
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new green-to-blue upconversion system with efficient photoredox catalytic properties†
Jorge
Castellanos-Soriano
 a,
Till J. B.
Zähringer
b,
Jorge C.
Herrera-Luna
a,
M. Consuelo
Jiménez
a,
Christoph
Kerzig
*b and
Raúl
Pérez-Ruiz
*a
a,
Till J. B.
Zähringer
b,
Jorge C.
Herrera-Luna
a,
M. Consuelo
Jiménez
a,
Christoph
Kerzig
*b and
Raúl
Pérez-Ruiz
*a
aDepartamento de Química, Universitat Politècnica de Valencia, Camino de Vera S/N, 46022, Valencia, Spain. E-mail: raupreru@upv.es
bDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55128, Mainz, Germany. E-mail: ckerzig@uni-mainz.de
First published on 5th April 2023
Abstract
The design and development of new triplet–triplet annihilation upconversion (TTA-UC) systems combining triplet sensitizers with acceptor compounds have attracted considerable interest. In this vein, sensitizers made from purely organic dyes rather than transition-metal complexes appear to be more convenient from an environmental point of view. BODIPYs are a very well-known class of dyes with applications in a widespread range of scientific areas. Owing to the versatility of BODIPYs, we present herein a new asymmetric BODIPY with excellent photophysical properties to be used as an appropriate sensitizer in a bimolecular TTA-UC system. Detailed spectroscopic measurements demonstrated the ability of this new design to sensitize TTA-UC by combination with a suitable acceptor such as 2,5,8,11-tetra-tert-butylperylene (TBPe), allowing a successful conversion of green to blue light. The singlet-excited TBPe so obtained is capable of activating aryl chlorides reductively which initiated the functionalization of N-methylpyrrole (Meerwein-type arylation) and formation of both substituted triarylethylenes (Mizoroki–Heck reaction) and heteroarene phosphonates (photo-Arbuzov reaction). Product yields reveal that our TTA-UC system behaved as a highly efficient photocatalytic entity.
Introduction
Photon upconversion based on triplet–triplet annihilation (TTA-UC) has attracted considerable interest in the last decade due to its applicability to a broad range of scientific areas such as anticounterfeiting,1 solar fuels,2,3 photovoltaics,4–7 biophotonics,8–12 and more recently in photoredox catalysis.13–15 The motivation for employing this phenomenon is mainly due to two reasons: (i) the development of numerous TTA-UC systems (from hybrid organic/inorganic to purely organic materials) that provide lots of chances for their applications,16 and (ii) the high efficiency at low excitation intensities with noncoherent light17,18 in comparison with other UC systems based on two-photon absorption.19Typical TTA-UC implies the association of multistep photochemical events (Fig. 1A). After absorption of low energy photons (hν1), the triplet excited state (T1) of the sensitizer (or a donor) is produced by intersystem crossing (ISC) from the singlet excited state (S1). Subsequently, triplets of the annihilator (or an acceptor) are populated through triplet–triplet energy transfer (TTEnT) from the triplets of the sensitizer (the Dexter mechanism). When two annihilator molecules at their triplet states are able to collide during their lifetimes, a higher singlet energy level is formed by means of triplet–triplet annihilation (TTA) and delayed upconverted fluorescence (hν2) is consequently generated. The emission energy of the new photon is greater than the energy of the initially absorbed photon, making this procedure one of the most attractive wavelength conversion technologies.
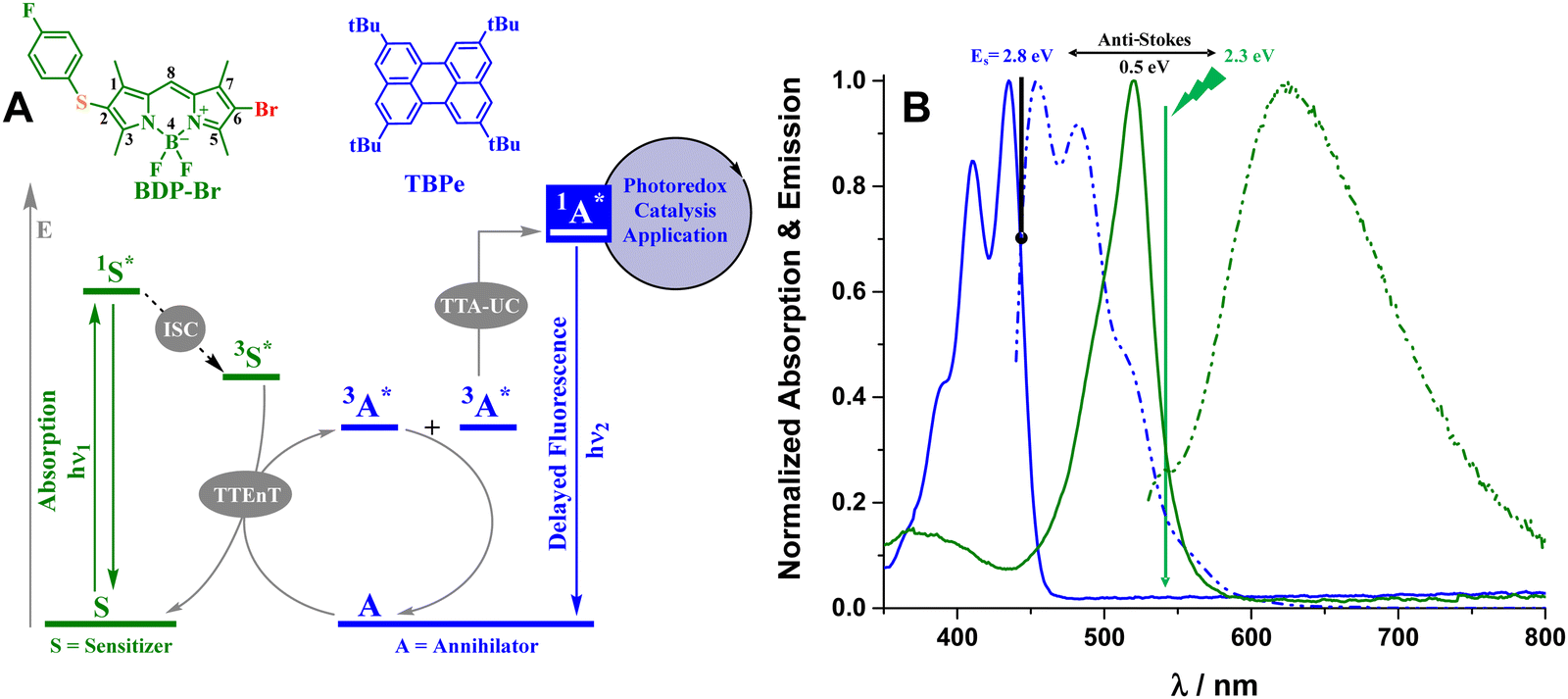 | ||
| Fig. 1 (A): Schematic representation of the corresponding photophysical events associated with the TTA-UC phenomenon enabled by the BDP-Br (sensitizer) and TBPe (annihilator) pair and its implementation in photoredox catalysis. (B): Normalized absorption (solid line) and emission (dashed dotted line) spectra of BDP-Br (green) and TBPe (blue) in 8 μM aerated acetonitrile/dimethylacetamide (4/1 v/v) solution. | ||
Based on the literature data, Pd and Pt porphyrins as sensitizers20 coupled with organic emitters like rubrene,21 perylene22 and diphenylanthracene23 are found to be the most common TTA-UC systems. The triplet formation of the sensitizers occurs efficiently via spin–orbital interactions promoted by the heavy atoms (noble metals such as Pd and Pt), permitting the UC luminescence of the system. Investigations regarding other sensitizers such as semiconductor nanocrystals,24 quantum dots,25 perovskites,26 lanthanide complexes,27 complexes with more earth-abundant metals,28,29 molecules showing TADF (thermally activated delayed fluorescence),30 and electron donor–acceptor dyads31 have also been reported. Most of these systems require expensive and complex sensitizers, presenting somehow several drawbacks including low photostability, structural instability, and a limited usable range of solvent conditions. Therefore, the employment of more readily available and organic sensitizers with high extinction coefficients in the appropriate absorption region, high photostability, and insensitivity to solvent environments appears to be more desirable.
In this context, boron dipyrromethene (BODIPY) derivatives fulfill the abovementioned criteria and the addition of halogen substituents (the so-called internal heavy-atom effect)32 directly onto the chromophore, not to the peripheral moieties, provides a direct effect on the ISC quantum yield and the population of triplets substantially enhances.33–35 Thus, the number of reports on the photosensitization of TTA-UC using BODIPY-based donors has increased over the last few years.36,37 Encouraged by this continuous evolution and on the basis of the viable and straightforward functionalization of the BODIPY framework, it would be of great interest to construct new BODIPY dyes with special properties to be used as suitable sensitizers in a bimolecular TTA-UC system.
As a matter of fact, we have herein prepared an asymmetric BODIPY (6-bromo-2-(4-fluorophenyl)thio-1,3,5,7-tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene, BDP-Br, Fig. 1A) in which the presence of heteroatoms such as sulfur atoms plus one bromine is expected to not only alter the energy level of the triplet state but also shift the UV-Vis absorption band to the green region. This molecular design permits a lower energetic light input (λexc > 530 nm) and milder reaction conditions, while avoiding iodine atoms that frequently result in rather photolabile dyes. The ability to successfully sensitize TTA-UC in solution has been demonstrated through spectroscopic methods, employing an adequate acceptor such as 2,5,8,11-tetra-tert-butylperylene (TBPe).
Moreover, the generation of highly energetic species by this phenomenon (Fig. 1B) seems useful for the application of photoredox catalysis via upconversion. Hence, the activation of an aryl chloride with a high-energy demanding bond using this novel TTA-UC couple has led to different chemical transformations such as N-methylpyrrole functionalization, the production of a triarylethylene compound and the formation of a new C–P bond. These three overall redox-neutral reactions have been investigated based on product analysis and spectroscopic measurements, supporting the relationship between the photophysical (ISC, TTEnT, and TTA) and photochemical (SET, radical trapping, and C–C or C–P couplings) events.
Results and discussion
Synthesis of BDP-Br
As stated above, one of the main goals in this study was to synthesize a new BODIPY-like sensitizer to be involved in a bimolecular system for TTA-UC purposes. Interestingly, this new entity possesses an asymmetric structure with a 4-fluorophenyl sulfide group attached to one side of the chromophore whereas a bromine atom was attached to the opposite side of the chromophore. This disposition conferred a particular electronic density with the occurrence of strong absorption of visible light, an efficient ISC, and a long-lived triplet state.Based on the reported data on sulfenylation of pyrroles catalyzed by copper,38 the addition of 4-fluorothiophenol to the starting material 3,5-dimethylpyrrole-2-carboxaldehyde was first carried out, obtaining a 50% yield of the corresponding product A (Fig. 2). The next step was crucial in the construction of an asymmetrical structure and the condensation of compound A with 2,4-dimethylpyrrole led to the formation of intermediate dipyrromethane B with 68% yield. Now, the treatment of B with a base such as triethylamine followed by boron trifluoride diethyl etherate39 yielded 66% of the BODIPY derivative BDP (2-(4-fluorophenyl)thio-1,3,5,7-tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene, Fig. 2) whose structure was confirmed by X-ray analysis. The last step consisted of the addition of a bromine atom to BDP by NBS (N-bromosuccinimide) treatment and the target compound BDP-Br was quantitatively obtained. The location of bromine at position 6 was successfully supported by X-ray analysis (Fig. 2). It should be noted that structural determination by single-crystal X-ray diffraction revealed not only that both BODIPY species crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) but also the planarity of the BODIPY core was confirmed which was extended through the bromine group in the case of BDP-Br. Thus, the design of BDP-Br could have a high impact on the ISC quantum yield. On the one hand, the presence of the 4-fluorophenyl sulfide group would give rise to a weakly emissive twisted intramolecular charge-transfer excited state (TICT)40 between the sulfur's lone electron pair and an excited electron-deficient BODIPY moiety. On the other hand, chromophore structures bearing heavy atoms such as Br would affect directly the triplet state lifetime of the molecule and would stabilize the ISC process.41 Therefore, BDP-Br appears to be the ideal choice to be applied to both TTA-UC and photoredox catalysis.
but also the planarity of the BODIPY core was confirmed which was extended through the bromine group in the case of BDP-Br. Thus, the design of BDP-Br could have a high impact on the ISC quantum yield. On the one hand, the presence of the 4-fluorophenyl sulfide group would give rise to a weakly emissive twisted intramolecular charge-transfer excited state (TICT)40 between the sulfur's lone electron pair and an excited electron-deficient BODIPY moiety. On the other hand, chromophore structures bearing heavy atoms such as Br would affect directly the triplet state lifetime of the molecule and would stabilize the ISC process.41 Therefore, BDP-Br appears to be the ideal choice to be applied to both TTA-UC and photoredox catalysis.
 | ||
| Fig. 2 (A): Synthetic route for the formation of BDP-Br. For more details on the synthetic procedures, see the ESI.† (B): ORTEP pictures of the X-ray structures of BDP and BDP-Br. | ||
General photophysical and photochemical properties of BDP-Br
Regarding optical characterization, we investigated the photophysical properties of BDP-Br, which include absorption/emission bands, molar absorption coefficients, Stokes shifts, fluorescence quantum yields, emission rate constants, singlet energies and lifetimes in different solvents (Table 1). The absorption spectra of BDP-Br in all solvents presented an absorption band (λmax.abs) in the green region (517–527 nm), being only weakly dependent on the solvent polarity (see Fig. S5 in the ESI†). It displayed molar extinction coefficients (ε), Stokes shifts (Δ![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) ) and fluorescence lifetimes in the same order of magnitude as found for similar BODIPYs.40 Interestingly, lower absolute fluorescence quantum yields (ΦF) were obtained in more polar solvents, suggesting that the intersystem crossing efficiency from the lowest singlet excited state to the triplet would be enhanced by the internal heavy-atom effect or the formation of a TICT state. Besides, the phosphorescence spectrum of BDP-Br in the ethanol matrix at 77 K was recorded at λmax of 780 nm (Fig. S6 in the ESI†), and was in accordance with those of halogen-substituted BODIPYs previously reported.42 Furthermore, the lowest triplet energy (ET) of BDP-Br was estimated to be 1.68 eV, which was found to be larger than that of TBPe (1.53 eV)15 and makes energy transfer possible from the T1 of BDP-Br to the T1 of TBPe.
) and fluorescence lifetimes in the same order of magnitude as found for similar BODIPYs.40 Interestingly, lower absolute fluorescence quantum yields (ΦF) were obtained in more polar solvents, suggesting that the intersystem crossing efficiency from the lowest singlet excited state to the triplet would be enhanced by the internal heavy-atom effect or the formation of a TICT state. Besides, the phosphorescence spectrum of BDP-Br in the ethanol matrix at 77 K was recorded at λmax of 780 nm (Fig. S6 in the ESI†), and was in accordance with those of halogen-substituted BODIPYs previously reported.42 Furthermore, the lowest triplet energy (ET) of BDP-Br was estimated to be 1.68 eV, which was found to be larger than that of TBPe (1.53 eV)15 and makes energy transfer possible from the T1 of BDP-Br to the T1 of TBPe.
| Solvent | λ max.abs | λ max.em | ε | Δc |
E S | Φ F | τ F | k F |
|---|---|---|---|---|---|---|---|---|
| a Maximum absorption/emission peak (in nm). b Molar extinction coefficient (in M−1 cm−1). c Stokes shifts (in cm−1). d Singlet energy (in eV). e Absolute fluorescence quantum yield. f Singlet lifetime (in ns). g Fluorescence rate constant (in s−1). MeOH: methanol; DMSO: dimethylsulfoxide; DCM: dichloromethane; DMA: N,N-dimethylacetamide; ACN: acetonitrile. | ||||||||
| MeOH | 517 | 612 | 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
3003 | 2.30 | 0.05 | 2.1 | 2.4 × 107 |
| ACN | 520 | 614 | 29500 |
2944 | 2.18 | 0.05 | 2.0 | 2.0 × 107 |
| DMSO | 522 | 630 | 25070 |
3284 | 2.25 | 0.04 | 2.5 | 1.6 × 107 |
| DMA | 525 | 625 | 66670 |
3048 | 2.21 | 0.04 | 2.2 | 1.8 × 107 |
| DCM | 525 | 609 | 26700 |
2628 | 2.27 | 0.10 | 2.3 | 4.3 × 107 |
| Hexane | 527 | 587 | 25070 |
1939 | 2.28 | 0.30 | 3.5 | 8.5 × 107 |
| ACN/DMA (4:1) |
524 | 625 | 36600 |
3084 | 2.26 | 0.04 | 2.5 | 1.6 × 107 |
To investigate the inherent photostability of the novel sensitizer, comparative stability measurements under monochromatic green light excitation with the widely used reference compound [Ru(bpy)3](PF6)2 were carried out.43 These semi-quantitative studies (Fig. S10, ESI†) revealed the outstanding photostability of BDP-Br – its photodegradation quantum yield is lower than that of the (already quite stable) [Ru(bpy)3]2+ by about two orders of magnitude.
ISC quantum yield of BDP-Br
One of the key parameters to determine the TTA-UC efficiency is the ISC quantum yield (ΦISC) of the sensitizer. This value is regularly estimated by singlet oxygen measurements in an indirect fashion, whereas a direct method can yield more reliable results. In this context, the ΦISC of BDP-Br was determined using laser flash photolysis actinometry44,45 with the well-defined ΦISC of [Os(phen)3](PF6)2 as the reference system. Briefly, this method is based on identical excitation conditions at a laser wavelength of 532 nm (Fig. 3A), adding a known concentration of TBPe (10 mM) to the sensitizers. Then, the triplet energy transfer efficiency ηTTEnT is compared after excitation (Fig. 3B and C) and the generation of the triplet-excited perylene46 is detected using transient absorption spectroscopy (vide infra for more detailed TTEnT measurements with the BDP-Br–TBPe pair). Since we are using the same acceptor, the relative concentration of the generated 3TBPe* is given by the ratio of at a selected wavelength (Fig. 3D). Considering the widely accepted ΦISC,Ref value for [Os(phen)3](PF6)2 as 1, a ΦISC of 0.16 for BDP-Br was estimated (see eqn (1) and the ESI† for more experimental details). Based on the literature data that unsubstituted BODIPY shows negligible ISC,47 herein it clearly appears that ISC is substantially accelerated for BDP-Br, allowing us to employ this BODIPY derivative as the sensitizer for triplet energy transfer-driven chemistry. The overall higher fluorescence quantum yield and a longer singlet lifetime of BDP compared to those of BDP-Br allow us to attribute the increased ISC quantum yield mainly to the heavy atom effect of the bromine atom rather than the fluorophenyl sulfide moiety (see Table 1 and Table S1, ESI†). The favorable absorption properties of BDP-Br with only one dominant absorption band in the green region and weak absorptivity in the blue region (vide supra) together with its high photostability make it a suitable candidate for a green-to-blue upconversion system.
at a selected wavelength (Fig. 3D). Considering the widely accepted ΦISC,Ref value for [Os(phen)3](PF6)2 as 1, a ΦISC of 0.16 for BDP-Br was estimated (see eqn (1) and the ESI† for more experimental details). Based on the literature data that unsubstituted BODIPY shows negligible ISC,47 herein it clearly appears that ISC is substantially accelerated for BDP-Br, allowing us to employ this BODIPY derivative as the sensitizer for triplet energy transfer-driven chemistry. The overall higher fluorescence quantum yield and a longer singlet lifetime of BDP compared to those of BDP-Br allow us to attribute the increased ISC quantum yield mainly to the heavy atom effect of the bromine atom rather than the fluorophenyl sulfide moiety (see Table 1 and Table S1, ESI†). The favorable absorption properties of BDP-Br with only one dominant absorption band in the green region and weak absorptivity in the blue region (vide supra) together with its high photostability make it a suitable candidate for a green-to-blue upconversion system.
 | ||
| Fig. 3 Data sets used for the ISC quantum yield determination of BDP-Br. (A): Absorption spectra of deaerated 7 μM BDP-Br and deaerated 76 μM [Os(phen)3](PF6)2 in ACN. (B): Kinetic traces of 3BDP-Br in the absence (upper panel) and presence (lower panel) of 10 mM TBPe. (C): Kinetic measurements of the [Os(phen)3](PF6)2 phosphorescence in the absence and presence of TBPe. (D): Transient absorption spectra of BDP-Br and [Os(phen)3](PF6)2 with 10 mM TBPe recorded 1 μs after excitation with 532 nm laser pulses. Laser pulses of constant energy (12 mJ) were used in this series of experiments. Inset: kinetic measurements at 484 nm. | ||
BDP-Br and TBPe pair for TTA-UC
Considering the triplet state energy of BDP-Br as 1.68 eV, TBPe was selected as the suitable annihilator owing to its rather low triplet state energy of 1.53 eV15 and high singlet-excited state energy of 2.81 eV, while the tert-butyl groups suppress unwanted excimer formation.46 Given that ultrafast photo-processes cannot be expected for our bimolecular UC system in the diluted solution,48–50 we believed that laser flash photolysis (LFP) experiments in the ns–μs timescale range was the appropriate choice to further investigate the TTA-UC system (Fig. 4). Thus, a pronounced ground state bleach and a broad absorption band centered at around 640 nm and 450 nm were observed right after the laser pulse (λexc = 532 nm and ∼10 ns pulse duration) (Fig. 4A). All signals decay to baseline with a natural lifetime of ∼100 μs (Fig. 4B) and we assigned them to the triplet-excited BDP-Br. Stern–Volmer experiments were performed using TBPe as the quencher, yielding a TTEnT rate constant of 4.1 × 109 M−1 s−1 in line with the reported triplet energies (Fig. 4B). With the addition of TBPe, new absorption bands are detected in the TA spectrum (red) after BDP-Br has been fully quenched and the kinetic traces in Fig. 4A (inset) clearly demonstrate that quenching and photoproduct formation occur with identical rates. The new transient species shows absorption peaks at 484 nm and 453 nm which can be safely ascribed to the triplet state of TBPe (Fig. 4A, red spectrum).46 | ||
| Fig. 4 Mechanistic LFP experiments using 532 nm laser pulses. If not stated otherwise, a solution of 9 μM BDP-Br and 100 μM TBPe in deaerated ACN was used. (A) TA spectra of BDP-Br in the presence (red) and absence (blue) of TBPe recorded after 2 μs (blue) or 30 μs (red). Inset: corresponding kinetic traces. (B) BDP-Br triplet quenching experiments with increasing TBPe concentrations. Inset: the corresponding Stern–Volmer plot. (C) Time-gated emission spectra of TBPe (green, integration time: 2–200 μs), BDP-Br (dark yellow, integration time: 0–100 ns) and BDP-BrwithTBPe (purple, integration time: 2–200 μs). For comparison, an emission spectrum of a strongly diluted (1 μM) TBPe solution (dotted grey line) after direct excitation using 355 nm laser pulses is shown. (D) Kinetic traces of 3TBPe at 484 nm and UC emission at 480 nm. Inset: the kinetic trace of 3TBPe on a longer timescale. | ||
Having established the TTEnT step, we then turned to the annihilation process. Individually, the sensitizer only shows fluorescence peaking at ∼610 nm while no emission is detected from the annihilator after pulsed 532 nm excitation (Fig. 4C). When combined, however, emission signals with a spectral upshift respective to the excitation wavelength with maxima at 480 nm and 461 nm are detected. The detection time window (2–200 μs after excitation) allowed us to exclude the prompt emission of TBPe and BDP-Br such that delayed fluorescence expected for TTA-UC can be selectively observed. A spectrum containing the prompt emission of TBPe in a highly diluted solution is shown in Fig. 4C for comparison. It should be noted that inner filter effects in the absorption–emission overlap region become apparent at concentrations >10 μM (Fig. S11, ESI†). Kinetic traces of both the 3TBPe absorption at 484 nm and the 1TBPe* emission concurrently reach a maximum at ∼11 μs (Fig. 4D). The faster increase and decrease of the UC emission compared to the triplet concentration is in line with the bimolecular nature of the annihilation process and provides further evidence for TTA-UC.51
In addition to the time-resolved studies that provided deep insights into the novel TTA-UC system, we investigated the UC efficiency using a 514 nm cw laser for excitation (Fig. 5). The UC emission peaking at 484 nm gradually enhanced with increasing laser intensity (10–100 mW). A spectral upshift of 0.40 eV was observed when the singlet-excited state energy of TBPe and the excitation wavelength (almost perfectly corresponding to the BDP-Br absorption maximum) were considered. To determine the UC quantum yield, ΦUC, [(Ru(bpy)3](PF6)2 was used as the reference system (see the ESI† for details of the method).52 As depicted in the inset of Fig. 5, the ΦUC for our metal-free system (with a theoretical limit of 50%) was found to reach a maximum of 2.7%. Despite the fact that this value was lower than others from quantum-yield-optimized systems, the performance of the current TTA-UC system to act as the photocatalyst to drive challenging photoredox reactions appeared to be sufficient since well-known bimolecular systems that show ΦUC well below 1% were able to trigger photochemical transformations such as reductive pollutant degradation.53 Furthermore, the moderate quantum yield is likely compensated by the very high sensitizer and annihilator photostability (vide supra).
 | ||
| Fig. 5 Photon upconversion studies of 28 μM BDP-Br and 100 μM TBPe in deaerated ACN using a 514 nm cw laser for excitation. Main plot: UC Emission spectra with increasing laser intensity. Inset: the external UC quantum yield plotted against the laser intensity. See the text and ESI† for details. | ||
Application of the BDP-Br/TBPe system to photoredox catalysis
Several biphotonic excitation strategies in photoredox catalysis have been developed over the last few years in order to address critical bond activations or electron transfers under mild conditions and using lower-energy visible light. Reactions such as two-photon absorption (TPA)54 and consecutive photoinduced electron transfer (conPET)55 can be carried out using a single photocatalyst. In the case of TTA-UC, a bimolecular system governs the catalytic cycle by the energy input of two photons where the primary photosensitizer transfers the absorbed energy to the co-catalyst (annihilator). Although the application of this technology to chemical transformations has been recently reviewed,13 there is still plenty of room concerning the employment of improved systems. Therefore, in view of the outstanding TTA-UC properties of the BDP-Br/TBPe system, this couple was embedded as the photocatalyst into different challenging intermolecular coupling reactions.To this end, we first explored the feasibility of activating 2-acetyl-5-chlorothiophene as the model reagent with visible light employing the BDP-Br/TBPe system. Quenching experiments were carried out by means of transient absorption spectroscopy (TAS) under conditions that are similar to those of the photocatalytic studies (Fig. 6A). Clearly, the delayed fluorescence intensity of TBPe gradually decreased in the presence of increasing amounts of the quencher. Indeed, the Stern–Volmer analysis of delayed emission quenching (Fig. 6A inset) revealed a KSV of 117 M−1; taking into account this value and the TBPe singlet lifetime (τF = 5 ns in ACN/DMA 4/1 v/v, see the ESI† for details), the fluorescence quenching rate constant, kq(S1), was found to be 2.3 × 1010 M−1 s−1, indicating that the target compound quenches the delayed 1TBPe* with a diffusion-controlled rate. A very similar Stern–Volmer constant (KSV = 135 M−1) was obtained for 1TBPe* quenching by 2-acetyl-5-chlorothiophene upon the direct excitation of TBPe (λexc = 405 nm) in neat ACN, supporting the UC-driven activation mechanism.
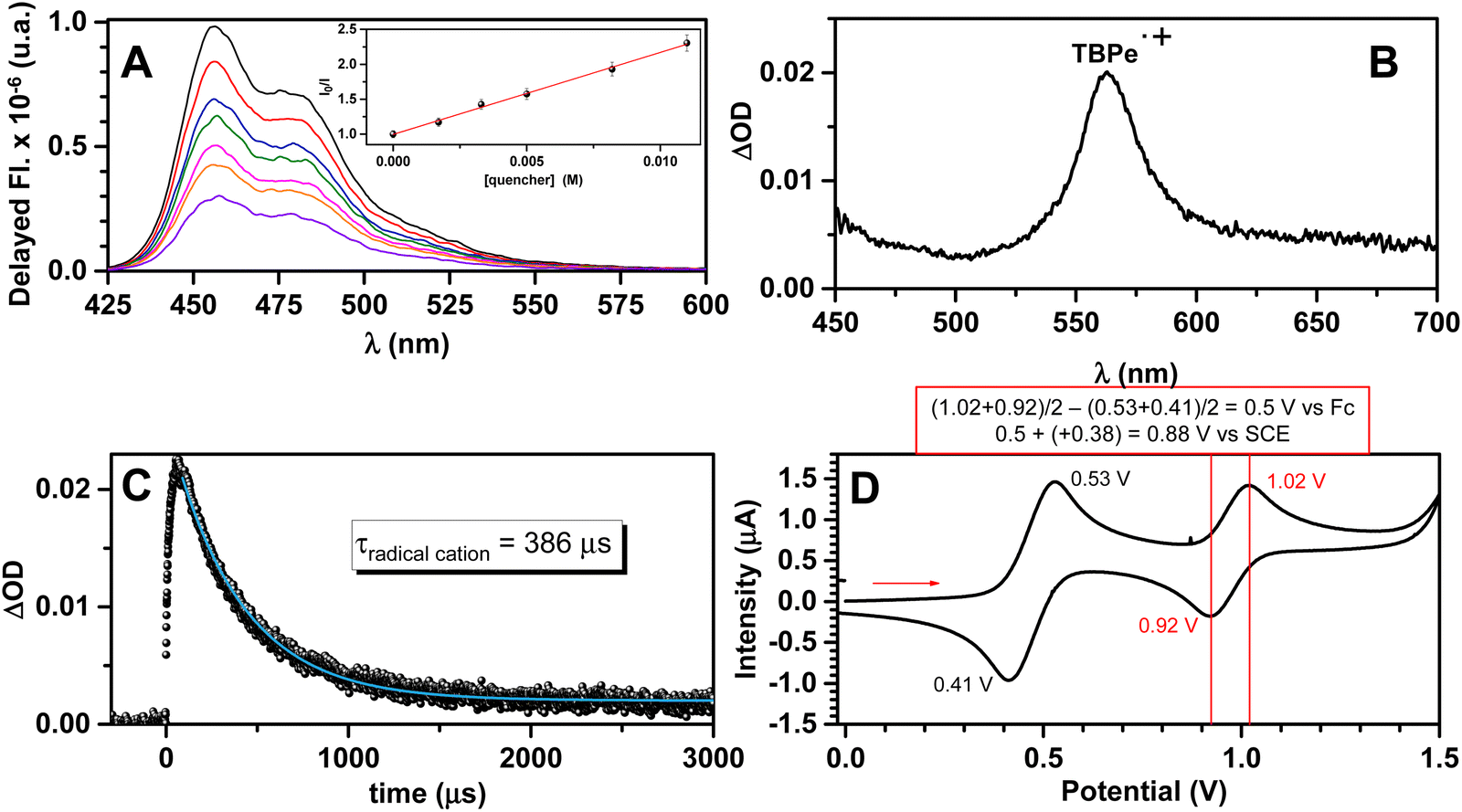 | ||
| Fig. 6 Activation of the model reagent by delayed fluorescence 1TBPe*. (A): Delayed fluorescence spectra of a mixture of BDP-Br (0.1 mM) and TBPe (1 mM) in anaerobic ACN/DMA (4/1 v/v) solution recorded 2 μs after the laser pulse (λexc = 532 nm) in the presence of increasing amounts of 2-acetyl-5-chlorothiophene (up to 11 mM); inset: the Stern–Volmer plot to obtain kq (S1); experimental errors were lower than 5% of the obtained values. (B): TAS measurements (λexc = 532 nm) of BDP-Br (0.1 mM) and TBPe (1 mM) in the anaerobic ACN solution in the presence of 10 mM of 2-acetyl-5-chlorothiophene recorded 140 μs after the laser pulse. (C): Temporal profile of the TBPe radical cation monitored at 560 nm. The blue line is the fit function used to estimate the lifetime. (D): Cyclic voltammogram for an ACN solution of TBPe (0.1 mM). | ||
To further demonstrate the interaction between TTA-generated 1TBPe* and 2-acetyl-5-chlorothiophene through an initial electron transfer step and to identify possible transient intermediates, additional time-resolved studies were performed (Fig. 6B). Hence, irradiation of the BDP-Br/TBPe system (λexc = 532 nm) in the presence of 2-acetyl-5-chlorothiophene was carried out. Interestingly, a new absorption band at ca. 560 nm was clearly observed which was safely ascribed to the TBPe radical cation (TBPe˙+) based on the literature data.56 From the temporal profile at 560 nm (Fig. 6C), a lifetime on the order of 400 μs was estimated for this intermediate.
In addition, thermodynamic analysis for ET processes strongly supported these data. The free energy change (ΔGET in kcal mol−1) associated with the ET process was estimated by the application of the Weller equation.57 The oxidation potential (E°(TBPe˙+/TBPe)) of TBPe (Fig. 6D) and its singlet and triplet energy (E*(S1 or T1), for details see the ESI†) were experimentally obtained. Their corresponding values are 0.88 V vs. SCE, 64.6 and 34.6 kcal mol−1, respectively. The resulting excited-state oxidation potential for the TBPe's S1 state is as high as −1.93 V vs. SCE, which should permit challenging photocatalytic reductions via TTA-UC.
Considering the reduction potential of the model reagent 2-acetyl-5-chlorothiophene (−1.66 V vs. SCE),58 the ΔGET (S1) and ΔGET (T1) values were estimated as −6.0 kcal mol−1 and +23.9 kcal mol−1, respectively. Therefore, the activation of 2-acetyl-5-chlorothiophene by delayed 1TBPe* would be an exergonic process, whereas the mechanism from 3TBPe* is thermodynamically unfeasible. Same calculations were indeed performed for BDP-Br with the question whether this species could act as an activator. Considering the experimental data of BDP-Br (Eox = + 1.23 V vs. SCE, E(S1) = 52.1 kcal mol−1 and E(T1) = 38.7 kcal mol−1, see the ESI† for details), both ΔGET (S1) and ΔGET (T1) were estimated as positive values (+13.8 kcal mol−1 and +27.2 kcal mol−1, respectively), ruling out any ET from the excited BDP-Br.
These results allowed us to conclude that the activation of 2-acetyl-5-chlorothiophene was successfully powered using the TTA-UC system through a SET process (Fig. 7). Thus, the conversion of low energy light (2.3 eV) into the higher energy delayed 1TBPe* (2.8 eV) allowed the one-electron injection, giving rise to the formation of the unstable radical anion of the model compound which rapidly released chloride to afford the corresponding heteroarene radical. This reactive species could be now trapped by adequate nucleophiles to produce complex compounds (Fig. 7). Therefore, we investigated the application of this cascade of processes (TTA-UC + SET + intermolecular coupling) to different important reactions such as the Meerwein arylation or the Mizoroki–Heck reaction in order to functionalize the thiophene ring by C–C coupling and the Arbuzov-like reaction to construct the corresponding thiophene phosphonate.
 | ||
| Fig. 7 Mechanism of TTA-UC enabled by the BDP-Br(S)/TBPe(A) pair and its adaptation to photoredox catalysis through a SET-initiated reductive activation of an aryl chloride. Involvement of sequential processes ISC, TTEnT, TTA and SET as key steps. ISC = intersystem crossing; TTEnT = triplet–triplet energy transfer; TTA = triplet–triplet annihilation; SET = single-electron transfer. | ||
The photocatalyzed Meerwein arylation is a classic reaction in which aryl diazonium salts were employed as aryl radical precursors.59 However, most of these species are not neither commercially available nor stable in the presence of air, being their employment sometimes detrimental. In this context, aryl halides were used as alternative electrophilic reagents in such reactions. Recently, a TTA-UC system based on the diiodoBOPHY-like derivative as the sensitizer and 9,10-diphenylanthracene as the annihilator photocatalyzed the coupling reaction between our model compound (2-acetyl-5-chlorothiophene) and N-methylpyrrole, yielding 62% of the final biaryl product.60 The instability of the sensitizer under the irradiation conditions (λexc = 450 nm) could be an important factor for the incomplete conversion of the starting material or the lack of more product formation. Thus, we focused our attention on the same reaction utilizing BDP-Br/TBPe as the TTA-UC photocatalytic system. Optimal conditions (see Table S2 for the optimization of the model reaction in the ESI†) involved catalytic amounts of BDP-Br (2% mol) and TBPe (10% mol), with pulsed laser irradiation in the green region at 532 nm in ACN/DMA (4/1 v/v) solution for 4 hours under anaerobic conditions. The result was markedly improved to that previously obtained with an 83% yield of the final product. It is worth mentioning that the ACN/DMA mixture as a solvent was found to be ideal not only for enhancing the selectivity of the process but also for favoring the complete dissolution of TBPe.
At the same shape, TTA-UC technology was very recently applied to the photocatalyzed Mizoroki–Heck reaction to obtain substituted triarylethylenes using aryl halides (especially bromides) as aryl radical precursors.61 With the optimizing conditions stated above, our model compound was subjected to green-light photolysis (λexc = 532 nm) in the presence of diphenylethylene as the trapping agent and catalytic amounts of the BDP-Br/TBPe system (for detailed information on the reaction conditions, see the ESI†). Full conversion of the starting material was observed after 4 hours of irradiation and an excellent 89% yield of the final product was obtained. This finding notably improved the previous example (i.e., 49%)61 despite the lower-energy light input and the shortened irradiation time, confirming the efficiency of the photocatalytic activity of the TTA-UC couple.
Biphotonic-based methods for the C–P bond formation of (hetero)aryl halides with trialkyl phosphite via the visible-light-induced photo-Arbuzov reaction have been barely explored.58,62 In particular, these studies followed a consecutive photoinduced electron transfer (ConPET) mechanism using organic dyes as photocatalysts together with a sacrificial agent as the base. As far as we are aware, the photo-Arbuzov reaction catalyzed using a TTA-UC system has not been attempted yet and we were interested whether this protocol could be a powerful strategy to forge new C(sp2)–P bonds under mild conditions. The phosphorylation of the model compound, 2-acetyl-5-chlorothiophene, was thus carried out in the presence of BDP-Br/TBPe as the TTA-UC photocatalytic system under the optimal conditions (for detailed information on the reaction conditions, see the ESI†). Interestingly, the targeted product was obtained with an excellent 90% yield, which was comparable to the previously reported value (91%)58 in spite of the absence of sacrificial agents.
Conclusions
In summary, we have developed a purely organic upconversion system that efficiently catalyzes challenging reductions under green-light excitation conditions. Our tailor-made BODIPY sensitizer shows efficient triplet formation and energy transfer to the selected annihilator, while possessing outstanding photostability properties compared to the most famous photoactive metal complex as the reference. The excited annihilator singlet obtained via upconversion is a strong photoreductant providing about 2 V of reductive power. Given the low price and durability of green light sources such as frequency-doubled Nd:YAG lasers (which are also the key components of most commercial laser pointers) and green diode-based lamps compared to blue lasers and diodes, our approach should be within reach for most photochemical laboratories to activate substrates in a selective fashion under metal-free conditions.Author contributions
All authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts of interest.Acknowledgements
We thank the Generalitat Valenciana (project CIDEGENT/2018/044), the Spanish Government (project PID2019-105391GB-C22 funded by MCIN/AEI/10.13039/501100011033), the German Federal Environmental Foundation (DBU, PhD fellowship to T. J. B. Z., grant number 20022/028) and the German Research Foundation (DFG, grant number KE 2313/3-1) for financial support. We also thank Prof. Julia Pérez-Prieto for spectroscopic facilities.References
- A. L. Hagstrom, H.-L. Lee, M.-S. Lee, H.-S. Choe, J. Jung, B.-G. Park, W.-S. Han, J.-S. Ko, J.-H. Kim and J. H. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 8985–8992 CrossRef CAS PubMed
.
- M. Barawi, F. Fresno, R. Pérez-Ruiz and V. A. de la Peña O’Shea, ACS Appl. Energy Mater., 2019, 2, 207–211 CrossRef CAS
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 RSC
- L. Frazer, J. K. Gallaher and T. W. Schmidt, ACS Energy Lett., 2017, 2, 1346–1354 CrossRef CAS
- T. Dilbeck and K. Hanson, J. Phys. Chem. Lett., 2018, 9, 5810–5821 CrossRef CAS PubMed
- Y. Zhou, C. Ruchlin, A. J. Robb and K. Hanson, ACS Energy Lett., 2019, 4, 1458–1463 CrossRef CAS
- H.-L. Lee, J. H. Park, H.-S. Choe, M.-S. Lee, J.-M. Park, N. Harada, Y. Sasaki, N. Yanai, N. Kimizuka, J. Zhu, S. H. Bhang and J.-H. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 26571–26580 CrossRef CAS PubMed
- Q. Liu, M. Xu, T. Yang, B. Tian, X. Zhang and F. Li, ACS Appl. Mater. Interfaces, 2018, 10, 9883–9888 CrossRef CAS PubMed
- S. H. C. Askes, M. S. Meijer, T. Bouwens, I. Landman and S. Bonnet, Molecules, 2016, 21, 1460 CrossRef PubMed
- Q. Liu, T. Yang, W. Feng and F. Li, J. Am. Chem. Soc., 2012, 134, 5390–5397 CrossRef CAS PubMed
- C. Wohnhaas, A. Turshatov, V. Mailänder, S. Lorenz, S. Baluschev, T. Miteva and K. Landfester, Macromol. Biosci., 2011, 11, 772–778 CrossRef CAS PubMed
- R. Pérez-Ruiz, Top. Curr. Chem., 2022, 380(23), 1–12 Search PubMed
- F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed
- S. Wen, J. Zhou, P. J. Schuck, Y. D. Suh, T. W. Schmidt and D. Jin, Nat. Photonics, 2019, 13, 828–838 CrossRef CAS
- M. Wu, T.-A. Lin, J. O. Tiepelt, V. Bulović and M. A. Baldo, Nano Lett., 2021, 21, 1011–1016 CrossRef CAS PubMed
- E. Radiunas, S. Raišys, S. Juršėnas, A. Jozeliūnaitė, T. Javorskis, U. Šinkevičiūtė, E. Orentas and K. Kazlauskas, J. Mater. Chem. C, 2020, 8, 5525–5534 RSC
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC
- Y. Y. Cheng, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66–71 RSC
- S. Hoseinkhani, R. Tubino, F. Meinardi and A. Monguzzi, Phys. Chem. Chem. Phys., 2015, 17, 4020–4024 RSC
- D. Dzebo, K. Moth-Poulsen and B. Albinsson, Photochem. Photobiol. Sci., 2017, 16, 1327–1334 CrossRef CAS PubMed
- R. Weiss, Z. A. VanOrman, C. M. Sullivan and L. Nienhaus, ACS Mater. Au, 2022, 2, 641–654 CrossRef CAS PubMed
- Z. A. VanOrman, C. R. Conti, G. F. Strouse and L. Nienhaus, Chem. Mater., 2021, 33, 452–458 CrossRef CAS
- Z. A. VanOrman, H. K. Drozdick, S. Wieghold and L. Nienhaus, J. Mater. Chem. C, 2021, 9, 2685–2694 RSC
- N. Kiseleva, P. Nazari, C. Dee, D. Busko, B. S. Richards, M. Seitz, I. A. Howard and A. Turshatov, J. Phys. Chem. Lett., 2020, 11, 2477–2481 CrossRef CAS PubMed
- M. Yang, S. Sheykhi, Y. Zhang, C. Milsmann and F. N. Castellano, Chem. Sci., 2021, 12, 9069–9077 RSC
- C. Wang, F. Reichenauer, W. R. Kitzmann, C. Kerzig, K. Heinze and U. Resch-Genger, Angew. Chem., Int. Ed., 2022, 61, e202202238(3 of 8) Search PubMed
- W. Chen, F. Song, S. Tang, G. Hong, Y. Wu and X. Peng, Chem. Commun., 2019, 55, 4375–4378 RSC
- Y. Hou, J. Liu, N. Zhang and J. Zhao, J. Phys. Chem. A, 2020, 124, 9360–9374 CrossRef CAS PubMed
- P. Yuster and S. I. Weissman, J. Chem. Phys., 1949, 17, 1182 CrossRef CAS
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162–12163 CrossRef CAS PubMed
- X. Xiao, W. Tian, M. Imran, H. Cao and J. Zhao, Chem. Soc. Rev., 2021, 50, 9686–9714 RSC
- J. M. Lee, J.-M. Park, J. Ho Yoon, J.-H. Kim and J. P. Kim, ChemPhotoChem, 2023, e202200326 Search PubMed
- K. Chen, Y. Dong, X. Zhao, M. Imran, G. Tang, J. Zhao and Q. Liu, Front. Chem., 2019, 7, 821 CrossRef CAS PubMed
- N. A. Bumagina, E. V. Antina, A. A. Ksenofontov, L. A. Antina, A. A. Kalyagin and M. B. Berezin, Coord. Chem. Rev., 2022, 469, 214684 CrossRef CAS
- D. Alves, R. G. Lara, M. E. Contreira, C. S. Radatz, L. F. B. Duarte and G. Perin, Tetrahedron Lett., 2012, 53(26), 3364–3368 CrossRef CAS
- E. V. Antina, M. B. Berezin, N. A. Dudina, S. L. Burkova and A. Y. Nikonovab, Russ. J. Inorg. Chem., 2014, 59(10), 1427–1434 CrossRef
- E. Palao, T. Slanina and P. Klán, Chem. Commun., 2016, 52, 11951–11954 RSC
- K. N. Solov'ev and E. A. Borisevich, Phys.-Usp., 2005, 48, 231–253 CrossRef
- X.-F. Zhang, X. Yang, K. Niu and H. Geng, J. Photochem. Photobiol., A, 2014, 285, 16–20 CrossRef CAS
- J. B. Bilger, C. Kerzig, C. B. Larsen and O. S. Wenger, J. Am. Chem. Soc., 2021, 143, 1651–1663 CrossRef CAS PubMed
- M.-S. Bertrams and C. Kerzig, Chem. Commun., 2021, 57, 6752–6755 RSC
- P. Müller and K. Brettel, Photochem. Photobiol. Sci., 2012, 11, 632–636 CrossRef PubMed
- C. Ye, V. Gray, K. Kushwaha, S. K. Singh, P. Erhart and K. Börjesson, Phys. Chem. Chem. Phys., 2020, 22, 1715–1720 RSC
- A. Schmitt, B. Hinkeldey, M. Wild and G. Jung, J. Fluoresc., 2009, 19, 755–758 CrossRef CAS PubMed
- C. Wang, M. Sun, H. Wang and G. Zhao, J. Phys. Chem. Lett., 2023, 14, 164–169 CrossRef CAS PubMed
- C. Wang, L. Ma, S. Wang and G. Zhao, J. Phys. Chem. Lett., 2021, 12, 12129–12134 CrossRef CAS PubMed
- C. Wang, Y. Liu, X. Feng, C. Zhou, Y. Liu, X. Yu and G. Zhao, Angew. Chem., Int. Ed., 2019, 59, 11642–11646 CrossRef PubMed
- Y. Murakami and K. Kamada, Phys. Chem. Chem. Phys., 2021, 23, 18268–18282 RSC
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishic and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC
- B. Pfund, D. M. Steffen, M. R. Schreier, M.-S. Bertrams, C. Ye, K. Börjesson, O. S. Wenger and C. Kerzig, J. Am. Chem. Soc., 2020, 142, 10468–10476 CrossRef CAS PubMed
- C. Kerzig, X. Guo and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS PubMed
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed
- C. A. Steren, H. van Willigen, L. BiczoÂk, N. Gupta and H. Linschitz, J. Phys. Chem., 1996, 100, 8920–8926 CrossRef CAS
- ΔGET (kcal mol−1) = 23.06 x (Eox – ERED) – E*(S1 or T1): A. Weller, Z. Phys. Chem., 1982, 133, 93–98 CrossRef CAS
- J. C. Herrera-Luna, D. Díaz Díaz, M. C. Jiménez and R. Pérez-Ruiz, ACS Appl. Mater. Interfaces, 2021, 13, 48784–48794 CrossRef CAS PubMed
- D. Prasad Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734–4743 CrossRef PubMed
- C. G. López-Calixto, M. Liras, V. A. de la Peña O’Shea and R. Pérez-Ruiz, Appl. Catal., B, 2018, 237, 18–23 CrossRef
- F. Garnes-Portolés, R. Greco, J. Oliver-Meseguer, J. Castellanos-Soriano, M. C. Jiménez, M. López- Haro, J. C. Hernández-Garrido, M. Boronat, R. Pérez-Ruiz and A. Leyva-Pérez, Nat. Catal., 2021, 4, 293–303 CrossRef
- R. S. Shaikh, S. J. S. Düsel and B. König, ACS Catal., 2016, 6, 8410–8414 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2236113 and 2236116. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cp00811h |
| This journal is © the Owner Societies 2023 |