Interaction of thimerosal with proteins—ethylmercury adduct formation of human serum albumin and β-lactoglobulin A
Stefan Trümpler
a,
Wiebke Lohmann
a,
Björn Meermann
a,
Wolfgang Buscher
a,
Michael Sperling
ab and
Uwe Karst
*a
aUniversity of Münster, Institute of Inorganic and Analytical Chemistry, Corrensstr. 30, 48149, Münster, Germany. E-mail: uk@uni-muenster.de; Fax: +49 251 83 36013; Tel: +49 251 83 33141
bEuropean Virtual Institute for Speciation Analysis, Mendelstr. 11, 48149, Münster, Germany
Abstract
The interaction of thimerosal, an ethylmercury-containing bactericide and fungicide used as preservative in vaccines and other drugs, with free thiols in proteins has been investigated using gradient reversed phase liquid chromatography (LC) with inductively coupled plasma mass spectrometry (ICP-MS) and electrospray mass spectrometry (ESI-MS) detection. As model proteins, β-lactoglobulin A (18.4 kDa) from bovine milk and human serum albumin (66.5 kDa) have been used. Physiological conditions upon an intravenous injection of thimerosal-containing drugs were mimicked. The formation of ethylmercury–protein adducts was proved and the identification of the binding site of ethylmercury, a free thiol residue in the peptide T13 was achieved after tryptic digestion of β-lactoglobulin A.
Introduction
Thimerosal (THI, Fig. 1) is a mercury-containing compound used as preservative in vaccines and other drugs in concentrations up to 100 mg L−1. In multi-dose ampullae, its antibacterial and antifungal properties prevent microbial contamination during repeated removal of single doses. It was introduced in 1931 and has been banned in the European Union since 2001 due to suspected neurotoxic effects, especially in children. A connection between THI-containing vaccines and an increased occurrence of autism could neither be proven nor excluded until recently.1–4 However, THI is still in use in the US and in developing countries. Current discussions about the toxicity of THI and other organomercury compounds are either centred around effects like autism or the exact functions of molecular mimicry, by which mercury species are suspected to be transported in the human body.5–8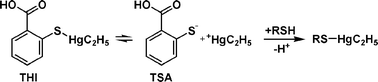 | ||
| Fig. 1 Hydrolysis of thimerosal (THI) in aqueous media to thiosalicylic acid (TSA) and ethylmercury (EtHg) and subsequent addition of EtHg to free thiols. | ||
THI decomposes into thiosalicylic acid (TSA) and ethylmercury (EtHg) in aqueous media (Fig. 1).9 The latter belongs to the dangerous organomercury species which are known for their high neurotoxicity.10 In contrast to inorganic mercury (Hg2+), the absorption, distribution, metabolism and excretion of EtHg from THI in the human body is still poorly understood. In many studies, the strong affinity of mercury species towards sulfur, especially in amino acids, peptides and proteins, is pointed out. Respective investigations have been already carried out since the 1970s, when a variety of proteins and their respective adducts formed with different mercury species were examined with radioactively labelled mercury and the equilibrium dialysis technique.11 In 1990, the effects of methylmercury on bovine serum albumin were studied with ultra-filtration and subsequent analysis of total mercury.12 More recent medical or toxicological studies of THI are based on in vivo experiments with mice13,14 or monkeys2 and mostly determine total mercury by atomic absorption spectrometry (AAS) in different tissues, body fluids or excrements. Speciation analysis by gas chromatography (GC) has been applied in these studies for supplementary purposes. Pichichero et al. obtained blood and faeces samples from newborn infants before and after vaccinations, but also monitored total mercury over time by AAS.3 Other modern separation techniques like capillary electrophoresis (CE) or liquid chromatography (LC) have been used in several recent studies for the speciation analysis of mercury with biogenic thiols, proteins and DNA. In publications by Li et al., the effects of different organomercury species on herring sperm DNA and human serum albumin (HSA) were investigated with CE-AAS.15,16 In a recent study, the adduct formation of methylmercury and inorganic mercury with biologically relevant low molecular weight thiols like cysteine (CYS) or glutathione (GSH) has been investigated.17 Guo et al. employed organomercury compounds as selective labels for GSH and β-lactoglobulin A (β-LGA) in order to count thiols by the number of mercury labels.18
To the best of our knowledge, to date, no data exist about the effects of THI on HSA, the most abundant serum protein in humans. Therefore, we focused on the interaction of EtHg from THI hydrolysis with HSA. β-LGA was used as a model protein to reduce the complexity of the investigated system, and because its general suitability for alkylmercury binding has been shown before.18 Both HSA and β-LGA contain only one free thiol function, which is the suspected binding site of interest. HSA (M ∼ 66.5 kDa) contains 35 thiol groups, 34 of which form a total of 17 disulfide bridges, and β-LGA (M ∼ 18.4 kDa) contains 5 thiol groups, four of which are present in two disulfide bridges.
In order to mimic an intravenous injection of a THI-containing drug, physiological conditions were simulated for the experiments. The respective work using liquid chromatography coupled to inductively-coupled plasma mass spectrometry (LC/ICP-MS), with the goal to obtain element-selective data, and to electrospray ionisation time-of-flight mass spectrometry (LC/ESI-ToF-MS), to receive exact mass data for the identification of molecular ions, is described in this manuscript.
Experimental
Reagents and chemicals
Thimerosal, human serum albumin, β-lactoglobulin A from bovine milk, trypsin from bovine pancreas (≥10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 BAEE units per mg protein) and formic acid were obtained from Sigma-Aldrich Chemie GmbH (Steinheim, Germany). Ethylmercury chloride (EtHgCl) and hexamethyl disilazane (HMDS) were purchased from ABCR (Karlsruhe, Germany). Acetonitrile (ACN) and methanol in HPLC grade, urea, ammonium formate and ammonium hydrogen carbonate were obtained from Fluka Chemie GmbH (Buchs, Switzerland). All chemicals were of the highest available purity. Water was purified using a Milli-Q Gradient A 10 system and filtered through a 0.22 μm Millipak 40 filter (Millipore, Billerica, MA, USA). Disposable PD-10 Sephadex® G-25M desalting columns were obtained from GE Healthcare (Buckinghamshire, UK).
000 BAEE units per mg protein) and formic acid were obtained from Sigma-Aldrich Chemie GmbH (Steinheim, Germany). Ethylmercury chloride (EtHgCl) and hexamethyl disilazane (HMDS) were purchased from ABCR (Karlsruhe, Germany). Acetonitrile (ACN) and methanol in HPLC grade, urea, ammonium formate and ammonium hydrogen carbonate were obtained from Fluka Chemie GmbH (Buchs, Switzerland). All chemicals were of the highest available purity. Water was purified using a Milli-Q Gradient A 10 system and filtered through a 0.22 μm Millipak 40 filter (Millipore, Billerica, MA, USA). Disposable PD-10 Sephadex® G-25M desalting columns were obtained from GE Healthcare (Buckinghamshire, UK).
An incubation solution with physiological pH of 7.4 was prepared by adjusting the pH of a 20 mM of ammonium formate solution with 25% aqueous ammonia. Stock solutions of THI, HSA and β-LGA were prepared daily by dissolving the solids in the incubation solution. EtHgCl was dissolved in a mixture of water and methanol (v/v) 80:20. Volumetric flasks were silanised to prevent adsorption of the analytes to container walls. For silanisation, 100 vials were heated in a 1 L flask together with 300 μL of HMDS for 3 h at 120 °C, rinsed once with water and dried before use.
Instrumentation and settings
Full scan mass spectra (m/z 50–3000) were recorded using ESI(+)-MS under the following conditions: end plate offset: −500 V, capillary: −4000 V, nebulizer gas (N2): 800 kPa, drying gas (N2): 8.0 L min−1, drying temperature: 200 °C, capillary exit: 150.0 V, skimmer 1: 50.0 V, skimmer 2: 26.5 V, hexapole 1: 23.0 V, hexapole 2: 21.4 V, hexapole RF: 350 V, transfer time: 80.0 μs, pre-pulse storage: 16.0 μs, detector: −1000 V. Internal calibration was performed using sodium formate clusters at the beginning of each data acquisition run.
An ELAN 6000 ICP-MS system from Perkin Elmer (Rodgau, Germany) was used as detector. The skimmer and sampler cones were made of platinum. We used a cooled cyclonic spray chamber (∼5 °C) PC3 from ESI (Elemental Scientific Inc., Omaha, NE, USA) and the sample introduction was performed using a Meinhard nebulizer TR-30-A3 (SPETEC GmbH, Erding, Germany). The nebulizer gas flow was 0.43 L min−1.
Two mercury isotopes were observed (m/z 200, 202), using the following settings: 1400 W (RF power), plasma gas flow was 15 L min−1, auxiliary gas flow was 1 L min−1, oxygen gas flow: 0.1 mL min−1, dwell time: 50 ms.
| Time/min | 0.0 | 2.0 | 12.0 | 19.0 | 21.0 | 23.0 |
| %B (ACN) | 25 | 25 | 60 | 60 | 25 | 25 |
Sample preparation procedures
Solutions of β-LGA and HSA (approx. 2 × 10−4 M) with a ten-fold excess of THI were prepared in the incubation solution containing 8 M urea and were incubated at 37 °C in a water bath for one hour. Non-spiked protein solutions were prepared similarly. After incubation, excess THI and urea were removed on a PD-10 desalting column with the incubation solution as mobile phase according to the manufacturer’s protocol. An aliquot of the solution was removed and diluted to approximately 2 × 10−5 M protein for direct measurement at the ESI-ToF-MS to control the adduct formation. 1 mL of spiked and non-spiked β-LGA solution each was subjected to tryptic digestion by adding 4 mg NH4HCO3 and an appropriate amount of trypsin solution in water (2 mg mL−1) to receive 5% (w/w) trypsin referring to the protein concentration. The digestion proceeded for two hours at 37 °C in a water bath and was then stopped by adding 500 μL methanol, containing 0.6% formic acid. The resulting solutions were subjected to LC/ESI-ToF-MS analysis subsequently.
Results and discussion
First, the proteins were incubated with THI at 37 °C and pH 7.4 for 1 h to simulate physiological conditions as closely as possible. A protein concentration corresponding to the natural abundance of HSA (30–60 mg mL−1)20 was selected and THI was added in a concentration (100 μg mL−1) which is present directly after intravenous injection, resulting in a 2.4-fold molar excess of protein. The concentration of the model protein β-LGA was equimolar to HSA for comparative purposes. To monitor the THI–protein adduct formation, LC/ICP-MS was used, which allows the acquisition element-selective information about mercury. The two most abundant mercury isotopes, 200Hg and 202Hg, were monitored, because an unnatural isotopic ratio of the two would have indicated possible interferences during detection. In Fig. 2, the chromatograms of EtHgCl and THI standards and the protein solutions are presented (only the m/z of 202Hg is shown for clarity reasons). EtHg elutes at 2 min in the void volume under these conditions, and by the respective EtHg signal in the THI run, it is obvious that the previously described hydrolysis has proceeded after the incubation time of 1 h (Fig. 1).9 Intact THI elutes at around 6 min under these conditions. However, in the protein solutions, no mercury signals are detected except those at 10 and 11 min for the β-LGA and HSA solution, respectively. The small peaks at 2 and 6 min in the protein runs were below the limit of detection of 3σ. That means that neither THI nor free EtHg were still present in the protein solutions after one hour incubation time. Comparative analysis by LC/ESI-ToF-MS showed that the signals were indeed the respective β-LGA–EtHg and HSA–EtHg adducts. Data of these runs are not shown, since similar results will be presented and discussed below. Regarding published data by which the elimination half-time of total mercury from human blood after THI administration is seven days, the selected incubation time shows a situation close to the time of injection.21 Apparently, the affinity for EtHg towards the proteins is strong enough to accelerate the hydrolysis and to shift the hydrolysis equilibrium (Fig. 1) completely towards the degradation products. This indicates that THI decomposes very quickly within the body after injection into the bloodstream and that it may bind to thiols from naturally abundant proteins.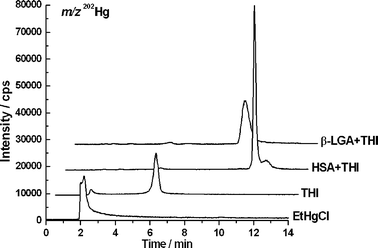 | ||
| Fig. 2 LC/ICP-MS chromatograms of EtHg and THI standard solutions and the THI–protein mixtures. Quantitative adduct formation has occurred, indicated by the absence of free THI or EtHg in the protein solutions. Chromatograms of different solutions are shifted from the baseline for clarity reasons. | ||
In the following experiments, ESI-ToF-MS was used to obtain molecular information. THI was added in tenfold molar excess over the proteins in order to maximise the formation of the EtHg–protein adducts observed in the physiological incubation experiment. In order to prevent precipitation of the proteins under these conditions, urea was added to the incubation solution (final concentration 8 M) to denature the proteins. In Fig. 3, the mass spectra of the incubated proteins after clean-up are shown. Corresponding m/z values are listed in Table 2. In Fig. 3A, the charge state 10+ of the β-LGA solutions is shown. The black spectrum shows the pure protein. In the grey curve, the pure protein signal is significantly reduced and another signal with a mass increase of 22.87 towards the pure protein is observed. This can be deconvoluted to the EtHg–β-LGA adduct . Fig. 3B shows analogous data for HSA. The mass spectrum shows the protein in the 43+ charge state. The largest signal in the centre belongs to the so-called non-mercaptalbumin. This is a modified form of HSA, in which its free thiol group forms a disulfide bond with an additional cysteine molecule.22 Since this fraction of HSA is not easily accessible for EtHg, this signal remains the most abundant one even after incubation with a tenfold excess of THI. The free mercaptalbumin, however, is significantly decreased. In addition, the signal of the EtHg–HSA adduct emerged. The mass increase in this case is 5.31 amu and it is less than the shift observed in β-LGA, due to the higher charge state of HSA compared to β-LGA. Nevertheless, the mass shifts in both β-LGA and HSA are deconvoluted to an absolute value of 228.7. This increase confirms the replacement of the proton at the free thiol group by an EtHg ion. Thus, “C2H4Hg” is effectively added to the protein, which is confirmed by the agreement of expected and experimental values in Table 2.
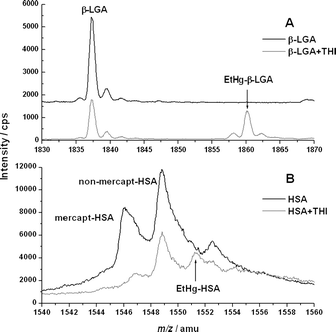 | ||
| Fig. 3 ESI-ToF-MS spectra of (A) β-LGA and its EtHg adduct in the charge state 10+; (B) HSA in its free mercaptalbumin and non-mercaptalbumin form and the HSA–EtHg adduct in the charge state 43+. m/z values are listed in Table 2. | ||
In the next step, a tryptic digest of β-LGA and the EtHg–β-LGA adduct was carried out in order to identify the binding site of the EtHg moiety. Trypsin cleaves peptide bonds after arginine and lysine, breaking β-LGA down into 18 peptides . Since we assumed that EtHg binds to sulfhydryl groups, only the peptides T5, T13 or T18 possess a possible binding site. In contrast to other protein treatment procedures,18 no reduction of disulfide bonds was performed in this case, because preliminary experiments showed that the attached EtHg group was removed upon reduction. Without reduction, the two disulfide bridges in the β-LGA and the respective EtHg adduct remain intact. One of those connects the peptides T5 (amino acids 61–69) and T18 (149–162) that contain one cysteine unit each. The other is located within T13 (102–124), which contains the three remaining cysteines. Fig. 4 shows the isotopic patterns of (A) the pure T13 and (B) the EtHg–T13 adduct after LC separation. Both compounds are doubly charged and their respective monoisotopic masses are labelled with an asterisk. The very good correlation of calculated (line spectra) and measured values (profile spectra) is visible in the figure, thus confirming that the location of the modification is the peptide T13. Deviation values are 0.9 ppm for the unmodified T13 and 4.3 ppm for the EtHg–T13 adduct .
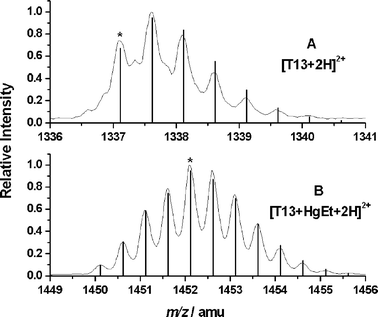 | ||
| Fig. 4 ESI-ToF-MS spectra of the peptide T13 after tryptic digestion of (A) β-LGA and (B) β-LGA with THI, both doubly charged. Profile spectra show the measured spectra and vertical lines the respective calculated values. A characteristic difference in isotopic patterns is visible between the free T13 and the mercury-containing one. Deviations between calculated and detected masses are 0.9 ppm for T13 and 4.3 ppm for EtHg+T13. Monoisotopic peaks are labelled with an asterisk. | ||
The natural isotopic pattern of mercury consists of seven stable isotopes with a very characteristic pattern of abundance. When introducing a mercury atom into an organic molecule with more than 100 carbon atoms (like in the peptide T13), the isotopic pattern of mercury can no longer be resolved. However, it is still possible to clearly distinguish between mercury-containing and mercury-free molecules by a significant and characteristic alteration of the peptide isotopic pattern. The recorded mass spectrum of the digest of β-LGA+THI was investigated for further mercury-containing peptides without success. Since the only EtHg that was found in the complete protein after incubation with THI (Fig. 3, Table 2) and attached to the T13 after digestion, the mercury atom must have been bound to the only free thiol group in the β-LGA.
With the selected model protein β-LGA, we were able to show the quantitative hydrolysis of THI with complete binding of EtHg under physiological conditions by LC/ICP-MS (Fig. 2) and prove the adduct formation with the denatured protein by exact mass measurements with ESI-ToF-MS (Fig. 3, Table 2). The subsequent tryptic digestion with intact disulfide bridges allowed the LC separation of the tryptic peptides and the identification of the EtHg–T13 adduct by ESI-ToF-MS (Fig. 4). With the selected experimental conditions, only one possible binding site in the T13 was left, thus indicating that the EtHg unit was bound to the free thiol group.
Experiments for the physiologically relevant protein HSA were carried out in parallel to those with β-LGA and the resulting data show very good agreement. The quantitative adduct formation with EtHg mimicking the intravenous injection of a THI-containing drug was shown (Fig. 2) as well as the binding of one EtHg unit to HSA (Fig. 3, Table 2). From the similar behaviour of β-LGA and HSA in the joint experiments, we postulate a correlating behaviour for the last step, meaning that the EtHg moiety binds to the one free thiol group in HSA.
Conclusions
The interactions of EtHg from THI with HSA and β-LGA were investigated by the comparative application of LC/ICP-MS and LC/ESI-ToF-MS. The evaluation of element selective and molecular data allowed an insight into physiological processes after THI administration in concordance with the β-LGA model system. Thus, the similarities to the HSA were proven during the course of experiments and led to equal conclusions for the final experiments. Evidence for the free thiol group in the used proteins as binding site for EtHg was found.For future research, the use of a dual LC/ESI-ToF-MS/ICP-MS system for simultaneous acquisition of element-selective and structural data would be promising. One main advantage of the use of a joint system would be the prevention of intersystem variations which occur during the use of different LC systems. Furthermore, to obtain data with even more relevance to physiological processes, model systems with increasing similarity to human blood should be investigated. Suitable models would have to take natural levels of cysteine and glutathione into account, which are also present in significant concentrations.
References
- L. K. Ball, R. Ball and R. D. Pratt, Pediatrics, 2001, 107, 1147–1154 Search PubMed.
- T. M. Burbacher, D. D. Shen, N. Liberato, K. S. Grant, E. Cernichiari and T. Clarkson, Environ. Health Perspect., 2005, 113, 1015–1021 CAS.
- M. E. Pichichero, A. Gentile, N. Giglio, V. Umido, T. Clarkson, E. Cernichiari, G. Zareba, C. Gotelli, M. Gotelli, L. Yan and J. Treanor, Pediatrics, 2008, 121, e208–e214 Search PubMed.
- J. G. Dórea, Am. J. Perinatol., 2007, 24, 387–400 CrossRef.
- C. C. Bridges and R. K. Zalups, Toxicol. Appl. Pharmacol., 2005, 204, 274–308 CrossRef CAS.
- R. E. Hoffmeyer, S. P. Singh, C. J. Doonan, A. R. S. Ross, R. J. Hughes, I. J. Pickering and G. N. George, Chem. Res. Toxicol., 2006, 19, 753–759 CrossRef CAS.
- C. C. Bridges and R. K. Zalups, Chem. Res. Toxicol., 2006, 19, 1117–1118 CrossRef CAS.
- R. E. Hoffmeyer, S. P. Singh, C. J. Doonan, A. R. S. Ross, R. J. Hughes, I. J. Pickering and G. N. George, Chem. Res. Toxicol., 2006, 19, 1118–1120 CrossRef CAS.
- X. Wu, H. Liang, K. A. O’Hara, J. C. Yalowich and B. B. Hasinoff, Chem. Res. Toxicol., 2008, 21, 483–493 CrossRef CAS.
- M. Horvat and D. Gibicar, in Handbook of Elemental Speciation II, ed. R. Cornelis, Wiley VCH, Weinheim, 2005, pp. 285–291 Search PubMed.
- S. C. Fang and E. Fallin, Bull. Environ. Contam. Toxicol., 1976, 15, 110–117 CrossRef CAS.
- A. Yasutake, K. Hirayama and M. Inoue, Arch. Toxicol., 1990, 64, 639–643 CrossRef CAS.
- G. Zareba, E. Cernichiari, R. Hojo, S. McNitt, B. Weiss, M. M. Mumtaz, D. E. Jones and T. W. Clarkson, J. Appl. Toxicol., 2007, 27, 511–518 CrossRef CAS.
- S. Havarinasab, E. Björn, J. Ekstrand and P. Hultman, Toxicology, 2007, 229, 23–32 CrossRef CAS.
- Y. Li, Y. Jiang and X.-P. Yan, Anal. Chem., 2006, 78, 6115–6120 CrossRef CAS.
- Y. Li, X.-P. Yan, C. Chen, Y.-L. Xia and Y. Jiang, J. Proteom. Res., 2007, 6, 2277–2286 CrossRef CAS.
- E. M. Krupp, B. F. Milne, A. Mestrot, A. A. Meharg and J. Feldmann, Anal. Bioanal. Chem., 2008, 390, 1753–1764 CrossRef CAS.
- Y. Guo, L. Chen, L. Yang and Q. Wang, J. Am. Soc. Mass Spectrom., 2008, 19, 1108–1113 CrossRef CAS.
- W. Lohmann, H. Hayen and U. Karst, Covalent Protein Modification by Reactive Drug Metabolites Using On-Line Electrochemistry/Liquid Chromatography/Mass Spectrometry, Anal. Chem. Search PubMed , accepted for publication.
- A. T. Høstmark, S. E. Tomten and J. E. Berg, J. Hypertens., 2005, 23, 725–730 CrossRef CAS.
- T. W. Clarkson and L. Magos, Crit. Rev. Toxicol., 2006, 36, 609–662 CrossRef CAS.
- T. P. King, J. Biol. Chem., 1961, 236, PC5 CAS.
| This journal is © The Royal Society of Chemistry 2009 |