Slow photoelectron velocity-map imaging of the CnH− (n = 5–9) anions
Etienne
Garand
a,
Tara I.
Yacovitch
a,
Jia
Zhou
a,
Sean M.
Sheehan
a and
Daniel M.
Neumark
*ab
aDepartment of Chemistry, University of California, Berkeley, California, USA. E-mail: dneumark@berkeley.edu; Fax: +1 (510) 642-3635; Tel: +1 (510) 642-3502
bChemical Science Division, Lawrence Berkeley National Laboratory, Berkeley, California, 94720, USA
First published on 25th May 2010
Abstract
High-resolution photoelectron spectra of the CnH− anions with n = 5–9 are acquired with slow electron velocity-map imaging (SEVI). Spectral features are assigned with the help of electronic structure calculations and Franck–Condon simulations. Well-resolved transitions to the linear ![[X with combining tilde]](https://www.rsc.org/images/entities/i_char_0058_0303.gif) 2Π and ã4Σ− neutral states are observed for species with an odd number of carbon atoms. For C6H− and C8H−, transitions to the 2Π neutral ground state and the low lying Ã2Σ+ excited state are observed. Precise electron affinities, term energies, fine structure splittings, and gas-phase vibrational frequencies are determined. The C5H−, C7H− and C9H− SEVI spectra are consistent with the anions having 3Σ− linear triplet ground states.
2Π and ã4Σ− neutral states are observed for species with an odd number of carbon atoms. For C6H− and C8H−, transitions to the 2Π neutral ground state and the low lying Ã2Σ+ excited state are observed. Precise electron affinities, term energies, fine structure splittings, and gas-phase vibrational frequencies are determined. The C5H−, C7H− and C9H− SEVI spectra are consistent with the anions having 3Σ− linear triplet ground states.
Introduction
The linear carbon monohydride radical chains, CnH, are important species in hydrocarbon combustion1 and in the interstellar medium.2 In both environments, these radicals serve as precursors for the formation of larger hydrocarbon chains and polycyclic species. The neutral chains with n = 2–8 have been observed in the circumstellar envelope of evolved stars,3 where they can be formed by reactions of C and C2 with unsaturated, closed-shell hydrocarbons.3–5 The negatively charged carbon monohydrides C4H−, C6H−, and C8H− were among the first anions to be detected in interstellar and circumstellar media,6–11 where they are formed by radiative attachment.12 The chemistry of these species is closely coupled to their spectroscopy, motivating the work described in this paper, in which we continue our investigation of the carbon monohydride chains via negative ion photodetachment.13–17 High resolution photoelectron spectra (PE) of the CnH− (n = 5–9) anions are measured using slow electron velocity-map imaging (SEVI). The SEVI spectra provide a detailed probe of the neutral and anionic states of these species, and yield insights into their geometries, vibronic structure, energetics, and the possibility of structural isomers.Carbon monohydride radical chains have been the subject of numerous experimental and theoretical studies. Maier and co-workers have measured the UV-visible electronic absorption spectra of several neutral2,18–23 and anionic24–27 CnH− (n = 5–9) species in the gas phase and in rare-gas matrices. Thaddeus and co-workers have used Fourier transformed microwave (FTMW) spectroscopy and millimetre-wavelength absorption spectroscopy to study the neutral C5–9H species28–36 as well as the C6H− and C8H− anions.6,37 PE spectra of CnH− (n = 5, 6, 8) have also been reported.13,16 Reaction kinetics and bond dissociation energies have been measured using flowing afterglow and guided ion beam mass spectrometry.38,39 Many electronic structure calculations at various levels of sophistication have been carried out on the neutral and anionic carbon monohydrides in the size-range of interest here.16,40–51
This body of experimental and theoretical work has revealed significant differences in CnH neutrals and anions depending on whether n is odd or even. In particular, there is evidence for multiple structural isomers for the odd-n anionic and neutral species. For example, theory and experiment indicate that the cyclic-C3H isomer lies slightly lower in energy than the linear-C3H isomer for both the anion and the neutral species.17,52 Both linear and cyclic isomers of C5H have been observed in microwave spectroscopy.28,35 High level ab initio calculations43,44 have identified the linear isomer of C5H to be lower in energy. Using these energetics and PE spectroscopy, Sheehan et al.16 recently suggested that a cyclic isomer was the lowest energy structure for the C5H− anion. Bowie and co-workers46,53 have also reported the synthesis of several anionic and neutral isomers of C5H and C7H using mass spectrometry techniques and precursors with the proper bond connectivity.
In contrast, it is well-established that the neutral and anionic even-carbon CnH species are linear. The anions have closed-shell 1Σ+ ground states with strong acetylenic character.48 The neutral species have low-lying 2Π and 2Σ+ states whose ordering switches with chain length.13,40 The C2H and C4H radicals have 2Σ+ ground-states, while the longer chains have 2Π ground-states; the Ã2Π − 2Σ+ splitting is only 213 cm−1 in C4H.15 The proximity of these two states in C2H and C4H leads to considerable spectral complexity attributed to strong vibronic coupling in both species,14,15,54,55 while vibronic coupling in the longer chains with 2Π ground states has not been investigated.
In this paper, we report high resolution PE spectra of the CnH− anions, n = 5–9, acquired with SEVI.56 These spectra are analyzed with the help of electronic structure calculations and Franck–Condon (FC) simulations. For all species, well-resolved vibrational and spin–orbit transitions to the linear 2Π ground state are observed. A second band of peaks is also observed for all species. It is assigned to the ã4Σ− excited state for C5H, C7H and C9H and to the low-lying Ã2Σ+state for C6H and C8H. Several vibrational frequencies of the neutral species are determined for the first time. This work also demonstrates that the C5H−, C7H− and C9H− anions have 3Σ− linear ground states, in contrast to the bent structures previously reported.16,46,53
Experimental
The SEVI apparatus has been described in detail elsewhere.57,58 SEVI is a high resolution variant of PE spectroscopy in which mass-selected anions are photodetached at a series of wavelengths.56 The resulting photoelectrons are collected by velocity-map imaging (VMI)59,60 using relatively low extraction voltages, with the goal of selectively detecting slow electrons with high efficiency and enlarging their image on the detector. At each photodetachment wavelength, one obtains a high resolution photoelectron spectrum over a limited range of electron kinetic energies.In this experiment, CnH− anions were produced from of a gas mixture comprising 1% acetylene and 1% propyne in a balance of Ar. The gas mixture, at a stagnation pressure of 300 psi, was expanded into the source vacuum chamber through an Even–Lavie pulsed valve61 equipped with a grid discharge described in detail elsewhere.62 Briefly, gas from the pulsed valve passed through a 2.5 mm × 23 mm channel made from Teflon and aluminium, within which were two fine grids made of stainless steel wire mesh and separated by 1 mm. The first grid was held to ground while the second was floated to around −500 VDC through a 1 kΩ resistor; the discharge was induced by passage of the expanding gas through the grids. Anions formed in the gas expansion were perpendicularly extracted into a Wiley–McLaren time-of-flight mass spectrometer and directed to the detachment region by a series of electrostatic lenses and pinholes. A pulse on the last ion deflector allowed only the desired mass into the interaction region.
Anions were photodetached between the repeller and the extraction plates of the VMI stack by the focused output of a Nd:YAG pumped tunable dye-laser. The photoelectron cloud formed was then coaxially extracted down a 50 cm flight tube and mapped onto a detector comprising a chevron-mounted pair of time-gated, imaging quality microchannel plates coupled to a phosphor screen, as is typically used in photofragment imaging experiments.59 Events on the screen were collected by a 1024 × 1024 charge-coupled device (CCD) camera and sent to a computer. Electron velocity-mapped images resulting from 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–100000 laser pulses were summed, quadrant symmetrized and inverse-Abel transformed.63 Photoelectron spectra were obtained via angular integration of the transformed images. The spectra presented here are plotted with respect to electron binding energy (eBE), defined as the difference between the energy of the photodetachment photon and the measured electron kinetic energy (eKE).
000–100000 laser pulses were summed, quadrant symmetrized and inverse-Abel transformed.63 Photoelectron spectra were obtained via angular integration of the transformed images. The spectra presented here are plotted with respect to electron binding energy (eBE), defined as the difference between the energy of the photodetachment photon and the measured electron kinetic energy (eKE).
The apparatus was calibrated by acquiring SEVI images of atomic Cl− and S− at several different photon energies, using the accurate electron affinities of these species reported previously.64 With the −350 V VMI repeller voltage used in this study, the full widths at half maximum of the chloride peaks were 7.5 cm−1 at 150 cm−1 eKE and 18 cm−1 at 715 cm−1. In the SEVI experiment, within the same image, all observed transitions have similar widths in pixels (Δr), so transitions observed further from threshold (larger r) are broader in energy. By varying the laser wavelength, a series of images in which the transitions of interest are close to the detachment threshold can be acquired, yielding a complete, high resolution photoelectron spectrum. Linewidths in the spectra presented here are limited by unresolved rotational structure. Since the origin of an unresolved rotational profile may not be aligned with the observed peak maximum, we report error bars of one Gaussian standard deviation for all energy determinations, typically 8 cm−1 for the highest resolution scans performed here.
SEVI also provides information on the photoelectron angular distribution (PAD). For one-photon detachment, the PAD is given by:65,66
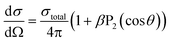
Results
SEVI spectra of the odd-carbon anions, C5H−, C7H− and C9H−, are shown in Fig. 1–3, while the SEVI spectra of the two even-carbon species, C6H− and C8H−, are shown in Fig. 4 and 5. The peak positions, shifts from the band origin, PADs, and assignments (see Analysis section) are summarized in Tables 1–5. Because the anisotropy parameter varies with photon energy,67 we reported PADs as “p” or “s+d” for features having β > 0 and β < 0, respectively.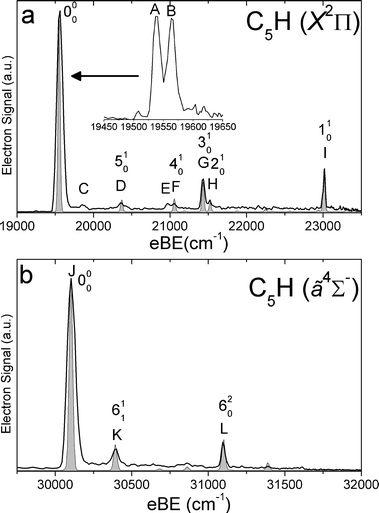 | ||
| Fig. 1 SEVI spectra of C5H− covering the electron binding energy ranges of 19000 to 23500 cm−1 (panel a) and 29500 to 32000 cm−1 (panel b). Franck–Condon simulations show as gray-shaded peaks. Inset shows high-resolution scan of indicated feature. | ||
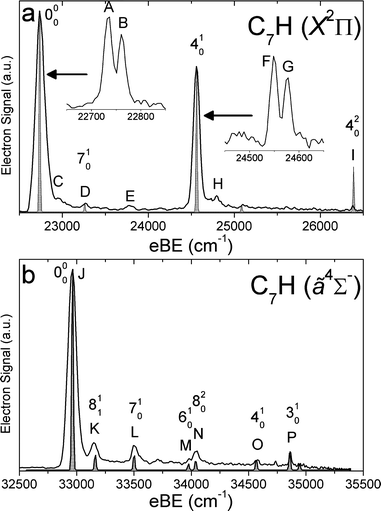 | ||
| Fig. 2 SEVI spectra of C7H− covering the electron binding energy ranges of 22500 to 26500 cm−1 (panel a) and 32500 to 35500 cm−1 (panel b). Franck–Condon simulations show as gray-shaded peaks. Insets show high-resolution scans of indicated features. | ||
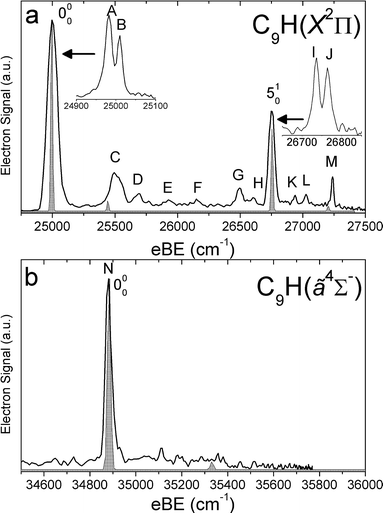 | ||
| Fig. 3 SEVI spectra of C9H− covering the electron binding energy ranges of 24750 to 27500 cm−1 (panel a) and 34500 to 35800 cm−1 (panel b). Franck–Condon simulations show as gray-shaded peaks. Insets show high-resolution scans of indicated features. | ||
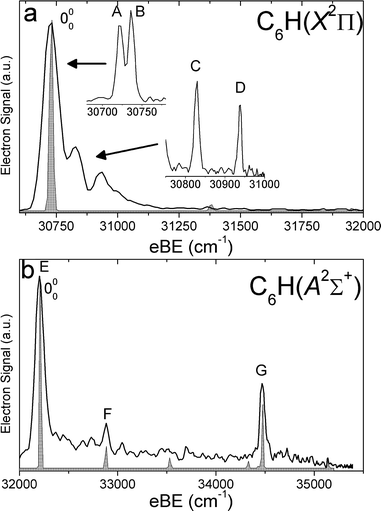 | ||
| Fig. 4 SEVI spectra of C6H− covering the electron binding energy ranges of 30600 to 32000 cm−1 (panel a) and 32000 to 35500 cm−1 (panel b). Franck–Condon simulations show as gray-shaded peaks. Inset shows high-resolution scan of indicated feature. | ||
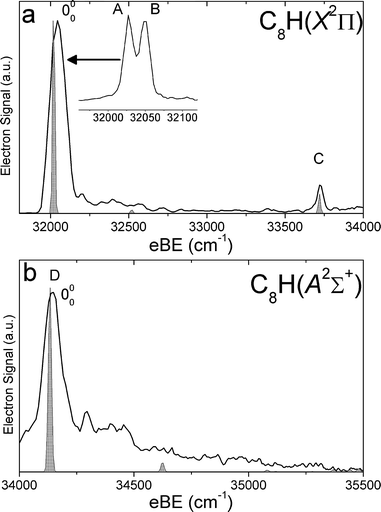 | ||
| Fig. 5 SEVI spectra of C8H− covering the electron binding energy ranges of 31750 to 34000 cm−1 (panel a) and 34000 to 35500 cm−1 (panel b). Franck–Condon simulations show as gray-shaded peaks. Inset shows high-resolution scan of indicated feature. | ||
| Peak | Position/cm−1 | Shift/cm−1 | PAD | Assignments (Vibs) | States |
|---|---|---|---|---|---|
| A | 19539 |
0 | s+d | 000 |
2Π½ ← 3Σ− |
| B | 19564 |
25 | s+d | 000 |
2Π![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) ← 3Σ− ← 3Σ− |
| C | 19851 |
312 | s+d | 611 |
2Π ← 3Σ− |
| D | 20372 |
833 | s+d | 510 |
2Π ← 3Σ− |
| E | 20960 |
1421 | s+d | 620 |
2Π ← 3Σ− |
| F | 21060 |
1521 | s+d | 410 |
2Π ← 3Σ− |
| G | 21434 |
1895 | s+d | 310 |
2Π ← 3Σ− |
| H | 21527 |
1988 | s+d | 210 |
2Π ← 3Σ− |
| I | 23014 |
3475 | s+d | 110 |
2Π ← 3Σ− |
| J | 30101 |
0 | p | 000 |
ã
4Σ− ← 3Σ− |
| K | 30391 |
290 | p | 611 |
ã
4Σ− ← 3Σ− |
| L | 31098 |
997 | p | 620 |
ã
4Σ− ← 3Σ− |
| Peak | Position/cm−1 | Shift/cm−1 | PAD | Assignments (Vibs) | States |
|---|---|---|---|---|---|
| A | 22734 |
0 | s+d | 000 |
2Π½ ←3Σ− |
| B | 22761 |
27 | s+d | 000 |
2Π←3Σ− |
| C | 22957 |
223 | s+d | 811 |
2Π←3Σ− |
| D | 23258 |
524 | s+d | 710 |
2Π←3Σ− |
| E | 23776 |
1042 | s+d | 820 |
2Π←3Σ− |
| F | 24549 |
1815 | s+d | 410 |
2Π½←3Σ− |
| G | 24576 |
1842 | s+d | 410 |
2Π←3Σ− |
| H | 24793 |
2059 | s+d | 410811 |
2Π ← 3Σ− |
| I | 26379 |
3645 | s+d | 420 |
2Π ← 3Σ− |
| J | 32969 |
0 | p | 000 |
ã
4Σ−← 3Σ− |
| K | 33149 |
180 | p | 811 |
ã
4Σ−← 3Σ− |
| L | 33506 |
537 | p | 710 |
ã
4Σ−← 3Σ− |
| M | 33980 |
1011 | p | 610 |
ã
4Σ−← 3Σ− |
| N | 34041 |
1072 | p | 820 |
ã
4Σ−← 3Σ− |
| O | 34572 |
1603 | p | 410 |
ã
4Σ−← 3Σ− |
| P | 34863 |
1894 | p | 310 |
ã
4Σ−← 3Σ− |
| Peak | Position/cm−1 | Shift/cm−1 | PAD | Assignments (Vibs) | States |
|---|---|---|---|---|---|
| A | 24980 |
0 | s+d | 000 |
2Π½ ←3Σ− |
| B | 25010 |
30 | s+d | 000 |
2Π←3Σ− |
| C | 25487 |
507 | p | ||
| D | 25671 |
691 | p | ||
| E | 25928 |
948 | p | ||
| F | 26157 |
1177 | p | ||
| G | 26495 |
1515 | p | ||
| H | 26603 |
1623 | p | ||
| I | 26735 |
1755 | s+d | 510 |
2Π½←3Σ− |
| J | 26766 |
1786 | s+d | 510 |
2Π←3Σ− |
| K | 26934 |
1954 | p | ||
| L | 27024 |
2044 | p | ||
| M | 27236 |
2256 | p | ||
| N | 34884 |
9904 | p | 000 | ã4Σ−←3Σ− |
| Peak | Position/cm−1 | Shift/cm−1 | PAD | Assignments (Vibs) | States |
|---|---|---|---|---|---|
| A | 30722 |
0 | s+d | 000 |
2Π←1Σ+ |
| B | 30737 |
15 | s+d | 000 |
2Π½←1Σ+ |
| C | 30828 |
106 | s+d |
2Π←1Σ+ |
|
| D | 30937 |
215 | s+d |
2Π ← 1Σ+ |
|
| E | 32214 |
0 | p | 000 |
Ã
2Σ+ ← 1Σ+ |
| F | 32880 |
651 | p | 610 |
Ã
2Σ+ ← 1Σ+ |
| G | 34474 |
2260 | p | 210 |
Ã
2Σ+ ← 1Σ+ |
| Peak | Position/cm−1 | Shift/cm−1 | PAD | Assignments (Vibs) | States |
|---|---|---|---|---|---|
| A | 32028 |
0 | s+d | 000 |
2Π←1Σ+ |
| B | 32049 |
21 | s+d | 000 |
2Π½←1Σ+ |
| C | 33725 |
1697 | s+d | 510 |
2Π ← 1Σ+ |
| D | 34140 |
2112 | p | 000 |
Ã
2Σ+ ← 1Σ+ |
For each anion, panels a and b illustrate lower and higher eBE regions, respectively, of the SEVI spectra. Higher resolution SEVI traces taken at photon energies ∼100–150 cm−1 above the feature at lowest eBE in the upper panels reveal each to be a closely spaced doublet, as shown in the insets in Fig. 1a–5a. These traces show doublet splittings of 25 cm−1, 15 cm−1, 27 cm−1, 21 cm−1 and 30 cm−1 for C5H− to C9H−, respectively. Doublets with the same splittings were also resolved for the other intense transition in the first band of C7H− (peaks F and G) and C9H− (I and J). The same splittings are presumably found on other features in Fig. 1a–5a. However, their weak intensities prevented the acquisition of SEVI spectra sufficiently close to their respective detachment thresholds to observe these small splittings. No doublets were found in high resolution traces of the more intense features in Fig. 1b–5b.
In the C5H−, C6H−, C7H− and C8H− spectra, all the features in panel a have “s+d” PADs while those in panel b have “p” PADs. In the C9H− spectra, peaks A, B, I and J in Fig. 3a have “s+d” PADs, while the remaining smaller peaks all have “p” PADs. The single peak N in Fig. 3b also has a “p” PAD. In the SEVI spectra of C5H−, C7H− and C9H−, the lowest eBE transitions (peak A) of the first band are found at eBE of 19539 cm−1, 22724 cm−1 and 24980 cm−1, respectively, while those of the second band are higher in energy by 10502 cm−1, 10235 cm−1 and 9904 cm−1, respectively. The large energy separation and different PADs indicate the features in panels a and b of Fig. 1–3 belong to different electronic states. Compared to the odd-carbon species, the SEVI spectra of C6H− and C8H− show higher electron binding energies for the lower energy band. The lower band origins are found at eBE of 30722 cm−1 and 32028 cm−1 in the C6H− and C8H− spectra, respectively, while the first bands in panel b are only 1492 cm−1 and 2112 cm−1 higher in energy. Although these relatively small energy intervals could represent vibrational frequencies, the different PADs for the features in panels a and b of Fig. 4–5 imply that the spectra in Fig. 4b and 5b are from transitions to low-lying excited electronic states of C6H and C8H, consistent with the assignment of earlier anion photoelectron spectra.13
For C5H−, C6H− and C8H−, the observed band structures and positions are consistent with the previously reported anion PE spectra.13,16 However, the improved resolution of SEVI over conventional PE spectroscopy reveals numerous new features, especially in the lower bands of C5H− and C6H−. The SEVI spectra of C7H− and C9H− are the first photodetachment spectra reported for these species.
Computational details
Electronic structure calculations were performed on the relevant neutral and anionic states of CnH. The current calculations serve to produce, at a uniform level of theory, all geometries, normal modes and vibrational frequencies necessary to perform Franck–Condon simulations and interpret the photoelectron spectra. Our calculations were carried out with density functional theory (DFT) using the Becke three-parameter Lee, Yang, and Parr exchange–correlation functional68,69 (B3LYP) and the augmented correlation consistent polarized valence triple-zeta basis set70 (AVTZ).All computations were performed using the GAUSSIAN03 program.71 Franck–Condon simulations were performed with the FCFgaus03 and PESCAL programs72,73 using the Sharp–Rosenstock–Chen method74 that treats all the modes as independent harmonic oscillators and relates the normal mode coordinates of the initial and final states via the Duschinsky transformation.75 The calculated geometries and harmonic vibrational frequencies were used as a starting point for the FC simulations. The neutral frequencies were adjusted to the experimental values and the normal mode displacements were adjusted to fit the experimental spectra.
For C5H− and C7H−, several low-lying isomers with singlet and triplet spin multiplicity have been previously reported.16,46,53 The carbon backbone of these isomers can be a single chain, a branched chain, or a branched three-membered ring. Consequently, we searched for the lowest energy isomers of C5H−, C7H− and C9H− by performing electronic structure calculations at the B3LYP/6-311+G level of theory on several different initial geometries for both the singlet and triplet surfaces. The lowest energy isomers were then re-optimized at the B3LYP/AVTZ level. We found that the isomer comprising a single carbon chain terminated by one hydrogen was lower in energy than the next-lowest isomer by 0.29 eV, 0.65 eV and 0.84 eV, for n = 5, 7, and 9, respectively. These lowest energy isomers were found to have a triplet ground state with the first singlet state lying higher in energy by 0.31 eV, 0.43 eV and 0.47 eV. The next-lowest isomers also consisted of a single carbon chain, but with the hydrogen located on the third carbon for n = 5, 7 and the fifth carbon for n = 9. Hence, only triplet states with H-terminated carbon chains were analyzed in more detail for the odd-carbon anions, For the even-carbon species, only the well-known linear 1Σ+ anion state and the accessible low-lying 2Π and 2Σ+ neutral states were considered.
Calculated geometries and relative energies of the anionic and neutral states of the CnH species are shown in Table 6, while harmonic vibrational frequencies are presented in Table 7. Vibrational mode labels for the neutral ground state are used throughout even if the ordering of the frequencies changes. For C5H−, the minimum energy structure is a 3A′ state with an almost linear carbon backbone and a CCH angle of 164.1°. However, the corresponding linear 3Σ− first-order transition state was found to be only 0.001 eV above the minimum energy structure. With such a small energy difference, the zero-point energy of the CCH bending mode certainly exceeds the barrier to linearity. Thus, the C5H− ground state will be considered to be the linear 3Σ− state with the expectation that this state is either quasilinear or has a very shallow CCH bending potential. This result differs from previous theoretical studies16,46,76 using the same B3LYP functional with smaller basis sets that yielded strongly bent structures for the triplet ground-state of C5H−. This basis set effect might also explain why Blanksby et al.46 did not find the chain isomer to be the lowest in energy by using single-point energy calculations at the RCCSD(T)/AVDZ level on the geometries obtained at the B3LYP/AVDZ level. For C7H− and C9H−, the truly linear 3Σ− state was found to be the minimum energy structure. This result is again in contrast with previous DFT studies53,76 using smaller basis sets that found slightly bent structures for the triplet ground states of C7H− and C9H−.
| Species | State | ΔE | Geometry | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Transition state. | |||||||||||
| C 5 H | C 1 –C 2 | C 2 –C 3 | C 3 –C 4 | C 4 –C 5 | C 5 –H | C 1 C 2 C 3 | C 2 C 3 C 4 | C 3 C 4 C 5 | C 4 C 5 H | ||
| Anion |
3A′ |
−2.56 | 1.284 | 1.302 | 1.305 | 1.25 | 1.061 | 180.0 | 179.5 | 176.9 | 164.1 |
| 3Σ−a | −2.56 | 1.285 | 1.301 | 1.308 | 1.245 | 1.058 | |||||
| Neutral |
2Π |
0.00 | 1.304 (1.308) | 1.264 (1.267) | 1.321 (1.329) | 1.221 (1.224) | 1.063 (1.055) | ||||
| ã 4Σ− | 1.35 | 1.253 | 1.287 | 1.311 | 1.23 | 1.061 | |||||
| C 6 H | C 1 –C 2 | C 2 –C 3 | C 3 –C 4 | C 4 –C 5 | C 5 –C 6 | C 6 –H | |||||
| Anion |
1Σ+ |
−3.64 | 1.252 (1.2575) | 1.342 (1.3518) | 1.231 (1.2313) | 1.348 (1.3581) | 1.218 (1.2189) | 1.059 (1.0594) | |||
| Neutral |
2Π |
0.00 | 1.281 (1.257) | 1.312 (1.332) | 1.24 (1.237) | 1.334 (1.341) | 1.215 (1.216) | 1.063 (1.056) | |||
| Ã 2Σ+ | 0.60 | 1.21 | 1.355 | 1.214 | 1.354 | 1.207 | 1.061 | ||||
| C 7 H | C 1 –C 2 | C 2 –C 3 | C 3 –C 4 | C 4 –C 5 | C 5 –C 6 | C 6 –C 7 | C 7 –H | ||||
| Anion |
3Σ− |
−2.95 | 1.275 | 1.309 | 1.28 | 1.271 | 1.321 | 1.233 | 1.059 | ||
| Neutral |
2Π |
0.00 | 1.295 (1.301) | 1.275 (1.273) | 1.298 (1.309) | 1.243 (1.243) | 1.332 (1.341) | 1.215 (1.217) | 1.062 (1.057) | ||
| ã 4Σ− | 1.31 | 1.241 | 1.304 | 1.281 | 1.259 | 1.326 | 1.221 | 1.061 | |||
| C 8 H | C 1 –C 2 | C 2 –C 3 | C 3 –C 4 | C 4 –C 5 | C 5 –C 6 | C 6 –C 7 | C 7 –C 8 | C 9 –H | |||
| Anion |
1Σ+ |
−3.79 | 1.255 (1.2600) | 1.334 (1.3440) | 1.236 (1.2360) | 1.332 (1.3435) | 1.230 (1.2284) | 1.347 (1.3592) | 1.216 (1.2166) | 1.059 (1.0598) | |
| Neutral |
2Π |
0.00 | 1.281 | 1.306 | 1.248 | 1.315 | 1.233 | 1.339 | 1.212 | 1.062 | |
| Ã 2Σ+ | 0.71 | 1.212 | 1.351 | 1.218 | 1.343 | 1.218 | 1.352 | 1.208 | 1.062 | ||
| C 9 H | C 1 –C 2 | C 2 –C 3 | C 3 –C 4 | C 4 –C 5 | C 5 –C 6 | C 6 –C 7 | C 7 –C 8 | C 8 –C 9 | C 9 –H | ||
| Anion |
3Σ− |
−3.25 | 1.271 | 1.312 | 1.268 | 1.284 | 1.294 | 1.256 | 1.330 | 1.225 | 1.059 |
| Neutral |
2Π |
0.00 | 1.290 | 1.280 | 1.287 | 1.254 | 1.311 | 1.235 | 1.338 | 1.212 | 1.062 |
| ã 4Σ− | 1.27 | 1.233 | 1.315 | 1.265 | 1.276 | 1.298 | 1.246 | 1.334 | 1.217 | 1.061 | |
| State | ν 1 | ν 2 | ν 3 | ν 4 | ν 5 | ν 6 | ν 7 | ν 8 | ν 9 | ν 10 | ν 11 | ν 12 | ν 13 | ν 14 | ν 15 | ν 16 | ν 17 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C 5 H | σ | σ | σ | σ | σ | π | π | π | π | ||||||||
|
3Σ− |
3473 | 1909 | 1790 | 1479 | 761 | 130i | 456 | 396 | 135 | ||||||||
|
2Π |
3449 | 2054 | 1952 | 1461 | 771 | 438/756 | 393/582 | 289/363 | 128/133 | ||||||||
| ã 4Σ− | 3453 | 1975 | 1721 | 1563 | 780 | 502 | 432 | 393 | 139 | ||||||||
| C 6 H | σ | σ | σ | σ | σ | σ | π | π | π | π | π | ||||||
|
1Σ+ |
3480 | 2222 | 2142 | 1979 | 1206 | 641 | 483 | 550 | 494 | 264 | 111 | ||||||
|
2Π |
3451 | 2101 | 2132 | 1898 | 1224 | 650 | 574/701 | 512/554 | 393/466 | 191/245 | 89/107 | ||||||
| Ã 2Σ+ | 3466 | 2285 | 2220 | 2114 | 1212 | 642 | 659 | 530 | 655 | 304 | 118 | ||||||
| C 7 H | σ | σ | σ | σ | σ | σ | σ | π | π | π | π | π | π | ||||
|
3Σ− |
3474 | 2023 | 1949 | 1769 | 1611 | 1071 | 564 | 334 | 488 | 470 | 387 | 198 | 81 | ||||
|
2Π |
3453 | 2139 | 2051 | 1908 | 1581 | 1085 | 569 | 531/725 | 475/612 | 436/536 | 312/367 | 185/191 | 78/79 | ||||
| ã 4Σ− | 3455 | 2056 | 1999 | 1677 | 1628 | 1093 | 569 | 569 | 503 | 430 | 376 | 203 | 81 | ||||
| C 8 H | σ | σ | σ | σ | σ | σ | σ | σ | π | π | π | π | π | π | π | ||
|
1Σ+ |
3478 | 2231 | 2200 | 2096 | 1982 | 1354 | 945 | 493 | 515 | 563 | 532 | 467 | 278 | 166 | 65 | ||
|
2Π |
3454 | 2189 | 2109 | 2044 | 1904 | 1373 | 959 | 499 | 604/694 | 543/595 | 508/528 | 399/447 | 248/266 | 145/162 | 62/63 | ||
| Ã 2Σ+ | 3463 | 2279 | 2241 | 2174 | 2109 | 1346 | 947 | 491 | 654 | 619 | 572 | 493 | 318 | 178 | 66 | ||
| C 9 H | σ | σ | σ | σ | σ | σ | σ | σ | σ | π | π | π | π | π | π | π | π |
|
3Σ− |
3473 | 2072 | 2055 | 1945 | 1735 | 1658 | 1245 | 860 | 446 | 434 | 532 | 498 | 464 | 377 | 230 | 133 | 52 |
|
2Π |
3454 | 2180 | 2130 | 2021 | 1850 | 1638 | 1255 | 869 | 448 | 583/706 | 563/663 | 485/580 | 438/506 | 322/364 | 218/224 | 126/131 | 49/50 |
| ã 4Σ− | 3457 | 2104 | 2048 | 2003 | 1735 | 1521 | 1260 | 869 | 448 | 600 | 588 | 498 | 420 | 348 | 235 | 134 | 51 |
All the neutral isomers considered here were found to have linear 2Π ground-states. The calculated bond lengths in the ground states of C5H, C6H and C7H are very similar to those obtained from microwave spectroscopy36 and previous high level electronic structure calculations.40,43 The calculations yield non-degenerate bending modes, as expected for vibrational modes of a 2Π state subject to Renner–Teller (RT) coupling. For the neutral odd-carbon species, a linear 4Σ− excited state lying ∼1.3 eV above the ground-state was found, similar to previous calculations on C3H17 and C5H.16 The calculated geometries for these 4Σ− states are similar to those of the 3Σ− anion state but with a shorter C1–C2 bond. For C6H and C8H, a low-lying 2Σ+ excited state was found, in accordance with previous experimental and theoretical work.13,40,47 The other observed or predicted excited states18–21,23,44,47 of the linear neutral CnH species are outside the energy range of the current study and thus were not considered theoretically here.
Analysis
In this section, detailed analyses and assignments of the SEVI spectral features are performed using the electronic structure calculations and FC simulations. Because of their different electronic character, leading to qualitatively different PE spectra, the odd- and even-carbon species will be considered separately. Peak assignments are shown in Tables 1–5, while experimentally determined electron affinities, term energies and vibrational frequencies are summarized in Table 8.| Species | State | Energy | ν 1 | ν 2 | ν 3 | ν 4 | ν 5 | ν 6 | ν 7 | ν 8 |
|---|---|---|---|---|---|---|---|---|---|---|
| C5H |
3Σ− |
208 | ||||||||
|
2Π |
EA = 2.4225 | 3462 | 1975 | 1882 | 1508 | 820 | ||||
| ã 4Σ− | T0 = 1.3095 | 498 | ||||||||
| C6H |
2Π |
EA = 3.8090 | ||||||||
| Ã 2Σ+ | T0 = 0.1751 | 2260 | 651 | |||||||
| C7H |
3Σ− |
356 | ||||||||
|
2Π |
EA = 2.8187 | 1815 | 510 | |||||||
| ã 4Σ− | T0 = 1.2690 | 1894 | 1603 | 1011 | 537 | 536 | ||||
| C8H |
2Π |
EA = 3.9701 | ||||||||
| Ã 2Σ+ | T0 = 0.2619 | 1697 | ||||||||
| C9H |
2Π |
EA = 3.0971 | 1755 | |||||||
| ã 4Σ− | T0 = 1.2279 | |||||||||
C5H−, C7H− and C9H−
In the C5H−, C7H− and C9H− SEVI spectra shown in Fig. 1–3, the splittings between the bands in the upper and lower panels are very similar to the calculated splittings in Table 6 between the neutral2Π and ã4Σ− states. Both states are accessible from the C2n + 1H− (n = 2–4) 3Σ− ground states, which have a […]π4π4σ2π2 molecular orbital (MO) configuration. Removal of an electron from the highest occupied π or σ MO produces the neutral 2Π or ã4Σ−state, respectively. As shown previously,14,15,62 photodetachment from π and σ MOs often proceeds via “s+d” and “p” scattering, respectively. We thus assign the lower energy band of peaks to the 2Π neutral ground state and the second band to the ã4Σ− first excited state, with both bands originating from the anion 3Σ− state.
In the 2Π bands, the splitting of the vibrational origin into two closely spaced peaks, labeled A and B in Fig. 1a–3a, is attributed to spin–orbit coupling in the neutral vibrational ground state, and peaks A and B are assigned as transitions to the Π1/2 and Π3/2 spin–orbit components of this state. In C5H and C7H, the doublet splittings are 25 cm−1 and 27 cm−1, respectively. These values are in good agreement with the spin–orbit splittings of 24.22 cm−1 and 26.17 cm−1 derived from FTMW and millimetre-wavelength absorption measurements33,34 as well as the splittings seen in the visible Ã2Δ←2Π bands of C5H and C7H.23 In C9H, peaks A and B are spaced by 30 cm−1; our spectrum is the first experimental determination of the spin–orbit splitting for this species. From the eBE of peak A, we obtain a refined electron affinity (EA) of C5H and the first experimental EA of C7H and C9H. These values are EA(C5H) = 2.4225 ± 0.0010 eV, EA(C7H) = 2.8187 ± 0.0010 eV and EA(C9H) = 3.0971 ± 0.0010 eV. The calculated EAs at the B3LYP/AVTZ level of theory, including zero-point energy corrections, are 2.56 eV, 2.95 eV and 3.25 eV for C5H, C7H and C9H, respectively, as indicated in Table 6. Thus, the calculations systematically overestimate the EA of these species by around 0.15 eV.
FC simulations of the 2Π bands are shown as gray-shaded peaks in Fig. 1a–3a. Only the σ vibrational modes were included in these simulations; inclusion of the bending modes would require incorporating Renner–Teller coupling into the simulations and is beyond the scope of this work. The good agreement between the simulated and experimental spectra allows us to assign most of the observed features. In C5H, peaks D, F, G and H are assigned to the 510, 410, 310 and 210 transitions, respectively. These four modes are all carbon backbone stretching vibrations. Peak I is assigned to the 110 transition in which the C–H stretching mode is excited. In C7H, peak D is assigned to the 710 transition while peaks F and G are assigned to the Π1/2 and Π3/2 spin–orbit components of the 410 transition, respectively. Similarly, in C9H, peaks I and J are assigned to the Π1/2 and Π3/2 spin–orbit components of the 510 transition, respectively. All of these modes are also carbon backbone stretching vibrations and their vibrational frequencies agree well with the calculated values.
Several smaller peaks are not reproduced by the FC simulations and are thus assigned to transitions involving bending modes. While the definitive assignments of these features would require a fuller understanding of RT coupling in these species, some tentative assignments can be made on the basis of the calculated harmonic bending vibration frequencies shown in Table 7. In C5H and C7H, the most likely active bending vibrations are the CCH bending modes (ν6 and ν8, respectively) because of the large changes in frequency between the anion and the neutral. This change in frequency would give rise to ν20 transitions as well as ν11 sequence bands located at higher eBE than the origin transition. For C5H, peaks C and E could arise from the 611 and 620 transitions, while peaks C and E in the C7H− spectrum could be the 811 and 820 transitions. These assignments yield ν6 frequencies (ignoring anharmonicities and RT effects) of 398 and 710 cm−1 in C5H− and C5H, respectively, and ν8 frequencies of 298 and 521 cm−1 for anionic and neutral C7H. While these values are within range of our calculated frequencies in Table 7, evaluation of these assignments and frequencies is difficult because the calculated bend frequencies in the neutral are non-degenerate, the experimental levels have unknown RT splittings, and the calculation for C5H− yields an imaginary ν6 frequency at the linear geometry.
The situation is different in the 2Π band of C9H, where all the weaker transitions that are not reproduced by the FC simulations have a different PAD than the main spectral features (peaks A, B, I, and J). While these weaker features could originate from a different C9H isomer, the lowest lying isomers of C9H− are calculated to lie more than 0.84 eV above the linear isomer and thus are unlikely to contribute to the spectrum. These features are more likely to be nominally forbidden transitions to the 2Π state, such as bending vibrations with odd Δν, which gain intensity through vibronic coupling. In this case, those vibronically allowed transitions normally have the same PAD as the transitions to the electronic state from which intensity is borrowed.14,77 The closest electronic state which can couple via bending modes with the 2Π state is the Ã2Δ state. This state is expected to lie 1.88 eV above the ground state based on electronic structure calculations and extrapolations of the corresponding Ã2Δ←2Π transitions in C5H and C7H.23 This term energy is 0.45 eV and 0.21 eV higher in C5H and C7H, respectively, providing a possible explanation as to why vibronic coupling effects appear stronger in C9H than in the shorter chain species.
We next consider the ã4Σ− bands in Fig. 1b–3b. The dominant peak in each panel, labeled J in the C5H− and C7H− spectra and N in the C9H− spectrum, is assigned to the 000 transition. The term energies of the ã4Σ− states of C5H, C7H and C9H are thus determined to be 1.3095 ± 0.0010 eV, 1.2690 ± 0.0010 eV and 1.2279 ± 0.0010 eV, respectively. These values are in excellent agreement with our calculations that predicted ã4Σ− term energies of 1.35 eV, 1.31 eV and 1.27 eV for these species. FC simulations, shown in shaded gray in Fig. 1b–3b, result in assignments of all the remaining features in the ã4Σ− bands of C5H and C7H.
In the C5H− spectrum, peaks K and L can be reproduced in the FC simulation by assigning them to the 611 and 620 transitions, respectively, by assigning a frequency of 208 cm−1 for the corresponding anion bending mode, and by setting the anion vibrational temperature to 150 K. This assignment of peak L yields a value of 498 cm−1 for the ν6 fundamental of the ã4Σ− state, close to our calculated value of 502 cm−1 (Table 6). The low ν6 anion frequency implied by this assignment is consistent with the very shallow CCH bending potential of C5H− inferred from our calculations. This anion frequency is preferable to that inferred from our assignment of peak C in the 2Π band, given the absence of RT splittings in the ã4Σ− band.
Similar CCH bending activity is found in the SEVI spectrum of C7H− where peaks K and N are assigned to the 811and 820 transitions, respectively. These assignments of peak yield 356 cm−1 and 536 cm−1 for the anion and neutral ν8 fundamentals (CCH bend), both of which are in reasonable agreement with calculated values in Table 7. The remaining peaks in the C7H− spectrum, labeled L, M, O and P, are assigned to the 710, 610, 410and 310 transitions, respectively. These modes are all stretching vibrations of the carbon backbone and their frequencies are in good agreement with the calculated values.
C6H− and C8H−
In the C6H−and C8H− SEVI spectra shown in Fig. 4 and 5, the lower energy band is assigned to the2Π neutral ground state and the second band to the Ã2Σ+ first excited state. Both states are accessible from the C6H− and C8H−1Σ+ ground states, which have a […]π4π4σ2π4 MO configuration. Removal of an electron from the highest occupied π or σ MO produces the neutral 2Π or Ã2Σ+ state, respectively. As discussed earlier, photodetachment from π and σ MOs often proceeds via “s+d” and “p” scattering, respectively, consistent with the observed PADs.
In the 2Π bands, the two closely spaced peaks, labeled A and B in Fig. 4a–5a, are assigned as transitions to the Π3/2 and Π1/2 spin–orbit components, respectively, of the vibrational ground state. The splittings between those two peaks are 15 cm−1 and 21 cm−1, in good agreement with the spin–orbit splittings of −15.11 cm−1 and −19.33 cm−1 derived from FTMW and millimetre wavelength absorption measurements33,34 on C6H and C8H, respectively. From the eBE of the peaks labeled A, we obtain refined EAs for these two species: EA(C6H) = 3.8090 ± 0.0010 eV and EA(C8H) = 3.9701 ± 0.0010 eV. These values are within the uncertainties of those previously determined by Taylor et al.13 They are also close to the calculated EAs, at the B3LYP/AVTZ level, of 3.64 eV and 3.79 eV for C6H and C8H, respectively.
The dominant peaks E and D in Fig. 4b and 5b, respectively, are assigned to the 000 transitions of the Ã2Σ+ state. The term energies of the first excited state are thus determined to be 0.1751 ± 0.0010 eV and 0.2619 ± 0.0010 eV, for C6H and C8H respectively. These values are within the uncertainties of the term energies reported by Taylor et al.13 They are smaller than our calculated term energies of 0.60 eV and 0.71 eV for these species. However, the C6H Ã2Σ+ state experimental term energy is in good agreement with the vertical transition energy of 0.22 eV calculated by Cao and Peyerimhoff.47
The C6H− and C8H− SEVI spectra display little vibrational activity. Some vibrational transitions can be assigned with the FC simulations, the results of which are shown in shaded gray in Fig. 4 and 5. In the Ã2Σ+ band of C6H, peaks F and G are assigned to the 610 and 210 transitions, respectively. In the 2Π band of C8H, peak C is assigned to the 510 transition.
Peaks C and D in the 2Π band of C6H are not reproduced by the FC simulations and are thus assigned to bending vibrational modes. Again, the definitive assignment of these features would require a fuller understanding of the RT coupling in C6H. Peaks C and D are shifted by only 106 cm−1 and 215 cm−1 from the origin. These positions roughly correspond to the calculated frequencies of the ν10 and ν11 modes. However, transitions to non-totally symmetric modes with odd Δν are nominally forbidden, even in the presence of RT coupling. These peaks could be the 1120 and 1020 transitions; such an assignment would imply that the ν11 and ν12 frequencies are much smaller than the calculated values or that the RT coupling for these modes is very strong.
Another possibility is that peaks C and D are Δν = 1 bending transitions that gain intensity through vibronic coupling with the nearby Ã2Σ+ state, similar to what was observed in the SEVI spectra14,15 of C2H− and C4H−. Usually, we would expect these features to have “p” PADs, similar to the features associated with the Ã2Σ+ state,14 and not the observed “s+d” PADs. However, in the SEVI spectrum of C4H−, features were assigned to vibronically allowed Δν = 1 bending transitions even though the PADs were the same as for nearby, fully allowed transitions.15
Discussion
This study addresses the key issue of the structure of CnH− anions with an odd number of carbon atoms. Previous DFT studies on C5H−, C7H−and C9H− have reported bent geometries for the triplet ground state of the isomer comprising a single carbon chain terminated by a hydrogen.16,46,53,76 In contrast, the electronic structure calculations presented here predict a quasilinear geometry for C5H− and linear geometries for C7H−and C9H−. The FC simulations involving transitions from these calculated linear 3Σ− anion ground electronic states to the2Π and ã4Σ− neutral states were found to be in excellent agreement with the well-resolved features of the SEVI spectra. The 2Π states of C5H, C7H and C9H, which can be unambiguously identified from the resolved spin–orbit splitting, are known to be linear from FTMW spectroscopy.33,36 The observed small vibrational activity in the 2Π band is therefore consistent with linear-to-linear transitions. If the anion had a bent geometry, extensive bending progressions would be expected.78 We thus conclude that the triplet ground states of the C5H−, C7H−and C9H− anions are linear, in accordance with our electronic structure calculations.
Another issue addressed here is the energy ordering of the various isomers of the odd-carbon anion species. For C5H−, a theoretical study by Blanksby et al.46 found two isomers to be more stable than the single chain with terminal hydrogen (l-C5H−) discussed here. One isomer has a hydrogen located on the middle of a single carbon chain (C2[CH]C2−, 1A1), and the other a branched three-membered ring with the hydrogen located on the ring ([c-C3H]C2−, 1A′); they were found to be 0.15 eV and 0.12 eV below l-C5H−, respectively. A third isomer, consisting of a branched three-membered ring with the hydrogen located at the end of the branch ([c-C3]C2H−, 1A1), was found to lie 0.13 eV above l-C5H−. A recent PE study of C5H− invoked the presence of both l-C5H− and [c-C3]C2H− isomers in order to assign all the observed spectral features. Based on their electron affinities and the calculated energetics of the neutral C5H isomers,43 the [c-C3]C2H− isomer was assigned to lie 0.16 eV below the l-C5H− isomer.
The electronic structure calculations and the C5H− SEVI spectra presented here point toward a somewhat different conclusion. First, the B3LYP/AVTZ calculations predict that l-C5H− is the most stable isomer by 0.29 eV. Secondly, only the l-C5H− isomer was necessary to assign the all the observed spectral features in the C5H− SEVI spectra. The main difference between the analysis of the current SEVI spectra and the previous PE spectra comes from the linear 3Σ−l-C5H− anion geometry used in the FC simulations instead of the bent 3A′ geometry. The large vibrational activity expected for a bent-to-linear transition was not compatible with some of the observed features in the PE spectrum and these were thus assigned to a cyclic isomer. Since the current study used short chain precursors (acetylene and propyne) that would not have hindered the formation of the various isomers, we conclude that the l-C5H− (3Σ−) isomer is the lowest energy structure on the C5H− potential energy surface. Similar conclusions can be reached for the l-C7H− (3Σ−) and l-C9H− (3Σ−) isomers on the same basis of the present DFT calculations and the analysis of the C7H− and C9H− SEVI spectra.
Our SEVI spectra also probe vibronic coupling in the carbon monohydrides. In C2H, the small 3600 cm−1 spacing between the 2Σ+ and Ã2Π states results in strong pseudo-Jahn–Teller coupling that is well-documented and understood.14 For C4H, the 2Σ+ state lies only 213 cm−1 below the Ã2Π state which presumably yields even larger perturbations of the vibronic levels,15 but these effects have not been theoretically addressed yet. For the larger even-carbon species studied here, the ordering of the two states is reversed and the splittings are 1492 cm−1 and 2112 cm−1 for C6H and C8H, respectively. While the appearance of peaks C and D in the C6H− SEVI spectra might be an indication of vibronic coupling, depending on their assignments, no indication of vibronic coupling is found in the C8H spectra. This suggests that the amplitude of pseudo-Jahn–Teller coupling between the low-lying 2Σ+ and 2Π states of the even-carbon is small in C6H and even smaller in the longer chains. The situation is reversed in the odd-carbon species; evidence of vibronic coupling is found in the SEVI spectra of C9H− but not in the spectra of the smaller chain species. If this coupling occurs between the 2Π and the Ã2Δ states, as proposed above, vibronic coupling could be increasingly important for the longer-chain odd-carbon species because the 2Π–Ã2Δ splitting decreases with chain-length.23
Conclusions
High-resolution photoelectron spectra of the C5–9H− species acquired with SEVI are reported. The spectra of C5H−, C7H−, and C9H− show well resolved transitions to the2Π and ã4Σ− neutral states. For C6H− and C8H−, transitions to the 2Π and Ã2Σ+ neutral states are observed. Most of the observed spectral features are assigned with the help of electronic structure calculations and FC simulations. Precise electron affinities and term energies are determined and several gas-phase vibrational frequencies are determined for the first time. For the odd-carbon species, the observed spectral features and electronic structure calculations are consistent with linear 3Σ− ground states for the anions, with no other isomers contributing to the spectra.
Acknowledgements
This work was supported by the Air Force Office of Scientific Research under Grant No. F49620-03-1-0085. E.G. and T.Y. both thank the National Science and Engineering Research Council of Canada (NSERC) for a postgraduate scholarship.Notes and references
- J. H. Kiefer, S. S. Sidhu, R. D. Kern, K. Xie, H. Chen and L. B. Harding, Combust. Sci. Technol., 1992, 82, 101–130 CrossRef.
- E. B. Jochnowitz and J. P. Maier, Mol. Phys., 2008, 106, 2093–2106 CrossRef CAS.
- L. M. Ziurys, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12274–12279 CrossRef CAS.
- X. B. Gu, Y. Guo, F. T. Zhang, A. M. Mebel and R. I. Kaiser, Faraday Discuss., 2006, 133, 245–275 RSC.
- R. I. Kaiser, C. Ochsenfeld, D. Stranges, M. Head-Gordon and Y. T. Lee, Faraday Discuss., 1998, 109, 183–204 RSC.
- M. C. McCarthy, C. A. Gottlieb, H. Gupta and P. Thaddeus, Astrophys. J., 2006, 652, L141–L144 CrossRef CAS.
- S. Brunken, H. Gupta, C. A. Gottlieb, M. C. McCarthy and P. Thaddeus, Astrophys. J., 2007, 664, L43–L46 CrossRef CAS.
- J. Cernicharo, M. Guelin, M. Agundez, K. Kawaguchi, M. McCarthy and P. Thaddeus, Astron. Astrophys., 2007, 467, L37–L40 CrossRef CAS.
- Y. Kasai, E. Kagi and K. Kawaguchi, Astrophys. J., 2007, 661, L61–L64 CrossRef CAS.
- A. J. Remijan, J. M. Hollis, F. J. Lovas, M. A. Cordiner, T. J. Millar, A. J. Markwick-Kemper and P. R. Jewell, Astrophys. J., 2007, 664, L47–L50 CrossRef CAS.
- N. Sakai, T. Sakai, Y. Osamura and S. Yamamoto, Astrophys. J., 2007, 667, L65–L68 CrossRef CAS.
- E. Herbst and Y. Osamura, Astrophys. J., 2008, 679, 1670–1679 CrossRef CAS.
- T. R. Taylor, C. S. Xu and D. M. Neumark, J. Chem. Phys., 1998, 108, 10018–10026 CrossRef CAS.
- J. Zhou, E. Garand and D. M. Neumark, J. Chem. Phys., 2007, 127, 114313 CrossRef.
- J. Zhou, E. Garand and D. M. Neumark, J. Chem. Phys., 2007, 127, 154320 CrossRef.
- S. M. Sheehan, B. F. Parsons, T. A. Yen, M. R. Furlanetto and D. M. Neumark, J. Chem. Phys., 2008, 128, 174301 CrossRef.
- S. M. Sheehan, B. F. Parsons, J. Zhou, E. Garand, T. A. Yen, D. T. Moore and D. M. Neumark, J. Chem. Phys., 2008, 128, 034301 CrossRef.
- P. Freivogel, J. Fulara, M. Jakobi, D. Forney and J. P. Maier, J. Chem. Phys., 1995, 103, 54–59 CrossRef CAS.
- M. Kotterer and J. P. Maier, Chem. Phys. Lett., 1997, 266, 342–346 CrossRef CAS.
- H. Linnartz, T. Motylewski and J. P. Maier, J. Chem. Phys., 1998, 109, 3819–3823 CrossRef CAS.
- H. Linnartz, T. Motylewski, O. Vaizert, J. P. Maier, A. J. Apponi, M. C. McCarthy, C. A. Gottlieb and P. Thaddeus, J. Mol. Spectrosc., 1999, 197, 1–11 CrossRef CAS.
- H. Ding, T. Pino, F. Guthe and J. P. Maier, J. Chem. Phys., 2001, 115, 6913–6919 CrossRef CAS.
- H. Ding, T. Pino, F. Guthe and J. P. Maier, J. Chem. Phys., 2002, 117, 8362–8367 CrossRef CAS.
- J. Fulara, D. Lessen, P. Freivogel and J. P. Maier, Nature, 1993, 366, 439–441 CrossRef CAS.
- M. Grutter, M. Wyss and J. P. Maier, J. Chem. Phys., 1999, 110, 1492–1496 CrossRef CAS.
- M. Tulej, F. Guthe, M. Schnaiter, M. V. Pachkov, D. A. Kirkwood, J. P. Maier and G. Fischer, J. Phys. Chem. A, 1999, 103, 9712–9716 CrossRef CAS.
- T. Pino, M. Tulej, F. Guthe, M. Pachkov and J. P. Maier, J. Chem. Phys., 2002, 116, 6126–6131 CrossRef CAS.
- C. A. Gottlieb, E. W. Gottlieb and P. Thaddeus, Astron. Astrophys., 1986, 164, L5–L6 CAS.
- J. C. Pearson, C. A. Gottlieb, D. R. Woodward and P. Thaddeus, Astron. Astrophys., 1988, 189, L13–L15 CAS.
- M. C. McCarthy, M. J. Travers, P. Kalmus, C. A. Gottlieb and P. Thaddeus, Astrophys. J., 1996, 467, L125–L127 CrossRef CAS.
- M. C. McCarthy, M. J. Travers, A. Kovacs, C. A. Gottlieb and P. Thaddeus, Astron. Astrophys., 1996, 309, L31–L33 CAS.
- M. J. Travers, M. C. McCarthy, C. A. Gottlieb and P. Thaddeus, Astrophys. J., 1996, 465, L77–L80 CrossRef CAS.
- M. C. McCarthy, M. J. Travers, A. Kovacs, C. A. Gottlieb and P. Thaddeus, Astrophys. J. Suppl., 1997, 113, 105–120 CrossRef CAS.
- M. C. McCarthy, W. Chen, A. J. Apponi, C. A. Gottlieb and P. Thaddeus, Astrophys. J., 1999, 520, 158–161 CrossRef CAS.
- A. J. Apponi, M. E. Sanz, C. A. Gottlieb, M. C. McCarthy and P. Thaddeus, Astrophys. J., 2001, 547, L65–L68 CrossRef CAS.
- M. C. McCarthy and P. Thaddeus, J. Chem. Phys., 2005, 122, 174308 CrossRef CAS.
- H. Gupta, S. Brunken, F. Tamassia, C. A. Gottlieb, M. C. McCarthy and P. Thaddeus, Astrophys. J., 2007, 655, L57–L60 CrossRef CAS.
- C. Barckholtz, T. P. Snow and V. M. Bierbaum, Astrophys. J., 2001, 547, L171–L174 CrossRef CAS.
- Y. Shi and K. M. Ervin, J. Phys. Chem. A, 2008, 112, 1261–1267 CrossRef CAS.
- D. E. Woon, Chem. Phys. Lett., 1995, 244, 45–52 CrossRef CAS.
- A. L. Sobolewski and L. Adamowicz, J. Chem. Phys., 1995, 102, 394–399 CrossRef CAS.
- S. Dua, J. C. Sheldon and J. H. Bowie, J. Chem. Soc., Chem. Commun., 1995, 1067–1068 RSC.
- T. D. Crawford, J. F. Stanton, J. C. Saeh and H. F. Schaefer, J. Am. Chem. Soc., 1999, 121, 1902–1911 CrossRef CAS.
- J. Haubrich, M. Muhlhauser and S. D. Peyerimhoff, J. Phys. Chem. A, 2002, 106, 8201–8206 CrossRef CAS.
- L. Pan, B. K. Rao, A. K. Gupta, G. P. Das and P. Ayyub, J. Chem. Phys., 2003, 119, 7705–7713 CrossRef CAS.
- S. J. Blanksby, S. Dua and J. H. Bowie, J. Phys. Chem. A, 1999, 103, 5161–5170 CrossRef CAS.
- Z. X. Cao and S. D. Peyerimhoff, Phys. Chem. Chem. Phys., 2001, 3, 1403–1406 RSC.
- P. Botschwina and R. Oswald, Int. J. Mass Spectrom., 2008, 277, 180–188 Search PubMed.
- P. Botschwina, R. Oswald, G. Knizia and H. J. Werner, Z. Phys. Chem., 2009, 223, 447–460 CrossRef CAS.
- F. Pichierri, J. Phys. Chem. A, 2008, 112, 7717–7722 CrossRef CAS.
- M. L. Senent and M. Hochlaf, Astrophys. J., 2010, 708, 1452–1458 CrossRef CAS.
- C. Ochsenfeld, R. I. Kaiser, Y. T. Lee, A. G. Suits and M. HeadGordon, J. Chem. Phys., 1997, 106, 4141–4151 CrossRef CAS.
- S. Dua, J. H. Bowie and S. J. Blanksby, Eur. J. Mass Spectrom., 1999, 5, 309–317 CrossRef CAS.
- R. F. Curl, P. G. Carrick and A. J. Merer, J. Chem. Phys., 1985, 82, 3479–3486 CrossRef CAS.
- K. M. Ervin and W. C. Lineberger, J. Phys. Chem., 1991, 95, 1167–1177 CrossRef CAS.
- D. M. Neumark, J. Phys. Chem. A, 2008, 112, 13287 CrossRef CAS.
- A. Osterwalder, M. J. Nee, J. Zhou and D. M. Neumark, J. Chem. Phys., 2004, 121, 6317–6322 CrossRef CAS.
- M. J. Nee, A. Osterwalder, J. Zhou and D. M. Neumark, J. Chem. Phys., 2006, 125, 014306 CrossRef.
- D. W. Chandler and P. L. Houston, J. Chem. Phys., 1987, 87, 1445–1447 CrossRef CAS.
- A. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477–3484 CrossRef CAS.
- U. Even, J. Jortner, D. Noy, N. Lavie and C. Cossart-Magos, J. Chem. Phys., 2000, 112, 8068–8071 CrossRef CAS.
- E. Garand, T. I. Yacovitch and D. M. Neumark, J. Chem. Phys., 2009, 130, 064304 CrossRef.
- E. W. Hansen and P. L. Law, J. Opt. Soc. Am. A, 1985, 2, 510–520 CrossRef.
- J. C. Rienstra-Kiracofe, G. S. Tschumper, H. F. Schaefer, S. Nandi and G. B. Ellison, Chem. Rev., 2002, 102, 231–282 CrossRef CAS.
- J. Cooper and R. N. Zare, J. Chem. Phys., 1968, 48, 942–943 CrossRef CAS.
- K. L. Reid, Annu. Rev. Phys. Chem., 2003, 54, 397–424 CrossRef CAS.
- E. P. Wigner, Phys. Rev., 1948, 73, 1002–1009 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- K. M. Erwin, FCFgauss03: Gaussian 03 output conversion program, 2004 Search PubMed.
- K. M. Erwin, PESCAL, Fortran program, 2008 Search PubMed.
- P. Chen, in Unimolecular and Bimolecular Reaction Dynamics, ed. T. Baer, C.-Y. Ng and I. Powis, John Wiley & Sons, Chichester, 1994, pp. 371–425 Search PubMed.
- F. Duschinsky, Acta Physicochim. URSS, 1937, 7, 551–566 Search PubMed.
- S. J. Blanksby, A. M. McAnoy, S. Dua and J. H. Bowie, Mon. Not. R. Astron. Soc., 2001, 328, 89–100 CrossRef CAS.
- E. Garand, T. I. Yacovitch and D. M. Neumark, J. Chem. Phys., 2008, 129, 074312 CrossRef.
- E. Garand, T. I. Yacovitch and D. M. Neumark, J. Chem. Phys., 2009, 131, 054312 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |