Speciation of selenomethionine metabolites in wheat germ extract†
Yasumitsu
Ogra
*,
Takashi
Kitaguchi
,
Kazuya
Ishiwata
,
Noriyuki
Suzuki
,
Toshihiko
Toida
and
Kazuo T.
Suzuki
Graduate School of Pharmaceutical Sciences, Chiba University, Inohana, Chuo, Chiba 260-8675, Japan. E-mail: ogra@p.chiba-u.ac.jp; Fax: +81 43 226 2866; Tel: +81 43 226 2866
First published on 18th November 2008
Abstract
Selenometabolites transformed from selenomethionine (SeMet) in wheat germ extract (WGE) were identified by complementary use of HPLC-ICP-MS and HPLC-ESI-MS/MS. Three selenium (Se)-containing peaks tentatively named WGE1, WGE2, and WGE3 were detected by HPLC-ICP-MS. WGE1 had [M]+ at m/z 212 on HPLC-ESI-MS analysis, and its fragment ions indicated that WGE1 is selenomethionine methylselenonium (MeSeMet). WGE2 and WGE3 exhibited absorption at 254 nm and molecular ions at m/z 433 and 447, respectively. Their fragment ions revealed that WGE2 and WGE3 are Se-adenosylselenohomocysteine (AdoSeHcy) and Se-adenosylselenomethionine (AdoSeMet), respectively. Their structures were coincident with the absorption of WGE2 and WGE3 at 254 nm. In addition, a trace amount of AdoSeMet was suggested to also exist in rabbit reticulocyte lysate, a mammalian in vitro translation system, because the transformation of AdoSeMet from SeMet was completely inhibited by (2S)-2-amino-4,5-epoxypentanoic acid (AEPA), a potent inhibitor of AdoMet synthetase. These results suggest that SeMet and methionine (Met) share a common metabolic pathway, i.e., SeMet is not only incorporated into proteins in place of Met but also metabolized to AdoSeMet in higher eukaryotes and MeSeMet in plants.
Introduction
Selenium (Se) is an essential micronutrient in animals because it promotes the activities of such selenoenzymes as glutathione peroxidases, iodothyronine 5′-deiodinase, and thioredoxin reductase, by existing in the form of selenocysteine (SeCys) in their active centers.1,2 In contrast, Se is not an essential element in plants; rather, it is a serviceable element for growth, similar to sodium and silicon. In spite of its non-essentiality, Se metabolism in plants has been attracting much interest for two reasons. One, is its used in phytoremediation for the removal of contaminating Se. Although Se is a useful material in the semiconductor and electronic industries, it is also an environmental contaminant.3 Phytoremediation is a low-cost and environmentally friendly technique to remove Se contaminants. Indeed, the Indian mustard, Brassica juncea, has been extensively studied for Se phytoremediation4–6 because of its high Se accumulating ability, fast growth, and high biomass . In this plant, absorbed inorganic Se, such as selenite (Se (IV)) and selenate (Se (VI)), is transformed into less toxic forms, i.e., selenoamino acids, such as Se-methylselenocysteine (MeSeCys) and γ-glutamylmethylselenocysteine (GluMeSeCys). The other reason is its use as a nutritional aid in the form of Se supplements or medicines for cancer prevention and other diseases. As inorganic Se salts exhibit substantial toxicities to animals, less toxic Se compounds from plants are useful for this purpose. Indeed, it has been reported that some selenoamino acid derivatives decrease the incidence of prostate, skin, and mammary cancers7–9 and exert cytotoxic effects on cultured tumor cells.10–12 However, Se metabolism in plants is not fully revealed, and novel selenopeptides and selenometabolites have been reported with the development of analytical techniques based on mass spectrometry.13,14Selenomethionine (SeMet) is one of the key metabolites in selenized yeasts and plants.15–18 In selenized yeast, SeMet biosynthesized from inorganic Se is incorporated into proteins and metabolized to other biomolecules such as Se-adenosylselenomethionine (AdoSeMet) derivatives.19,20 As already suggested, the replacement of methionine (Met) with SeMet did not significantly alter protein structure in bacteria and fungi.21,22 In addition, we have also reported that SeMet, which could not be discriminated from Met, was incorporated into proteins in an in vitro translation system using wheat germ extract (WGE),23 also, the fluorescence of green fluorescent protein (GFP) and the activity of JNK stimulatory phosphatase-1 (JSP-1) were not affected by the substitution of Met with SeMet by in vitro translation.23 Thus, the biosynthesis of SeMet-containing proteins is responsible for the detoxification of Se in plants in addition to the biosynthesis of MeSeCys and GluMeSeCys.24 On the other hand, although Met is used not only for protein synthesis but also metabolism into coenzymes, such as methylmethionine (MeMet) and S-adenosylmethionine (AdoMet, SAM), as shown in yeast, the metabolism of SeMet into such coenzymes in higher eukaryotes, i.e., plants and animals, has yet to be revealed.
Although the inductively coupled plasma mass spectrometer (ICP-MS) is a superior instrument for elemental speciation owing to its high sensitivity and specificity when used in combination with separation instruments such as a high-performance liquid chromatograph (HPLC), the identification of Se compounds by HPLC-ICP-MS is accomplished by matching the retention times of samples with those of authentic standards.25,26 Thus, identification by HPLC-ICP-MS is limited to the situation where authentic Se standards are available. As a complementary method to HPLC-ICP-MS, HPLC coupled with electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI)-tandem mass spectrometry (MS/MS) is used to identify unknown selenometabolites. Indeed, novel Se-containing metabolites, such as selenosugars, selenoamino acid derivatives, and AdoSeMet derivatives, have been successfully identified with this method.20,27–31
In this study, we intend to reveal the biological transformation of SeMet into SeMet derivatives in a model system of higher eukaryotes, i.e., WGE, by identifying Se-containing molecules with HPLC-ICP-MS and HPLC-ESI-MS/MS. As mentioned above, WGE is a good model system to reveal the metabolism of SeMet. In addition, the in vitrometabolism of SeMet was also evaluated in rabbit reticulocyte lysate (RRL), a mammalian in vitro translation system. Moreover, we also intend to depict the entire metabolic pathway of SeMet in higher eukaryotes by assigning Se metabolites on the basis of selenometabolomics.
Experimental
Chemicals
L-Selenomethionine (SeMet) and Wakogel® 100C18 were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). S-Adenosylhomocysteine (AdoHcy), S-adenosylmethionine (AdoMet), and 3-chloroperoxybenzoic acid (mCPBA) were purchased from Sigma (St. Louis, MO, USA). Methionine methylsulfonium chloride (MeMet) and methyl iodide were purchased from Tokyo Chemical Industry, Co. Ltd. (Tokyo, Japan). A cell-free protein synthesis kit consisting of WGE and GFP mRNA for in vitro translation was kindly provided by ZOEGENE Co. (Yokohama, Japan). RRL for in vitro translation was purchased from Promega (Madison, WI, USA). All other reagents used in this study were of the highest or analytical grade. Deionized water (18.3 MΩ cm−1) was used throughout.Synthesis of selenomethionine methylselenonium
SeMet (9.8 mg, 0.05 mmol) was dissolved in deionized water (400 μL), and methyl iodide (92 μL, 1.5 mmol) and acetonitrile (800 μL) were added. The reaction mixture was gently stirred at room temperature for three days in the dark. Then, the reaction mixture was evaporated in vacuo and the residue was dissolved in D2O to confirm the structure by 1H-NMR (400 MHz, JEOL 400α). Selenomethionine methylselenonium (MeSeMet) was purified on an HPLC equipped with a multi-mode size exclusion column (Shodex Asahipak GS-320HQ, 7.5 i.d. × 300 mm with a guard column; Showa Denko, Tokyo, Japan). The HPLC column was eluted with 10 mM ammonium acetate, pH 6.5, at a flow rate of 0.6 mL min−1.Synthesis of potent AdoMet synthetaseinhibitor
(2S)-2-Amino-4,5-epoxypentanoic acid (AEPA), a potent inhibitor of AdoMet synthetase, was synthesized in-house according to the literature with slight modifications.32 (2S)-Allylglycine (Sigma, St. Louis, MO, USA) (250 mg), triethylamine (1.02 mL), and di-tert-butyldicarbonate ((Boc)2O) (1.096 mL) were mixed in 8.8 mL of 50 : 50 v/v% H2O and tetrahydrofuran (THF) under a nitrogen atmosphere. Then, the reaction mixture was stirred at room temperature for 4 h. The reaction mixture was evaporated in vacuo and the residue was dissolved in ethyl acetate (30 mL). The pH of this solution was adjusted to 2–3 with citric acid solution on ice. The organic phase was collected and the reactant was extracted twice from the aqueous phase with ethyl acetate (15 mL). The combined organic phase was washed three times with deionized water and once with saturated sodium chloride solution, then dried with anhydrous sodium sulfate, filtered, and evaporated. The residue was dissolved and purified on a silica gel column (eluate: 5% methanol and 95% methylene chloride) to afford Boc-protected allylglycine (yield 331.3 mg, 1.55 mmol, 44%). The chemical purity of Boc-protected allylglycine was confirmed by 1H-NMR measurements in CDCl3. Boc-protected allylglycine (40 mg) and mCPBA (84 mg) were mixed in methylene chloride (5 mL) for 4 h at room temperature under a nitrogen atmosphere. Then, sodium metabisulfite (100 mg) in deionized water (500 μL) was added to stop the reaction, and the reactant was evaporated in vacuo. The residue was purified on a silica gel column (eluate: 5% methanol, 1% acetic acid, and 94% methylene chloride) to afford Boc-protected AEPA. The eluate containing Boc-protected AEPA was reacted with 90% formic acid (1 mL) for 20 min at room temperature to cleave a Boc group. The reaction mixture was evaporated in vacuo and lyophilized. The residue (4.4 mg, 3.36 μmol, 17.9%) was dissolved in deionized water and purified on an ODS column (Wakogel® 100C18) to afford AEPA. Chemical purity of AEPA was confirmed on a 1H-NMR instrument in D2O and a fast atom bombardment mass spectrometer (FAB-MS, JEOL JMS-AX500).Apparatus
An Agilent7500ce ICP-MS (Agilent Technologies, Hachiouji, Japan) equipped with an octopole reaction system was used in the deuterium (D2) reaction mode to detect Se without interference originating from polyatomic ions.33 Important parameters for operation were as follows: plasma RF power, 1500 W; plasma gas flow, 15.0 L min−1; auxiliary gas flow, 1.15 L min−1; nebulizer gas flow, 1.05 L min−1; D2 gas flow, 2.0 mL min−1; m/z monitored, 77, 78, 80, and 82; dwell time, 100 ms; and point per peak, 1. An API3000 triple quadrupole mass spectrometer with an electrospray ionization source (ESI-MS/MS; Applied Biosystems, Tokyo, Japan) was used to assign the structures of unknown Se compounds. The ESI-MS/MS was operated in the positive ion mode under the following conditions: ion spray voltage, 5000 V; gas temperature, 450 °C; nebulizer gas, 8 units; curtain gas, 8 units; declustering potential, 20 V; focusing potential, 200 V; and entrance potential, −10 V.In vitro translation of GFP mRNA
GFP mRNA was translated in vitro according to our previous report.23 Briefly, a 50 μL aliquot of the reaction mixture consisted of 10 μL of WGE, 0.5 μL of creatine kinase, 30 U of RNase inhibitor, 25 μg of mRNA, Cys- and Met-free amino acid mixture (each 0.3 mmol L−1), RNase-free water, 0.3 mmol L−1 Cys, and 0.3 mmol L−1 SeMet. The reaction mixture was incubated at 26 °C for 8 h, and the reaction was stopped by cooling at 4 °C. The reaction mixture was subjected to fluorescence measurement and speciation of Se in SeMet-containing GFP, as mentioned below.Double detection of fluorescence and Se in SeMet-containing GFP by HPLC-FD-ICP-MS
A 20 μL aliquot of the reaction mixture was applied to an HPLC coupled with a fluorescence detector (FD, Shimadzu RF-530, Shimadzu) and an ICP-MS to analyze the fluorescence intensity of GFP detected at 395 nm excitation wavelength and 510 nm emission wavelength, and the Se distribution detected at m/z 80, respectively. The HPLC system (Prominence, Shimadzu) consisted of an online degasser, an HPLC pump, a Rheodyne six-port injector, and a multi-mode size exclusion column (Shodex Asahipak GS-520HQ, 7.5 i.d. × 300 mm with a guard column; Showa Denko). The multi-mode size exclusion column (GS-520HQ, exclusion size: >300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) was eluted with 50 mmol L−1 Tris–HCl, pH 7.4, at a flow rate of 0.6 mL min−1. The eluate was introduced into FD and ICP-MS in this sequence.
000 Da) was eluted with 50 mmol L−1 Tris–HCl, pH 7.4, at a flow rate of 0.6 mL min−1. The eluate was introduced into FD and ICP-MS in this sequence.
In vitro metabolism of SeMet in WGE or RRL
SeMet was incubated with the reaction mixture for the in vitro translation mentioned above minus GFP mRNA. To efficiently metabolize SeMet that was not incorporated into GFP protein, GFP mRNA was removed from the reaction mixture of WGE. RRL was also used to compare the in vitrometabolism of SeMet in animals with that in plants, i.e., WGE. A 50 μL aliquot of the reaction mixture consisted of 35 μL of RRL, 10 μL of Met-free amino acid mixture (each 1.0 mmol L−1), 4 μL of 2.5 mmol L−1SeMet, and 1 μL of 125 mmol L−1AEPA or deionized water. The reaction mixture was incubated at 30 °C for 30 min, and the reaction was stopped by cooling at 4 °C.Speciation of Se compounds by HPLC-PDA-ICP-MS
A 20 μL aliquot of the reaction mixture for in vitrometabolism using WGE or RRL was applied to an HPLC coupled with a photodiode array (PDA, Shimadzu SPD-M20A, Shimadzu) and an ICP-MS to analyze absorptions at 200–900 nm and the distribution of Se at m/z 80, respectively. The HPLC system was equipped with a multi-mode size exclusion column (Shodex Asahipak GS-320HQ, 7.5 i.d. × 300 mm with a guard column; Showa Denko). The multi-mode size exclusion column (GS-320HQ, exclusion size: >40000 Da) was eluted with 50 mmol L−1ammonium acetate, pH 6.5, at a flow rate of 0.6 mL min−1. The eluate was introduced into the PDA and ICP-MS in this sequence.
HPLC-ESI-MS/MS analysis
A 2.0 μL aliquot of the reaction mixture for in vitrometabolism was applied to an HPLC equipped with a multi-mode size exclusion column of narrow bore size (Shodex GS320A-2E, 2.0 i.d. × 250 mm; Showa Denko) to scan Se-containing ions and to perform tandem MS analysis. The column was eluted with 10 mmol L−1ammonium acetate, pH 6.5, at a flow rate of 40 μL min−1, and the eluate was introduced into the ionization spray of ESI-MS/MS without splitting. The introduction into ESI-MS/MS was time-separated for Se elution using a four-way valve, i.e., the eluate was introduced from 24.9 to 26.7, from 31.7 to 33.0, and from 49.0 to 51.4 min for unknown Se compounds tentatively named WGE1, WGE2, and WGE3, respectively. These retention times of the Se compounds, WGE 1, WGE2 and WGE3, were estimated by HPLC-ICP-MS equipped with the same column as HPLC-ESI-MS/MS prior to the analyses by HPLC-ESI-MS/MS. The dissociation of each Se-containing molecular ion of the unknown Se compounds at m/z 212, 433, and 447 was induced with 25 eV collision energy, and fragment ions were detected with the second mass spectrometer. Mass calibration and optimization of ESI-MS/MS parameters were performed daily prior to use in accordance with the manufacturer’s instructions.Results and discussion
Detection of low molecular weight SeMet metabolites in WGE
The Se peak that appeared at the retention time of 15.0 min was accompanied by a fluorescence peak, suggesting that this peak corresponded to GFP translated in vitro in WGE, and it contained the Se of SeMet (Fig. 1). On the other hand, the peak that appeared at the retention time of 21.6 min was that of intact SeMet because its retention time matched that of the authentic standard. In addition to these two peaks, two other Se peaks were detected at 27.8 and 32.5 min (Fig. 1). Since compounds corresponding to the two additional peaks were more slowly eluted than SeMet on the multi-mode size exclusion column (GS-520HQ), these Se-containing compounds seemed to be low molecular weight compounds. Therefore, to realize better separation of low molecular weight compounds on GS-520HQ, GS-320HQ (normal bore column) or GS320A-2E (narrow bore column) with a smaller exclusion size than GS-520HQ were used for later experiments. On GS-320HQ, three Se peaks were detected after the SeMet peak in the reaction mixture for in vitrometabolism of SeMet (Fig. 2). However, as Se standards whose retention times matched those of the three Se compounds were not available, the three unknown peaks at retention times of 30.5, 42.7, and 59.0 min were tentatively named WGE1, WGE2, and WGE3, respectively, according to their elution order. The percentage concentration of Se in these Se-containing compounds at 8 h after incubation were 33.2, 24.9, 24.9, and 14.5% for SeMet, WGE1, WGE2, and WGE3, respectively, as calculated from their peak areas. As GFP mRNA was not added to the reaction mixture, Se in GFP protein was not detected. WGE2 and WGE3, but not WGE1, showed absorptions at 254 nm (Fig. 2, upper). WGE3 spontaneously changed to WGE2 during storage (data not shown). These results suggest that WGE2 and WGE3 have closely related structures.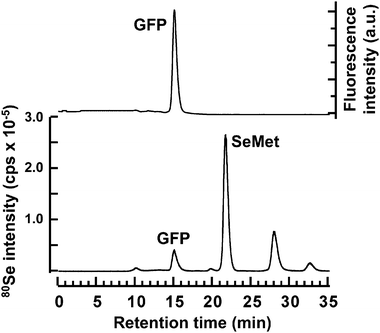 | ||
| Fig. 1 Elution profiles of fluorescence and Se in the reaction mixture of wheat germ extract for the translation of SeMet-containing GFP in a cell-free system. A 20 μL aliquot of the reaction mixture was subjected to HPLC coupled with FD and ICP-MS to measure the fluorescence intensity of GFP detected at an excitation wavelength of 395 nm and an emission wavelength of 510 nm (upper panel) and the Se distribution at m/z 80 (lower panel), respectively. The multi-mode size exclusion column (GS-520HQ) was eluted with 50 mmol L−1 Tris–HCl, pH 7.4, at a flow rate of 0.6 mL min−1. | ||
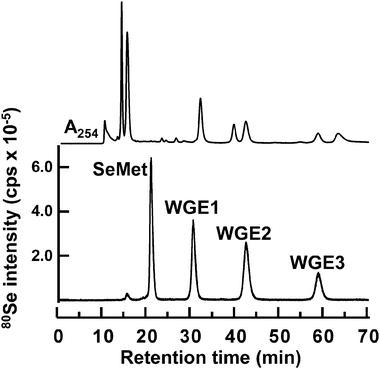 | ||
| Fig. 2 Elution profile of Se in the reaction mixture of wheat germ extract for the in vitrometabolism of SeMet. A 20 μL aliquot of the reaction mixture was subjected to HPLC (GS-320HQ) coupled with PDA and ICP-MS to measure absorbance at 254 nm (upper) and Se distribution at m/z 80 (lower), respectively. The column was eluted with 10 mmol L−1ammonium acetate, pH 6.5, at a flow rate of 0.6 mL min−1. | ||
Identification of unknown SeMet metabolites in WGE
Se consists of six isotopes, 74Se (0.89%), 76Se (9.36%), 77Se (7.63%), 78Se (23.8%), 80Se (49.6%), and 82Se (8.73%). The signal showing the isotope pattern of Se was observed at around m/z 212.0 in the eluate introduced from 24.9 to 26.7 min on GS320A-2E, and corresponded to WGE1, despite the fact that the 74Se-containing molecular ion was not detected due to trace abundance (Fig. 3a). It has already been reported that the Se-containing compound in plant samples showed m/z 212 as its molecular ion,26,34i.e., selenomethionine methylselenonium (MeSeMet) is detected as [M]+. GS320A-2E is a multi-mode size exclusion column that shows cation-exchange properties under the operating conditions. Therefore, it is reasonable that MeSeMet was eluted more slowly than SeMet despite the fact that its molecular mass is larger than that of SeMet, and its molecular ion was detected as [M]+. The chromatographic behavior of the molecular ion at m/z 212 on GS320A-2E was similar to that of Se in WGE1 detected by ICP-MS on GS-320HQ (Fig. 4). To confirm the structure of WGE1, the precursor ion at m/z 212 was subjected to MS/MS analysis. All fragment ions, m/z 56, 74, 84, and 102 of WGE1 were completely identical to those of the synthesized MeSeMet (Fig. 5a and a′), and could be assigned to the fragments of MeSeMet depicted in Fig. 5a. Furthermore, serial fragment ions at m/z 56, 74, 84, and 102 are commonly found in Se compounds having an [–Se(CH2)2CH(NH2)COOH] moiety, such as selenohomocysteine (SeHCys), SeMet, selenoethionine, and selenohomolanthionine.14,35 In addition to these spectrometric data, spiked synthesized MeSeMet was co-eluted with the reaction mixture of WGE on GS-320HQ. The spike specifically increased the intensity of the peak assigned to WGE1 (data not shown). Consequently, the identity of WGE1 was confirmed to be MeSeMet.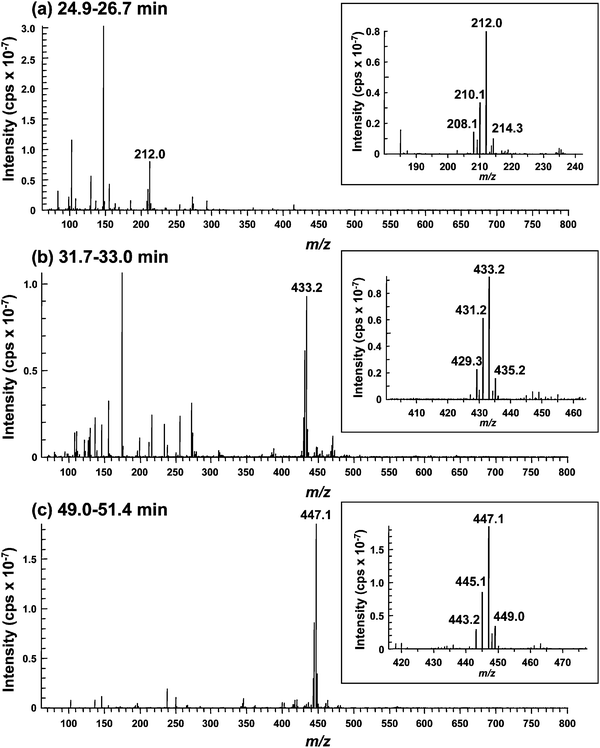 | ||
| Fig. 3 ESI-MS spectra of unknown Se compounds in the eluate of the reaction mixture of wheat germ extract for the in vitrometabolism of SeMet. A 2.0 μL aliquot of the reaction mixture was applied to an HPLC equipped with a multi-mode size exclusion column of narrow bore size (GS320A-2E). The column was eluted with 10 mmol L−1ammonium acetate, pH 6.5, at a flow rate of 40 μL min−1, and the eluate was introduced into the ionization spray of ESI-MS/MS without splitting. The introduction into ESI-MS was time-separated for Se elution using a four-way valve, i.e., the eluate was introduced from 24.9 to 26.7 min (A, WGE1), from 31.7 to 33.0 min (B, WGE2), and from 49.0 to 51.4 min (C, WGE3) min. Part of the spectrum containing the characteristic isotope pattern of Se is enlarged in the inset. | ||
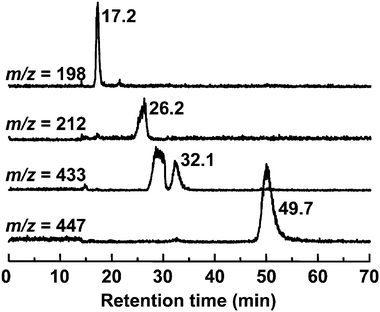 | ||
| Fig. 4 Selected ion monitoring chromatograms of the reaction mixture of wheat germ extract for the in vitrometabolism of SeMet. A 2.0 μL aliquot of the reaction mixture was applied to an HPLC equipped with a multi-mode size exclusion column of narrow bore size (GS320A-2E). The column was eluted with 10 mmol L−1ammonium acetate, pH 6.5, at a flow rate of 40 μL min−1, and the eluate was introduced into the ionization spray of ESI-MS/MS without splitting. Se-containing ions shown in Fig. 3 were monitored at m/z 198 (SeMet), 212 (WGE1), 433 (WGE2), and 447 (WGE3). | ||
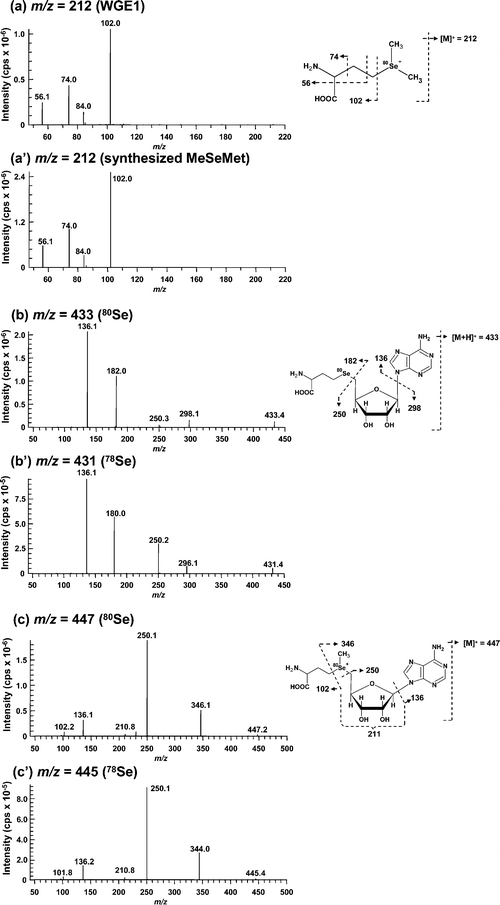 | ||
| Fig. 5 Collision-induced dissociation mass spectra (ESI-MS/MS) of Se-containing positive molecular ions in unknown Se compounds. The dissociation of each Se-containing molecular ion of WGE1 (a), synthesized MeSeMet (a′), WGE2 containing 80Se (b), WGE2 containing 78Se (b′), WGE3 containing 80Se (c) and WGE3 containing 78Se (c′) at m/z 212 (a and a′), 433 (b), 431 (b′), 447 (c) and 445 (c′) was induced with 25 eV collision energy, and fragment ions were detected with the second mass spectrometer. The assignments of the fragments obtained are shown on the right of each MS/MS spectrum. | ||
In the eluate introduced from 31.7 to 33.0 min containing WGE2, the Se isotope pattern was detected around m/z 433 (Fig. 3b). The chromatographic behavior of the molecular ion at m/z 433 on GS320A-2E was similar to that of Se in WGE2 despite the fact that the broad peak was eluted more rapidly than that of WGE2 (Fig. 4). However, as the broad peak did not contain Se detected by ICP-MS on GS-320HQ (Fig. 2), this peak originated from sample matrices. The Se-containing compound with m/z 433 as [M + H]+ was found in selenized yeast and its structure was suggested to be Se-adenosylselenohomocysteine (AdoSeHcy).20,36 As shown in Fig. 5b and 5b′, fragment ions commonly generated from m/z 433 of 80Se and 431 of 78Se, i.e., m/z 136 and 250 were suggested to be the moiety not containing Se in AdoSeHcy. On the other hand, fragment ions having 2 atomic mass unit differences between Fig. 5b and b′, i.e., m/z 182 and 180, and m/z 298 and 296 were suggested to be the moiety containing Se in AdoSeHcy. Thus, all fragment ions originated from m/z 433, i.e., m/z 136, 182, 250, and 298, were assigned to the fragments of AdoSeHcy depicted in Fig. 5b. In addition, the absorption at 254 nm is not contradictory to the fact that WGE2 may have a purine base moiety. The chromatographic behavior of WGE2 on GS-320HQ was similar to that of commercially available AdoHcy, a sulfur analog of AdoSeHcy (data not shown).
The Se isotope pattern was observed around m/z 447 in the eluate introduced from 49.0 to 51.4 min containing WGE3 (Fig. 3c). The molecular ion at m/z 447 was actually detected at a retention time of 49.7 min on GS320A-2E (Fig. 4). Fragment ions commonly generated from m/z 447 of 80Se and 445 of 78Se, i.e., m/z 102, 136, 210 and 250 were suggested to be the moiety not containing Se in AdoSeMet (Fig. 5c and c′). On the other hand, fragment ions having 2 atomic mass unit differences between Fig. 5c and c′, i.e., m/z 346 and 344 were suggested to be the moiety containing Se in AdoSeMet (Fig. 5c and c′). Thus, all fragment ions originated from m/z 447, m/z 102, 136, 211, 250, and 346, could be assigned and suggest that the precursor ion at m/z 447 is Se-adenosylselenomethionine (AdoSeMet) detected as [M]+ (Fig. 5c right). As these fragments of AdoSeMet were quite similar to those of AdoMet, we speculated that WGE3 is AdoSeMet.37,38 The slower elution of WGE3 than WGE2 on GS-320 columns supports our speculation that WGE3 is AdoSeMet because AdoSeMet, a selenonium ion, is more strongly retained than WGE2, a neutral compound, on GS-320 columns that show cation-exchange properties. The chromatographic behavior of WGE3 on GS-320HQ was similar to that of commercially available AdoMet, a sulfur analog of AdoSeMet (data not shown). Wróbel et al. reported that AdoSeMet was rapidly demethylated to AdoSeHcy in yeast extract.36 This is consistent with our observation that WGE3 was unstable and changed to WGE2 in WGE. In addition, WGE3 also showed an absorption at 254 nm, further evidence that WGE3 is AdoSeMet (Fig. 2). The presence of AdoSeMet in selenized yeast has been suggested by the indirect method, i.e., matching of retention times on HPLC-ICP-MS between unknown Se-containing compounds in selenized yeast extract and biologically synthesized AdoSeHcy and AdoSeMet in E. coli.36 Although the identification of unknown Se-containing compounds by HPLC-ICP-MS is a well established method, other mass spectrometry techniques that directly afford molecular information are more reliable for the identification of novel Se-containing compounds.35,39–41 There are no studies of the identification of AdoSeMet in biological samples by direct methods. This study demonstrates for the first time that AdoSeMet is a metabolite of SeMet in a plant sample, through measurements with the direct method, HPLC-ESI-MS/MS.
In vitro metabolism of SeMet in RRL
RRL is another cytosolic source for in vitromRNA translation.42 Contrary to WGE that is used as a plant system, RRL is used as a mammalian system. Thus, we evaluated whether SeMet was also metabolized in this mammalian system. Although more than 95% of SeMet was unchanged in the reaction mixture of RRL containing no mRNA, three metabolites of SeMet were eluted at retention times of 15.8, 19.8, and 57.0 min (Fig. 6a). An Se peak appearing at 29.7 min was a contaminant of SeMet we used because the same peak was also detected in the SeMet solution (data not shown). The Se peak appearing at the retention time of 57.0 min was similar to that of WGE3, AdoSeMet (59.0 min on GS-320HQ). However, as only 1.5% of SeMet was transformed to this metabolite, the amount of this selenometabolite was too small for use in ESI-MS/MS. Hence, we used AEPA, a potent inhibitor of AdoMet synthetase, to inhibit the transformation of SeMet to AdoSeMet.43 As expected, the in vitrometabolism of SeMet to AdoSeMet was completely inhibited by AEPA (Fig. 6b). This suggests that SeMet is also transformed to AdoSeMetin vitro in the mammalian system. In addition, although the identities of two peaks with short retention times are still unknown because their amounts were lower than the detection level of ESI-MS/MS, the latter peak appearing at the retention time of 19.8 min showed a decrease in intensity with AEPA treatment, suggesting that the selenocompound corresponding to this peak might be an AdoSeMet derivative specifically formed in the mammalian system. In contrast, the former peak appearing at the retention time of 15.8 min did not show any change in intensity with AEPA treatment and thus, this peak might correspond to a unique SeMetmetabolite or SeMet-binding factor(s).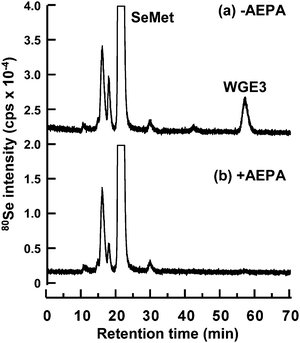 | ||
| Fig. 6 Elution profile of Se in the reaction mixture of rabbit reticulocyte lysate for the in vitrometabolism of SeMet. The reaction mixture was incubated with (b) or without (a) AEPA, a potent inhibitor of AdoMet synthetase. A 20 μL aliquot of the reaction mixture was subjected to HPLC (GS-320HQ) coupled with ICP-MS to measure Se distribution at m/z 80. The column was eluted with 10 mmol L−1ammonium acetate, pH 6.5, at a flow rate of 0.6 mL min−1. | ||
In this study, we identified three SeMet metabolites in WGE, i.e., MeSeMet, AdoSeHcy, and AdoSeMet, and one of them, i.e., AdoSeMet, was also detected in RRL. MeSeMet was previously identified in the leaves of B. juncea.44 It was reported that MeSeMet is a precursor of a volatile Se compound, dimethylselenide (DMSe), and the formation of DMSe results in the decrease in Se content in the plant.45 From these results, we suggest that the formation of MeSeMet is the mechanism underlying Se detoxification. In addition, the thioanalog of MeSeMet, MeMet, is formed by methyl group exchange between Met and AdoMet mediated by methionine S-methyltransferase (MMT). MeMet acts as a methyl group donor to homocysteine.46 Thus, MeSeMet is also expected to act as a methyl group donor (Fig. 7).
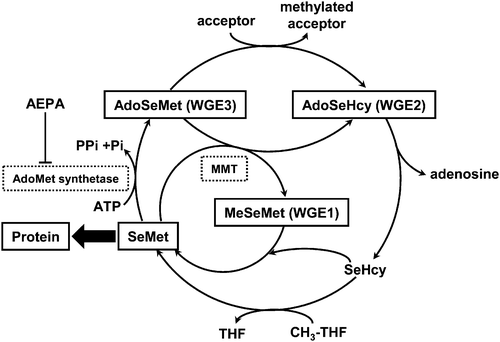 | ||
| Fig. 7 Proposed extended pathway for SeMetmetabolism in wheat germ extract. SeMet and methionine may share a common metabolic pathway, as suggested by Kocsis et al.49AdoSeHcy, Se-adenosylselenohomocysteine; AdoSeMet, Se-adenosylselenomethionine; AEPA, (2S)-2-amino-4,5-epoxypentanoic acid; MeSeMet, selenomethionine methylselenonium; MMT, methionine S-methyltransferase; SeHcy, selenohomocysteine; SeMet, selenomethionine; THF, tetrahydrofolate. | ||
AdoMet also acts as a methyl group donor to participate in the methylation of biomolecules, such as DNA, proteins, and metabolites. It was reported recently that mutated yeast lacking AdoMet synthetase showed tolerance to SeMet.47 In other words, AdoMet synthetase may induce SeMet toxicity by transforming SeMet to more toxic metabolite(s) of SeMet. In addition, AdoSeMet has higher reactivity as a methyl group donor than AdoMet.48 Taken together, AdoSeMet is suggested to disrupt the physiological methylation mediated by AdoMet. Thus, the formation of AdoSeMet may be the mechanism underlying SeMet toxicity.
Conclusions
As depicted in Fig. 7, we indicated that SeMet and Met share a common metabolic pathway, as suggested by Kocsis et al.49 It is known that SeMet and Met share a common pathway for protein synthesis. Furthermore, SeMet is suggested to participate in other metabolic pathways, such as transformation to MeSeMet and AdoSeMet, in the plant system by methods based on molecular mass spectrometry.Acknowledgements
We would like to acknowledge Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (Nos. 19390033 to Y. O. and 16209004 to K. T. S.), and financial support from the Agilent Technologies Foundation, USA. We also wish to thank ZOEGENE Co. for the in vitro translation kit and control mRNA, and Showa Denko for providing the narrow bore column and the HPLC system.References
- M. P. Rayman, Lancet, 2000, 356, 233–241 CrossRef CAS.
- M. Birringer, S. Pilawa and L. Flohe, Nat. Prod. Rep., 2002, 19, 693–718 RSC.
- Z. Q. Lin and N. Terry, Environ. Sci. Technol., 2003, 37, 606–615 CrossRef CAS.
- D. L. LeDuc, M. AbdelSamie, M. Montes-Bayón, C. P. Wu, S. J. Reisinger and N. Terry, Environ. Pollut., 2006, 144, 70–76 CrossRef CAS.
- G. Banuelos, N. Terry, D. L. LeDuc, E. A. Pilon-Smits and B. Mackey, Environ. Sci. Technol., 2005, 39, 1771–1777 CrossRef CAS.
- D. L. LeDuc, A. S. Tarun, M. Montes-Bayón, J. Meija, M. F. Malit, C. P. Wu, M. AbdelSamie, C. Y. Chiang, A. Tagmount, M. deSouza, B. Neuhierl, A. Bock, J. Caruso and N. Terry, Plant Physiol., 2004, 135, 377–383 CrossRef CAS.
- A. M. Nomura, J. Lee, G. N. Stemmermann and G. F. J. Combs, Cancer Epidemiol. Biomarkers Prev., 2000, 9, 883–887 CAS.
- K. E. Burke, J. Clive, G. F. J. Combs and R. M. Nakamura, J. Am. Acad. Dermatol., 2003, 49, 458–472 CrossRef.
- C. Ip, M. Birringer, E. Block, M. Kotrebai, J. F. Tyson, P. C. Uden and D. J. Lisk, J. Agric. Food Chem., 2000, 48, 2062–2070 CrossRef CAS.
- R. Sinha, S. C. Kiley, J. X. Lu, H. J. Thompson, R. Moraes, S. Jaken and D. Medina, Cancer Lett., 1999, 146, 135–145 CrossRef CAS.
- U. Jung, X. Zheng, S. O. Yoon and A. S. Chung, Free Radic. Biol. Med., 2001, 31, 479–489 CrossRef CAS.
- Y. Ogra, K. Ishiwata, Y. Iwashita and K. T. Suzuki, J. Chromatogr., A, 2005, 1093, 118–125 CrossRef CAS.
- M. Dernovics, P. Giusti and R. Łobinski, J. Anal. At. Spectrom., 2007, 22, 41–50 RSC.
- Y. Ogra, T. Kitaguchi, K. Ishiwata, N. Suzuki, Y. Iwashita and K. T. Suzuki, J. Anal. At. Spectrom., 2007, 22, 1390–1396 RSC.
- Y. Ogra, K. Ishiwata, J. Ruiz-Encinar, R. Łobinski and K. T. Suzuki, Anal. Bioanal. Chem., 2004, 379, 861–866 CrossRef CAS.
- V. Díaz Huerta, M. L. Fernández Sánchez and A. Sanz-Medel, Anal. Bioanal. Chem., 2006, 384, 902–907 CrossRef.
- V. Gergely, K. M. Kubachka, S. Mounicou, P. Fodor and J. A. Caruso, J. Chromatogr., A, 2006, 1101, 94–102 CrossRef CAS.
- T. Kitaguchi, Y. Ogra, Y. Iwashita and K. T. Suzuki, Eur. Food Res. Technol., 2008, 277, 1455–1460 CrossRef.
- J. Ruiz-Encinar, L. Ouerdane, W. Buchmann, J. Tortajada, R. Łobinski and J. Szpunar, Anal. Chem., 2003, 75, 3765–3774 CrossRef.
- S. McSheehy, J. Szpunar, V. Haldys and J. Tortajada, J. Anal. At. Spectrom., 2002, 17, 507–514 RSC.
- G. N. Schrauzer, J. Nutr., 2000, 130, 1653–1656 CAS.
- G. N. Schrauzer, Adv. Food Nutr. Res., 2003, 47, 73–112 Search PubMed.
- Y. Ogra, T. Kitaguchi, N. Suzuki and K. T. Suzuki, Anal. Bioanal. Chem., 2008, 390, 45–51 CrossRef CAS.
- P. D. Whanger, J. Am. Coll. Nutr., 2002, 21, 223–232 Search PubMed.
- Z. Pedrero, Y. Madrid and C. Cámara, J. Agric. Food Chem., 2006, 54, 2412–2417 CrossRef CAS.
- O. Cankur, S. K. V. Yathavakilla and J. A. Caruso, Talanta, 2006, 70, 784–790 CrossRef CAS.
- Y. Ogra, K. Ishiwata, H. Takayama, N. Aimi and K. T. Suzuki, J. Chromatogr., B: Analyt. Technol. Biomed. Life Sci., 2002, 767, 301–312 CrossRef CAS.
- L. Bendahl and B. Gammelgaard, J. Anal. At. Spectrom., 2004, 19, 950–957 RSC.
- H. Goenaga Infante, G. OConnor, M. Rayman, R. Hearn and K. Cook, J. Anal. At. Spectrom., 2006, 21, 1256–1263 RSC.
- M. Dernovics and R. Łobinski, Anal. Chem., 2008, 80, 3975–3984 CrossRef CAS.
- C. Gabel-Jensen, K. Lunøe, K. Grimstrup Madsen, J. Bendix, C. Cornett, S. Stürup, H. Rüsz Hansen and B. Gammelgaard, J. Anal. At. Spectrom., 2008, 23, 727–732 RSC.
- D. Guillerm, K. Lavrador and G. Guillerm, Synth. Commun., 1995, 25, 877–882 CAS.
- Y. Ogra, K. Ishiwata and K. T. Suzuki, Anal. Chim. Acta, 2005, 554, 123–129 CrossRef CAS.
- S. V. Ketavarapu Yathavakilla, M. Shah, S. Mounicou and J. A. Caruso, J. Chromatogr., A, 2005, 1100, 153–159 CrossRef CAS.
- T. Lindemann and H. Hintelmann, Anal. Bioanal. Chem., 2002, 372, 486–490 CrossRef CAS.
- K. Wróbel, K. Wróbel and J. A. Caruso, J. Anal. At. Spectrom., 2002, 17, 1048–1054 RSC.
- H. Gellekink, D. van Oppenraaij-Emmerzaal, A. van Rooij, E. A. Struys, M. den Heijer and H. J. Blom, Clin. Chem., 2005, 51, 1487–1492 CrossRef CAS.
- H. Fu, L. Xu, Q. Lv, J. Z. Wang, H. Z. Xiao and Y. F. Zhao, Rapid. Commun. Mass Spectrom., 2000, 14, 1813–1822 CrossRef CAS.
- M. Montes-Bayón, E. G. Yanes, C. Ponce de León, K. Jayasimhulu, A. Stalcup, J. Shann and J. A. Caruso, Anal. Chem., 2002, 74, 107–113 CrossRef CAS.
- J. Szpunar, Analyst, 2005, 130, 442–465 RSC.
- Y. Ogra, Anal. Bioanal. Chem., 2008, 390, 1685–1689 CrossRef CAS.
- H. R. Pelham and R. J. Jackson, Eur. J. Biochem., 1976, 67, 247–256 CrossRef CAS.
- K. Lavrador, B. Allart, D. Guillerm and G. Guillerm, J. Enzyme Inhib., 1998, 13, 361–367 Search PubMed.
- S. V. K. Yathavakilla, M. Shah, S. Mounicou and J. A. Caruso, J. Chromatogr., A, 2005, 1100, 153–159 CrossRef CAS.
- A. Tagmount, A. Berken and N. Terry, Plant. Physiol., 2002, 130, 847–856 CrossRef CAS.
- F. James, K. D. Nolte and A. D. Hanson, J. Biol. Chem., 1995, 270, 22344–22350 CrossRef CAS.
- M. G. Malkowski, E. Quartley, A. E. Friedman, J. Babulski, Y. Kon, J. Wolfley, M. Said, J. R. Luft, E. M. Phizicky, G. T. DeTitta and E. J. Grayhack, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6678–6683 CrossRef CAS.
- D. F. Iwig and S. J. Booker, Biochemistry, 2004, 43, 13496–13509 CAS.
- M. G. Kocsis, P. Ranocha, D. A. Gage, E. S. Simon, D. Rhodes, G. J. Peel, S. Mellema, K. Saito, M. Awazuhara, C. Li, R. B. Meeley, M. C. Tarczynski, C. Wagner and A. D. Hanson, Plant Physiol., 2003, 131, 1808–1815 CrossRef CAS.
Footnote |
| † This paper is dedicated to our co-author, the late Emeritus Professor Kazuo T. Suzuki, Chiba University, suddenly passed away on July 15, 2008. |
| This journal is © The Royal Society of Chemistry 2009 |