Hydrogen storage and carbon dioxide capture in an iron-based sodalite-type metal–organic framework (Fe-BTT) discovered via high-throughput methods†
Kenji
Sumida
a,
Satoshi
Horike
a,
Steven S.
Kaye
a,
Zoey R.
Herm
a,
Wendy L.
Queen
bc,
Craig M.
Brown
b,
Fernande
Grandjean
d,
Gary J.
Long
e,
Anne
Dailly
f and
Jeffrey R.
Long
*a
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: jrlong@berkeley.edu
bNational Institute of Standards and Technology, Center for Neutron Research, Gaithersburg, MD 20899, USA
cDepartment of Materials Science and Engineering, University of Maryland, College Park, MD 20742, USA
dDepartment of Physics, B5, University of Liège, B-4000, Sart-Tilman, Belgium
eDepartment of Chemistry, Missouri University of Science and Technology, University of Missouri, Rolla, MO 65406, USA
fChemical and Environmental Sciences Laboratory, General Motors Company, Warren, Michigan 48090, USA
First published on 18th June 2010
Abstract
Using high-throughput instrumentation to screen conditions, the reaction between FeCl2 and H3BTT·2HCl (BTT3− = 1,3,5-benzenetristetrazolate) in a mixture of DMF and DMSO was found to afford Fe3[(Fe4Cl)3(BTT)8]2·22DMF·32DMSO·11H2O. This compound adopts a porous three-dimensional framework structure consisting of square [Fe4Cl]7+ units linked via triangular BTT3− bridging ligands to give an anionic 3,8-net. Mössbauer spectroscopy carried out on a DMF-solvated version of the material indicated the framework to contain high-spin Fe2+ with a distribution of local environments and confirmed the presence of extra-framework iron cations. Upon soaking the compound in methanol and heating at 135 °C for 24 h under dynamic vacuum, most of the solvent is removed to yield Fe3[(Fe4Cl)3(BTT)8(MeOH)4]2 (Fe-BTT), a microporous solid with a BET surface area of 2010 m2 g−1 and open Fe2+ coordination sites. Hydrogen adsorption data collected at 77 K show a steep rise in the isotherm, associated with an initial isosteric heat of adsorption of 11.9 kJ mol−1, leading to a total storage capacity of 1.1 wt% and 8.4 g L−1 at 100 bar and 298 K. Powder neutron diffraction experiments performed at 4 K under various D2 loadings enabled identification of ten different adsorption sites, with the strongest binding site residing just 2.17(5) Å from the framework Fe2+ cation. Inelastic neutron scattering spectra are consistent with the strong rotational hindering of the H2 molecules at low loadings, and further reveal the catalytic conversion of ortho-H2 to para-H2 by the paramagnetic iron centers. The exposed Fe2+ cation sites within Fe-BTT also lead to the selective adsorption of CO2 over N2, with isotherms collected at 298 K indicating uptake ratios of 30.7 and 10.8 by weight at 0.1 and 1.0 bar, respectively.
Introduction
Metal–organic frameworks have recently come under intense investigation for gas storage and separation applications, owing to their high internal surface areas, convenient modular synthesis, and chemical tunability.1 In the area of cryogenic hydrogen storage, gravimetric and/or volumetric capacities that approach the U.S. Department of Energy targets2 for mobile hydrogen storage systems have been demonstrated within the highest-surface area materials.1e,3 However, at room temperature, these materials typically exhibit little or no improvement over the density of pure compressed hydrogen gas as a result of the weak dispersion-type (physisorptive) interactions that predominate as the mode of gas uptake. Indeed, the isosteric heat of adsorption within these materials usually lies within a range of 5–7 kJ mol−1, which is far below the estimated optimal value of ca. 15 kJ mol−1 for storage at 298 K.1j,4 This value applies to the whole adsorption range for an adsorbent operating between 1.5 and 100 bar, highlighting the need for materials that not only possess binding sites of the appropriate adsorption enthalpy, but also have a high concentration of these sites in order to maximize the quantity of usable hydrogen.One strategy for improving the room temperature hydrogen storage performance of metal–organic frameworks is through the generation of exposed metal cation sites on the framework surface.5,6 Previously, we have reported a number of metal–organic frameworks in which removal of a solvent molecule from the coordination sphere of the framework metal cations yields strong adsorption sites for H2. Indeed, zero-coverage isosteric heats of H2 adsorption of 10.1 and 9.5 kJ mol−1 were observed in the cubic sodalite-type frameworks Mn3[(Mn4Cl)3(BTT)8(MeOH)10]2 (Mn-BTT, BTT3− = 1,3,5-benzenetristetrazolate)5f and HCu[(Cu4Cl)3(BTT)8]·3.5HCl (Cu-BTT),5h which remarkably retain their structures upon desolvation. One possibility for improving the isosteric heat of adsorption within this structure type would be through the replacement of the Mn2+ or Cu2+ ions in the structure with M2+ cations having a smaller radius. The greater charge density of the exposed metal cations on the framework surface should then be more effective at inducing a dipole in H2, leading to stronger binding.
Metal–organic frameworks with unsaturated metal cation sites are also of potential utility as separation materials for the capture of CO2 from flue gas streams.5j,7 While some frameworks have been shown to have tremendous capacities for CO2 at high pressures,1d,i most of these materials exhibit low uptake in the pressure region of interest of 0.1–0.2 bar for flue gas separations. Perhaps most importantly, for a framework to be viable as a separation material, a high selectivity for CO2 over the other components of the gas stream, primarily N2, is required. Exposed metal cations can provide polarizing sites that interact selectively with CO2, which has a greater polarizability than N2. Thus, tuning of the pore surface charge through variation of the metal cation may be one route toward optimized CO2 capture materials that exhibit reversible capture at minimal energy cost.
From a fundamental perspective, systematic studies of the gas sorption properties of isostructural materials containing unsaturated coordination sites are currently limited to a small number of systems, such as M2(DOBDC) (M = Mg, Mn, Co, Ni, Zn; H4DOBDC = 2,5-dihydroxyterephthalic acid).5j,7,8 The study of a larger number of systems is needed in order to understand more fully the effects of metal substitution on crucial gas adsorption properties, such as the isosteric heat of adsorption for H2, or the separation factor for CO2 over N2. Moreover, the extent to which other metal cations can adopt the Mn- and Cu-BTT structure type is currently unknown, which prompted our attempts to expand the library of these materials.
While a number of Fe3+-based porous metal–organic frameworks have been reported to date,9 the preparation of the Fe2+-based frameworks has been relatively rare.10 Most importantly, to our knowledge, no compounds with accessible, open Fe2+ coordination sites have been studied for gas adsorption. Herein, we report the synthesis and characterization of the iron-based sodalite-type metal–organic framework Fe3[(Fe4Cl)3(BTT)8(MeOH)4]2 (Fe-BTT), in which exposed Fe2+ cations indeed serve as strong adsorption sites for H2 and CO2.
High-throughput methodology
The modular synthesis of metal–organic frameworks, wherein a metal salt and an organic bridging ligand are combined under solventothermal conditions, is a tremendously versatile approach for the discovery of new materials. While convenient, the optimal synthetic procedures affording a desired porous phase in pure crystalline form are frequently discovered only following the survey of a large number of reaction conditions. This is in part due to the considerable impact that subtle changes in the reaction parameters can have on the resulting products. Indeed, for a given combination of metal salt and bridging ligand, numerous different crystalline phases are often possible. Furthermore, the undesired side-products are frequently extended network solids that are insoluble in organic solvents, and prove to be inseparable from the desired phase by conventional solution techniques. Thus, the identification and optimization of the synthetic conditions for a new metal–organic framework typically involves the systematic variation of many reaction parameters, including the metal salt, metal-to-ligand ratio, solvent composition, acid/base content, reaction temperature, and reaction time.A thorough survey of so many different reaction parameters means that the total number of screening reactions quickly becomes unwieldy. In such cases, high-throughput methodologies, which have been successfully implemented in the synthesis and characterization of a variety of inorganic materials,11 are of potential benefit. In fact, very recently, a number of metal–organic framework systems have been studied by high-throughput approaches, showing that the development of solventothermal syntheses of this class of materials can be well-suited to automated methods.9b,12
The high-throughput methodology employed in this work was performed using an automated solid- and liquid-dispensing robotic module (Fig. S1,† manufactured by Symyx Technologies,‡ Sunnyvale, CA) mounted within a glove box under a dinitrogen atmosphere. The system allows rapid dispensing of reagents and solvents into individual reaction vials (1, 2, 4, 8, or 20 mL capacity), and depending on the vial size, up to 96 reactions can be performed on a single vial plate (see Fig. S2†). The composition of each reaction mixture is programmed through a computer interface, that also allows the experimental variables, such as reaction temperature, ramping and cooling rate, and reaction time, to be precisely set and monitored. Once the individual reactions are prepared and sealed, the synthesis is performed under static conditions on the robot deck. The product formed within each reaction vial can be imaged using a camera installed on the robot deck, allowing for rapid identification of the reaction conditions leading to sizable single crystals. Additionally, the solids precipitated within each reaction vial can be isolated and automatically dispensed onto a glass plate for high-throughput powder X-ray diffraction measurements. The experimental data, such as the actual quantities of reagents dispensed, powder X-ray diffraction patterns, and image data, are collectively recorded within a centralized database for analysis and comparison.
The following section describes the optimized synthesis and characterization of Fe-BTT. Note that although this iron-based metal–organic framework is isostructural with Mn- and Cu-BTT, a large number of screening reactions were required in order to obtain the material in pure form. Significantly, initial attempts to synthesize Fe-BTT using reaction conditions similar to those under which the other analogues could be prepared proved unsuccessful. Thus, the optimized conditions were developed following successive refinements of the reaction parameters using small-scale reactions (typically 1 or 2 mL-scale reactions). Most importantly, the resulting optimized conditions were vastly different from those for Mn-BTT and Cu-BTT, highlighting the benefit of systematic combinatorial screening performed using an automated high-throughput system.
Synthesis and characterization
The reaction between FeCl2 and H3BTT in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio in a mixture of DMF and DMSO leads to the formation of Fe3[(Fe4Cl)3(BTT)8]2·22DMF·32DMSO·11H2O (solvated Fe-BTT) as pale-yellow cubic crystals. X-ray structural analysis of a single crystal revealed the expected porous cubic (space group: Pm
:1 mole ratio in a mixture of DMF and DMSO leads to the formation of Fe3[(Fe4Cl)3(BTT)8]2·22DMF·32DMSO·11H2O (solvated Fe-BTT) as pale-yellow cubic crystals. X-ray structural analysis of a single crystal revealed the expected porous cubic (space group: Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, a = 18.8235(11) Å) framework structure consisting of chloride-centered [Fe4Cl]7+ square units connected via triangular BTT3− ligands to form a 3,8-net (Fig. 1). Here, six [Fe4Cl]7+ fragments and eight BTT3− ligands combine to form a truncated octahedral, sodalite-like cage with an internal diameter of approximately 10.3 Å (based on van der Waals radii). Adjacent cages share square faces to form a cubic, anionic framework with a three-dimensional channel system with pore openings of approximately 9 Å in diameter. Despite significant residual electron density observed within the sodalite cages and in the channels of the framework, the X-ray data did not allow a clear determination of the location of the charge-balancing Fe2+ cations within the pores of the solvated structure. However, their presence within the pores could be identified by Mössbauer spectroscopy (see below).
m, a = 18.8235(11) Å) framework structure consisting of chloride-centered [Fe4Cl]7+ square units connected via triangular BTT3− ligands to form a 3,8-net (Fig. 1). Here, six [Fe4Cl]7+ fragments and eight BTT3− ligands combine to form a truncated octahedral, sodalite-like cage with an internal diameter of approximately 10.3 Å (based on van der Waals radii). Adjacent cages share square faces to form a cubic, anionic framework with a three-dimensional channel system with pore openings of approximately 9 Å in diameter. Despite significant residual electron density observed within the sodalite cages and in the channels of the framework, the X-ray data did not allow a clear determination of the location of the charge-balancing Fe2+ cations within the pores of the solvated structure. However, their presence within the pores could be identified by Mössbauer spectroscopy (see below).
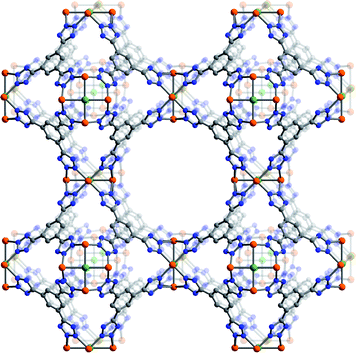 | ||
| Fig. 1 A portion of the single-crystal X-ray structure of Fe-BTT. Orange, green, gray, and blue spheres represent Fe, Cl, C, and N atoms, respectively. Solvent molecules, H atoms, and charge-balancing cations are omitted for clarity. Selected interatomic distances (Å) and angles (°): Fe–Cl 2.649(1), Fe–N 2.132(2), Fe⋯Fe 3.746(2), Fe–Cl–Fe 90.0, N–Fe–N 87.4(1), 91.7(1), Fe–N–N 124.7(1), 125.6(1). | ||
Activation of Fe-BTT for gas adsorption experiments was performed by first exchanging the solvent molecules within the pores and bound to the Fe2+ metal centers with methanol, followed by heating of the solid at 135 °C under dynamic vacuum for 24 h. While the outgas rate after this time was <2 mTorr min−1, powder neutron diffraction data (discussed below) indicate that approximately 30% of the framework Fe2+ centers retain a bound methanol molecule. The ensuing formulation of Fe3[(Fe4Cl)3(BTT)8(MeOH)4]2 for Fe-BTT is indeed consistent with elemental analysis and infrared spectroscopy. Unfortunately, attempts to utilize higher activation temperatures to remove all of the residual solvent resulted in decomposition of the framework. This observation is consistent with thermogravimetric analysis data for methanol-exchanged Fe-BTT (Fig. S14 in the ESI†), which display a rapid drop in mass above 150 °C.
Low-pressure N2 adsorption measurements performed on Fe-BTT at 77 K revealed a type-I adsorption isotherm characteristic of a microporous solid. Fits to the data yielded a BET surface area13 of 2010 m2 g−1, and a Langmuir surface area of 2200 m2 g−1. These values are close to the accessible surface area of 2195 m2 g−1 calculated from the fully-desolvated crystal structure.14 Note that the BET surface area of Fe-BTT lies slightly below the corresponding value for Mn-BTT (2100 m2 g−1) and above that obtained for Cu-BTT (1710 m2 g−1). The differences correlate with the unit cell volumes of the three isostructures (Mn-BTT, 6985(1) Å3; Fe-BTT, 6670(1) Å3; Cu-BTT: 6430(4) Å3). The unit cell volume is directly related to the ionic radii of the three metal ions (high-spin Mn2+: 0.83 Å; Fe2+: 0.78 Å; Cu2+: 0.73 Å) and the resulting distances for the metal–chloride and metal–nitrogen bonds.
Mössbauer spectra
The sample used for Mössbauer spectroscopy was prepared by immersing the as-synthesized form of Fe-BTT in DMF, thereby generating a material in which all of the DMSO and water molecules within the pores had been replaced by DMF molecules. Selected Mössbauer spectra of this sample obtained between 10 and 295 K are shown in Fig. 2, and the corresponding hyperfine parameters are given in Table S2.† The Mössbauer spectrum at 10 K was used to determine the optimal stoichiometry of the DMF-solvated material by identifying a component doublet, shown in red in Fig. 2, that is assigned to [Fe(DMF)6]3+ complexes within the pores. The relative areas of the spectral components then lead to the formulation [FeIII(DMF)6]0.63[FeII(DMF)6]0.56[(FeII4Cl)3(BTT)8]·nDMF. The relative areas of the spectral components were constrained to be consistent with this stoichiometry at all temperatures. Relaxation of this constraint led to at most minor changes in the relative areas. In these fits, the hyperfine parameters of the doublet assigned to [Fe(DMF)6]2+, shown in black in Fig. 2, have been constrained to the parameters reported for [Fe(DMF)6](ClO4)2 in the solid state.15 Note that while the preparation of Fe-BTT utilized Fe2+ ions, we envisage the origin of the [FeIII(DMF)6]3+ signal to be a result of oxidation during sample work-up or transfer to the spectrometer.![Mössbauer spectra collected between 10 and 295 K for a sample of DMF-solvated Fe-BTT. The four green doublets are attributed to the high-spin Fe2+ ions in the [Fe4Cl]7+ units of the framework, and the black and red doublets to charge-balancing high-spin Fe2+ and Fe3+ ions within the pores.](/image/article/2010/SC/c0sc00179a/c0sc00179a-f2.gif) | ||
| Fig. 2 Mössbauer spectra collected between 10 and 295 K for a sample of DMF-solvated Fe-BTT. The four green doublets are attributed to the high-spin Fe2+ ions in the [Fe4Cl]7+ units of the framework, and the black and red doublets to charge-balancing high-spin Fe2+ and Fe3+ ions within the pores. | ||
The first unusual feature of the Mössbauer spectra is the broad nature of the spectral component associated with the Fe2+ ions in the [Fe4Cl]7+ units of the framework. Crystallographically, the four Fe2+ centers are equivalent, but a broad absorption profile, rather than the expected single quadrupole doublet is observed. This broadening occurs because of a distribution in the positions of the DMF, [Fe(DMF)6]3+, and [Fe(DMF)6]2+ species that, at least at lower temperatures, are frozen within the pores. This distribution of local environments has been fit with the four high-spin Fe2+ quadrupole doublets shown in green.
The second unusual feature found in the spectra is the narrowing at 225 K and above of the distribution of local environments of the Fe2+ ions in the framework. This narrowing occurs because at 225 K and above the mobility of the DMF molecules in the pores increases dramatically, consistent with the melting point of 212 K for bulk DMF. Relatedly, there should also be an increase in the mobility of the [FeIII(DMF)6]3+ and [FeII(DMF)6]2+ complexes within the pores at some temperature above 155 K, leading to an averaging of the local environments. This increased mobility within the pores averages the local structural perturbations at an Fe2+ site and the framework Fe2+ ions therefore become electronically and structurally more similar.
Hydrogen storage properties
Low-pressure H2 adsorption isotherms for an activated sample of Fe-BTT were collected at 77 and 87 K, as shown in Fig. 3. The adsorption of 2.3 wt% at 77 K, and 1.6 wt% at 87 K, is completely reversible, and the steep initial portion of each isotherm is indicative of the presence of strongly-polarizing binding sites with a high affinity for H2. A virial type fitting to the data recorded at the two temperatures allowed the calculation of the isosteric heat of adsorption as a function of H2 coverage, as shown in Fig. 4. Indeed, the steep rise in the isotherms translates to a zero-coverage isosteric heat of adsorption of 11.9 kJ mol−1, which is among the largest recorded for H2 binding in a metal–organic framework.16 As the loading of H2 is increased, the isosteric heat decreases in magnitude, corresponding to the strongest binding sites becoming saturated and subsequent population of weaker binding sites. At an uptake of 1 wt%, the isosteric heat reaches 7 kJ mol−1, whereafter more typical dispersive interactions dominate as the binding mode.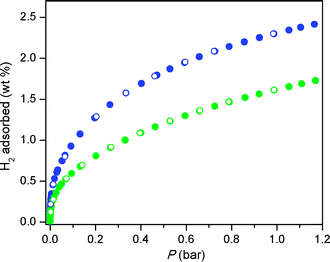 | ||
| Fig. 3 Low-pressure H2 adsorption isotherms in Fe-BTT recorded at 77 K (blue) and 87 K (green). Closed and open symbols represent adsorption and desorption, respectively. | ||
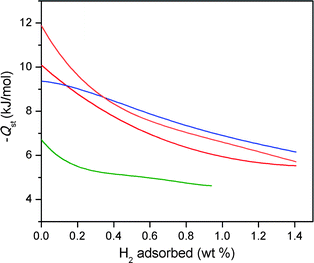 | ||
| Fig. 4 Plots of the isosteric heat of adsorption as a function of hydrogen uptake in Fe-BTT (orange), Mn-BTT (red),5f Cu-BTT (blue),5h and Zn4O(1,4-benzenedicarboxylate)3 (green).3b | ||
Comparison of the initial isosteric heat of adsorption for Fe-BTT to that observed for Mn-BTT (10.1 kJ mol−1) reveals a significant increase as a result of metal substitution. This is consistent with the higher charge-to-radius ratio for Fe2+ compared to Mn2+, leading to a more polarizing binding site at the metal ion. Furthermore, in the case of Cu-BTT, although Cu2+ is a smaller ion than Fe2+, it has an anisotropic electron distribution that reduces the charge at the open coordination site, leading to a longer Cu2+–H2 interaction distance.5e This longer distance translates to a lower initial isosteric heat of adsorption of 9.5 kJ mol−1. However, beyond 0.4 wt% uptake, the isosteric heat of adsorption for Cu-BTT overtakes that of Fe-BTT. This is likely due to the greater number of desolvated M2+ ions in Cu-BTT, whereas Fe-BTT retains some bound methanol molecules.
The higher-pressure adsorption isotherms recorded at 77 K and 298 K are shown in Fig. 5. At 77 K, Fe-BTT exhibits a maximum excess adsorption of 3.7 wt%. The total adsorption was calculated using the experimental pore volume measured by argon porosimetry, which yielded a value of 0.715 cm3 g−1. This result is close to the idealistic pore volume calculated from the crystallographic data (0.724 cm3 g−1). The total uptake, which is more relevant for assessment of the system sorption properties, reaches 4.1 wt% and 35 g L−1 at 95 bar. Surprisingly, these values are below the corresponding total adsorption values recorded for Mn-BTT (6.9 wt%, 60 g L−1 at 90 bar),5f and Cu-BTT (5.7 wt%, 53 g L−1 at 90 bar).5h Nevertheless, the total uptake in Fe-BTT at 95 bar constitutes a 16% increase in volumetric density compared to that of pure H2 gas at the same pressure.
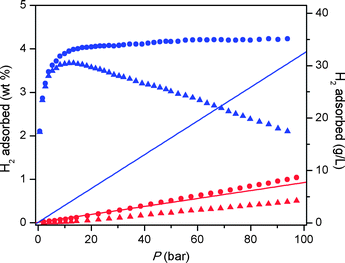 | ||
| Fig. 5 High-pressure H2 adsorption isotherms in Fe-BTT recorded at 77 K (blue) and 298 K (red). Triangles and circles represent excess and total uptake, while the solid lines show the density of pure H2 gas. | ||
At 298 K, the total H2 uptake in Fe-BTT reaches 1.0 wt% and 8.4 g L−1 owing to the high isosteric heat of adsorption at the unsaturated metal centers. This is still somewhat lower than the uptake observed for Mn-BTT, which is likely due to the differences in the unit cell, in particular the position of the extra-framework cations, observed for the desolvated structures. The volumetric uptake still, however, ranks among the highest observed at room temperature for metal–organic frameworks, highlighting the importance of open coordination sites for high-density H2 storage.1j
Powder neutron diffraction data
Powder neutron diffraction experiments using the high resolution diffractometer BT1 at the National Institute of Standards and Technology Center for Neutron Research (NCNR) have directly confirmed that the large isosteric heat of adsorption for H2 in Fe-BTT is associated with the presence of open Fe2+ coordination sites associated with the [Fe4Cl]7+ clusters of the framework. Prior to D2 loadings, the neutron diffraction pattern of the evacuated form of Fe-BTT was collected using the Ge(311) monochromator and an in-pile collimation of 15 min of arc corresponding to a wavelength of 2.0878 Å. The subsequent structure determination and Rietveld analysis17 allowed two important structural features of the evacuated material to be elucidated. Firstly, the position of the charge-balancing Fe2+ ions could be refined at the expected occupancy of 1/8 for a position approximately 4.23 Å directly above the chloride anion of the [Fe4Cl]7+ cluster in a bowl-like binding site (see Fig. S3†). Interestingly, this is in contrast to the desolvated structures of Mn-BTT and Cu-BTT, wherein the extra-framework cations were observed between two tetrazolate rings in a chelated coordination geometry. Detailed analysis of the neutron diffraction data for Fe-BTT indicated an absence of nuclear scattering density at the analogous chelate site. Secondly, a partial occupancy of approximately 30% of methanol was detected bound to the framework Fe2+ ions. Note that throughout the diffraction experiments, there was no evidence for any superlattice or magnetic Bragg reflections and the data could be entirely described from the nuclear structure indicating lack of long-range magnetic order.The binding sites for D2 within Fe-BTT were located by taking the nuclear density Fourier difference maps between the loaded and bare materials, and allowing the positions and occupancies of D2 to freely refine while fixing the methanol location and composition. The evacuated framework was sequentially dosed with quantities of approximately 4, 8, 20, 33, 65, and 98 D2 molecules per formula unit. A portion of the neutron diffraction pattern obtained at 4 K at a loading of 8 D2 molecules per formula unit is shown in Fig. 6. The first position in the structure to be occupied by D2, labeled I in Fig. 7, corresponds to the strongest adsorption site and lies a remarkably short 2.17(5) Å from the framework Fe2+ ion. This is indeed consistent with the higher initial isosteric heat of adsorption for H2 compared to Mn-BTT and Cu-BTT, which exhibited metal–D2 distances of 2.27 and 2.47 Å, respectively.5e,g To our knowledge, this constitutes the shortest metal–D2 distance yet observed in a metal–organic framework. The close approach of the hydrogen molecules to the surface is essential for achieving a high storage density. Interestingly, the extra-framework cations do not appear to act as high-affinity binding sites, since no nuclear density was observed in the proximity of these sites at low loadings. This demonstrates that the greater isosteric heat of adsorption observed at zero-coverage is primarily provided by the unsaturated metal sites of the [Fe4Cl]7+ cluster.
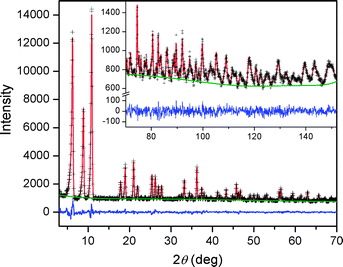 | ||
| Fig. 6 Powder neutron data for Fe-BTT loaded with 8 D2 molecules per formula unit. Green lines, crosses, and red lines represent the background, experimental, and calculated diffraction patterns, respectively. The blue line represents the difference between experimental and calculated patterns. The final Rietveld goodness-of-fit parameter was χ2 = 1.063. | ||
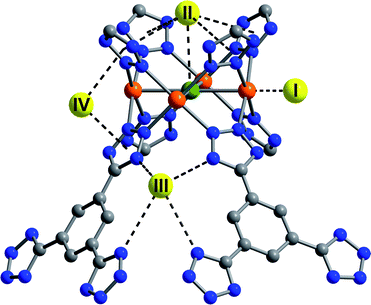 | ||
| Fig. 7 The first four D2 binding sites within Fe-BTT determined from Rietveld analysis of the powder neutron diffraction data. Yellow, orange, green, blue, and gray spheres represent D2, iron, chlorine, nitrogen, and carbon, respectively. Hydrogen atoms have been omitted for clarity. | ||
At higher D2 loadings, additional adsorption sites become more prominent. The next strongest binding position, labeled II in Fig. 7, is situated approximately 3.44 Å above the chloride ion of the [Fe4Cl]7+ units. Four tetrazolate rings are also within 3.60 Å of this site, forming a bowl-like adsorption pocket with a negative surface charge. Up to a loading of 8 D2 molecules per formula unit, only sites I and II are occupied, suggesting that the binding enthalpy at these two sites are considerably higher than the subsequently occupied sites. At a loading of 20 D2 molecules per formula unit, two further sites, labeled III and IV, become occupied. Site III places the D2 molecules at the openings of the sodalite-type cages, allowing interactions with the N2 atoms of four tetrazolate ligands at a distance of approximately 3.45 Å. Site IV is located in the channels of the structure, with the D2 centroid 3.16 Å from the plane of two tetrazolate rings. All subsequently occupied sites, while localized, are typical for physisorbed hydrogen, as reflected in a framework–D2 distance of greater than 3.6 Å.
Comparison of the D2 adsorption sites of Fe-BTT and its analogues reveals a number of differences. Most notably, site III is not occupied within Mn-BTT and Cu-BTT until much higher D2 loadings. The third binding site in the other frameworks corresponds to a position analogous to that of site IV in Fe-BTT, while the position of the fourth occupied site, which places the D2 molecule in close contact with the aromatic ring of the BTT3− ligand, is not occupied in Fe-BTT until higher loadings. These differences presumably reflect changes associated with the slightly differing unit cell dimensions and the distinct position of the extra-framework cations in Fe-BTT compared to Mn-BTT and Cu-BTT. Full details of the position of the binding sites, and the loading characteristics thereof are presented in Tables S3–10.†
Inelastic neutron scattering spectra
The H2 loading characteristics of Fe-BTT have been further probed by inelastic neutron scattering5l,18 using the FANS spectrometer at the NCNR.19 Spectra were obtained by subjecting the compound to a collimated and monochromated neutron beam, and determining the energy transferred to the sample at a bank of 3He detectors after passing the scattered neutrons through a low-energy band-pass filter consisting of polycrystalline Bi, Be, and graphite. Data were collected at approximately the same loading levels as for the neutron diffraction experiments to allow the best opportunity for correlation of the FANS spectra with the binding sites observed crystallographically. Routine data reduction and subtraction of the evacuated Fe-BTT spectrum was performed,20 and selected spectra are shown in Fig. 8.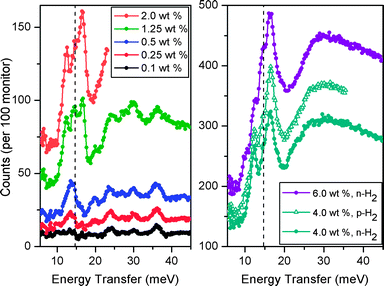 | ||
| Fig. 8 Inelastic neutron spectra at several hydrogen loadings following subtraction of the spectrum of evacuated Fe-BTT. Filled circles and open triangles represent data for normal hydrogen (n-H2) and para-hydrogen (p-H2), respectively. Left: lowest n-H2 data up to 2.0 wt%. Right: higher n-H2 loadings up to 6 wt%, and p-H2 at 4 wt%. In both panels, the dashed line represents the expected energy transfer of 14.7 meV corresponding to the first rotational transition (J = 0→1) for free hydrogen. | ||
In the lowest loading data (<0.5 wt%), there is an absence of a rotational line at 14.7 meV corresponding to the first rotational transition of free hydrogen. This is indicative of a strong rotational hindering potential of the binding sites occupied at these levels, consistent with the large initial isosteric heat of H2 adsorption. At 0.25 wt%, a loading at which sites I and II are populated, two lines are observed at approximately 13.2 and 19.7 meV. These peaks gain intensity upon increasing the loading to 0.5 wt% of H2, and can be assigned as rotational lines that correspond to a rotational barrier of approximately 15 meV. Upon increasing the loading to 1.25 wt%, sites III and IV become occupied at approximately 50% occupancy. The rotational lines associated with these sites presumably give rise to the new distinct peaks around the energy of free H2, implying a lower rotational barrier. Further increases in the loading to 2.0 wt% increase the intensities of these lines, which corresponds to increasing the population of these sites, as well as the population of an additional site (site V in Fig. S3†) that is a dispersive H2–framework interaction. The highest loading data of 4.0 and 6.0 wt% exhibit very broad indistinct features at higher energy (>20 meV), adopting the character of molecular recoil from weakly bound hydrogen. The overall intensity of the spectral features scale well with the loading level for all spectra. Furthermore, at a loading of 6 wt%, there is the additional appearance of a shoulder on the most intense peak, which is at a frequency characteristic of bulk hydrogen as would be typical of weakly adsorbed hydrogen on a carbon surface.
Comparison of the spectra for normal H2 (n-H2) and para-H2 (p-H2) at a loading of 4 wt% reveals a scaling factor of 1.2. The composition of n-H2 at room temperature is 25% p-H2, and 75% ortho-H2 (o-H2). No significant change in this composition is expected upon lowering of the temperature of the gas, unless the conversion between the two forms is catalyzed by a paramagnetic material. Indeed, the small scaling factor between the two spectra is indicative of the conversion of o-H2 to p-H2 by Fe-BTT. This is presumably due to the H2 coming into close proximity with a paramagnetic high-spin Fe2+ center following dosing of H2 at approximately 50 K, initiating interconversion between the two forms. However, as the sample is cooled to the final temperature of 4 K, the hydrogen mobility and access to the Fe2+ centers becomes sufficiently hindered such that the para–ortho ratio becomes frozen. Evidence for catalytic conversion by solely the Fe2+ centers also comes from the dependence of the scaling factor on the loading level. Indeed, as the loading level of p-H2 is decreased, the scaling factor approaches 1, consistent with a greater fraction of H2 molecules being able to interact with the Fe2+ centers. At a loading of 0.1 wt%, the p-H2 and n-H2 become virtually superimposable due to the H2 being completely accommodated at the unsaturated coordination site.
Selective CO2 adsorption
Fig. 9 displays the CO2 and N2 adsorption isotherms obtained for Fe-BTT at 298 K. The initial steep portion of the CO2 isotherm likely corresponds to the strong binding of CO2 to the exposed Fe2+ cation sites. The uptake of approximately 3.8 wt% reached in this steep portion is consistent with the interaction of 0.4 CO2 molecules per available Fe2+ cation. At 1 bar, the CO2 uptake reaches 13.5 wt%. The near-linear adsorption profile for N2 is indicative of its low affinity for the open metal sites, as expected from its relatively low polarizability. Indeed, this leads to an N2 uptake of just 1.0 wt% at 1 bar. This is also reflected in the uptake ratio by weight for CO2 over N2, which reaches 30.7:1 at 0.1 bar, and 10.8:1 at 1.0 bar.
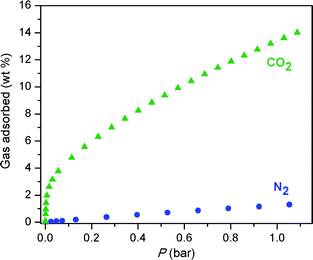 | ||
| Fig. 9 Adsorption isotherms for Fe-BTT for the uptake of CO2 (green triangles) and N2 (blue circles) recorded at 298 K. | ||
Assessing the utility of an adsorbent for capture of CO2 using single-component isotherms requires that the relative pressures of the gases in a flue gas be taken into account. A typical flue gas has a total pressure of approximately 1 bar, and consists of primarily N2 (70–75 wt%), CO2 (15–16 wt%), and H2O (5–7 wt%).21 This converts to partial pressures of approximately 0.15 and 0.75 bar for CO2 and N2, respectively. The separation factor calculated from the experimental uptakes at these partial pressures is approximately 5.5:1. For comparison with other metal–organic frameworks investigated for CO2 capture, we note that the uptake of 4.8 wt% at 0.1 bar of CO2 is surpassed only by the compounds M2(DOBDC) (M = Mg, Co, Ni Zn), which possess a higher concentration of open metal cation sites.5j,7 While these compounds are also favored for their exceptional water stability,22 it is possible that the larger pore openings within Fe-BTT could provide an advantage in terms of gas diffusion rates.
Outlook
The foregoing results demonstrate the successful use of a high-throughput methodology for the synthesis of a new sodalite-type metal–organic framework, Fe-BTT, featuring open Fe2+ cation sites. Gas adsorption measurements combined with neutron diffraction data and inelastic neutron scattering spectroscopy show that these sites provide a strong interaction with H2, drawing it close to the surface to afford an enhanced storage capacity at 298 K. The exposed cation sites further lead to selective adsorption of CO2 over N2 at 298 K, rendering it of potential interest for CO2 capture from flue gas. We anticipate that this powerful synthetic methodology will now provide access to a broad range of isostructural materials, for which the greater polarizing power of cations such as Mg2+, Co2+, and Ni2+ can be expected to give rise to improved H2 storage and CO2 capture properties. In addition, compounds of this type incorporating triazolate or pyrazolate groups in place of tetrazolate can be expected to exhibit enhanced thermal and chemical stability.23 Such improvements in stability are deemed essential for enabling the complete desolvation of the metal cation sites, and improving the suitability of these metal–organic frameworks for real-world applications.Acknowledgements
This research was funded by the General Motors Company, the US Defense Logistics Agency, and the Center for Gas Separations Relevant to Clean Energy Technologies, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under award number DE-SC0001015. Work at NIST was partially supported by the U.S. Department of Energy within the Hydrogen Sorption Center of Excellence. We acknowledge Fulbright New Zealand for partial support of K.S. F.G. acknowledges support from the Fonds National de la Recherche Scientifique, Belgium (grants 9.456595 and 1.5.064.05). We also thank Dr M. T. Sougrati for assistance in obtaining some of the Mössbauer spectra, Dr T. Boussie, Dr C. Yuan, and Dr R. Rosen of Symyx Technologies for assistance with setting up the high-throughput apparatus, and Dr A. G. DiPasquale for helpful discussions.Notes and references
- (a) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef; (b) S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS; (c) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata and Y. Kawazoe, Nature, 2005, 436, 238 CrossRef CAS; (d) A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998 CrossRef CAS; (e) H. Furukawa, M. A. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17, 3197 RSC; (f) G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC; (g) S. Ma, D. Sun, J. M. Simmons, C. D. Collier, D. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130, 1012 CrossRef CAS; (h) R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966 CrossRef; (i) P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. De Weireld, J.-S. Chang, D.-Y. Hong, Y. K. Hwang, S. H. Jhung and G. Férey, Langmuir, 2008, 24, 7245 CrossRef; (j) L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC; (k) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 Search PubMed.
- EERE: Hydrogen, Fuel Cells, & Infrastructure Technologies Program, http://www.eere.energy.gov/hydrogenandfuelcells/, (accessed January 2010).
- (a) M. Latroche, S. Surblé, C. Serre, C. Mellot-Draznieks, P. L. Llewellyn, J.-H. Lee, J.-S. Chang, S. H. Jhung and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 8227 CrossRef CAS; (b) S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176 CrossRef CAS; (c) K. Sumida, M. R. Hill, S. Horike, A. Dailly and J. R. Long, J. Am. Chem. Soc., 2009, 131, 15120 CrossRef CAS; (d) K. Koh, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2009, 131, 4184 CrossRef CAS.
- S. K. Bhatia and A. L. Myers, Langmuir, 2006, 22, 1688 CrossRef CAS.
- (a) P. D. C. Dietzel, Y. Morita, R. Blom and H. Fjellvag, Angew. Chem., Int. Ed., 2005, 44, 6354 CrossRef CAS; (b) M. Dincă and J. R. Long, J. Am. Chem. Soc., 2005, 127, 9376 CrossRef CAS; (c) N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504 CrossRef CAS; (d) A. Vimont, J.-M. Goupil, J.-C. Lavalley, M. Daturi, S. Surblé, C. Serre, F. Millange, G. Férey and N. Audebrand, J. Am. Chem. Soc., 2006, 128, 3218 CrossRef CAS; (e) H. R. Moon, N. Kobayashi and M. P. Suh, Inorg. Chem., 2006, 45, 8672 CrossRef CAS; (f) M. Dincă, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876 CrossRef CAS; (g) P. D. C. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellvag, Chem. Commun., 2006, 959 RSC; (h) M. Dincă, W. S. Han, Y. Liu, A. Dailly, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2007, 46, 1419 CrossRef CAS; (i) M. Dincă and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766 CrossRef CAS; (j) S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870 CrossRef CAS; (k) P. D. C. Dietzel, R. Blom and H. Fjellvag, Eur. J. Inorg. Chem., 2008, 3624 CrossRef CAS; (l) Y. Liu, H. Kabbour, C. M. Brown, D. A. Neumann and C. C. Ahn, Langmuir, 2008, 24, 4772 CrossRef CAS.
- Unsaturated metal sites within inorganic and hybrid materials have previously been shown to facilitate hydrogen adsorption: (a) P. M. Forster, J. Eckert, J.-S. Chang, S.-E. Park, G. Férey and A. K. Cheetham, J. Am. Chem. Soc., 2003, 125, 1309 CrossRef CAS; (b) M. R. Hartman, V. K. Peterson, Y. Liu, S. S. Kaye and J. R. Long, Chem. Mater., 2006, 18, 3221 CrossRef CAS; (c) P. M. Forster, J. Eckert, B. D. Heiken, J. B. Parise, J. W. Yoon, S. H. Jhung, J.-S. Chang and A. K. Cheetham, J. Am. Chem. Soc., 2006, 128, 16846 CrossRef CAS.
- (a) P. D. C. Dietzel, R. E. Johnsen, H. Fjellvag, S. Bordiga, E. Groppo, S. Chavan and R. Blom, Chem. Commun., 2008, 5125 RSC; (b) P. D. C. Dietzel, V. Besikiotis and R. Blom, J. Mater. Chem., 2009, 19, 7362 RSC; (c) D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637 CrossRef CAS; (d) A.Ö. Yazaydin, R.Q. Snurr, T.-H. Park, K. Koh, J. Liu, M. D. LeVan, A. I. Benin, P. Jakubczak, M. Lanuza, D. B. Galloway, J. J. Low and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 18198 CrossRef CAS.
- W. Zhou, H. Wu and T. Yildirim, J. Am. Chem. Soc., 2008, 130, 15268 CrossRef CAS.
- (a) T. R. Whitfield, X. Wang, L. Liu and A. J. Jacobson, Solid State Sci., 2005, 7, 1096 CrossRef CAS; (b) S. Bauer, C. Serre, T. Devic, P. Horcajada, J. Marrot, G. Férey and N. Stock, Inorg. Chem., 2008, 47, 7568 CrossRef CAS; (c) C. Volkringer, D. Popov, T. Loiseau, G. Férey, M. Burghammer, C. Riekel, M. Haouas and F. Taulelle, Chem. Mater., 2009, 21, 5695 CrossRef CAS; (d) P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172 CrossRef CAS.
- (a) G. J. Halder, K. W. Chapman, S. M. Neville, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2008, 130, 17552 CrossRef CAS; (b) S. Ma, D. Yuan, J.-S. Chang and H.-C. Zhou, Inorg. Chem., 2009, 48, 5398 CrossRef CAS.
- (a) K. Choi, D. Gardner, N. Hilbrandt and T. Bein, Angew. Chem., Int. Ed., 1999, 38, 2891 CrossRef CAS; (b) J. Klein, C. W. Lehmann, H.-W. Schmidt and W. F. Maier, Angew. Chem., Int. Ed., 1998, 37, 3369 CrossRef CAS; (c) L. Zhang, J. Yao, C. Zeng and N. Xu, Chem. Commun., 2003, 2232 RSC; (d) H. Koinuma and I. Takeuchi, Nat. Mater., 2004, 3, 429 CrossRef CAS; (e) A. Corma, M. Moliner, J. M. Serra, P. Serna, M. J. Díaz-Cabañas and L. A. Baumes, Chem. Mater., 2006, 18, 3287 CrossRef CAS.
- (a) R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939 CrossRef CAS; (b) A. Sonnauer, F. Hoffmann, M. Fröba, L. Kienle, V. Duppel, M. Thommes, C. Serre, G. Férey and N. Stock, Angew. Chem., Int. Ed., 2009, 48, 3791 CrossRef CAS.
- K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552 CrossRef CAS.
- T. Düren, F. Millange, G. Férey, K. S. Walton and R. Q. Snurr, J. Phys. Chem. C, 2007, 111, 15350 CrossRef.
- W. Linert, V. Gutmann, O. Baumgartner, G. Wiesinger and H. Kirchmayr, Inorg. Chim. Acta, 1983, 74, 123 CrossRef CAS.
- The greatest isosteric heat of adsorption at zero-coverage yet observed for a metal–organic framework is 15.1 kJ mol−1 in Co4(H2O)4(MTB)2 (H4MTB = methanetetrabenzoic acid), which features exposed Co2+ cations: Y. E. Cheon and M. P. Suh, Chem. Commun., 2009, 2296 Search PubMed . Moreover, a site-specific binding enthalpy of 13.5 kJ mol−1 has been observed in Ni2(DOBDC) (H4DOBDC = 2,5-dihydroxyterephthalic acid), which possesses exposed Ni2+ cations: J. G. Vitillo, L. Gegli, S. Chavan, G. Ricchiardi, G. Spoto, P. D. C. Dietzel, S. Bordiga and A. Zecchina, J. Am. Chem. Soc., 2008, 130, 8386 RSC.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210 CrossRef CAS.
- A number of inelastic neutron scattering studies of hydrogen adsorption in metal–organic frameworks have been reported recently: (a) J. Rowsell, J. Eckert and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 14904 CrossRef CAS; (b) Y. Liu, C. M. Brown, D. A. Neumann, V. K. Peterson and C. J. Kepert, J. Alloys Compd., 2007, 446–447, 385 CrossRef CAS; (c) F. M. Mulder, T. J. Dingemans, H. G. Schimmel, A. J. Ramirez-Cuesta and G. J. Kearley, Chem. Phys., 2008, 351, 72 CrossRef CAS; (d) C. M. Brown, Y. Liu, T. Yildirim, V. K. Peterson and C. J. Kepert, Nanotechnology, 2009, 20, 204025 CrossRef.
- T. J. Udovic, C. M. Brown, J. B. Leao, P. C. Brand, R. D. Jiggetts, R. Zeitoun, T. A. Pierce, I. Peral, J. R. D. Copley, Q. Huang, D. A. Neumann and R. J. Fields, Nucl. Instrum. Methods Phys. Res., Sect. A, 2008, 588, 406 CrossRef CAS.
- R. T. Azuah, L. R. Kneller, Y. Qiu, P. L. W. Tregenna-Piggott, C. M. Brown, J. R. D. Copley and R. M. Dimeo, J. Res. Natl. Inst. Stand. Technol., 2009, 114, 241.
- M. C. Trachtenberg, R. M. Cowan, and D. A. Smith, in Proceedings of the Sixth Annual Conference on Carbon Capture & Sequestration, Pittsburgh, USA, 2007 Search PubMed.
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834 CrossRef CAS.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, full description of the high-throughput instrumentation, powder X-ray diffraction patterns, thermogravimetric analyses, nitrogen and hydrogen adsorption data. CCDC reference number 766493. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00179a |
| ‡ Identification of commercial equipment or products in the text is not intended to imply any recommendation or endorsement by the National Institute of Standards and Technology. |
| This journal is © The Royal Society of Chemistry 2010 |