometry (EIS), as a powerful tool for the analysis of interface properties, was utilized to investigate electron transfer properties of modified electrodes. In general, the typical Nyquist plot of EIS includes a semicircle and a linear portion, respectively corresponding to the electron transfer limited process and diffusion limited process. The value of electron transfer resistance was estimated according to the diameter of the semicircle of the Nyquist plots, which controlled the electron transfer kinetics of redox probe at the electrode surface and reflected the interfacial electron transfer ability. Z′ and Z′′ are the real variable and the negative value of the imaginary variable of impedance. Fig. 1D presents EIS results from 5 mmol L−1 of [Fe(CN)6]3−/4− (in 0.1 mol L−1 of KCl) at GCE, ASiG/GCE, G/GCE and ASiG/G/GCE with the frequency range swept from 104 to 10−2 Hz. It could be seen that the biggest well-defined semi-circle at higher frequencies was obtained at the bare GCE, indicating the biggest interface impedance. When ASiG was deposited on the surface of GCE, there was only a tiny change of the semi-circle. However, the interface electron resistance decreased remarkably after G was immobilized on the electrode. The results indicated that the presence of G nanosheets could facilitate the electron transfer between [Fe(CN)6]3−/4− probe and the electrode. For ASiG/G/GCE, the smallest value of electron transfer resistance was achieved, which was attributed to the synergistic effects of G and ASiG. G nanosheets served with high surface area, excellent conductivity and provide excellent interfacial contact between ASiG and G for fast electron transport. Also architectures of ASiG offer high surface area and porous structure for charge transfer and reduce ion diffusion length. So ASiG/G/GCE can significantly enhance the conductivity of the electrode interface and accelerate the electron transfer rate.
Electrochemical response of rutin on different modified electrodes
The structure of rutin and the electrode reaction equation is expressed in Fig. 2. The electro-oxidation reaction of rutin on the ASiG–G modified GCE was a two-electron two-proton process. The mechanism of electrooxidation for rutin was that firstly a proton dissociated to give the mono-anionic species, which was then oxidized to form a radical anion. And then, a second reversible one-electron oxidation was occurred and the radical anion was change to form dehydro-rutin. Finally, the latter species was rapidly dehydrated to yield the final product of 3′,4′-diquinone.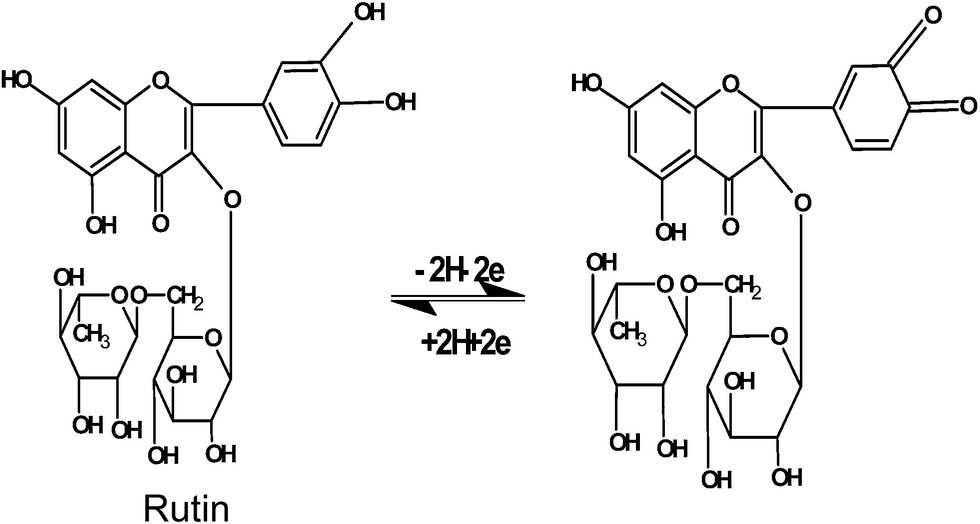 | ||
| Fig. 2 The electrode-reaction equation of rutin. | ||
The electrochemical behaviors of rutin on different electrodes were studied by cyclic voltammetry (CV). Fig. 3 shows the CVs of 1.0 μmol L−1 rutin on different modified electrodes in pH 3.0 PBS. It's obvious that a pair of well-defined redox peaks appeared on each electrode, which was the typical electrochemical response of rutin and indicating that the electrochemical reaction of rutin had took place on the working electrode. Rutin was an electroactive substance with four hydroxyl groups present on its molecular structure and its electrochemistry had been elucidated with a two-electron and two-proton redox process. It could be seen that a pair of peaks appeared with the smallest redox peak currents and the peak-to-peak separation (ΔEp) as 127 mV (curve d), revealing the slowest electron transfer rate on the GCE. On the ASiG/GCE (curve c), the redox peak currents increased with the ΔEp as 97 mV, which should be attributed to the presence of ASiG on the electrode surface, which provided a porous structure for rutin to exchange electrons. On the G/GCE (curve b), a great increasement of the redox peak current was achieved with the ΔEp as 86 mV. The result indicated that G on the electrode surface could act as an effective mediator to promote the electrochemical reaction of rutin, which could be attributed to the specific characteristics of G, such as good conductivity, high surface area and inherent electrocatalytic ability towards rutin. On the ASiG/G/GCE (curve a) the biggest redox peak currents appeared with the ΔEp as 77 mV, demonstrating a more reversible process than the electrodes G/GCE and ASiG/GCE, and indicating that the ASiG/G composite on the electrode surface exhibited the best electrocatalytic ability and facilitated the fast electron transfer rate. On the basis of results mentioned above, the great electrochemical activity of ASiG/G/GCE for rutin was confirmed adequately.
 | ||
| Fig. 3 CVs of rutin (1.0 μmol L−1) in PBS buffer solution (0.1 mol L−1, pH 3.0) on different electrodes at 100 mV s−1: ASiG/G/GCE (a), G/GCE (b), ASiG/GCE (c), and GCE (d). | ||
Optimization of conditions
To achieve a better electrochemical performance of the fabricated ASiG/G/GCE for the detection of rutin, different mass ratios of ASiG:G (1:1, 2:1, 3:1 and 1:2) were investigated by CV. As shown in Table 1, the modified electrode with different mass ratios of ASiG:G displayed different responses towards the detection of rutin. The largest peak current was observed at the ratio of 1:1, which could be attributed to that with the increasement in the proportion of G in the silica, a lot of single sheet of graphene get together and lead to a decline in conductive ability. Hence, the referred ratio of 1:1 was considered as the optimum mass ratio of ASiG:G.
| Mass ratio (ASiG:G) |
Ipa (μA) |
|---|---|
| 1:1 |
115.2 |
| 2:1 |
66.2 |
| 3:1 |
60.2 |
| 1:2 |
75.7 |
Effects from scan rate under the used range from 60 to 300 mV s−1 on the redox of rutin at the ASiG/G/GCE were investigated to analyze relevant reaction kinetics. As shown in Fig. 4, with the increase of scan rate, the redox peak current increased regularly, accompanied by a slight shift of redox potential. In particular, both the anodic peak current (Ipa) and cathodic peak current (Ipc) increased linearly with the scan rate from 60 to 300 mV s−1 (inset of Fig. 4), and the corresponding linear regression equations were Ipa (μA) = 2.042ν (mV s−1) − 87.84 (R2 = 0.995) and Ipc (μA) = −1.566ν (mV s−1) + 69.49 (R2 = 0.995), indicating an adsorption-controlled process. In addition, as shown in Fig. 4, the anodic (Epa) and cathodic (Epc) peak potentials have a linear relationship with the Napierian logarithm of scan rate (lnν) at higher scan rates. According to Laviron's equation,30 the slope of the line for Epa and Epc could be expressed as RT/(1 − α)nF and −RT/αnF, respectively.
|
Epa = E0′ + RTlnν/((1 − α)nF)
| (1) |
|
Epc = E0′ − RTlnν/(αnF)
| (2) |
|
lgks = αlg(1 − α) + (1 − α)lgα − lg(RT/nFν) − α(1 − α)(nFΔEp)/(2.303RT)
| (3) |
lnν (mV s−1) (R = 0.992) and Epc = 0.615 − 0.048lnν (mV s−1) (R = 0.996). Therefore, the electron-transfer coefficient (α) electron-transfer number (n) and electrode reaction standard rate constant (ks) were calculated as about 0.52, 2, and 1.68 s−1, respectively, with a relative standard deviation (RSD) of 2.9%. The adsorbed amount of rutin on the surface of ASiG/G/GCE was further calculated by the following equation: ip = n2F2AΓν/4RT. The value of the surface concentration of the rutin (Γ) was obtained with the result as 8.02 × 10−9 mol cm−2 based on the relationship of ip with ν. The results indicated a good adsorptivity and a fast electron-transfer process of rutin at the ASiG/G composite film.
 | ||
| Fig. 4 CVs of ASiG/G modified GCE in PBS (pH 3.0) containing 1 μmol L−1 rutin at different scan rates from 300, 280, 260, 240, 220, 200, 180, 160, 140, 120, 100, 80 to 60 mV s−1 (from a to m). Inset: the plot of the redox peak current versus scan rate; the relationship between the redox peak potential and the Napierian logarithm of scan rate (lnν). | ||
As shown in Fig. 5, when the pH was changed from 2.0 to 5.0, the anodic peak potential moved towards the negative direction, which indicated that the proton had participated in the redox reaction of rutin. With the increase of pH, the peak current increased firstly (at pH ≤ 3.0), and then became gradual decrease (at pH > 3.0). Thus, the maximum value of Ipa appeared at pH = 3.0. When the pH was 5.0, the Ipa became very low, because of the proton involved in the electrochemical reaction. Additionally, rutin became an anion at a high pH, and then gave rise to the electrostatic repulsion between rutin and G, thus resulting in the decrease of peak current. In this experiment, pH 3.0 was considered as the optimal pH for the detection of rutin. Moreover, Fig. 5 exhibited a good linear relationship between the anodic peak potential (Epa) and pH and corresponding regression equation was Epa = −0.057 pH + 0.640 (R = 0.997). The equation showed a slope value of −58 mV pH−1 was close to the theoretical value of −59 mV pH−1, indicating that the ratio of electron and proton (taking part in the electrode reaction) was 1:1.
 | ||
| Fig. 5 (A) Calibration plot of oxidation peak current (Ipa) to different pHs obtained at cyclic voltammograms of ASiG/G composite modified electrode in different pH solutions (2–5) containing 1.0 μmol L−1 rutin. Scan rate: 100 mV s−1. (B) In same conations, the calibration plot for anodic peak potential (Epa) to different pHs. | ||
The determination of rutin
To evaluate analytical performance of the novel electrochemical sensor for rutin, differential pulse voltammetry (DPV) was explored for the quantitative determination of rutin. As inserted in Fig 6, the logarithm of peak currents had a good linear relationship with the logarithm of rutin concentration in the range of 0.001 to 1.2 μmol L−1 under the optimal conditions. Corresponding linear regression equation was expressed as I (μA) = 113.71c (μmol L−1) + 5.02 with R2 of 0.993. Based on the signal-to-noise ratio of 3 (S/N), the detection limit was estimated to be 3.3 nmol L−1, with the RSD of detected results to be 4.1%. In comparison with previously reported electrochemical methods using different modified electrodes (Table 2) for rutin detection, the as-made ASiG/G/GCE showed both a broader range and a lower detection limit, which may be attributed to the combined advantages of the strong accumulation efficiency of ASiG for rutin, the large specific surface area and upstanding electric conductivity of G. Through the above analysis and comparison, it is not difficult to draw the conclusion that the ASiG/G/GCE exhibits an enhanced electrocatalytic performance. The main reason is that such sensor combines the advantages of the ASiG and G, including of the porous structure, large surface area, excellent conductivity and good catalytic activity. | ||
| Fig. 6 DPVs of ASiG/G/GCE in PBS (0.10 mol L−1, pH 3.0) versus rutin of different concentrations (a–i): 0.001, 0.005, 0.01, 0.05, 0.2, 0.4, 0.6, 0.8, 1.2 μmol L−1. Inset: the corresponding calibration plot of DPV currents versus rutin concentrations. | ||
| Modified electrode | Linear range (μmol L−1) | Detection limit (μmol L−1) | References |
|---|---|---|---|
| SWCNTS/CILE | 0.1–800.0 | 0.01 | 31 |
| Nafion/β-cyclodextrin/CRG | 0.006–50.0 | 0.002 | 32 |
| β-CDEP/GCE | 0.13–2.0 | 0.079 | 33 |
| GR/CILE | 0.07–100.0 | 0.024 | 34 |
| PAO-GR/CILE | 0.03–800.0 | 0.0083 | 35 |
| GR-MnO2/CILE | 0.01–500.0 | 0.0027 | 36 |
| Nafion-GO-IL/CILE | 0.08–100.0 | 0.016 | 37 |
| ASiG/G/GCE | 0.001–1.2 | 0.0003 | This work |
Reproducibility, stability and anti-interference performance
The repeatability and stability of modified electrodes were significant properties for analytical measurement, and thus were investigated in this work. For 1.0 μmol L−1 of rutin, five respective measurements were performed with the identical ASiG/G/GCE, and the measured results of peak current showed 3.7% of RSD. Under the same condition, 1.0 μmol L−1 rutin was measured with five freshly prepared ASiG/G/GCEs. Similar current responses were obtained with 4.5% of RSD. These results demonstrated that the fabricated ASiG/G/GCE was highly repeatable.In addition, the steady-state response current of 1.0 μmol L−1 of rutin was detected by the ASiG/G/GCE every day after preparation to examine the detection stability. The RSD of detected results was calculated to be 4.3%, indicating a high stability of rutin detection by using the ASiG/G/GCE.
As well known, many foreign substances exist in real samples. Therefore, the influence of potential coexistent substances was estimated to verify the feasibility of the proposed method for applications. As a result, there was no obvious interference in the response of rutin, showing a constant amount of 1.0 μmol L−1 rutin in 0.1 M PBS solution (pH of 2.0) after the addition of various foreign species, including quercetin (the metabolites of rutin), glucose, ascorbic acid and amino acids of 500-fold concentrations, metal ions of 1000-fold concentrations. The tolerance limit was defined as the concentration ratio of additive to rutin, producing a low relative error of less than ±5.0% (Table 3). These results demonstrate that the as-prepared electrochemical sensor has an excellent anti-interference ability.
| Coexisting substances | Concentration (mM) | Relative error (%) |
|---|---|---|
| Glucose | 0.5 | −2.12 |
| Ascorbic acid | 0.5 | 1.35 |
| Quercetin | 0.5 | 3.12 |
| L-Arginine | 0.5 | 3.22 |
| Glutamic acid | 0.5 | −2.31 |
| L-Tryptophan | 0.5 | 3.76 |
| L-Phenylalanine | 0.5 | 2.48 |
| Mg2+ | 1.0 | 2.34 |
| K+ | 1.0 | −1.86 |
| Cu2+ | 1.0 | 3.28 |
| Zn2+ | 1.0 | 3.04 |
| Na+ | 1.0 | 2.97 |
| Ca2+ | 1.0 | 2.85 |
Analytical application
To further evaluate the practicability of this electrochemical sensor, the rutin content in compound rutin tablets (20 mg per tablet) was analyzed by the proposed method. The recovery was performed with the standard addition method to evaluate the accuracy of the sensor. Measured results were satisfactory with the recovery of 97.4–101.2%, suggesting that this method could be applicable for the determination of rutin in pharmaceutical samples.We also examined the electrocatalytic oxidation of rutin in three human blood serum samples. The plasma samples were pretreated which had shown in the Section 2.5. Rutin at the ASiG/G modified electrode was not detected in human plasma. And when the diluted serum sample was spiked with various concentrations of rutin, the differential voltammograms was recorded at the surface of modified electrode. The results were shown in Table 4. As shown in Table 4, the recoveries for rutin from the serum samples ranged from 94.0% to 105.0% and the RSD value was less than 5.0%, indicating the new composite modified electrode could be applied for the determination of rutin in complex biological samples.
| Samples | Added (μmol L−1) | Founded (μmol L−1) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| Human plasma 1 | 0.020 | 0.021 | 105.0 | 4.1 |
| 0.060 | 0.058 | 96.7 | 2.8 | |
| 0.100 | 0.104 | 104.0 | 3.1 | |
| Human plasma 2 | 0.200 | 0.196 | 98.0 | 4.3 |
| 0.400 | 0.385 | 96.3 | 4.8 | |
| 0.600 | 0.590 | 98.3 | 2.4 | |
| Human plasma 3 | 0.800 | 0.805 | 100.6 | 1.8 |
| 1.000 | 0.965 | 96.5 | 3.6 | |
| 1.200 | 1.178 | 98.2 | 3.8 |
Conclusions
In summary, a novel composite ASiG/G was prepared using layer G nanosheets and nano-particle ASiG and applied for the electrochemical detection of rutin. ASiG offered high surface area and porous structure for rutin accumulation and permeation. G nanosheets provide high surface area, excellent conductivity and excellent interfacial contact between ASiG and G for fast electron transport. As combining the excellent properties of ASiG and G, an excellent analytical performance was achieved for the detection of rutin. This work offers a promising candidate for electrochemical interface construction and electrochemical biosensing. Moreover, in practical application investigations, a simple electroanalytical method to determine rutin in plasma has been developed, broadening the analytical applications of functionalized graphene in the pharmaceutical research.Acknowledgements
We really appreciate the financial support from the National Natural Science Foundation of China (21405086, 21275082, 81102411, 21203228 and 21475071), the National Key Basic Research Development Program of China (973 special preliminary study plan, Grant no. 2012CB722705), the Taishan Scholar Program of Shandong Province and the Natural Science Foundation of Qingdao (13-1-4-128-jch and 13-1-4-202-jch).Notes and references
- K. S. Novoselov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- Y. Wang, L. H. Tang, Z. H. Li, Y. H. Lin and J. H. Li, Nat. Protoc., 2014, 9, 1944–1955 CrossRef CAS PubMed.
- L. H. Tang, Y. Wang, Y. Liu and J. H. Li, ACS Nano, 2011, 5, 3817–3822 CrossRef CAS PubMed.
- K. P. Liu, J. J. Zhang, G. H. Yang, C. M. Wang and J. J. Zhu, Electrochem. Commun., 2010, 12, 402–405 CrossRef CAS PubMed.
- Q. Zhang, S. Y. Wu, L. Zhang, J. Lu, F. Verproot, Y. Liu, Z. Q. Xing, J. H. Li and X. M. Song, Biosens. Bioelectron., 2011, 26, 2632–2637 CrossRef CAS PubMed.
- G. F. Wang, X. P. He, L. Chen, Y. H. Zhu and X. J. Zhang, Colloids Surf., B, 2014, 116, 714–719 CrossRef CAS PubMed.
- Z. G. Liu, A. Zhang and Y. J. Guo, Biosens. Bioelectron., 2014, 58, 242–248 CrossRef CAS PubMed.
- C. Srikanth, K. Chelladurai and S. M. Chen, Sci. Adv. Mater., 2014, 8, 1760–1768 Search PubMed.
- S. L. Yang, J. S. Liang and S. L. Luo, Anal. Chem., 2013, 85, 7720–7725 CrossRef CAS PubMed.
- C. Boesch-Saadatmandi, A. E. Wagner, S. Wolffram and G. Rimbach, Pharmacol. Res., 2012, 65, 523–530 CrossRef CAS PubMed.
- J. Tao, Q. X. Hu, J. Yang, R. R. Li, C. Y. Chen, L. Wang, R. Shattock and K. L. Ben, Antiviral Res., 2007, 75, 227–233 CrossRef CAS PubMed.
- J. L. He, Y. Yang, X. Yang, Y. L. Liu, Z. H. Liu, G. L. Shen and R. Q. Yu, Sens. Actuators, B, 2006, 114, 94–100 CrossRef CAS PubMed.
- Y. M. Temerk, H. S. M. Ibrahim and W. Schuhmann, Microchim. Acta, 2006, 153, 7–13 CrossRef CAS.
- Q. H. Lu, C. D. Ba and D. Y. Chen, J. Pharm. Biomed., 2008, 47, 888–891 CrossRef CAS PubMed.
- G. Chen, H. W. Zhang and J. N. Ye, Anal. Chim. Acta, 2000, 423, 69–76 CrossRef CAS.
- H. T. Duan, Y. Chen and G. Chen, J. Chromatogr. A, 2010, 1217, 4511–4516 CrossRef CAS PubMed.
- Z. H. Song and S. Hou, Talanta, 2002, 57, 59–67 CrossRef CAS.
- W. Sun, M. X. Yang, Y. Z. Li, Q. Jiang, S. F. Liu and K. Jiao, J. Pharm. Biomed., 2008, 48, 1326–1331 CrossRef CAS PubMed.
- C. H. Wang, Y. X. Wang and H. J. Liu, J. Pharm. Biomed., 2011, 4, 291–296 CrossRef PubMed.
- Z. Legnerová, D. Šatinský and P. Solich, Anal. Chim. Acta, 2003, 497, 165–174 CrossRef PubMed.
- A. C. Oliveira and L. H. Mascaro, Int. J. Electrochem. Sci., 2011, 6, 804–818 CAS.
- X. Q. Lin, J. B. He and Z. G Zha, Sens. Actuators, B, 2006, 119, 608–614 CrossRef CAS PubMed.
- G. P. Jin, J. B. He, Z. B. Rui and F. S. Meng, Electrochim. Acta, 2006, 51, 4341–4346 CrossRef CAS PubMed.
- J. H. Jin, C. Kwon, W. Park, S. Kim and S. Jung, J. Electroanal. Chem., 2008, 623, 142–146 CrossRef CAS PubMed.
- K. P. Liu, J. P. Wei and C. M. Wang, Electrochim. Acta, 2011, 56, 5189–5194 CrossRef CAS PubMed.
- H. S. Yin, Y. L. Zhou, L. Cui, T. Liu, P. Ju, L. S. Zhu and S. Y. Ai, Microchim. Acta, 2011, 173, 337–345 CrossRef CAS.
- G. Q. Alonso, H. Francisco, S. Torres, M. Emilia and M. Francisco, Electrocatalysis, 2013, 4, 259–266 CrossRef.
- F. Soponar, A. C. Mot and C. Sarbu, J. AOAC Int., 2010, 93, 804–810 CAS.
- Z. H. Wang, Q. H. Han, J. F. Xia, L. Xia, S. Bi, F. F. Zhang, Y. Z. Xia, Y. H. Li and L. H. Xia, J. Electroanal. Chem., 2014, 726, 107–111 CrossRef CAS PubMed.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
- Z. H. Zhu, X. Y. Sun, X. M. Zhuang, Y. Zeng, W. Sun and X. T. Huang, Thin Solid Films, 2010, 519, 928–933 CrossRef CAS PubMed.
- K. P. Liu, J. P. Wei and C. M. Wang, Electrochim. Acta, 2011, 56, 5189–5194 CrossRef CAS PubMed.
- C. P. da Silva, A. C. Franzoi, S. C. Fernandes, J. Dupont and I. C. Vieira, Enzyme Microb. Technol., 2013, 52, 296–301 CrossRef CAS PubMed.
- F. Gao, X. W. Qi, X. L. Cai, Q. X. Wang, F. Gao and W. Sun, Thin Solid Films, 2012, 520, 5064–5069 CrossRef CAS PubMed.
- W. Sun, Y. H. Wang, S. X. Gong, Y. Cheng, F. Shi and Z. F. Sun, Electrochim. Acta, 2013, 109, 298–304 CrossRef CAS PubMed.
- W. Sun, X. Z. Wang, H. H. Zhu, X. H. Sun, F. Shi, G. N. Li and Z. F. Sun, Sens. Actuators, B, 2013, 178, 443–449 CrossRef CAS PubMed.
- S. Hu, H. H. Zhu, S. Y. Liu, J. Xiang, W. Sun and L. Q. Zhang, Microchim. Acta, 2012, 178, 211–219 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |