
Excellent hydrogen evolution by a multi approach via structure–property tailoring of titania†
Abstract
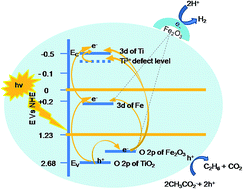
Photocatalytic water splitting by solar energy is an ideal economic method for hydrogen generation. An attempt has been made to overcome the main barriers of photocatalytic hydrogen evolution (these being rapid recombination of electron–hole pairs, the process of back reaction and poor activation of titania by visible light) by the usage of Fe-induced titania. The method facilitates a cage like mesoporous structure and an effective surface level doping. The induction of dopants and formation of coupled semiconductor oxides of well matched band energy contribute favorably to the rise of mid bands and the perfect alignment of their energy levels, leading to a shallow trapping and detrapping of electrons, necessary for efficient charge separation and transfer. Decrease in the band gap energy to ∼2.0 eV and hence the extended light absorption in modified titania, together with a shift in band edge potentials (caused by pH variation), results in an activated titania, desirable for enhanced visible light hydrogen evolution. The modified nanostructure shows an excellent hydrogen evolution of 255 mmol g−1 h−1, one of the highest reported so far. Sodium acetate acts as a good sacrificial agent in preventing the back reaction. Characterization techniques like spectroscopy (XRD, UV, reflectance, EDX, XPS and PL) and microscopy (FESEM and HRTEM) have been used to evidence the presence of Fe as dopant/as oxide and to study the structure–property tailoring that occurs during chemical modification.
 Please wait while we load your content...
Please wait while we load your content...

