DOI:
10.1039/C5RA06716B
(Paper)
RSC Adv., 2015,
5, 50783-50789
Rapid synthesis of rGO–MoO3 hybrids and mechanism of enhancing sensing performance to H2S
Received
14th April 2015
, Accepted 2nd June 2015
First published on 2nd June 2015
Abstract
Hybrids of reduced graphene oxide (rGO)–MoO3 nanorods were synthesized successfully by a facile in situ solution growth method under a relatively low temperature of 150 °C for 1 h. The structure and properties of the hybrids have been characterized by XRD, SEM, TEM, Raman, PL and XPS analysis. The sensing performance of pure MoO3 and rGO–MoO3 hybrids to H2S were examined, the results indicate that the hybrids exhibit higher response and lower operating temperature compared with the pure MoO3 nanorods, especially, the 2.5 wt% rGO–MoO3 hybrid exhibits the highest sensitivity and the fastest response to H2S, which makes them promising candidates in the field of gas sensors for detection of H2S gas. The sensing mechanism for MoO3 to H2S which is enhanced is also discussed in detail from rGO action in the hybrid and formation of a hetero-junction at the interface of the hybrid.
1 Introduction
With the development of industry, various toxic gases are increasingly released into the air, resulting in serious environmental pollution.1 As a colorless and “rotten egg smell” substance, hydrogen sulfide (H2S) is very harmful to both the human body and the environment.2 Therefore, it is very important to develop H2S sensors with good sensing performances for the monitoring and control of H2S gas. Gas sensors based on metal oxide semiconductors, such as SnO2, ZnO, WO3 and MoO3, etc. have attracted much attention because of their structure stability, low cost, and facile fabrication, and are well known to be effective devices in detecting flammable and toxic gases with enough sensitivity.3–7 MoO3 has been recognized to be promising sensing material for detection of H2S, but pure MoO3 usually exhibits lower sensitivity and higher operating temperatures. Therefore, many attempts have been employed to improve the gas sensing performance of MoO3, for example, loading of noble metal, doping of element or forming of composites, etc.8–12 Recently, graphene has attracted a great deal of attention owing to its good conductivity, superior chemical stability and high specific surface area, as well as its potential applications in photocatalysis, energy storage and sensor fields.13,14 The reduced graphene oxide (rGO) can be obtained by reducing of the oxide graphite (GO) that results from oxidizing of graphite using modified Hummer's method.15 More recently, incorporating of rGO in metal oxides to construct the hybrids have attracted widely interest, which will help the generation of the novel sensing materials that sensing performance over the constituent counterparts due to their synergetic or complementary effect.16–20 Choi et al.21 reported the SnO2/rGO mixtures that exhibited response of 34 to 5 ppm H2S at operating temperature of 200 °C, and the response and recovery times were about 100 s. Jiang et al.22 synthesized Fe2O3/rGO composites via a super critical CO2-assisted thermal method and the lowest detect limit of 10 ppm H2S was obtained using chemiluminescence measurement at 130 °C. However, the hybrids of rGO–MoO3 are still rarely reported, and application for detection of H2S is much more less, even if for pure MoO3. There have been development two hybrids related to MoO3 with rGO, that is, Yang et al.23 prepared MoO3–rGO composites by hydrothermal method at 180 °C for 24 h, but did not involve the application of the materials. Noerochim et al.24 prepared MoO3–rGO composites via a two-step microwave hydrothermal and the composites were applied to cathode materials of lithium batteries. Therefore, the development of facile and practical methods to structure sensing hybrids still remains a challenging issue. Herein, we assembled the MoO3 nanorods on the rGO nanosheets via a facile in situ microwave hydrothermal method and enhanced sensitivity and reduced the operating temperature for detection of H2S. Table 1 listed the response of MoO3–rGO hybrid to H2S in this work compared with that of other metal oxides/rGO hybrids reported in literatures.
Table 1 Response of rGO–MoO3 hybrid to H2S in this work compared with the responses of other metal oxides/rGO composites reported in literatures
| Materials |
Synthesis method |
Operating temp. (°C) |
Response/H2S concentration |
Tresponse/Trecovery |
| rGO–MoO3 hybrids |
In situ microwave hydrothermal method |
110 |
44.7/40 ppm |
109/36 s |
| Fe–MoO3 (ref. 8) |
Hydrothermal method |
270 |
184.1/100 ppm |
15/34 s |
| α-MoO3/CuO9 |
Hydrothermal method |
270 |
272/10 ppm |
40/40 s |
| α-MoO3/ZnO10 |
Hydrothermal method |
270 |
30/10 ppm |
13/29 s |
| RGO–SnO2 (ref. 21) |
Electrospinning |
200 |
34/5 ppm |
3.3/1.9 min |
| Fe2O3/graphene22 |
Super critical CO2-assisted thermal method |
190 |
Intensity change of chemiluminescence/15 ppm |
0.5/30 s |
| Graphene–MoO3 (ref. 23) |
Hydrothermal method |
— |
— |
— |
| MoO3–graphene24 |
Two-step microwave hydrothermal method |
— |
— |
— |
2 Experimental
2.1 Synthesis of reduced graphene oxide (rGO)
Under the condition of stirring and ice bath, a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of concentrated H2SO4/H3PO4 (90:10 mL) was added to a mixture of 1.0 g of graphite flakes and 6.0 g of KMnO4 in three-neck flask. After stirring for 20 min, the reaction was then ultrasonicated for 2 h at a power level of 250 W. The reaction was cooled to room temperature and poured onto ice-water of 100 mL with 30% H2O2 (1.5 mL). The dispersion was centrifuged under 5000 rpm for 10 min, and the supernatant was decanted away. The remaining precipitate was then washed several times with 30% HCl solution in a cycle of centrifugation and decantation, and finally washed with ethanol and ether. The product was allowed to dry at room temperature for 12 h. Exfoliation of graphene oxide (GO) was achieved by ultrasonication of the dispersion on an ultrasonic bath. For preparation of rGO, 0.8 g of glucose was added to 40 mL of homogeneous exfoliated GO dispersion (1.0 mg mL−1), followed by stirring for 30 min. Then 0.4 of aqueous ammonia solution (25%, w/w) was added to the resulting dispersion. After being vigorously shaken for a few minutes, the mixture was kept in a microwave oven (WX-8000 microwave system) for 15 min at 120 °C. The resulting black dispersion was then filtered and washed with distilled water several times, and the obtained rGO was re-dispersed in water for further use.
:1 mixture of concentrated H2SO4/H3PO4 (90:10 mL) was added to a mixture of 1.0 g of graphite flakes and 6.0 g of KMnO4 in three-neck flask. After stirring for 20 min, the reaction was then ultrasonicated for 2 h at a power level of 250 W. The reaction was cooled to room temperature and poured onto ice-water of 100 mL with 30% H2O2 (1.5 mL). The dispersion was centrifuged under 5000 rpm for 10 min, and the supernatant was decanted away. The remaining precipitate was then washed several times with 30% HCl solution in a cycle of centrifugation and decantation, and finally washed with ethanol and ether. The product was allowed to dry at room temperature for 12 h. Exfoliation of graphene oxide (GO) was achieved by ultrasonication of the dispersion on an ultrasonic bath. For preparation of rGO, 0.8 g of glucose was added to 40 mL of homogeneous exfoliated GO dispersion (1.0 mg mL−1), followed by stirring for 30 min. Then 0.4 of aqueous ammonia solution (25%, w/w) was added to the resulting dispersion. After being vigorously shaken for a few minutes, the mixture was kept in a microwave oven (WX-8000 microwave system) for 15 min at 120 °C. The resulting black dispersion was then filtered and washed with distilled water several times, and the obtained rGO was re-dispersed in water for further use.
2.2 Synthesis of MoO3–rGO hybrids
A certain amount of rGO was dispersed in 20 mL of deionized water and sonicated for 2 h to yield rGO nanosheets solution A. 1.21 g of Na2MoO4·2H2O was dissolved in 40 mL of deionized water to form solution B. Then the solution B was added to the solution A and kept stirring for 20 min. After the mixture was gently stirred for 10 min, 4 mL of HCl (6 M) was added slowly into the solution with stirring at room temperature. The reaction solution was then transferred into a 100 mL Teflon-lined autoclave and kept in a microwave oven (WX-8000 microwave system) at 150 °C for 1 h. The autoclave was allowed to cool to room temperature naturally, the obtained precipitate was collected by centrifugation, washed several times with deionized water and ethanol, and dried at 70 °C under vacuum overnight. For comparison, a series of rGO–MoO3 hybrids were synthesized by changing the rGO content. Pure MoO3 nanorods were also prepared through a similar procedure only in the absence of rGO.
2.3 Characterization
Crystallographic information for the samples was collected using powder X-ray diffraction (XRD, Shimadzu XRD-600 diffractometer, copper Kα radiation with λ = 0.154 nm), operated at 45 kV and 40 mA. Scanning rate of 10° min−1 was applied to record the patterns in range of 3–70° (2θ). The product morphology was examined by field emission scanning electron microscopy (FESEM, Hitachi S-4700, 20.0 kV), a transmission electron microscope (TEM). PL spectra were recorded from 345 to 630 nm at room temperature with a 325 nm excitation (RF-5301PC spectrometer). X-ray photoelectron spectroscopy (XPS) spectra were recorded on an X-ray photoelectron spectrometer (VG ESCALAB-MK) with aluminum Kα radiation. The Raman spectroscopy was performed on a laser Raman spectrometer (Renishaw inVia) using a visible laser (λ = 532 nm) at room temperature.
2.4 Sensor fabrication and gas-sensing test
The pure MoO3 and rGO–MoO3 samples were mixed respectively with ethanol to form paste, and then drop-coated onto the surface of a ceramic tube with Au electrodes and a Ni–Cr heating coil that was inserted through the ceramic tube to construct a sensor. The sensor was aged at the certain temperature for several days in order to improve the structure stability of the sensing material and the stability of sensor resistance. The sensing responses of aged samples were measured to different concentrations of H2S (5–80 ppm) using a JF02E gas sensor test system. The operating temperature of the sensor was adjusted by varying the voltage through an electric heating system. The resistance of the sensor in air (Rair) and in the air-test gas mixture (Rgas) was recorded, respectively. The response for reducing gas H2S is defined as the ratio of Rair/Rgas. The response time and recovery time of the sensor were measured as the time taken for the sensor output to reach 90% of its saturation after applying or switching off the gas in a step function.
3 Results and discussion
3.1 Structure and morphology of MoO3 and rGO–MoO3 hybrids
The X-ray diffraction (XRD) spectra of graphite, GO, rGO, α-MoO3 and rGO–MoO3 hybrids are shown in Fig. 1. The characteristic diffraction peak of as-prepared GO is at 10.1° corresponding to an interlayer distance of 8.9 Å as shown in Fig. 1b.25,26 For XRD, the interlayer spacing of the materials is proportional to the degree of oxidation. Also, the spectrum of GO had no peak at around 26° with an interlayer distance of 3.4 Å, indicating that the starting material (pristine graphite, 2θ = 26.5, Fig. 1a) were not present in the sample.27 Based on the XRD patterns of products in Fig. 1d–g, all the peaks in the spectra of as-prepared MoO3 and rGO–MoO3 hybrid can be indexed to the orthorhombic phase of MoO3 (JCPDS no. 05-0508) with lattice constants: a = 3.962 Å, b = 13.858 Å and c = 3.697 Å.7,23 For the as-prepared rGO–MoO3 hybrid, no typical peaks belonging to graphene oxide in 10.1°, indicating that the successful reduction of graphene oxide. In addition, the characteristic broad diffraction peak with low intensity around 24.6° corresponding to rGO (Fig. 1c) are not observed besides these representative diffraction peaks of MoO3, it can be ascribed to the low amount and low diffraction intensity of rGO, compared to the characteristic peaks of MoO3 between 20 and 30°.20,24,28
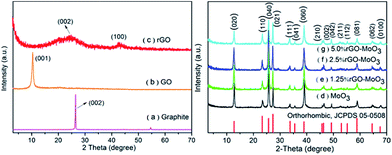 |
| | Fig. 1 XRD patterns of (a) graphite, (b) GO, (c) rGO, (d) as-prepared MoO3 and (e–g) rGO–MoO3 hybrids. | |
Fig. 2a presents the representative FESEM image of MoO3 nanorods, showing the prepared MoO3 nanorods with length of several micrometers and diameters in the range of 100–200 nm. FESEM and HRTEM Images (Fig. 2b and d) reveal further information about the morphology and structure of the rGO–MoO3 hybrid. The rGO shows a typical wrinkled morphology in the TEM image and the MoO3 nanorods with a typical length of about several micrometers and a diameter of about 100 nm are overlapped and surrounded by rGO. The distribution of random MoO3 nanorods on the rGO sheet and their intimate contact can also be confirmed by the TEM image in Fig. 2d. From typical FESEM image of prepared rGO nanosheets shown in Fig. 2c, a crumpled layered structure with several stacking layers is obviously seen.
 |
| | Fig. 2 FESEM images of α-MoO3 (a), rGO–MoO3 (b), rGO (c) and TEM image of rGO–MoO3 hybrid (d). | |
Raman spectra of GO, rGO, rGO–MoO3 and MoO3 are shown in Fig. 3. Two characteristic bands were observed in Raman spectrum of GO (Fig. 3a), 1352 cm−1 (D band) is attributed to the local defects/disorders and 1602 cm−1 (G band) can be assigned to the sp2 graphitized structure.17,23,27 After its reduction with glucose in the hydrothermal process, the Raman spectrum of as-prepared rGO (Fig. 3b) also exhibits the presence of D and G bands at 1344 and 1585 cm−1, respectively. Raman spectrum of the rGO–MoO3 hybrid (Fig. 3c) reveals that the D and G bands appear at about 1344 and 1585 cm−1, which confirms the existence of rGO.23 It should be noted that the frequency of the G and D bands in the rGO–MoO3 are very similar to that observed in the rGO. However, the decrease in the D/G intensity ratio, compared to GO, indicates an increase in the size of the in-plane sp2 domains and lower defects and disorders of the graphitized structures following the hydrothermal process.19,29 In addition, the peaks at 243, 284, 336, 375, 665, 817 and 994 cm−1 of the rGO–MoO3 hybrid curve and MoO3 curve fit well with the structure of MoO3 reported in previous report.24,30 The Raman bands at 994 cm−1 and 817 cm−1 can be assigned to the asymmetrical and symmetrical stretching vibrations of the terminal Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds while the band at 665 cm−1 is attributed to the asymmetrical stretching vibration of O–Mo–O bonds.24 Peaks observed in the range of 100–400 cm−1 correspond to various bending modes of α-MoO3 crystal.
O bonds while the band at 665 cm−1 is attributed to the asymmetrical stretching vibration of O–Mo–O bonds.24 Peaks observed in the range of 100–400 cm−1 correspond to various bending modes of α-MoO3 crystal.
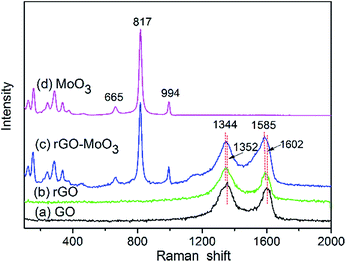 |
| | Fig. 3 Raman spectra of GO (a), rGO (b), rGO–MoO3 (c) and MoO3 (d). | |
Fig. 4 shows the PL spectra observed with an excitation wavelength of 325 nm for the rGO–MoO3 hybrids with different rGO content, respectively. It is well known that MoO3 can absorb photons with energy exceeding the forbidden energy gap of MoO3 and generate excited electrons. The excited electrons jump from basic state to excitation state, and then fluorescence emission is produced when the excited electrons come back to basic state. As observed in Fig. 4, the PL intensity of α-MoO3/2.5 wt% rGO is lower than that of rGO–MoO3 with rGO content of 0 wt%, 1.25 wt% and 5 wt%, because the PL emission originate from the recombination of excited electrons and holes, the lower PL intensity indicates a lower recombination rate of photo-generated electrons and holes.31 The lower PL intensity was explained as the surface nanometer carbon materials which can show collective polarization modes and sensitize semiconductors. The rGO has a high charge-carrier mobility of 20000 cm2 V−1 s−1, so it is very possible that the incorporation of rGO might enhance the charge separation,19 which effectively promote the sensing response and photocatalysis of rGO–MoO3.
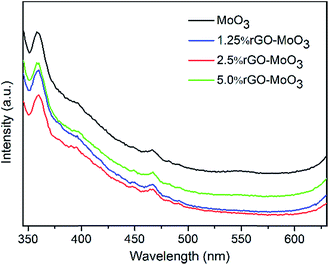 |
| | Fig. 4 PL spectra of MoO3 and rGO–MoO3 hybrids with different rGO content. | |
The composition and surface chemical states of as-prepared GO, rGO and rGO–MoO3 composites can be further provided by XPS. Deconvolution of the C 1s peak of GO (Fig. 5a) shows the presence of different oxygen-containing functional groups with binding energies at about 284.8, 286.7, 287.9, and 288.6 eV, which can attributable to the sp2 hybridized carbon (CC), C–O, CO, and O–CO species, respectively.27,32 This shows that high levels of oxidation of the GO sheets have occurred during the exfoliation. The C 1s spectra of rGO (Fig. 5b) were compared by deconvoluting each spectrum into three peaks that correspond to the following functional groups: carbon sp2 (CC, 284.8 eV), epoxy/hydroxyls (C–O, 286.7 eV), and carboxylates (O–CO, 289.1 eV). After the reduction, the CO signal disappeared and the C–O (286.7 eV) signal decreased markedly in intensity, suggesting the effective elimination of the oxygen-containing group in rGO. The ratio of peak intensity for C–O and C–C bonds in rGO is dramatically decreased compared to that of the as-synthesized GO, suggesting the effective reduction of GO. For rGO–MoO3 (Fig. 5c), the ratio of peak intensity for C–O and C–C bonds in rGO–MoO3 is further decreased compared to that of the as-synthesized rGO after microwave hydrothermal treatment process. The XPS of the Mo 3d shows the presence of two well-defined peaks (Mo 3d5/2 and Mo 3d3/2) at 232.6 and 235.7 eV (Fig. 5d), respectively.32,33 The binding energy and spin–orbit split-ting (ΔMo 3d = 3.1 eV) are in good agreement with those reported for Mo6+, which are for α-MoO3, suggesting that the α-MoO3 nanorods were located on the rGO sheets.
 |
| | Fig. 5 XPS spectra: C 1s peaks of GO (a), rGO (b) and rGO–MoO3 hybrids (c); Mo 3d spectra of rGO–MoO3 hybrids (d). | |
3.2 Gas sensing properties
Fig. 6 shows the sensor responses of the rGO–MoO3 hybrids in different rGO content to 40 ppm H2S gas at different operating temperatures. Many reports have demonstrated that the response of a semiconductor sensor is greatly affected by the operating temperature.4,5,12 As shown in Fig. 6, the response of pure α-MoO3 increases with increasing the operating temperature when the operating temperature is under 110 °C, which can be attributed to the fact that the thermal energy is high enough to overcome the activation energy barrier of chemisorption and surface reactions. When the operating temperature increases to 110 °C, the response reaches a maximum value of 21.8 and the temperature at which the response exhibits a maximum value is termed as optimum operating temperatures. If the temperature is further enhanced is under 110 °C, the amount of adsorbed gas on material surface will decrease with temperature, leading to the decrease of sensor response owing to decrease of reaction rate. When the desorption rate of gas is equal to the adsorption rate of gas, the maximum loading of chemisorbed ions is reached at the optimum temperature, resulting in the highest response.
 |
| | Fig. 6 Sensing responses to 40 ppm H2S of rGO–MoO3 hybrids with different rGO content. | |
Fig. 6 also shows the responses of rGO–MoO3 hybrids with different rGO content to 40 ppm H2S at the operating temperature of 70 to 220 °C. It also indicates that the H2S sensing properties are strongly influenced by rGO content in hybrid. It can be seen that the sensor response at 110 °C continuously increases when the rGO content varies from 1.25 to 2.5 wt%, and then decreases from 2.5 to 5 wt%. Accordingly, 2.5 wt% is believed to be the optimum rGO content and is chosen to test the transient gas-sensing properties herein after. Maximum response of 44.7 obtained with 2.5 wt% rGO–MoO3 to 40 ppm H2S is observed at 110 °C, which is 2.1 times of that obtained with pure α-MoO3.
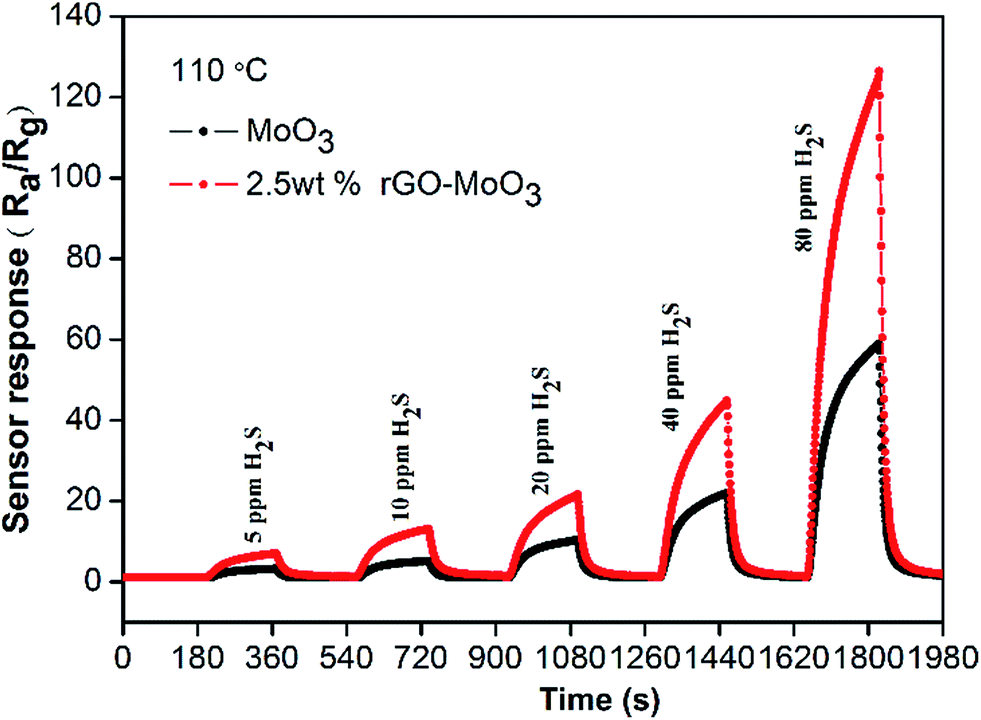
Fig. 7 shows the sensor responses of pure α-MoO3 (black curves) and 2.5 wt% rGO–MoO3 hybrid (red curves) to H2S gas with different concentrations at the same operating temperature. It is found that the sensor response values of the hybrid to 5–80 ppm H2S gases are obviously higher than those of pure α-MoO3. Furthermore, the hybrid can detect H2S gas concentration down to 5 ppm at operating temperature of 110 °C, however, the pure α-MoO3 nanorods have almost no response to H2S gas with a concentration less than 20 ppm.
 |
| | Fig. 7 Transient responses of α-MoO3 and 2.5 wt% rGO–MoO3 hybrid to different H2S concentrations. | |
The selectivity is one of important gas sensing properties for sensing materials. The cross sensing between H2S and other harmful reducing gases such as methanol, ethanol, acetone, NH3 and CO is still one of the major problems for practical H2S sensor. Fig. 8 shows the selectivity of H2S (20 ppm) to 100 ppm of ethanol, NH3, acetone, methanol and CO for the sensor based on the 2.5 wt% rGO–MoO3 hybrid at operating temperature of 110 °C. This result clearly demonstrates that the sensor based on the annealed hybrid not only has high response to H2S but also exhibits high selectivity to reducing gases, such as ethanol, acetone, NH3, methanol and CO. So, the hybrid synthesized by microwave hydrothermal treatment process is a promising sensing material for efficient and selective detection of H2S.
 |
| | Fig. 8 2.5 wt% rGO–MoO3 hybrid based sensor response for different gases. | |
The stability study is extremely important for the practical gas sensor. In the experimental process, we have attend to the stability of sensing response. Owing to the lower optimum temperature, the stability is rather good as shown in Fig. 9.
 |
| | Fig. 9 Stability of the sensor based on 2.5 wt% rGO–MoO3 hybrid to 40 ppm H2S at 110 °C. | |
3.3 Sensing mechanism of 2.5 wt% rGO–MoO3 hybrid
The reasons enhanced gas sensing for 2.5 wt% rGO–MoO3 hybrid to H2S can be understood from following two factors: firstly, according to the gas sensing mechanism of semiconducting metal oxide, when the MoO3 nanorods were exposed to air, a low conductance state of depletion layer will form on the surface of MoO3 and form chemisorbed oxygen ions at surface of α-MoO3 nanorods.34 Moreover,| | |
Ο2(ads) + e− → Ο2(ads)− T < 100 °C
| (2) |
| | |
Ο2(ads)− + e− → 2Ο(ads)− 100 °C ≤ T ≤ 300 °C
| (3) |
the n-type semiconductor MoO3 direct contact with metallic rGO, the electrons will migrate from the rGO to the MoO3 due to the difference of their work function until to the equilibrium is achieved at interface between the MoO3 and the rGO,35 that is, the Fermi level becomes equal in the entire hybrid, which makes the depletion layer becomes thick, leading to the resistance of hybrid in air is further increased. That is, renders the surface of the MoO3 nanorods makes more n-type. However, once the MoO3 is exposed to the reducing gas of H2S, the adsorbed H2S molecules at surface of the material reacts with adsorbed oxygen ions and release electrons into the conducing band of MoO3, leading to resistance decrease of MoO3 (Fig. 10). The equations of oxidizing reaction are written as following:8,10| | |
H2S + 3Ο(ads)− → SΟ2 + H2Ο + 3e−
| (5) |
 |
| | Fig. 10 Adsorption and reaction model of the sensing process on α-MoO3/rGO hybrid sensor surface. | |
Meantime, the rGO as an important role in hybrid facilitates the electrons transfer from the H2S molecules to MoO3, which also makes the hybrid resistance in H2S gas further decrease. In brief, the incorporating of rGO increases the resistance of hybrid in air and decreases the resistance of hybrid in H2S gas. According to response definition to reducing gas of H2S, the sensing response is significantly enhanced for the hybrid under relatively low operating temperature. Secondly, graphene as a support material is mainly due to that the ultrathin flexible graphene layers not only provide a support for anchoring well-dispersed metal oxides but also work as a highly conductive matrix for enabling good contact between the MoO3 and rGO. When MoO3 nanorods were loaded on the surface of the rGO nanosheets, the sensitization effect is attributed to increased surface area due to the large specific surface area of the graphene nanosheets and to enhanced electron transport in hybrid structures, which will facilitate gas adsorption, diffusion and reaction on material surface, leading to the sensing properties is significantly enhanced for the hybrid under relatively low operating temperature. However, detailed understanding of the role of rGO in the sensing mechanism of hybrid remains still lacking.
4 Conclusions
The rGO–MoO3 hybrids were prepared by the MoO3 nanorods in situ grew on the surface of rGO. The sensing test results indicate that the response of the 2.5 wt% rGO–MoO3 hybrid is 2.1 times higher than that of the pure MoO3 sample at 110 °C. The hybrid not only has high response and low operating temperature but also exhibits good selectivity, fast response and recovery that are 109 s and 36 s, respectively, to 40 ppm of H2S. The possible mechanism of the enhanced gas sensing characteristics is attributed to the potential synergic or complementary effect of heterojunction. Our present work provides a facile and effective method for efficient and selective detection of H2S.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant nos 21177007 and 51372013), the Fundamental Research Funds for the Central Universities (YS1406), Beijing Engineering Center for Hierarchical Catalysts and Beijing Key Laboratory of Environmentally Harmful Chemicals Analysis.
Notes and references
- Y. Li, W. Luo, N. Qin, J. Dong, J. Wei, W. Li, S. Feng, J. Chen, J. Xu, A. A. Elzatahry, M. H. Es-Saheb, Y. Deng and D. Zhao, Angew. Chem., Int. Ed., 2014, 53, 9035–9040 CrossRef CAS PubMed
 .
. - Q. Xu and T. Townsend, Chemosphere, 2014, 96, 105–111 CrossRef CAS PubMed .
- J. Zhang, J. Guo, H. Xu and B. Cao, ACS Appl. Mater. Interfaces, 2013, 5, 7893–7898 CAS .
- S. Bai, S. Chen, Y. Zhao, T. Guo, R. Luo, D. Li and A. Chen, J. Mater. Chem. A, 2014, 2, 16697–16706 CAS .
- S. Bai, K. Zhang, L. Wang, J. Sun, R. Luo, D. Li and A. Chen, J. Mater. Chem. A, 2014, 2, 7927–7934 CAS .
- L. Wang, P. Gao, G. Zhang, G. Chen, Y. Chen, Y. Wang and D. Bao, Eur. J. Inorg. Chem., 2012, 5831–5836 CrossRef CAS PubMed .
- N. Illyaskutty, H. Kohler, T. Trautmann, M. Schwotzer and V. P. M. Pillai, J. Mater. Chem. C, 2013, 1, 3976–3984 RSC .
- Q. Y. Ouyang, L. Li, Q. S. Wang, Y. Zhang, T. S. Wang, F. N. Meng, Y. J. Chen and P. Gao, Sens. Actuators, B, 2012, 169, 17–25 CrossRef CAS PubMed .
- T. S. Wang, Q. S. Wang, C. L. Zhu, Q. Y. Ouyang, L. H. Qi, C. Y. Li, G. Xiao, P. Gao and Y. J. Chen, Sens. Actuators, B, 2012, 171–172, 256–262 CrossRef CAS PubMed .
- H. L. Yu, L. Li, X. M. Gao, Y. Zhang, F. Meng, T. S. Wang, G. Xiao, Y. J. Chen and C. L. Zhu, Sens. Actuators, B, 2012, 171–172, 679–685 CrossRef CAS PubMed .
- P. G. Su and Y. T. Peng, Sens. Actuators, B, 2014, 193, 637–643 CrossRef CAS PubMed .
- S. Bai, K. Zhang, J. Sun, D. Zhang, R. Luo, D. Li and C. Liu, Sens. Actuators, B, 2014, 197, 142–148 CrossRef CAS PubMed .
- J. Luo, J. Liu, Z. Zeng, C. F. Ng, L. Ma, H. Zhang, J. Lin, Z. Shen and H. J. Fan, Nano Lett., 2013, 13, 6136–6143 CrossRef CAS PubMed .
- S. Gilje, S. Han, M. Wang, K. L. Wang and R. B. Kaner, Nano Lett., 2007, 7, 3394–3398 CrossRef CAS PubMed .
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed .
- J. H. Lee, N. Park, B. G. Kim, D. S. Jung, K. Im, J. Hur and J. W. Choi, ACS Nano, 2013, 7, 9366–9374 CrossRef CAS PubMed .
- Z. Gao, J. Wang, Z. Li, W. Yang, B. Wang, M. Hou, Y. He, Q. Liu, T. Mann, P. Yang, M. Zhang and L. Liu, Chem. Mater., 2011, 23, 3509–3516 CrossRef CAS .
- N. Chen, X. Li, X. Wang, J. Yu, J. Wang, Z. Tang and S. A. Akbar, Sens. Actuators, B, 2013, 188, 902–908 CrossRef CAS PubMed .
- J. Guo, Y. Li, S. Zhu, Z. Chen, Q. Liu, D. Zhang, W. J. Moon and D. M. Song, RSC Adv., 2012, 2, 1356–1363 RSC .
- M. Zhou, J. Yan and P. Cui, Mater. Lett., 2012, 89, 258–261 CrossRef CAS PubMed .
- S. J. Choi, B. H. Jang, S. J. Lee, B. K. Min, A. Rothschild and I. D. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 2588–2597 CAS .
- Z. Jiang, J. Li, H. Aslan, Q. Li, Y. Li, M. Chen, Y. Huang, J. P. Froning, M. Otyepka, R. Zboril, F. Besenbacher and M. Dong, J. Mater. Chem. A, 2014, 2, 6714–6717 CAS .
- X. Yang, C. Lu, J. Qin, R. Zhang, H. Tang and H. Song, Mater. Lett., 2011, 65, 2341–2344 CrossRef CAS PubMed .
- L. Noerochim, J. Z. Wang, D. Wexler, Z. Chao and H. K. Liu, J. Power Sources, 2013, 228, 198–205 CrossRef CAS PubMed .
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed .
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS PubMed .
- C. Zhu, S. Guo, Y. Fang and S. Dong, ACS Nano, 2010, 4, 2429–2437 CrossRef CAS PubMed .
- X. An, J. C. Yu, Y. Wang, Y. Hu, X. Yu and G. Zhang, J. Mater. Chem., 2012, 22, 8525–8531 RSC .
- H. Park, P. R. Brown, V. Bulovic and J. Kong, Nano Lett., 2012, 12, 133–140 CrossRef CAS PubMed .
- Y. Dong, S. Li, H. Xu, M. Yan, X. Xu, X. Tian, Q. Liu and L. Mai, Phys. Chem. Chem. Phys., 2013, 15, 17165–17170 RSC .
- D. Li, J. Hu, F. Fan, S. Bai, R. Luo, A. Chen and C. C. Liu, J. Alloys Compd., 2012, 539, 205–209 CrossRef CAS PubMed .
- J. Hu, A. Ramadan, F. Luo, B. Qi, X. Deng and J. Chen, J. Mater. Chem., 2011, 21, 15009–15014 RSC .
- X. Xia, Q. Hao, W. Lei, W. Wang, H. Wang and X. Wang, J. Mater. Chem., 2012, 22, 8314–8320 RSC .
- S. Bai, K. Zhang, R. Luo, D. Li, A. Chen and C. C. Liu, J. Mater. Chem., 2012, 22, 12643–12650 RSC .
- J. Yang, R. Li, N. Huo, W. L. Ma, F. Lu, C. Fan, S. Yang, Z. Wei, J. Li and S. S. Li, RSC Adv., 2014, 4, 49873–49878 RSC .
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.