DOI:
10.1039/C5RA06043E
(Paper)
RSC Adv., 2015,
5, 50790-50800
Magnetic iron/carbon nanorods derived from a metal organic framework as an efficient heterogeneous catalyst for the chemical oxidation process in water†
Received
5th April 2015
, Accepted 4th June 2015
First published on 4th June 2015
Abstract
By one-step carbonization, metal organic frameworks (MOFs) can be conveniently turned into hierarchical hybrid materials which exhibit versatile functionalities. Even though theoretically all MOFs can be carbonized to yield carbon-based hybrids, iron-based MOFs seem to be suitable precursors owing to the abundance and low-toxicity of iron. While many iron-based MOFs have been developed, most of these MOFs are synthesized using DMF, a carcinogenic solvent. Thus, an iron-based MOF, MIL-88A, appears to be an ideal precursor because it can be prepared just in water. MIL-88A-derived carbonaceous material consists of iron oxide nanoparticles and porous carbon, making it a magnetic porous support/adsorbent. However, its iron content and porosity in fact can enable it to be a promising heterogeneous and magnetic catalyst for chemical oxidation, which, however, has not been investigated. Herein, MIL-88A was used to prepare a magnetic iron/carbon nanorod (MICN). The MICN was characterized and then evaluated as a heterogeneous catalyst to activate oxidants, including hydrogen peroxide and sodium persulfate to decolorize Rhodamine B (RB) dye in water. While RB could not be removed via the adsorption to MICN, and degradation by oxidants, the combination of MICN and oxidants successfully decolorized RB owing to the iron content of MICN which activated peroxide and persulfate through a Fenton-like reaction. In addition, MICN loading was found to be a more critical factor than the oxidant dosage for RB decolorization. Elevated temperature also improved the RB decolorization, whereas basic conditions were not favorable for MICN-activated oxidative processes. Ultrasonication represented a useful external facilitator to enhance the decolorization while the addition of ascorbic acid greatly inhibited the activation process. The recyclability of MICN was also demonstrated, showing that MICN could be reused for multiple cycles without regeneration treatment. These features enable MICN to be an effective and easy-to-recover chemical oxidative catalyst.
1. Introduction
As one of the most intriguing materials nowadays, metal organic frameworks (MOFs) have attracted great attention both from industry and academia.1–4 MOFs, consisting of metal ions as “nodes” and organic ligands as “bridges”, can be designed using various metal species and organics to exhibit promising characteristics and versatile functionalities.5–7 These features have enabled MOFs to be attractive materials for phase separation,8–11 gas adsorption/storage,12–15 sensing devices,16 drug release,15,17 and photoluminescence,18 as well as catalysis.3,19,20
While various types of MOFs are continuously developed and their applications are extensively experimented, using MOFs as precursors to prepare carbonaceous materials has also drawn enormous attention. Since MOFs can be designed to be three-dimensional structures, MOFs can be easily converted to hierarchical carbonaceous derivatives via carbonization. For instance, a common zinc-based MOF, MOF-5, had been turned into porous carbon materials using 2-furylmethanol as an additional carbon source.21 Lately, a number of studies have also directly carbonized MOFs to obtain carbonaceous materials without using any additional carbon sources.22–26 This one-step carbonization of MOFs represents a novel and convenient route to prepare carbon materials. Considering that the resultant carbonaceous materials comprise carbon and metals/metal oxides, this preparation method can also be an alternative means to synthesize porous functional hybrid materials, which have successfully been employed in many applications such as phase separation,26 analytic chemistry,27 supercapacitors,22 energy storage,28,29 and catalysis.25
Even though theoretically all MOFs can be carbonized to prepare carbonaceous materials, non-noble-metal MOFs are much more favorable precursors owing to their availability and less environmental concerns. These non-noble-metal MOFs include iron-based,30 cobalt-based,31 copper-based,8 and zinc-based32 MOFs. In particular, the iron-based MOFs seem to be ideal MOFs as precursors to prepare MOF-derived carbonaceous materials because of abundance and low-toxicity of iron.
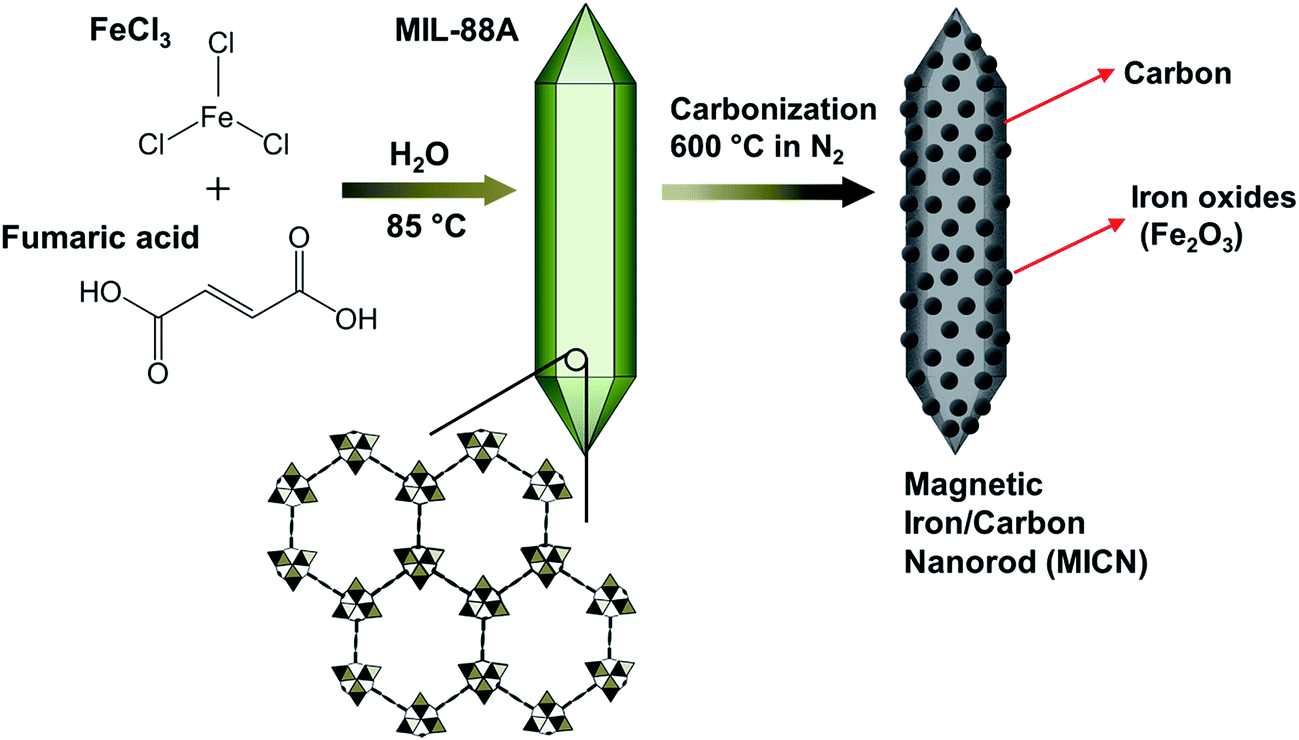
Up to date, several iron-based MOFs have been synthesized, such as MOF-53,33–35 MOF-74,36 MIL-88A,17,37 MIL-88B,38 MIL-88C,38 MIL-88D38 and MIL-101.39 While these iron-based MOFs can be synthesized in several solvents, a lot of these iron-based MOFs are typically prepared in N,N-dimethylformamide (DMF), which is carcinogenic according to the International Agency for Research on Cancer (IARC). During the synthesis, if these DMF-involved iron-based MOFs cannot be well-activated and undergone solvent exchange properly, DMF can remain in these MOFs, causing secondary pollution. Therefore, a DMF-free iron-based MOF should be a suitable MOF to prepare MOF-derived iron-based carbonaceous materials. To this end, MIL-88A is selectively chosen in this study because it can be synthesized using FeCl3 and fumaric acid just in water. MIL-88A-derived iron–carbon material can also exhibit magnetic and porous characteristics owing to its iron-oxide and carbon contents, respectively.40,41 This magnetic porous material has been demonstrated as a porous support to anchor gold and palladium40 and an adsorbent to adsorb dye in water.41 Nevertheless, the iron content and porosity of this MIL-88A-derived material in fact can enable this material a promising heterogeneous catalyst for chemical oxidation, which has not been thoroughly investigated so far. Thus in this study, MIL-88A was directly carbonized to prepare an iron-contained carbonaceous material. Considering the special hexagonal rod-like morphology of MIL-88A, the resultant magnetic hybrid was termed as magnetic iron/carbon nanorod (MICN).
The as-synthesized MICN was characterized and evaluated as a heterogeneous to activate oxidants (i.e., hydrogen peroxide (H2O2) and sodium persulfate (Na2S2O8)) to chemically oxidize a model pollutant, Rhodamine B (RB). Factor influencing the RB decolorization were investigated including oxidant dosage, MICN loading, temperature, pH, UV, ultrasonication and chemical inhibitors. MICN was also evaluated for its recyclability to catalytically activate oxidants to decolorize RB. Considering the chemical composition of MICN, a scheme was also proposed to possibly explain the mechanism of MICN-activated oxidative processes for RB decolorization.
2. Experimental
2.1. Materials
Chemicals involved in this study are all commercially available and used directly without purification. Fumaric acid, Rhodamine B (RB) dye and sodium persulfate were purchased from Sigma-Aldrich (USA). Hydrogen peroxide solution (30 wt% in H2O) was obtained from Union Chemicals (Taiwan). Iron chloride (FeCl3·H2O) and ascorbic acid were purchased from Merck (Germany). Methanol and tert-butyl alcohol (TBA) were obtained from Acros Organics (USA). Deionized (D.I.) water was prepared to exhibit less than 18 MOhm cm.
2.2. Synthesis of magnetic iron/carbon nanorod (MICN)
The synthesis procedure for MICN can be illustrated in Fig. 1. To obtain MICN, its precursor, MIL-88A, was first prepared based on the reported protocol.17,37 In a typical synthesis, a mixture of 10 g of fumaric acid and 1.0 g of FeCl3·H2O in 100 mL of D.I. water was added into a media bottle and stirred for 1 h to completely dissolve FeCl3 and fumaric acid. Next, the media bottle was capped and placed in an oven at 85 °C for 24 h. The precipitate was then collected, washed with ethanol and water, and dried at 100 °C under reduced pressure to obtain MIL-88A. MIL-88A nanocrystals were subsequently carbonized at 600 °C in nitrogen for 4 h to yield black powder, which was washed thoroughly with ethanol and dried at 85 °C to obtain MICN. The carbonization temperature (i.e., 600 °C) was selected according to the reported studies involving the carbonization of MOFs.42,43
 |
| | Fig. 1 Schematic illustration for the synthesis of MICN. | |
2.3. Characterization of MICN
To visualize morphology of MICN and its precursor, transmission electronic microscopy (TEM) (JEOL JEM-2010, Japan) was employed. The powder X-ray diffraction (XRD) pattern of MICN was obtained using an X-ray diffractometer (PANalytical, the Netherlands) with copper as an anode material (40 mA, 45 kV). Thermogravimetric (TG) curves of MICN and its precursor were measured using a thermogravimetric analyzer (ISI TGA i1000, USA) at a heating rate of 10 °C min−1 from 25 to 800 °C in air/nitrogen. Magnetization of MICN was measured in a gelatin capsule using a superconducting quantum interference device (SQUID) vibrating sample magnetometer (Quantum Design MPMS SQUID VSM, USA) at 27 °C. To characterize surface area and pore volume, N2 sorption/desorption isotherms were measured at a relative pressure (P/P0) ranging from 0.0001–0.99 using a volumetric sorption analyzer (Micromeritics ASAP 2020, USA).
2.4. Decolorization of RB using MICN-activated oxidative processes
RB decolorization using MICN-activated oxidative processes was investigated by batch-type experiments. In a typical experiment, 150 mg of oxidant (i.e., peroxide and persulfate) was first added to a 500 mL of RB solution with an initial concentration (C0) of 10 mg L−1, followed by addition of 150 mg of MICN. During the decolorization, sample aliquots were withdrawn from the mixture at pre-set times and the residual oxidant was quenched by potassium iodide. MICN was immediately separated from the solution by a permanent magnet and the remaining concentration of RB (Ct) was measured by a UV-Vis spectrophotometer at 554 nm (e-ChromTech CT-2000, Taiwan). To study effects of oxidant dosage and MICN loading, oxidant dosage and MICN loading both were varied from 100 to 500 mg L−1. The RB decolorization was also conducted at 25, 50 and 80 °C to investigate effect of temperature. Initial pH of RB solutions was also changed to 3 and 11 by adding 0.1 M sulfuric acid and sodium hydroxide solutions in order to investigate effects of acidic and basic conditions on the decolorization. Besides, we also examined several effects of facilitators and inhibitors on the RB decolorization. For studying effect of UV irradiation, a 9 W UV-A lamp (Philips PL-S9W, Netherland) was placed in the center of the batch reactor to facilitate MICN-activated oxidative processes. The enhancement of ultrasonication on the RB decolorization was assessed by placing the batch reactor in a temperature-controllable ultrasonication batch (Yeong-Hsin LEO-1002S, Taiwan). For evaluating effects of inhibitors, ascorbic acid, methanol and TBA were added to RB solution during the decolorization experiments.
The recyclability of MICN to activate oxidants for the RB decolorization was also investigated. The spent MICN was collected by a permanent magnet and used without regeneration treatment for subsequent tests. RB decolorization experiments in this study were duplicated and repeated at least twice.
3. Results and discussion
3.1. Characterization of MICN
MICN was derived from the carbonization of MIL-88A which can be seen in Fig. 2(a). Similar to other reported MIL-88A,37,44 the as-prepared MIL-88A crystals also exhibit a hexagonal rod-like morphology. The size (length) distribution of MIL-88A nanocrystals ranges from 50 to 600 nm with an average size of 200 nm (Fig. S1, see ESI†). Once MIL-88A was carbonized to MICN, the hexagonal rod-like morphology was still preserved as shown in Fig. 2(b). However, there are many dark-color nanoparticles surrounding the surface of MICN. These dark-color nanoparticles are considered as iron oxides derived from the iron–oxygen coordination between Fe3+ and di-carboxylic group of fumaric acid. The size range of these nanoparticles was approximately from 5 to 10 nm. To further determine the composition of these dark-color nanoparticles, the XRD pattern of MICN can be seen in Fig. 3(a) which reveals that iron content of MICN was not limited to a single phase of iron oxide. In fact, both α-Fe2O3 and γ-Fe2O3 can be observed in MICN. A few studies have indicated that the iron–oxygen cluster with MIL-88A or MIL-88B MOFs is in the form of Fe2O3.45 The appearance of α-Fe2O3 and γ-Fe2O3 in MICN indicates that the carbonization did not alter the iron–oxygen coordination. Additionally, we also observed the existence of Fe3C, confirming the presence of carbon and the bonding between iron and carbon in MICN.
 |
| | Fig. 2 TEM images of (a) MIL-88A and (b) the as-prepared MICN. The scale bar is 100 nm. | |
 |
| | Fig. 3 Characteristics of MICN: (a) XRD pattern of MICN ○: α-Fe2O3, □: γ-Fe2O3, △: Fe3C (b) magnetization curve of MICN at 27 °C, (c) coercivity of MICN% and (d) magnetic separation of MICN from water. | |
The magnetic property of MICN can be seen in Fig. 3(b), which displays the magnetization of MICN as a function of magnetic field. Under a varying magnetic field, MICN exhibited superparamagnetic characteristic with insignificant histeresis (see Fig. 3(c)) and coercivity was relatively low (∼50 Oe). Fig. 3(b) also reveals the saturation magnetization of MICN, which is 33 emu g−1. The manipulation of MICN in water is demonstrated in Fig. 3(d), showing that MICN can be well-dispersed in water and recovered from water using a permanent magnet. Fig. S2 (see ESI†) also demonstrates that MICN can be collected from water readily and completely within 10 s.
Considering that MICN was derived from the carbonization of MIL-88A, it is important to determine the yield from the carbonization. Fig. 4(a) shows the thermogravimetric (TG) analysis of MIL-88A in nitrogen with the derivative TG (DTG) signal. In the DTG result, a small peak can be found below 100 °C, possibly owing to moisture adsorbed from air to MIL-88A. Subsequently, a significant peak appears at around 250 °C, where the ligand (i.e., fumaric acid) started thermal-decomposition through 400 °C (ref. 46) and then the weight of MIL-88A remained almost unchanged through 800 °C. After the thermal-decomposition in nitrogen, the residual weight of MIL-88A was found to be 45 wt%, indicting the yield of MICN derived from MIL-88A. The as-prepared MICN was further calcined in air in order to estimate carbonaceous and iron-based fractions of MICN. The bottom figure of Fig. 4(a) shows the TG curve and DTG signal of MICN in air. After the calcination, the remaining weight of MICN was 78% and the weight loss was 22%, corresponding to the fractions of iron-based components (i.e., ferric and oxygenic compounds) and the carbonaceous fraction, respectively. The TGA result of MICN also demonstrates a superior thermal stability of MICN at relatively high temperature (i.e., 300–800 °C).
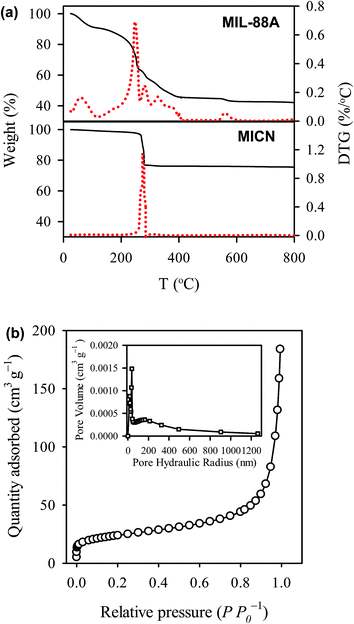 |
| | Fig. 4 Physical properties of MICN: (a) thermogravimetric analyses (TGA) of MIL-88A and MICN, and (b) N2 sorption and desorption isotherm. The inset shows pore size distribution of MICN. | |
Fig. 4(b) shows N2 sorption/desorption isotherms of MICN, which is considered to be a combination of the type II and III sorption isotherms. The BET surface area and pore volume of MICN were 84 m2 g−1 and 0.32 cm3 g−1 (single-point adsorption), respectively. The pore size distribution (see an inset in Fig. 4(b)) suggests that pores within MICN are mesoporous with a dominant range from 50 to 500 nm.
3.2. Decolorization of RB using MICN-activated oxidative processes
Before evaluating MICN as a catalyst to activate oxidants for RB decolorization, we first examined whether MICN can adsorb RB from water. Fig. 5 shows the RB decolorization (Ct/C0) via the adsorption as a function of time. In the end of 100 min adsorption test, there was almost no RB removal via the adsorption. This indicates that the surface of MICN did not exhibit affinity to attract RB molecule, possibly because RB was a cationic dye and the surface of MICN might be also positively charged owing to iron oxides.47 Moreover, RB decolorization using oxidants alone was also examined as references in comparison with MICN-activated oxidative processes. When peroxide (H2O2) was added to a RB solution, even though a slight amount (<5%) of RB was decolorized at the end of 100 min test, peroxide alone seemed to be incapable to decolorize RB. A similar result was observed when persulfate alone was used to decolorize RB, showing that oxidants alone were quite ineffective to decolorize RB. However, once MICN was combined with peroxide, the remaining concentration of RB (Ct/C0) was considerably decreased to 0.58. As we had revealed that MICN and peroxide alone were almost incapable of removing RB, the combination of MICN and peroxide led to successful decolorization of RB. This could be owing to that peroxide was activated in the presence of MICN. As discussed earlier, MICN consisted of α-Fe2O3, γ-Fe2O3 and Fe3C. Fe3+ in Fe2O3 and Fe2+ in Fe3C are expected to react with peroxide according to the following equations (eqn (1) and (2)):48–50| | |
Fe2+ + H2O2 → Fe3+ + OH− + OH˙
| (1) |
| | |
Fe3+ + H2O2 → Fe2+ + HO2˙− + H+
| (2) |
 |
| | Fig. 5 RB decolorization using oxidants and MICN-activated oxidations (oxidant = 300 mg L−1, MICN = 300 mg L−1, RB = 10 mg L−1, T = 25 °C, PSF = persulfate). | |
These reactions can be illustrated schematically in Fig. 6(a). When Fe2+ and Fe3+ react with peroxide to generate HO–O˙ and OH˙ radicals to decolorize RB, simultaneously Fe2+ and Fe3+ are oxidized and reduced to Fe3+ and Fe2+, respectively, which can initiate subsequent cycle of peroxide activation. On the other hand, the combination of MICN with persulfate was also evaluated and the result is also shown in Fig. 5. It can be readily noted that Ct/C0 rapidly decreased and dropped to 0.1 in 100 min. Even though persulfate alone was incapable of decolorizing RB, the combination of MICN and persulfate also led to significant removal of RB from water. This suggests that the existence of MICN could facilitate activation of persulfate considering the ferric and ferrous content (i.e., Fe3+ and Fe2+) in MICN. These Fe3+ and Fe2+ ions can react with persulfate to produce radicals as illustrated in Fig. 6(b) based on the following equations (eqn (3) and (4)):51
| | |
Fe2+ + S2O82− → Fe3+ + SO4˙− + SO42−
| (3) |
| | |
Fe3+ + S2O82− → Fe2+ + S2O8˙−
| (4) |
 |
| | Fig. 6 A proposed mechanism for the RB decolorization using MICN-activated oxidative processes. | |
As stated in eqn (3), Fe2+ has been recognized as one of the most common activators to facilitate the decomposition of persulfate.50 Nevertheless, lately a number of studies have demonstrated the activation of persulfate involving Fe3+. Liu et al. reported that the presence of Fe3+ can initiate the decomposition of persulfate via one-electron reduction of Fe3+ to Fe2+ which subsequently activate S2O82− as shown in eqn (4).51 By investigating the reaction between S2O82− and SO4˙−, McElroy et al. and Yu et al. also found the existence of S2O8˙− derived from the one-electron reduction of S2O82−.52,53 Besides, Liang et al. also confirmed that Fe3+ could activate persulfate to degrade trichloroethylenen (TCE) as Fe3+ was initially reduced to Fe2+, which subsequently activated persulfate to yield sulfate radicals.54
Despite the fact that peroxide and persulfate, combined with MICN, both successfully decolorized RB, the Ct/C0 of MICN-activated peroxide was noticeably different from that of MICN-activated persulfate. This could due to that the activation of peroxide using ferric and ferrous ions (so-called Fenton reaction) is highly pH-dependent compared to the activation of persulfate and typically its efficiency for degrading organics can be enhanced in acidic conditions.48,49 Since in this particular test, the initial pH of RB solutions was not adjusted (pH = 6.7), the decolorization extent of MICN-activated peroxide was not significant.
3.3. Effects of oxidant dosage and MICN loading on RB decolorization
As discussed in the earlier section, the presence of MICN greatly facilitated the activation of oxidants. Herein, we further investigated effects of two critical parameters on the RB decolorization, oxidant dosage and MICN loading. To examine the effects of oxidant dosage and MICN loading, both parameters were varied from 100 to 500 mg L−1. Fig. 7(a) shows that when peroxide was increased from 100 to 200 mg L−1 and MICN loading was fixed to 100 mg L−1, the removal efficiency (i.e., (C0 − Ct)/C0%) was only increased from 13 to 17%. When peroxide was raised to 500 mg L−1, the enhancement in the removal efficiency was still very limited (i.e., from 13 to 26%). Even though MICN was fixed to a higher loading (>200 mg L−1), the effect of peroxide dosage became even more insignificant, indicating the minor role of peroxide dosage in this MICN-activated peroxide process. Nevertheless, while peroxide dosage was fixed, regardless of the peroxide concentration, change in MICN loading considerably affected the removal efficiency. When MICN loading increased from 100 to 200 mg L−1, the removal efficiency rose from 13 to 31%, almost a two-fold enhancement. Similar enhancements can be observed once MICN loading became even higher (i.e., >200 mg L−1), indicating that MICN loading was a much more critical parameter in this oxidative process.
 |
| | Fig. 7 Effects of oxidant dosage and MICN loading on the decolorization efficiency: (a) peroxide and (b) persulfate (RB = 10 mg L−1, T = 25 °C, PSF = persulfate). | |
Fig. 7(b) reveals the effects of persulfate dosage and MICN loading, which were found to be similar to those observed in the case of peroxide. Although the removal efficiency of MICN-activated persulfate was inherently higher that of MICN-activated peroxide, increase in persulfate dosage was not effective to improve the removal efficiency either. However, once persulfate dosage was fixed and MICN loading was raised up, a considerable enhancement in the removal efficiency could be observed, indicating the important role of MICN loading during the activation of persulfate. These results suggest that maximization of RB decolorization can be achieved by increasing MICN loading instead of spend of oxidants. This also enables this MICN-activated oxidative process an attractive and cost-effective approach to decolorize dyes because the addition of MICN, regardless of the loading, can be recovered using a magnetic field, unlike oxidants which are non-reusable.
3.4. Effects of temperature and pH on the RB decolorization
In order to examine other important factors affecting the MICN-activated oxidative processes, we further evaluated the effects of temperature and pH. To distinguish the effect of temperature, we intentionally selected 25, 50 and 80 °C. Fig. 8(a) displays the RB decolorization as a function of time using MICN-activated peroxide at different temperatures. As the temperature was elevated, Ct/C0 was found to decrease significantly, showing the positive effect of elevated temperature. Besides, we noted that the increase in temperature also improved the decolorization kinetics. To quantitatively analyze the effect of temperature on the RB decolorization, the pseudo first order rate law was adopted to model the kinetic data. The pseudo first order rate law can be expressed in the following equation (eqn (5)):| |
Ct = C0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−kt) exp(−kt)
| (5) |
where k represents the apparent first order rate constant of RB decolorization. The k values for all tested temperatures are calculated and summarized in Table 1. The k value was found to increase from 0.100 to 0.565 min−1 as the temperature rose from 25 to 80 °C.
 |
| | Fig. 8 Effect of temperature on the decolorization efficiency using MICN-activated oxidants: (a) peroxide and (b) persulfate (RB = 10 mg L−1, oxidant = 300 mg L−1, MICN = 300 mg L−1, T = 25 °C). | |
Table 1 The pseudo first order rate constant (k) of the RB decolorization (C0 = 10 mg L−1) under different conditions using MICN activated oxidative processes
| MICN-activated oxidative process |
T (°C) |
k (min−1) |
R2 |
| Peroxide |
− |
25 |
0.100 |
0.951 |
| − |
50 |
0.242 |
0.962 |
| − |
80 |
0.565 |
0.986 |
| +UV irradiation |
25 |
0.176 |
0.952 |
| +Ultrasonication |
25 |
0.218 |
0.989 |
| Persulfate |
− |
25 |
0.045 |
0.981 |
| − |
50 |
0.118 |
0.980 |
| − |
80 |
0.477 |
0.983 |
| +UV irradiation |
25 |
0.095 |
0.985 |
| +Ultrasonication |
25 |
0.545 |
0.931 |
It can be also noted that most of correlation coefficients (R2) of the pseudo first rate law for describing the kinetic data are higher than 0.95, suggesting that the pseudo first rate law seemed to be a proper model to describe the decolorization kinetics. As the k value increased at elevated temperatures, the Arrhenius equation was adopted to determine the activation energy using the following equation (eqn (6)):
| |
 | (6) |
where
Ea represents the activation energy (kJ mol
−1);
k0 means the temperature-independent factor (g mg
−1 min
−1);
T denotes the solution temperature in Kelvin (K) and
R is the universal gas constant. The Arrhenius plot of MICN-activated peroxide process at different temperatures can be seen in Fig. S3(a) (see ESI
†). The data points are properly fitted using the linear regression (
R2 = 0.996), showing that the relationship between
k and temperature of the decolorization can be described by the Arrhenius equation. The corresponding
Ea was found to be 27.3 kJ mol
−1. On the other hand, we evaluated the effect of temperature on the decolorization using MICN-activated persulfate (
Fig. 8(b)). Although most of RB had been removed at 25 °C, the increase in temperature still noticeably improved the decolorization extent. More notably, the decolorization kinetics was considerably faster at elevated temperatures.
Table 1 also lists
k values of each temperature and the
k value was increased from 0.045 to 0.477 min
−1 as the temperature changed from 25 to 80 °C, revealing the importance of temperature effect. The Arrhenius plot of MICN-activated persulfate process was also shown in Fig. S3(b)
† with a correlation coefficient of 0.988. The corresponding
Ea value was found to be 37 kJ mol
−1.
Next, the effect of initial pH on the RB decolorization was also examined. Fig. 9(a) reveals the decolorization kinetics using MICN-activated peroxide under acidic, neutral (unadjusted) and basic conditions. Compared to the kinetics under the neutral (pH unadjusted) condition, the decolorization under the acidic condition led to a much lower Ct/C0, showing its positive effect on the decolorization. Nevertheless, an adverse effect was found when the initial pH of RB solution became 11. This indicates the basic environment was unfavorable for MICN-activated peroxide process. As discussed in the earlier section, the activation of peroxide to degrade organics is highly pH-dependent and the acidic environment is considered as a typical condition to maximize efficiency of such a process.48,49
 |
| | Fig. 9 Effect of pH on the decolorization efficiency using MICN-activated (a) peroxide and (b) persulfate (RB = 10 mg L−1, oxidant = 300 mg L−1, MICN = 300 mg L−1, T = 25 °C). | |
In MICN-activated persulfate process (Fig. 9(b)), variation in initial pH also led to significant change on the decolorization. Under the acidic condition, while the Ct/C0 at equilibrium was slightly higher than that under the unadjusted condition, the decolorization curve was actually similar to the curve obtained under the unadjusted condition. The reason that the decolorization at pH = 3 was not as effective as that at pH = 6.3 could be attributed to relatively high stability of persulfate under acidic conditions.55 In contrast, the decolorization at pH = 11 was significantly hindered, leading to a high Ct/C0 at equilibrium. This might be due to the self-decomposition of persulfate under basic conditions.55–57
3.5. Effects of UV, ultrasonication and inhibitors on the decolorization of RB in water
Since the activation of peroxide and persulfate can be facilitated by UV irradiation and ultrasonication,55,58–62 these two facilitators were introduced to MICN-activated oxidative processes to evaluate the effects of UV and ultrasonication irradiation. Fig. 10(a) shows the RB decolorization using MICN-activated peroxide with UV and US irradiation. The RB decolorization indeed was improved in the presence of UV irradiation. To rule out that such an improvement was derived from the photolysis of RB by UV, we performed RB decolorization by UV irradiation alone. The result is revealed in Fig. S4 (see ESI†) and almost no significant amount of RB was removed by the UV photolysis within the testing time frame. Therefore, this improvement could result from more hydroxyl radicals generated from activated peroxide induced by UV irradiation. The kinetics was also analyzed and the corresponding k was determined and listed in Table 1. Moreover, the effect of US irradiation was also shown in Fig. 10(a). Compared to the effect of UV, US irradiation appeared to be more effective to facilitate MICN-activated peroxide to decolorize RB. The corresponding k value was also calculated and shown in Table 1, which is much higher than the k value in the absence of US irradiation. It has been reported that the energy derived from US irradiation facilitates the generation of radicals, thereby improving the RB decolorization.55 In the case of persulfate, similar results can be obtained (Fig. 10(b)) and the corresponding k values are also summarized in Table 1. The US irradiation appeared to be a more effective means to facilitate the MICN-activated oxidative processes.
 |
| | Fig. 10 Effects of UV and ultrasonication (US) irradiation on the decolorization efficiency using MICN-activated oxidative processes: (a) peroxide and (b) persulfate (RB = 10 mg L−1, oxidant = 300 mg L−1, MICN = 300 mg L−1, T = 25 °C). | |
In addition to the facilitators, the effects of inhibitors were also examined to obtain insights into the mechanism of decolorization of MICN-activated oxidative processes. To inhibit MICN-activated peroxide for RB decolorization, we first employed ascorbic acid, a common radical scavenger. Fig. 11(a) shows the Ct/C0 of RB decolorization using MICN-activated peroxide in the presence of ascorbic acid. Only a slight decrease in Ct/C0 even after 100 min reaction, showing that ascorbic acid tremendously inhibited the RB decolorization. This result indicates the critical role of radicals generated from MICN-activated for decolorizing RB. Apart from ascorbic acid, methanol and TBA were also used to inhibit the decolorization. It can be seen that both methanol and TBA were able to inhibit the decolorization, possibly by suppressing the generation of radicals. Methanol is recognized as a universal radical scavenger which can inhibit several types of radicals, including hydroxyl and sulfate ones.63,64 The addition of methanol indeed suppressed the RB decolorization efficiency using MICN-activated peroxide, showing that a part of hydroxyl radicals were scavenged. On the other hand, TBA, a scavenger mainly for hydroxyl radicals,63,64 was also able to inhibit the decolorization, indicating the importance of hydroxyl radicals to decolorize RB decolorization. Nevertheless, the inhibition effect by TBA was found to be less significant than that by methanol in Fig. 11(a). This could be owing to that the additions of methanol and TBA were equal-concentration in mg L−1, rather than in mol L−1. Thus, the molar concentration of methanol added was in fact much higher than that of TBA.
 |
| | Fig. 11 Effect of inhibitors on the decolorization efficiency using MICN-activated MICN-activated oxidative processes: (a) peroxide and (b) persulfate (inhibitor = 300 mg L−1, RB = 10 mg L−1, oxidant = 300 mg L−1, MICN = 300 mg L−1, T = 25 °C). | |
In the case of MICN-activated persulfate (Fig. 11(b)), we observed similar results when ascorbic acid was used as a scavenger. The Ct/C0 became to 0.68 in the presence of ascorbic acid, showing its significant inhibition on MICN-activated persulfate process. When methanol was used, a similar result (Ct/C0 = 0.67 at equilibrium) was obtained and the decolorization was greatly suppressed. Since the decomposition of persulfate may also induce the generation of hydroxyl radicals, these result indicate that most of radicals (sulfate and hydroxyl ones) had been scavenged. On the other hand, when TBA was used, the inhibit effect was much less significant. Considering that TBA was mainly for scavenging hydroxyl radicals, this result suggests that radicals generated from MICN-activated persulfate process were mainly sulfate radicals with a small amount of hydroxyl radicals.
3.6. Recyclability of MICN to activate oxidants for the RB decolorization
As a heterogeneous catalyst, MICN had exhibited its magnetic controllability which can enable MICN easy-to-recover after the oxidative process. Herein, we further examine its recyclability to activate oxidants for the multiple-cycle RB decolorization. In this study, MICN was recovered magnetically and directly re-used in a subsequent decolorization experiment without any regeneration treatment. As shown in Fig. 12(a), the spent MICN was still able to activate peroxide to decolorize RB even after 4 cycles. Even though the Ct/C0 at equilibrium slightly increased after this 4-cycle test, catalytic activity of MICN still remained. In the case of persulfate, a similar result was observed (Fig. 12(b)). MICN was also able to activate persulfate to decolorize RB for multiple cycles, demonstrating that MICN can be re-used even without regeneration treatment.
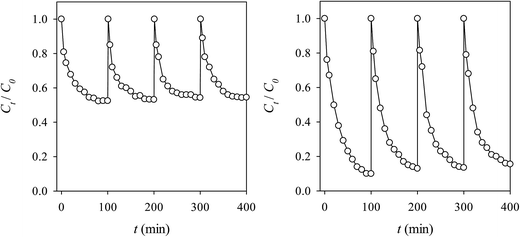 |
| | Fig. 12 Recyclability of MICN to activate (a) peroxide and (b) persulfate for the RB decolorization (RB = 10 mg L−1, oxidant = 300 mg L−1, MICN = 300 mg L−1, T = 25 °C). | |
4. Conclusion
In this study, magnetic iron/carbon nanorod (MICN) was prepared from the one-step carbonization of a DMF-free iron-based MOF, MIL-88A. MICN was used as a heterogeneous catalyst to activate common oxidants, including hydrogen peroxide and sodium persulfate to decolorize RB dye in water. While RB could not be removed via the adsorption to MICN, and degradation by peroxide/persulfate, the combination of MICN and oxidants successfully decolorized RB owing to the iron contents of MICN (i.e. Fe2+ and Fe3+), which activated peroxide and persulfate through the fenton-like reaction. Even though the combinations of MICN and peroxide and persulfate both could decolorize RB, the MICN combined with persulfate appeared to be more effective, possibly because the MICN combined with peroxide was highly-pH dependent. Besides, MICN loading was found to be a more critical factor than the oxidant dosage on the RB decolorzation. Elevated temperature also significantly improved the RB decolorization, whereas the basic condition was not favorable for both MICN-activated peroxide and persulfate processes. The recyclability of MICN was also demonstrated, showing that MICN could be reused for multiple-cycles without regeneration treatment. These features enable MICN an effective and easy-to-recover chemical oxidative catalyst.
References
- H.-C. J. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- C. Petit and T. J. Bandosz, Adv. Mater., 2009, 21, 4753–4757 CAS.
- N. Stock and S. Biswas, Chem. Rev., 2011, 112, 933–969 CrossRef PubMed.
- C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388 RSC.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626–636 RSC.
- K.-Y. A. Lin, H. Yang, C. Petit and F.-K. Hsu, Chem. Eng. J., 2014, 249, 293–301 CrossRef CAS PubMed.
- S. H. Jhung, N. A. Khan and Z. Hasan, CrystEngComm, 2012, 14, 7099–7109 RSC.
- I. Ahmed and S. H. Jhung, Mater. Today, 2014, 17, 136–146 CrossRef CAS PubMed.
- N. A. Khan, Z. Hasan and S. H. Jhung, J. Hazard. Mater., 2013, 244–245, 444–456 CrossRef CAS PubMed.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- J. W. Yoon, S. H. Jhung, Y. K. Hwang, S. M. Humphrey, P. T. Wood and J. S. Chang, Adv. Mater., 2007, 19, 1830–1834 CrossRef CAS PubMed.
- J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- L.-G. Qiu, Z.-Q. Li, Y. Wu, W. Wang, T. Xu and X. Jiang, Chem. Commun., 2008, 3642–3644, 10.1039/b804126a.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2011, 112, 1126–1162 CrossRef PubMed.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2013, 361–378, DOI:10.1021/cs400959k.
- J.-K. Sun and Q. Xu, Chem. Commun., 2014, 50, 13502–13505 RSC.
- J. Hu, H. Wang, Q. Gao and H. Guo, Carbon, 2010, 48, 3599–3606 CrossRef CAS PubMed.
- A. Banerjee, R. Gokhale, S. Bhatnagar, J. Jog, M. Bhardwaj, B. Lefez, B. Hannoyer and S. Ogale, J. Mater. Chem., 2012, 22, 19694–19699 RSC.
- G. Zhang, C. Li, J. Liu, L. Zhou, R. Liu, X. Han, H. Huang, H. Hu, Y. Liu and Z. Kang, J. Mater. Chem. A, 2014, 2, 8184–8189 CAS.
- G. Yu, J. Sun, F. Muhammad, P. Wang and G. Zhu, RSC Adv., 2014, 4, 38804–38811 RSC.
- N. L. Torad, M. Hu, S. Ishihara, H. Sukegawa, A. A. Belik, M. Imura, K. Ariga, Y. Sakka and Y. Yamauchi, Small, 2014, 10, 2096–2107 CrossRef CAS PubMed.
- C. Wei, X. Li, F. Xu, H. Tan, Z. Li, L. Sun and Y. Song, Anal. Methods, 2014, 6, 1550–1557 RSC.
- Z. Xiang, Z. Hu, D. Cao, W. Yang, J. Lu, B. Han and W. Wang, Angew. Chem., Int. Ed., 2011, 50, 491–494 CrossRef CAS PubMed.
- F. Zou, X. Hu, Z. Li, L. Qie, C. Hu, R. Zeng, Y. Jiang and Y. Huang, Adv. Mater., 2014, 26(38), 6622–6628 CrossRef CAS PubMed.
- S.-H. Huo and X.-P. Yan, J. Mater. Chem., 2012, 22, 7449–7455 RSC.
- J. Shao, Z. Wan, H. Liu, H. Zheng, T. Gao, M. Shen, Q. Qu and H. Zheng, J. Mater. Chem. A, 2014, 2, 12194–12200 CAS.
- D. J. Tranchemontagne, J. R. Hunt and O. M. Yaghi, Tetrahedron, 2008, 64, 8553–8557 CrossRef CAS PubMed.
- C. Zhang, F. Ye, S. Shen, Y. Xiong, L. Su and S. Zhao, RSC Adv., 2015, 5, 8228–8235 RSC.
- J.-J. Du, Y.-P. Yuan, J.-X. Sun, F.-M. Peng, X. Jiang, L.-G. Qiu, A.-J. Xie, Y.-H. Shen and J.-F. Zhu, J. Hazard. Mater., 2011, 190, 945–951 CrossRef CAS PubMed.
- L. Ai, C. Zhang, L. Li and J. Jiang, Appl. Catal., B, 2014, 148–149, 191–200 CrossRef CAS PubMed.
- S. Bhattacharjee, J.-S. Choi, S.-T. Yang, S. B. Choi, J. Kim and W.-S. Ahn, J. Nanosci. Nanotechnol., 2010, 10, 135–141 CrossRef CAS PubMed.
- W.-T. Xu, L. Ma, F. Ke, F.-M. Peng, G.-S. Xu, Y.-H. Shen, J.-F. Zhu, L.-G. Qiu and Y.-P. Yuan, Dalton Trans., 2014, 3792–3798 RSC.
- C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828–1831 CrossRef CAS PubMed.
- T. A. Vu, G. H. Le, C. D. Dao, L. Q. Dang, K. T. Nguyen, P. T. Dang, H. T. K. Tran, Q. T. Duong, T. V. Nguyen and G. D. Lee, RSC Adv., 2014, 4, 41185–41194 RSC.
- Z. Dong, X. Le, Y. Liu, C. Dong and J. Ma, J. Mater. Chem. A, 2014, 2, 18775–18785 CAS.
- H. J. Lee, W. Cho, E. Lim and M. Oh, Chem. Commun., 2014, 50, 5476–5479 RSC.
- Y. Li, T. Ben, B. Zhang, Y. Fu and S. Qiu, Sci. Rep., 2013, 3, 1–6 Search PubMed.
- B. Liu, H. Shioyama, H. Jiang, X. Zhang and Q. Xu, Carbon, 2010, 48, 456–463 CrossRef CAS PubMed.
- T. Chalati, P. Horcajada, R. Gref, P. Couvreur and C. Serre, J. Mater. Chem., 2011, 21, 2220–2227 RSC.
- K.-Y. A. Lin, H.-A. Chang and C.-J. Hsu, RSC Adv., 2015, 5, 32520–32530 RSC.
- E. Y. Ionashiro, F. J. Caires, A. B. Siqueira, L. S. Lima and C. T. Carvalho, J. Therm. Anal. Calorim., 2012, 108, 1183–1188 CrossRef CAS PubMed.
- M. Sastry, in Nanoparticles, ed. V. Rotello, Springer, US, 2004, ch. 9, pp. 225–250, DOI:10.1007/978-1-4419-9042-6_9.
- R. J. Bigda, Chem. Eng. Prog., 1995, 91(12), 62–66 CAS.
- S.-Y. Oh and D.-S. Shin, J. Chem. Technol. Biotechnol., 2013, 88, 145–152 CrossRef CAS PubMed.
- S. R. Pouran, A. A. A. Raman and W. M. A. Wan Daud, J. Cleaner Prod., 2014, 64, 24–35 CrossRef PubMed.
- H. Liu, T. A. Bruton, F. M. Doyle and D. L. Sedlak, Environ. Sci. Technol., 2014, 48, 10330–10336 CrossRef CAS PubMed.
- W. J. McElroy and S. J. Waygood, J. Chem. Soc., Faraday Trans., 1990, 86, 2557–2564 RSC.
- X.-Y. Yu, Z.-C. Bao and J. R. Barker, J. Phys. Chem. A, 2003, 108, 295–308 CrossRef.
- C. Liang, C.-P. Liang and C.-C. Chen, J. Contam. Hydrol., 2009, 106, 173–182 CrossRef CAS PubMed.
- W. Guo, S. Su, C. Yi and Z. Ma, Environ. Prog. Sustainable Energy, 2013, 32, 193–197 CrossRef CAS PubMed.
- A. Rastogi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 85, 171–179 CrossRef CAS PubMed.
- J. Sun, X. Li, J. Feng and X. Tian, Water Res., 2009, 43, 4363–4369 CrossRef CAS PubMed.
- J. Sun, M. Song, J. Feng and Y. Pi, Environ. Sci. Pollut. Res., 2012, 19, 1536–1543 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Appl. Catal., B, 2004, 54, 155–163 CrossRef CAS PubMed.
- E. R. Bandala, M. A. Peláez, D. D. Dionysiou, S. Gelover, J. Garcia and D. Macías, J. Photochem. Photobiol., A, 2007, 186, 357–363 CrossRef CAS PubMed.
- H. Shemer, Y. K. Kunukcu and K. G. Linden, Chemosphere, 2006, 63, 269–276 CrossRef CAS PubMed.
- T. T. H. Pham, S. K. Brar, R. D. Tyagi and R. Y. Surampalli, Ultrason. Sonochem., 2010, 17, 38–45 CrossRef CAS PubMed.
- F. Ji, C. Li, X. Wei and J. Yu, Chem. Eng. J., 2013, 231, 434–440 CrossRef CAS PubMed.
- F. Gong, L. Wang, D. Li, F. Zhou, Y. Yao, W. Lu, S. Huang and W. Chen, Chem. Eng. J., 2015, 267, 102–110 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06043e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.