Glucose lowering activity by oral administration of bis(allixinato)oxidovanadium(IV) complex in streptozotocin-induced diabetic mice and gene expression profiling in their skeletal muscles
Makoto
Hiromura
ab,
Yusuke
Adachi
b,
Megumi
Machida
b,
Masakazu
Hattori
c and
Hiromu
Sakurai
*bd
aMetallomics Imaging Research Unit, RIKEN CMIS, 6-7-3, Minatojima-minamimachi, Chuo-ku, Kobe, Hyogo 650-0047, Japan
bDepartment of Analytical and Bioinorganic Chemistry, Kyoto Pharmaceutical University, 5 Nakauchi-cho, Misasagi, Yamashina-ku, Kyoto 607-8414, Japan
cDivision of Diabetes, Clinical Research Institute for Endocrine and Metabolic Diseases, National Hospital Organization, Kyoto Medical Center, 1-1 Mukouhata-cho, Fushimi-ku, Kyoto 612-8555, Japan
dFaculty of Pharmaceutical Sciences, Suzuka University of Medical Science, 3500-3 Minami-Tamagaki-cho, Suzuka, Mie 513-8670, Japan. E-mail: sakuraih@suzuka-u.ac.jp; Fax: +81-59-340-0569
First published on 18th November 2008
Abstract
Vanadyl(IV) complexes are anti-diabetogenic agents. Intra-peritoneal administration of bis(allixinato)oxidovanadium(IV) [VO(alx)2] lowers high blood glucose levels in animal models of type 1 and type 2 diabetes. We have examined whether oral administration of VO(alx)2 restores impaired activation in signaling cascades related to glucose metabolism and insulin action, and alters gene expression in the skeletal muscles of streptozotocin (STZ)-induced diabetic mice (STZ-diabetic mice). We report here that daily oral administration of VO(alx)2 lowered high blood glucose levels in the STZ-diabetic mice. The oral administration of VO(alx)2 enhanced phosphorylation of Akt and glycogen synthase kinase-3β (GSK3β), located downstream of the insulin receptor cascade in the skeletal muscles. We analyzed gene expression in the muscles of the diabetic mice before and after insulin or VO(alx)2 treatment. Treating the diabetic mice with insulin or VO(alx)2 normalized the gene expression levels of 152 down-regulated and 11 up-regulated genes , and especially the up-regulation of Cyp2E1 and FoxO1 in the muscles of the diabetic mice. The insulin-mimetic effects of VO(alx)2 in the STZ-induced diabetic mice may be due to the enhancement of protein phosphorylation leading to the activation or inactivation of the transcriptional machinery. Our findings suggest that the insulin-mimetic effects of VO(alx)2 in diabetes may be due to changes in the protein phosphorylations and their gene expression levels.
Introduction
In the 21st century, the prevalence of metabolic diseases, including diabetes mellitus, is markedly increasing worldwide.1 Diabetes mellitus is classified into two major types: type 1, insulin-dependent diabetes; and type 2, noninsulin-dependent diabetes.2 Type 1 diabetes mellitus is a result of autoimmune destruction of pancreatic β cells leading to a lack of intrinsic insulin secretion, and sufferers consequently require daily insulin injections for survival. Type 2 diabetes, accompanied by obesity, impaired glucose metabolism, and insulin resistance, requires treatment with hypoglycemic or anti-diabetogenic synthetic compounds together with diet control and exercise.3,4 We have developed a novel chemical compound that can enhance the lowering of blood glucose and we have examined the action mechanisms of the compound in diabetic animals.Vanadium, a trace element in animals and humans, has a wide variety of biological and physiological functions.5Vanadyl (VO2+, +4 oxidation state) and vanadate (H2VO4−) complexes have insulin-mimetic, anti-tumorigenic and anti-osteogenic activities.6–9 In particular, vanadyl complexes with several coordinating environments around the vanadyl ion are candidate agents to treat the hyperglycemic state in animals and humans.6,9Vanadyl is known to be less toxic than vanadate.5,6Vanadyl complexes, bis(picolinate)oxidovanadium(IV) [VO(pa)2] and bis(maltolato)oxidovanadium(IV) [VO(ma)2] given by intra-peritoneal injection, show the ability to reduce high blood glucose levels in streptozotocin (STZ)-induced diabetic animals.6,9–11
Vanadium enhances tyrosine phosphorylation of the insulin receptor β-subunit (IRβ) and the insulin receptor substrate (IRS) by inhibiting protein tyrosine phosphatase 1B (PTP1B), which in turn activates the signaling pathways of phosphatidylinositol 3-kinase (PI3K)-Akt (also known as protein kinase B).12 Both Akt and GSK3β are important transmitters of the insulin signaling that regulates glucose metabolism.13 The activation of these signals stimulates glucose uptake, glycogen synthesis and lipogenesis, but inhibits lipolysis and gluconeogenesis.14
Glucose transporter 4 (GLUT4) is mainly expressed in insulin-responsive tissues, such as skeletal muscles and adipose tissues.13 The activation of the translocation of GLUT4 to the cell surface is important for glucose utilization. In the diabetic state, both protein and mRNA levels of GLUT4 are down-regulated.15,16 Oral administration of VO(ma)2 increased GLUT4 expression level in the skeletal muscles of STZ-rats.16
Insulin controls gene transcription by regulating the activation or suppression of transcription factors.17 Among them, the forkhead box transcription factor class O (FoxO) family and the sterol-response-element-binding protein (SREBP) family regulate transcriptional activities in the presence of insulin.17 These transcription factors regulate genes involved in glucose and lipid metabolism. Genes associated with β-oxidation, lipid saturase, and oxidative stress increased expression levels in the skeletal muscles or kidneys of diabetic mice and rats,15,18–22 while GLUT4, hexokinase II and the genes of the mitochondrial electron transport chain in the insulin action pathways were down-regulated.15 Efficacy of the vanadyl complexes on gene expression, however, remains unclear, especially when they are given orally.
Bis(allixinato)oxidovanadium(IV) [VO(alx)2] (Fig. 1A) is one of the most effective vanadyl complexes for lowering hyperglycemia by intra-peritoneal administration in both the STZ-induced diabetic and the obese type 2 diabetic KKAy mouse models.23,24VO(alx)2 enhanced the level of phospho-protein in the insulin signaling and induced the GLUT4 to the cell surfacein vitro.25 The oral administration of this complex not only improved hyperglycemia, but also normalized hypertension and leptin level in the KKAy mice. Thus, it indicates that VO(alx)2 pharmacologically improves both diabetes and metabolic diseases. In the present study, we have aimed to examine whether the oral administration of VO(alx)2 lowers high blood glucose levels in the STZ-diabetic mice and stimulates the insulin signaling pathway in vivo leading to improvement of diabetes. Furthermore, we have examined gene expression profiles to compare the effects of insulin and VO(alx)2 treatments on the skeletal muscles of the diabetic mice.
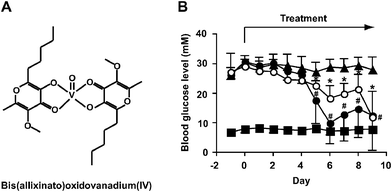 | ||
| Fig. 1 Improvement of hyperglycemia in STZ-diabetic mice following oral administration of VO(alx)2. (a) Structure of VO(alx)2. (b) Changes in blood glucose level in STZ-diabetic mice treated with insulin (1 U kg−1 body weight) by injection or VO(alx)2 (7 mg (137 μmol) V kg−1 body weight) by oral administration for 9 days (n = 4 to 7 mice/group). The symbols indicate the following: closed squares, non-diabetic control mice; closed triangles, STZ-diabetic mice; closed circles, insulin-treated STZ-diabetic mice; open circles, VO(alx)2-treated STZ-diabetic mice. Data are expressed as means ± SD. Significance: *P < 0.01 versus before treatment. | ||
Experimental
Materials and methods
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, νCC), 992 cm−1 (νVO). UV/Vis (H2O): λmax = 277 (ε = 13
O, νCC), 992 cm−1 (νVO). UV/Vis (H2O): λmax = 277 (ε = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 M−1 cm−1), 327 (5300), 819 (27) nm. HRMS (m/z): [M]+ calcd. for C24H34O9V, 517.1643; found, 517.1635. Analysis (calcd, found for C24H34O9V): C (55.71, 55.49%), H (6.62, 6.47%).
600 M−1 cm−1), 327 (5300), 819 (27) nm. HRMS (m/z): [M]+ calcd. for C24H34O9V, 517.1643; found, 517.1635. Analysis (calcd, found for C24H34O9V): C (55.71, 55.49%), H (6.62, 6.47%).
Methods
The animals were subjected to daily monitoring for measurements of physiological data (blood glucose, body weight, food intake, and water consumption) between 10 am and noon before treatment of insulin or VO(alx)2. Blood samples were obtained from the tail vein of mice and subjected to measuring blood glucose levels using the glucose oxidase method (Glucocard, Arkray, Kyoto, Japan). The animals were fasted for 16 h and sacrificed under anesthesia (etherization) for dissecting tissues.
650 different mouse genes including 12 probes per gene ) from GeneFrontier Corp. (Tokyo, Japan). The gene expression level was defined as significant when it significantly increased or decreased for diabetic (STZ) versus non-diabetic, diabetic versusinsulin-treated diabetic, and diabetic versusVO(alx)2-treated diabetic, as determined using the NANDEMO Analysis version 1.0 software (GeneFrontier Corp.), including perfectly matched (PM) and mismatched (MM) control oligonucleotides. The output criteria from each microarray were used to select genes , whose expression levels changed greater than 1.5-fold. A T-test was performed with P values <0.01 being considered significant to identify the genes that were differentially expressed across conditions. Genes were annotated using the GenBank.26 Protein metabolic functions and pathways were identified using the eukaryotic orthologous groups (KOG) database.27,28
Results
Lowering hyperglycemia in STZ-induced diabetic mice by oral administration of VO(alx)2
We examined the effect of oral administration of VO(alx)2 on hyperglycemia in the STZ-diabetic mice. The blood glucose concentrations in the STZ-diabetic mice were significantly higher than those in the non-diabetic control mice before starting the insulin or VO(alx)2 treatment (Fig. 1B, closed squares). The daily oral administration of VO(alx)2 with a single dose (137 μmol V kg−1 body weight) reduced the high blood glucose levels in the STZ-diabetic mice (Fig. 1B, open circles). Insulin-treatment as well as VO(alx)2-treatment improved hyperglycemia significantly in the STZ-diabetic mice (Fig. 1B, open circles for VO(alx)2 and closed circles for insulin). The STZ-diabetic mice consumed more food and fluid than the non-diabetic control mice (Table 1). The STZ-diabetic mice with VO(alx)2-treatment decreased the intake of food and fluid significantly in comparison with the untreated STZ-diabetic mice (Table 1), although the VO(alx)2-treated STZ-diabetic mice did not change body weight significantly.| Group | n | Body weight/g | Fluid intake/ml day−1 | Food intake/g day−1 | |||
|---|---|---|---|---|---|---|---|
| Before treatment | After treatment | Before treatment | After treatment | Before treatment | After treatment | ||
| 1 Data are expressed as means ± SD. Significance: *P<0.01 versus before treatment. | |||||||
| Non-diabetic control | 7 | 43.5 ± 1.8 | 42.2 ± 1.6 | 6.8 ± 2.2 | 5.3 ± 0.8 | 5.0 ± 0.6 | 5.0 ± 0.5 |
| STZ-mice non treatment | 7 | 39.2 ± 1.1 | 38.7 ± 1.6 | 37.9 ± 7.6 | 42.6 ± 7.4 | 8.6 ± 0.7 | 12.2 ± 5.1 |
| STZ-mice + insulin | 7 | 38.7 ± 2.2 | 40.8 ± 1.9 | 38.2 ± 10.8 | 7.4 ± 2.2* | 9.1 ± 1.4 | 5.5 ± 1.1* |
| STZ-mice + VO(alx)2 | 7 | 37.9 ± 1.3 | 35.5 ± 2.4 | 40.0 ± 7.8 | 14.7 ± 13.8* | 9.1 ± 1.3 | 4.8 ± 2.6* |
Effect of VO(alx)2 on signal transduction states and the level of GLUT4 protein in skeletal muscle
After nine days of treatment with VO(alx)2, the biceps femoris muscles of the fasted mice were isolated and used for biochemical and gene expression experiments. We analyzed the phosphorylation levels of the major signal transductionproteins in the skeletal muscles. Results from the analysis of the phosphorylation of Akt, glycogen synthase kinase-3β (GSK3β) and AMP-activated protein kinase (AMPK) in the cytosolic fractions are shown in Fig. 2. The phosphorylations of Akt and GSK3β were significantly decreased in the STZ-diabetic mice without the insulin or VO(alx)2 treatment (Fig. 2A and B). The oral administration of VO(alx)2 or insulin injections enhanced the phosphorylation of these proteins (Fig. 2A and B). The oral administration of VO(alx)2 enhanced the phosphorylation of AMPK in comparison with that in the untreated STZ-diabetic mice (Fig. 2C), indicating action of VO(alx)2 not only on the insulinsignaling cascade but also on AMPK signaling in the muscle cells.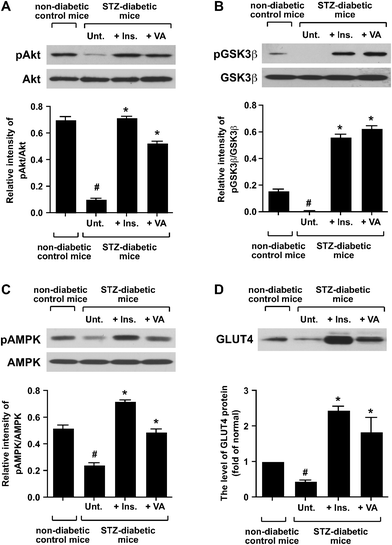 | ||
| Fig. 2 Effect of VO(alx)2 on the phosphorylation of Akt (a), GSK3β (b) and AMPK (c), and the level of GLUT4 protein (d) in skeletal muscle. Twenty μg of the cytosolic proteins was resolved on a 9% SDS-PAGE gel , transferred to a nitrocellulose membrane, and immunoblotted with anti-phospho-Ser473 Akt, anti-Akt, anti-phospho-Ser9 GSK3β, anti-GSK3β, anti-phospho-Thr172 AMPKα, and anti-AMPKα antibodies. Five μg of the crude muscle cell membranes was resolved on a 12% SDS-PAGE gel , transferred to a nitrocellulose membrane, and immunoblotted with the anti-GLUT4 antibody . The intensity of immunoblots, which indicates the phosphorylation state, was measured using NIH ImageJ software. Data are expressed as means ± SD. Significance: #P < 0.01 compared with non-diabetic control mice and STZ-diabetic mice. *P < 0.01 compared with STZ-diabetic mice and insulin-, or VO(alx)2-treated STZ-diabetic mice. | ||
The expression level of GLUT4 protein was examined in the skeletal muscles of the STZ-diabetic mice. The expression levels of GLUT4 protein in the insulin- and VO(alx)2-treated diabetic mice were approximately 3 to 5-fold higher than that in the untreated STZ-diabetic mice (Fig. 2D). The results indicate that the oral administration of VO(alx)2 as well as insulin injection recovered the insulin signaling pathway, leading to the enhancement of GLUT4 protein expression in the skeletal muscles.
Changes in gene expression levels of skeletal muscle in diabetic mice and insulin- and VO(alx)2-treated diabetic mice
We compared the gene expression levels in the skeletal muscles between the STZ-diabetic mice and the non-diabetic ddY control mice. The output criterion of the differential gene expression was selected to be a 1.5-fold change in the expression levels of up- or down-regulated genes when the significance value was set at P < 0.01 from 32650 mouse genes . The expression levels of 569 genes were found to be significantly different between STZ-diabetic mice and non-diabetic mice. The expression levels of these genes were further compared with the gene expression levels in the insulin-treated and VO(alx)2-treated mice. Finally, 163 genes of differential gene expression were selected. Among them, 152 were down-regulated, and 11 were up-regulated by treatment with insulin or VO(alx)2 in the STZ-diabetic mice as compared with the expression levels without treatment. Functions of the proteins encoded by the 163 genes were identified using the KOG database (P < 0.0001).27,28 From this identification, 19 genes were associated with metabolism, 61 genes associated with cellular processes and signaling, 19 genes associated with information storage and processing, and 46 genes were uncharacterized. The representative genes are listed in Table 2.
| Accession no. | Gene Symbol | Gene description | STZ:ND | STZ:Ins | STZ:VA |
|---|---|---|---|---|---|
| Upregulated by diabetes | Up (fold) | Down (fold) | Down (fold) | ||
| 1 The first column shows the GenBank accession numbers for the genes . The second column shows the Gene Symbol for the genes . The third column shows the names of the gene products. The fourth, fifth and sixth columns shows the fold changes in the gene expression level of STZ-diabetic mice versus non-diabetic control mice (STZ:ND), STZ-diabetic mice versusinsulin-treated STZ-diabetic mice (STZ:Ins) and STZ-diabetic mice versusVO(alx)2-treated STZ-diabetic mice (STZ:VA), respectively. | |||||
| Metabolism | |||||
| Carbohydrate transport and metabolism | |||||
| BC012720 | Fbp2 | Fructose bisphosphatase 2 | 2.0 | −1.5 | −2.5 |
| NM_173021 | Phka1 | Phosphorylase kinase alpha 1 | 1.9 | −1.7 | −2.9 |
| AK047095 | Ogt | O-linked N-acetylglucosamine (GlcNAc) transferase | 1.7 | −1.5 | −2.9 |
| Lipid transport and metabolism | |||||
| AK012088 | Acsl3 | Acyl-CoA synthetase long-chain family member 3 | 1.7 | −2.8 | −4.3 |
| NM_080555 | Ppap2b | Phosphatidic acid phosphatase type 2B | 1.6 | −2.0 | −1.8 |
| BC002082 | Fabp3 | Fatty acid binding protein 3 | 1.5 | −2.2 | −3.7 |
| Nucleotide transport and metabolism | |||||
| NM_025647 | Cmpk1 | Cytidylate kinase | 1.7 | −2.0 | −2.3 |
| NM_199446 | Phkb | Phosphorylase kinase beta | 1.6 | −1.9 | −5.3 |
| Energy production and conversion | |||||
| AK075673 | Etfdh | Electron transferring flavoprotein, dehydrogenase | 1.5 | −1.7 | −1.9 |
| Secondary metabolites biosynthesis, transport and catabolism | |||||
| NM_021282 | Cyp2e1 | Cytochrome P450, family 2, subfamily e, polypeptide 1 | 4.0 | −4.7 | −3.5 |
| Inorganic ion transport and metabolism | |||||
| NM_022885 | Slc30a5 | Solute carrier family 3, member 5 | 1.5 | −1.6 | −1.7 |
| BC027262 | Mt1 | Metallothionein 1 | 2.8 | −5.6 | −6.4 |
| AK075853 | Cat | Catalase | 1.8 | −1.5 | −2.8 |
| CELLULAR PROCESSES AND SIGNALING | |||||
| Cytoskeleton | |||||
| NM_033268 | Actn2 | Actinin alpha 2 | 2.1 | −1.5 | −2.9 |
| AF307855 | Actr3 | ARP3 actin-related protein 3 homolog | 1.8 | −1.7 | −2.5 |
| BC052186 | Finc | Filamin C, gamma (actin binding protein 280) | 1.7 | −1.8 | −3.0 |
| NM_153399 | Syne1 | Synaptic nuclear envelope 1 | 6.9 | −1.8 | −10.8 |
| NM_009448 | Tuba1c | Tubulin, alpha 6 | 2.7 | −2.7 | −2.1 |
| CELLULAR PROCESSES AND SIGNALING | |||||
| Post-translational modification, protein turnover, chaperones | |||||
| NM_022310 | Hspa5 | Heat shock 70 kD protein 5 (glucose-regulated protein) | 1.6 | −3.0 | −3.2 |
| NM_011631 | Hsp90b1 | Heat shock protein 90 kDa beta (Grp94), member 1 | 1.5 | −1.9 | −2.3 |
| BC037643 | Txnrd1 | Thioredoxin reductase 1 | 1.5 | −1.7 | −2.3 |
| Signal transduction mechanisms | |||||
| NM_207655 | Egfr | Epidermal growth factor receptor | 1.8 | −2.0 | −2.0 |
| AK044776 | Itpr1 | Inositol 1,4,5-triphosphate receptor 1 | 1.6 | −2.8 | −2.2 |
| NM_011058 | Pdgfra | Platelet derived growth factor receptor, alpha polypeptide | 1.6 | −1.7 | −2.0 |
| BC006708 | Mapk1 | Mitogen activated protein kinase 1 | 1.5 | −1.8 | −3.9 |
| INFORMATION STORAGE AND PROCESSING | |||||
| Transcription | |||||
| BC037688 | Stat3 | Signal transducer and activator of transcription 3 | 1.6 | −1.6 | −1.9 |
| NM_019739 | FoxO1 | Forkhead box O1 | 1.7 | −3.1 | −3.2 |
| Translation, ribosomal structure and biogenesis | |||||
| NM_013557 | Elf2ak1 | Eukaryotic translation initiation factor 2 alpha kinase 1 | 1.5 | −1.9 | −2.5 |
| NM_144958 | Elf4a1 | Eukaryotic translation initiation factor 4A1 | 1.6 | −2.0 | −1.9 |
| Accession No. | Gene Symbol | Gene description | STZ:ND | STZ:Ins | STZ:VA |
|---|---|---|---|---|---|
| Downregulated by diabetes | Down (fold) | Up (fold) | Up (fold) | ||
| Metabolism | |||||
| Lipid transport and metabolism | |||||
| NM_009127 | Scd1 | Stearoyl-coenzyme A desaturase 1 | −3.3 | 4.4 | 3.3 |
| X13135 | Fasn | Fatty acid synthase | −2.3 | 2.7 | 1.7 |
| Energy production and conversion | |||||
| BC005533 | Acly | ATP citrate lyase | −2.2 | 2.1 | 1.6 |
In lipid metabolism, the gene expression for the fatty acid bindingprotein 3 (Fabp3) and acyl-CoA synthetase long-chain family member 3 (Acsl3) were increased 1.7- and 1.5-fold in the diabetic mice in comparison with those of non-diabetic control mice, respectively. The genes were down-regulated by both insulin (2.2-fold for Fabp3 and 2.8-fold for Acsl3) and VO(alx)2 (3.7-fold for Fabp3 and 4.3-fold for Acsl3). On the other hand, mRNA of stearoyl-coenzyme A desaturase 1 (Scd1), which increased in diabetic mice, was up-regulated by both insulin (4.4-fold) and VO(alx)2 (3.3-fold) administration.
VO(alx)2 affected the alteration in the gene expression of carbohydrate transport and metabolism. The genes of fructose bisphosphatase 2 (Fbp2), phosphrorylase kinase alpha 1 (Phak1), and O-linked N-acetylglucosaminetransferase were up-regulated by 2.0-, 1.9-, and 1.7-fold in the diabetic mice, respectively. These genes were also down-regulated by insulin- and VO(alx)2 treatment in the diabetic mice.
We also detected changes in gene expression for the endoplasmic reticulum (ER)-Golgi proteins. The levels of stress-responsive heat shock proteins, such as Grp94 and Grp74, were up-regulated by 1.6-fold in the STZ-diabetic mice, and down-regulated by 3.0 and 3.2-fold in the insulin- and VO(alx)2-treated mice, respectively (Table 2). The expression of synaptic nuclear envelope 1 (Syne-1) was up-regulated by 6.9-fold in the STZ-diabetic mice, and down-regulated by 1.8 and 10.8-fold in the insulin- and VO(alx)2-treated mice, respectively (Table 2).
In these categories, the treatment of diabetes with VO(alx)2 restored not only alterations in the metabolism of carbohydrate and lipid but also signal transduction, and the levels of transcription factors, stress proteins, and others by affecting gene expression levels. Insulin treatment also restored alteration of expression levels of the same genes .
Recovery of GLUT4, Cyp2E1, and FoxO1 mRNA expression levels by oral administration of VO(alx)2 in the skeletal muscles of the diabetic mice
We found that the level of GLUT4 protein was recovered by VO(alx)2-treatment in STZ-diabetic mice (Fig. 2D). Hence, we examined whether the level of GLUT4 mRNA was also improved by treatment with VO(alx)2. The level of GLUT4 mRNA was significantly decreased, by approximately 2.6-fold, in the STZ-diabetic mice, and increased by insulin (5.7-fold) and VO(alx)2 treatment (2.5-fold) in STZ-diabetic mice (Fig. 3A). The up-regulation of GLUT4 mRNA by VO(ax)2 showed a similar effect to insulin on the activation of insulin signal pathways.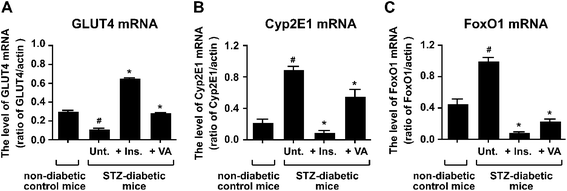 | ||
| Fig. 3 RT-PCR analysis of GLUT4 (a), Cyp2E1 (b) and FoxO1 (c). Reverse-transcription reactions were carried out as described in “Materials and Methods”. First-strand cDNA from the skeletal muscle was amplified by PCR using specific primer sets, and the PCR products were then separated by electrophoresis on a 2% agarosegel containing ethidium bromide. The mRNA expression levels of GLUT4, Cyp2E1, and FoxO1 were measured using the NIH ImageJ software. Data are expressed as means ± SD. Significance: #P < 0.01 compared with non-diabetic control mice and STZ-diabetic mice. *P < 0.01 compared with STZ-diabetic mice and insulin-, or VO(alx)2-treated STZ-diabteic mice. | ||
In the DNA microarray experiment, the STZ-diabetic mice showed a 4-fold up-regulation in the altered-Cyp2E1 gene expression in comparison with the non-diabetic control mice, while the insulin- and VO(alx)2-treated diabetic mice showed a 4.7- and 3.5-fold down-regulation, respectively (Table 2). In the RT-PCR analysis, the STZ-diabetic mice showed approximately 4-fold up-regulation of Cyp2E1 mRNA expression in comparison with the non-diabetic control mice, that was suppressed by the treatment of insulin (9.4-fold) or VO(alx)2 (1.6-fold) (Fig. 3B).
In addition, our DNA microarray data showed that the expression level of FoxO1 was also up-regulated by 1.7-fold in the untreated diabetic mice, and down-regulated by 3.1- and 3.2-fold in the insulin- and VO(alx)2-treated mice, respectively (Table 2). We confirmed the expression level of this gene using RT-PCR (Fig. 3C). The expression level of the FoxO1 gene was increased approximately 2.2-fold in the diabetic mice. In contrast, treating diabetes with insulin and VO(alx)2 decreased the expression level of this gene by approximately 11.2- and 4.3-fold, respectively.
Discussion
In this study, we have examined the effect of VO(alx)2 treatment in STZ-induced diabetic mice, was followed by analysis of phosphorylation of Akt, GSK3β and AMPK and the gene expression profiles in the skeletal muscles. VO(alx)2 was effective in lowering high blood glucose levels in the STZ-diabetic mice not only by intra-peritoneal injection,23 but also by oral administration. VO(alx)2 also lowers blood glucose levels in type 2 diabetic KKAy mice.24 Together with the present observation, VO(alx)2 may be a candidate drug for treating both type 1 and type 2 diabetes by oral administration despite their etiological differences.The chemical structure of VO(alx)2 is similar to that of VO(ma)2. However, the lipophilicity of VO(alx)2 is two-fold higher than that of VO(ma)2 (log P = 1.02 ± 0.06 for VO(alx)2 and 0.46 ± 0.11 for VO(ma)2).23 This different chemical character supports the fact that VO(alx)2 is incorporated and accumulated in cells or tissues more than VO(ma)2 at low concentrations.23,25 Hence, VO(alx)2 is concluded to be more potent than other vanadyl complexes already reported as anti-diabetogenic compounds.
In our study, VO(alx)2 enhanced the phosphorylation of AMPK in the skeletal muscles of STZ-induced diabetic mice. AMPK is phosphorylated and activated by an up-stream protein kinase, LKB1, which is activated by adiponectin, leptin or intracellularAMP/ATP ratio, leading to regulation of energy balance in the body.29,30 The function of this kinase is to produce energy by stimulating glucose uptake via the activation of GLUT4 translocation and lipid oxidation.29Vanadate induces the phosphorylation of AMPK in the DU145 human prostate carcinoma cell line , leading to the stimulation of the incorporation of glucose into the cells and the induction of the mRNA of hypoxia-inducible factor 1α (HIF1α).31 In our DNA array data, VO(alx)2 did not up-regulate HIF1α in the skeletal muscles of STZ-diabetic mice (data not shown). Our present data suggest that VO(alx)2 stimulates not only the insulinsignaling cascade but also energy-balance signaling in the skeletal muscles. The mechanism of the activation of AMPK by VO(alx)2 still remains to be clarified.
Based on the observations in the present study, we predict that VO(alx)2 affects gene expressions in the skeletal muscles. Stearoyl-CoA desaturase (SCD) is a rate-limiting enzyme catalyzing the synthesis of monounsaturated fatty acids, such as oleate and palmitate.32SCD deficiency results in an increase in energy expenditure and up-regulation of genes associated with fatty acid oxidation.33,34SCD knockout mice also increased β-oxidation and insulin sensitivity in the skeletal muscles.35 In our data, the expression of SCD1 decreased in the skeletal muscles of STZ-diabetic mice, and this reduction was restored by the insulin- and VO(alx)2 treatments in STZ-diabetic mice (Table 2). The expression of SCD1 is regulated by insulin, dietary factors, and environmental factors.35 Therefore, the down-regulation of SCD1 by VO(alx)2 might be associated with the activation of the insulin signaling pathway.
The expression of O-linked N-acetylglucosaminetransferase (OGT) was increased in the skeletal muscles of STZ-diabetic mice, but decreased by insulin- and VO(alx)2-treated STZ-diabetic mice (Table 2). OGT transfers N-acetylglucosamine (GlcNAc) to the serine or threonine residues on the nuclear and cytosolic proteins.36,37 This post-translational modification attenuated the insulin-response in vivo and in vitro.38 OGT modifies the proteins located downstream of the insulin receptor, such as IRS, Akt, glycogen synthase, and FoxO1, leading to suppression of the protein functions that involve the phosphorylation state and enzyme activities.36,39,40 The transcriptional mechanism of OGT is yet unknown, although the suppression of this gene function regulates the response to the signal proteins in the insulin-activation system. Furthermore, vanadate directly inhibits OGT enzymatic activity.41 Hence, VO(alx)2 may not only restore OGT expression by its insulin-mimetic activity, but also reduce the levels of O-GlcNAc-proteins to inhibit this enzyme activity.
VO(pa)2 and vanadate regulate transcription factors, such as NFκB and signal transducer and activator of transcription (STAT).42,43 STAT is involved in the leptin signaling that regulates food intake and energy balance.44 The expression of this gene was up-regulated by 1.6-fold in the STZ-diabetic mice, and down-regulated by 1.6 and 1.9-fold in the insulin- and VO(alx)2-treated mice, respectively (Table 2). Moreover, the expression of the transcription factor FoxO1 was also induced in the diabetic mice, and decreased in the insulin- and VO(alx)2-treated diabetic mice. FoxO1 is up-regulated by fasting, glucocorticoids, and diabetes.45,46 An increase in the FoxO1 gene expression induces muscle atrophy, lipid oxidation, and suppression of glucose oxidation in the skeletal muscles. Therefore, FoxO1 may regulate the metabolism of the skeletal muscles. The expression of FoxO1 is suppressed by insulin.47 Therefore, the down-regulation of FoxO1 by VO(alx)2 seems to contribute similarly to insulin action.
Not only the expression levels of Cyp2E1 mRNA and the protein but also its enzymatic activity increased in STZ-diabetic rat and human diabetic subjects.48,49 Cyp2E1 is known to metabolize endogenous compounds such as fatty acids, lipid hydroperoxides, and ketone bodies into aldehydes and several types of xenobiotics.50 We previously reported that administration of bis(6-methyl-picolinato)oxidovanadium(IV) (VO(6mpa)2) improved the altered enzymatic function and the level of cytochrome P450 2E1 (Cyp2E1) protein in STZ-diabetic rats.48 The gene expression of Cyp2E1 is directly controlled by insulin.51 This effect was reversed by the addition of a PI3Kinhibitor. VO(alx)2 enhanced the levels of phospho-proteins of Akt and GSK3β, and tyrosine-phosphorylation of insulin receptor β-subunit (IRβ) and insulin receptor substrate (IRS).25 Based on our results the restoration of Cyp2E1 and FoxO1 gene expression by VO(alx)2 was proposed, however, the detailed mechanism which relates to enhancement of the insulin signaling pathway is yet to be clarified.
The ER is the place where protein quality is controlled, and the secreted protein transported to the Golgi apparatus. Grp94 and Grp78 are the marker proteins of ER stress.52 Grp94 and Grp78 are increased by ER stress, such as misfolded proteins, hypoxia, and tumors.53,54 In our experiment, the mRNA of the induced Grp94 and Grp78 in the STZ-diabetic mice were suppressed by VO(alx)2. ER stress is associated with type 2 diabetes in the animal models of high fat diet-induced obesity and ob/ob animal models showing an increase in the expression levels of Grp94 and Grp78.54 In these animal models, the expression level of X-box-binding protein-1 (XBP-1) was a major inducer of ER stress in type 2 diabetes. In our present experiment, a significant change was undetectable in the gene expression level of XBP-1. The STZ-induced diabetic animals in the present study may have different action mechanisms of VO(alx)2, which suppresses the expression of Grp94 and Grp78, from the obese type 2 diabetic animals.
Syne-1 is identified as a Golgi- and nuclear envelope-localized protein in skeletal muscle cells.55 Syne-1 has multi-spectrin homology domains, which bind to the Golgi and nuclear envelope. One of the Golgi binding domains on Syne-1 acts as a dominant negative inhibitor for altering the structure of the Golgi complex. This effect impairs retrograde vesicle transport from the Golgi to the ER.56,57 Therefore, Syne-1 regulates vesicle transport from the Golgi to the ER. The up-regulation of Syne-1 in diabetic mice may abolish this retrograde transport and be linked to ER-stress. The amelioration of ER-stress or vesicle transport of ER-Golgi by VO(alx)2 are considered to be new target sites for improving diabetes. Abrogation of ER stress and the restoration of transport of ER-Golgi with VO(alx)2 need to be examined.
Recently, it has been reported that oral administration of vanadyl sulfate (VOSO4) corrected diabetes-altered gene expressions, which relate to the genes of glucose, lipid, and oxidative stress metabolism, in the skeletal muscles of STZ-diabetes rats.58 Treatment of diabetes rats with VOSO4corrected the genes of glucose, lipid metabolism, and oxidative stress metabolism. Diabetes has been shown to increase reactive oxygen species, and develop insulin resistance.59VO(alx)2 also corrected the gene expression levels of oxidative stress metabolism, such as metallothionein and catalase. Increased expression of antioxidantproteins and enzymes, such as catalase and superoxide were also reported in diabetic rats after treatment with vanadate.60 Hence, enhancement of antioxidantprotein and enzyme levels by VO(alx)2 support the reduction of reactive oxygen species, leading to the improvement of insulin resistance in diabetes.
In conclusion, oral administration of VO(alx)2 lowered hyperglycemia in STZ-diabetic mice, and restored the activation of, not only the insulinsignaling cascade but also the AMPK signaling. This is the first study to analyze the altered gene expression profiles of the skeletal muscles in diabetes after treating with an insulin-mimetic and anti-diabetogenic vanadyl complex, VO(alx)2. Although, vanadyl complexes, such as VO(pa)2, improved the levels of blood glucose and glycated hemoglobin, VO(alx)2 exhibited not only a hypoglycemic effect but also the improvement of hyperinsulinemia, hypercholesterolemia, and hypertension. Therefore, we propose that VO(alx)2 is a candidate anti-diabetogenic compound for treating both type 1 and type 2 diabetes. We expect that VO(alx)2 will be the subject of clinical trials in the future.
| IRβ | Insulin receptor β-subunit |
| IRS | Insulin receptor substrate |
| PI3K | Phosphatidylinositol 3-kinase |
| GLUT4 | Glucose transporter 4 |
| FoxO | Forkhead transcription factor class O |
| GSK3β | Glycogen synthase kinase-3β |
| STZ | Streptozotocin |
| VO(alx)2 | Bis(allixinato)oxidovanadium(IV) |
| VO(pa)2 | Bis(picolinato)oxidovanadium(IV) |
| VO(ma)2 | Bis(maltolato)oxidovanadium(IV) |
Acknowledgements
This study was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of the Japanese Government (Grants-in-Aid for Scientific Research (B), Scientific Research on Priority Areas, and Specially Promoted Research) to H. S. Our thanks are due to Dr M. Ogawa and Professor N. Gotoh (Department of Microbiology, Kyoto Pharmaceuticaal University) for their advice and suggestions on DNA micoarray data analysis and Dr S. Enomoto (Metallomics Imaging Research Unit, RIKEN) for his encouragement to prepare the manuscript.References
- S. Wild, G. Roglic, A. Green, R. Sicree and H. King, Diabetes Care, 2004, 27, 1047–1053 CrossRef.
- T. Kuzuya, S. Nakagawa, J. Satoh, Y. Kanazawa, Y. Iwamoto, M. Kobayashi, K. Nanjo, A. Sasaki, Y. Seino, C. Ito, K. Shima, K. Nonaka and T. Kadowaki, Diabetes Res. Clin. Pract., 2002, 55, 65–85 CrossRef.
- K. Alexandraki, C. Piperi, C. Kalofoutis, J. Singh, A. Alaveras and A. Kalofoutis, Ann. N. Y. Acad. Sci., 2006, 1084, 89–117 CrossRef CAS.
- D. M. Muoio and C. B. Newgard, Annu. Rev. Biochem., 2006, 75, 367–401 CrossRef CAS.
- D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, Chem. Rev., 2004, 104, 849–890 CrossRef CAS.
- H. Sakurai, A. Katoh and Y. Yoshikawa, Bull. Chem. Soc. Jpn., 2006, 70, 1645–1664 CrossRef.
- A. B. Daniel, A. M. W. Patricia, M. C. Ana and B. E. Susana, JBIC, JBIC, J. Biol. Inorg. Chem., 2003, 8, 459–468 Search PubMed.
- E. G. Ferrer, M. V. Salinas, M. J. Correa, L. Naso, D. A. Barrio, S. B. Etcheverry, L. Lezama, T. Rojo and P. A. Williams, JBIC, J. Biol. Inorg. Chem., 2006, 11, 791–801 CrossRef CAS.
- T. Storr, H. H. Thompson and C. Orvig, Chem. Soc. Rev., 2006, 35, 534–544 RSC.
- H. Sakurai, Y. Kojima, Y. Yoshikawa, K. Kawabe and H. Yashui, Coord. Chem. Rev., 2002, 226, 187–198 CrossRef CAS.
- L. Marzban, R. Rahimian, R. W. Brownsey and J. H. McNeill, Endocrinology, 2002, 143, 4636–45 CrossRef CAS.
- W. Basuki, M. Hiromura, Y. Adachi, K. Tayama, M. Hattori and H. Sakurai, Biochem. Biophys. Res. Commun., 2006, 349, 1163–1170 CrossRef CAS.
- M. A. Herman and B. B. Kahn, J. Clin. Invest., 2006, 116, 1767–1775 CrossRef CAS.
- A. K. Srivastava and M. Z. Mehdi, Diabetic Med., 2005, 22, 2–13 CrossRef CAS.
- V. K. Yechoor, M. E. Patti, R. Saccone and C. R. Kahn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10587–10592 CrossRef CAS.
- A. Mohammad, V. Sharma and J. H. McNeill, Mol. Cell. Biochem., 2002, 233, 139–143 CrossRef CAS.
- C. Mounier and B. I. Posner, Can. J. Physiol. Pharmacol., 2006, 84, 713–724 CrossRef CAS.
- Y. H. Suh, Y. Kim, J. H. Bang, K. S. Choi, J. W. Lee, W. H. Kim, T. J. Oh, S. An and M. H. Jung, J. Mol. Endocrinol., 2005, 34, 299–315 Search PubMed.
- H. Lan, M. E. Rabaglia, J. P. Stoehr, S. T. Nadler, K. L. Schueler, F. Zou, B. S. Yandell and A. D. Attie, Diabetes, 2003, 52, 688–700 CrossRef CAS.
- R. Sreekumar, P. Halvatsiotis, J. C. Schimke and K. S. Nair, Diabetes, 2002, 51, 1913–1920 CrossRef CAS.
- L. Hansen, M. Gaster, E. J. Oakeley, K. Brusgaard, E. M. Damsgaard Nielsen, H. Beck-Nielsen, O. Pedersen and B. A. Hemmings, Biochem. Biophys. Res. Commun., 2004, 323, 685–695 CrossRef CAS.
- K. H. S. Wilson, S. E. Eckenrode, Q. Z. Li, Q. G. Ruan, P. Yang, J. D. Shi, A. Davoodi-Semiromi, R. A. McIndoe, B. P. Croker and J. X. She, Diabetes, 2003, 52, 2151–2159 CrossRef CAS.
- Y. Adachi, J. Yoshida, Y. Kodera, A. Katoh, J. Takada and H. Sakurai, J. Med. Chem., 2006, 49, 3251–3256 CrossRef CAS.
- Y. Adachi, Y. Yoshikawa, J. Yoshida, Y. Kodera, A. Katoh, J. Takada and H. Sakurai, Biochem. Biophys. Res. Commun., 2006, 345, 945–950 CrossRef CAS.
- M. Hiromura, A. Nakayama, Y. Adachi, M. Doi and H. Sakurai, JBIC, J. Biol. Inorg. Chem., 2007, 12, 1275–1287 CrossRef CAS.
- D. A. Benson, I. Karsch-Mizrachi, D. J. Lipman, J. Ostell and D. L. Wheeler, Nucleic Acids Res., 2007, 35, D21–25 CrossRef CAS.
- E. V. Koonin, N. D. Fedorova, J. D. Jackson, A. R. Jacobs, D. M. Krylov, K. S. Makarova, R. Mazumder, S. L. Mekhedov, A. N. Nikolskaya, B. S. Rao, I. B. Rogozin, S. Smirnov, A. V. Sorokin, A. V. Sverdlov, S. Vasudevan, Y. I. Wolf, J. J. Yin and D. A. Natale, Genome Biol., 2004, 5, R7 CrossRef.
- R. L. Tatusov, N. D. Fedorova, J. D. Jackson, A. R. Jacobs, B. Kiryutin, E. V. Koonin, D. M. Krylov, R. Mazumder, S. L. Mekhedov, A. N. Nikolskaya, B. S. Rao, S. Smirnov, A. V. Sverdlov, S. Vasudevan, Y. I. Wolf, J. J. Yin and D. A. Natale, BMC Bioinf., 2003, 4, 41 CrossRef.
- Y. C. Long and J. R. Zierath, J. Clin. Invest., 2006, 116, 1776–1783 CrossRef CAS.
- G. Fruhbeck, Biochem. J., 2006, 393, 7–20 CrossRef CAS.
- J. T. Hwang, M. Lee, S. N. Jung, H. J. Lee, I. Kang, S. S. Kim and J. Ha, Carcinogenesis, 2004, 25, 2497–2507 CrossRef CAS.
- A. Dobrzyn and P. Dobrzyn, J. Physiol. Pharmacol., 2006, 57, 31–42 Search PubMed.
- J. M. Ntambi, M. Miyazaki, J. P. Stoehr, H. Lan, C. M. Kendziorski, B. S. Yandell, Y. Song, P. Cohen, J. M. Friedman and A. D. Attie, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11482–11486 CrossRef CAS.
- P. Dobrzyn, A. Dobrzyn, M. Miyazaki, P. Cohen, E. Asilmaz, D. G. Hardie, J. M. Friedman and J. M. Ntambi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6409–6414 CrossRef CAS.
- J. M. Ntambi and M. Miyazaki, Prog. Lipid Res., 2004, 43, 91–104 CrossRef CAS.
- G. W. Hart, M. P. Housley and C. Slawson, Nature, 2007, 446, 1017–1021 CrossRef CAS.
- N. E. Zachara and G. W. Hart, Biochim. Biophys. Acta, 2006, 1761, 599–617 CAS.
- X. Yang, P. P. Ongusaha, P. D. Miles, J. C. Havstad, F. Zhang, W. SO. Venus, J. E. Kudlow, R. H. Michell, J. M. Olefsky, S. J. Field and R. M. Evans, Nature, 2008, 451, 964–970 CrossRef CAS.
- K. Vosseller, L. Wells, M. D. Lane and G. W. Hart, Proc. Natl. Acad. Sci. U. S. A., 99, 5313–5318 Search PubMed.
- M. P. Housley, J. T. Rodgers, N. D. Udeshi, T. J. Kelly, J. Shabanowitz, D. F. Hunt, P. Puigserver and G. W. Hart, J. Biol. Chem., 2008, 283, 16283–16292 CrossRef CAS.
- S. Marshall and R. Okuyama, Biochem. Biophys. Res. Commun., 2004, 318, 911–915 CrossRef CAS.
- A. Mukhopadhyay, S. K. Manna and B. B. Aggarwal, J. Biol. Chem., 2000, 275, 8549–8555 CrossRef CAS.
- A. Kita, S. Uotani, H. Kuwahara, R. Takahashi, K. Oshima, H. Yamasaki, H. Mizuguchi, T. Hayakawa, Y. Nagayama, Y. Yamaguchi and K. Eguchi, Biochem. Biophys. Res. Commun., 2003, 302, 805–809 CrossRef CAS.
- Y. Minokoshi, Y. B. Kim, O. D. Peroni, L. G. Fryer, C. Muller, D. Carling and B. B. Kahn, Nature, 415, 339–343 Search PubMed.
- D. V. Gross, A. P. J. Van Den Huevel and M. J. Birnbaum, Oncogene, 2008, 27, 2320–2336 CrossRef CAS.
- Y. Kamei, J. Mizukami, S. Miura, M. Suzuki, N. Takahashi, T. Kawada, T. Taniguchi and O. Ezaki, FEBS Lett., 2003, 536, 232–236 CrossRef CAS.
- A. Barthel, D. Schmoll, K. D. Kruger, G. Bahrenberg, R. Walther, R. A. Roth and H. G. Joost, Biochem. Biophys. Res. Commun., 2001, 285, 897–902 CrossRef CAS.
- S. Kataoka, H. Yasui, M. Hiromura and H. Sakurai, Life Sci., 2005, 77, 2814–2829 CrossRef CAS.
- Z. Wang, S. D. Hall, J. F. Maya, L. Li, A. Asghar and J. C. Gorski, Br. J. Clin. Pharmacol., 2003, 55, 77–85 CrossRef CAS.
- C. S. Lieber, Physiol. Rev., 1997, 77, 517–544 CAS.
- K. J. Woodcroft, M. S. Hafner and R. F. Novak, Hepatology, 2002, 35, 263–273 CrossRef CAS.
- A. S. Lee, Trends Biochem. Sci., 2001, 26, 504–510 CrossRef CAS.
- L. Zhao and S. L. Ackerman, Curr. Opin. Cell Biol., 2006, 18, 444–452 CrossRef CAS.
- U. Ozcan, Q. Cao, E. Yilmaz, A. H. Lee, N. N. Iwakoshi, E. Ozdelen, G. Tuncman, C. Gorgun, L. H. Glimcher and G. S. Hotamisligil, Science, 2004, 306, 457–461 CrossRef.
- E. D. Apel, R. M. Lewis, R. M. Gray and J. R. Sanes, J. Biol. Chem., 2000, 275, 31986–31995 CrossRef CAS.
- L. L. Gough, J. Fan, S. Chu, S. Winnick and K. A. Beck, Mol. Biol. Cell, 2003, 14, 2410–2424 CrossRef CAS.
- L. L. Gough and K. A. Beck, Biochim. Biophys. Acta, 2004, 1693, 29–36 CAS.
- G. R. Willsky, L. H. Chi, Y. Liang, D. P. Gaile, Z. Hu and D. C. Crans, Physiol. Genomics, 2006, 26, 192–201 Search PubMed.
- A. R. Saltiel and C. R. Kahn, Nature, 2001, 414, 799–806 CrossRef CAS.
- S. Genet, R. K. Kale and N. Z. Baquer, Mol. Cell. Biochem., 2002, 236, 7–12 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |