
Synthesis of novel 1,4-disubstituted 1,2,3-triazolo-bosentan derivatives – evaluation of antimicrobial and anticancer activities and molecular docking†
K. Easwaramoorthiac,
A. Jeya Rajendran*a,
K. Chennakesava Raobc,
Y. Arunb,
C. Balachandrand,
P. T. Perumalb,
Nobuhiko Emid,
S. M. Mahalingame,
V. Duraipandiyanfg and
N. A. Al-Dhabig
aDepartment of Chemistry, Loyola College, Chennai-600034, India. E-mail: jeyaadmaterial@gmail.com; Tel: +91-944-411-6528
bOrganic Chemistry Division, CSIR-CLRI, Chennai-600020, India
cR&D Centre, Malladi Drugs & Pharmaceuticals Ltd., Chennai-600124, India
dDepartment of Hematology, Fujita Health University, 1-98, Dengakugakubo, Kutsukake-cho, Toyoake, Aichi 470-1192, Japan
eCentre for Drug Discovery and Department of Chemistry, Purdue University, Indiana, USA
fDivision of Ethnopharmacology, Entomology Research Institute, Loyola College, Chennai-600034, India
gDepartment of Botany and Microbiology, Addiriyah Chair for Environmental Studies, College of Science, King Saudi University, P.O.Box.2455, Riyadh-11451, Saudi Arabia
First published on 18th November 2015
Abstract
Novel 1,4-disubstituted 1,2,3-triazolo bosentan derivatives 1a–n from bosentan 2 were synthesized in good yields by sequential chlorination, azidation followed by Cu(I) catalyzed 1,3-dipolar cycloaddition. All obtained compounds 1a–n were evaluated for their antimicrobial and in vitro anticancer activities and by in silico docking studies. Among all tested compounds 1e,f and 1h–j show better antimicrobial activities against the tested bacteria and fungi. When subjected to anticancer testing, compounds 1g–j and 1n show significant activities against both A549 and SKOV-3 cell lines with IC50 values at 7.81 μg mL−1 and among them compound 1i exhibited very potent activity. In addition, no toxicity was calculated up to 2 mg mL−1 in Vero cells. In silico studies were conducted to investigate the possible bonding modes of 1a–n with target receptors namely DNA topoisomerase IV (4 EMV) and anaplastic lymphoma kinase (2XP2). Among them, compounds 1e and 1h show maximum binding energies with 4EMV and 2XP2 receptors, respectively which also exhibited good antimicrobial and potent anticancer activities.
Introduction
As per the WHO, multidrug-resistant organisms (MDRO) are an increasing threat to human life as well as the animal kingdom.1–5 Microorganisms gain resistance by mutation of their DNA and tolerate the effect of standard antimicrobial drugs resulting in uncured diseases/infections. Cancer is another major threat to human life. When in the early years of a person’s life, normal body cells divide faster to allow him/her to grow well. The division of cell rate decreases when the person becomes an adult and it happens only to replace exhausted cells or to heal injuries. Abnormal growth of cells results in the formation of cancers which are very difficult to cure and end with death in most cases. Most anticancer drugs have side effects on normal cells and result in some abnormal health conditions. Hence, this situation stresses the need for the recurrent development of new drugs that are more efficient and less expensive6 than the existing drugs.Endothelin A (ETA) and B (ETB) receptors, have been implicated in the development of several cancers through activation of pathways involved in cell proliferation, migration, invasion, osteogenesis and angiogenesis.7 Targeting the ET receptor and its antagonism constitute an attractive and challenging approach for cancer therapy.8,9 A large number of ET antagonists have been developed and studied in clinical trials for chronic heart failure, hypertension, cancer and fibrosis.10–13 Bosentan, a dual ETA/ETB receptor antagonist is approved for the treatment of pulmonary arterial hypertension14–16 and used in combination chemotherapy with cisplatin17 and paclitaxel for ovarian cancer.18 The development of a potent bosentan derivative as a specific, selective and dual ET receptor antagonist represents an effective treatment for cancer as a mono drug. Structural modification of bosentan may lead to an increase in selective dual activity with the ET receptor and may cause less or no hepatotoxic (damage to liver – a side effect of bosentan) and anemia.19,20 The current research activity focuses on the preparation of various 1,4-disubstituted 1,2,3-triazoles linked with the pyrimidine ring of bosentan to give triazolo-bosentan derivatives in order to test their anticancer and antimicrobial activities and to carry out molecular docking studies.
Generally, having two is better than one and the same concept is applicable to hybrid drugs which can have enhanced biological activity and can address issues of drug resistance.21–23 Many hybrid drugs are known to have advantages, as they can potentially overcome pharmacokinetic drawbacks better than their precursors and conventional drugs.24–26 These can be prepared by adjoining to two different drugs by a covalent bond or by an adequate fusion.27 Hybrid drugs such as NOSH-aspirin,28 artemisinin-quinine,29 reversed chloroquine30 are well known (Fig. 1).
 | ||
| Fig. 1 Hybrid drugs. | ||
Hybrid compounds of pyrimidines which are associated with other heterocycles exhibit good biological activities.31 Presently, the interest in developing new hybrid molecules from pyrimidine derivatives is increasing with vast applications in medicinal chemistry.32–37 Click chemistry is a brilliant concept in organic chemistry which involves synthesizing complex molecules in high yields with simple isolation techniques.38,39 Various 1,4-disubstituted 1,2,3-triazole derivatives have been reported in the literature using this well known click chemistry approach by Cu(I) catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC)40–45 which have broad applications in medicinal chemistry.46–49
Results and discussion
Chemistry
The key intermediate azide 4 was prepared by heating bosentan 2 with thionyl chloride in chloroform followed by treating with sodium azide in N,N-dimethylacetamide (DMA) (Scheme 1). The targeted new bis-pyrimidinyl linked 1,2,3-triazolo-bosentan derivatives 1a–n were prepared via one pot synthesis by reacting azide 4 with various terminal alkynes 5a′–k′ and 5l–n in the presence of CuI catalyst and N,N-diisopropylethylamine (DIPEA) in aqueous n-butanol medium at ambient temperature using a regular click chemistry approach (Scheme 2). Various conditions have been tried to optimize the method (Table 1). Terminal alkynes 5a′–f′ were obtained in situ by heating their corresponding amines 5a–f with propargyl chloride in the presence of a base (Scheme 2). Terminal alkynes 5g′–k′ were also made in situ by converting their corresponding acids 5g–k to acid chlorides followed by treatment with propargyl alcohol (Scheme 2). Terminal alkynes 5l–n were procured commercially and used as such. About 78–95% of overall yields (Scheme 4 and Table 2) were obtained when the optimized reaction conditions mentioned in this article were followed. Compounds 1a–n were obtained as pale brown to brown amorphous solids and further purification also resulted in amorphous solids hence single crystal XRD studies were not performed.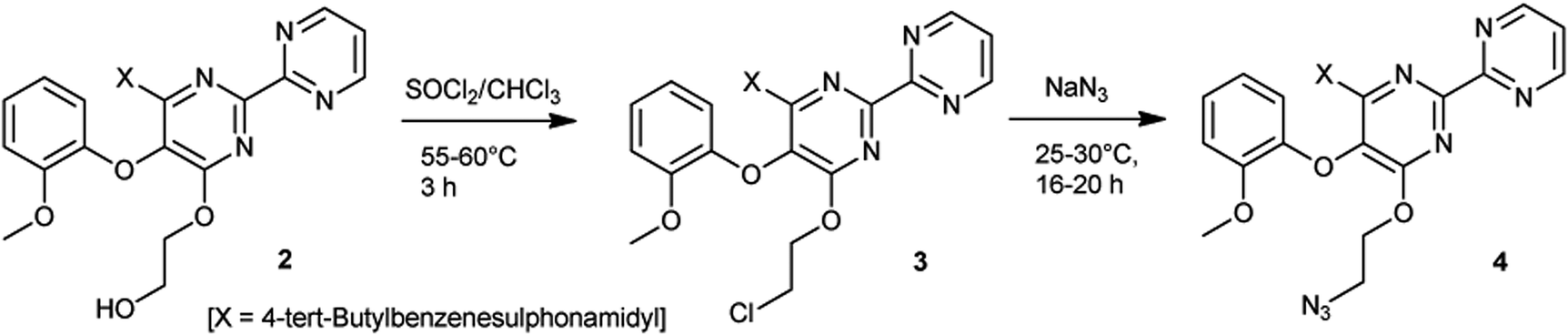 | ||
| Scheme 1 Synthesis of azide 4 from bosentan 2. | ||
 | ||
| Scheme 2 Overview of the synthesis of novel 1,4-disubstituted 1,2,3-triazoles 1a–n. | ||
 | ||
| Scheme 3 Synthesis of 1l – reaction optimization. | ||
| Entry | Base | Catalyst (mol%) | Solvent | Temp (°C) | Time (h) | Yieldb (%) |
|---|---|---|---|---|---|---|
| a DIPEA – diisopropylethylamine, TEA – triethylamine.b Isolated yields. | ||||||
| 1 | DIPEA | CuI (10) | n-BuOH | 25–30 | 10 | 75 |
| 2 | DIPEA | CuBr (10) | n-BuOH | 25–30 | 10 | 15 |
| 3 | DIPEA | CuCl (10) | n-BuOH | 25–30 | 10 | Trace |
| 4 | DIPEA | CuI (5) | n-BuOH | 25–30 | 10 | 46 |
| 5 | DIPEA | CuI (20) | n-BuOH | 25–30 | 10 | 93 |
| 6 | DIPEA | CuI (10) | H2O | 25–30 | 10 | 48 |
| 7 | DIPEA | CuI (10) | n-BuOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
25–30 | 10 | 95 |
| 8 | DIPEA | CuI (10) | EtOH | 25–30 | 10 | Trace |
| 9 | DIPEA | CuI (10) | EtOH | 45–50 | 20 | 15 |
| 10 | DIPEA | CuI (10) | EtOH/H2O (1:1) |
45–50 | 20 | 27 |
| 11 | DIPEA | CuI (10) | THF | 25–30 | 20 | 60 |
| 12 | TEA | CuI (10) | n-BuOH | 25–30 | 10 | 31 |
| 13 | TEA | CuI (10) | THF | 25–30 | 10 | 46 |
| 14 | KOH | CuI (20) | n-BuOH | 25–30 | 10 | Trace |
| 15 | KOH | CuI (20) | n-BuOH | 50–55 | 20 | Trace |
 | ||
| Scheme 4 One pot synthesis of 1,4-disubstituted 1,2,3-triazoles 1a–n from azide 4 using click chemistry. | ||
| Compound | Terminal alkynes prepared from | R | Mpa (°C) | Yieldb (%) |
|---|---|---|---|---|
| a Uncorrected melting points.b Obtained yields without any purification.c Obtained from commercial sources and used as such. | ||||
| 1a | Ephedrine | [N-Methyl-(1-hydroxy-1-phenyl)-2-propylamino] | 122–125 | 88 |
| 1b | Norephedrine | [(1-Hydroxy-1-phenyl)-2-propylamino] | 140–143 | 78 |
| 1c | Selegiline | [1-Phenyl-2-propylamino] | 99–102 | 90 |
| 1d | Phenylephrine | [N-Methyl-2-(3-hydroxyphenyl)-2-hydroxyethylamino] | 148–150 | 90 |
| 1e | 1-Aminoindane | [2,3-Dihydro-1H-inden-1-amino] | 102–105 | 88 |
| 1f | Noralfentanil | [4-(Methoxymethyl)-4-(N-phenyl-N-propionamide)piperidino] | 180–181 | 91 |
| 1g | Benzoic acid | [Benzoyloxy] | 95–98 | 92 |
| 1h | 2-Bromo-5-methoxybenzoic acid | [2-Bromo-5-methoxybenzoyloxy] | 160–164 | 84 |
| 1i | 4-Nitrobenzoic acid | [4-Nitrobenzoyloxy] | 168–171 | 88 |
| 1j | Veratroic acid | [3,4-Dimethoxybenzoyloxy] | 128–132 | 90 |
| 1k | 3-Methylbenzoic acid | [4-Methylbenzoyloxy] | 140–144 | 87 |
| 1l | Propargyl alcoholc | [Hydroxymethyl] | 168–170 | 95 |
| 1m | 1-Hexynec | [n-Butyl] | 101–103 | 89 |
| 1n | 1-Ethynyl-1-cyclohexanolc | [(1-Hydroxy)cyclohexyl] | 105–107 | 87 |
Optimization of reaction conditions
Various solvents, bases, catalysts, reaction temperature and time have been explored to optimize the reaction conditions during this research work (Table 1) using Scheme 3 as a model experiment. During the study, it was noticed that better results were obtained when CuI (10 mol%), DIPEA and n-BuOH/water mixture were used at room temperature for about 10 h (Table 1, entry 6). Usage of excess CuI did not improve the yield (Table 1, entry 5, 14 & 15) and usage of other copper salts resulted in very poor conversions (Table 1, entry 2 & 3). It can be seen that reaction can proceed in other polar solvents but did not go to completion (Table 1, entries 6–11 & 12). The reaction did not proceed when other bases such as triethylamine and potassium hydroxide were used instead of DIPEA (Table 1, entries 12–15).The structures of all novel 1,2,3-triazole 1a–n compounds were elucidated with the help of IR, 1H NMR, 13C NMR and mass spectra as exemplified for compound 1e. Characteristic IR bands of 1e: the broad band at 3422 cm−1 represents the –NH stretching. The medium band at 2963 cm−1 indicates the aliphatic stretching. The sharp bands that appear at 1674, 1605 & 1564 cm−1 show the presence of benzenoid rings. The band present at 1393 cm−1 confirms the presence of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching. The bands at 1252, 1219 & 1173 cm−1 support the presence of –C–N and –C–O stretching. The characteristic bands at 864, 762 & 694 cm−1 represent the existence of para- & ortho-disubstituted and monosubstituted aromatic rings respectively. The signals of 1H & 13C NMR of compound 1e are explained in Fig. 2. A distinguishing peak observed at m/z: 748 in the mass spectrum of 1e corresponds to the protonated molecular ion [M + H]+.
O stretching. The bands at 1252, 1219 & 1173 cm−1 support the presence of –C–N and –C–O stretching. The characteristic bands at 864, 762 & 694 cm−1 represent the existence of para- & ortho-disubstituted and monosubstituted aromatic rings respectively. The signals of 1H & 13C NMR of compound 1e are explained in Fig. 2. A distinguishing peak observed at m/z: 748 in the mass spectrum of 1e corresponds to the protonated molecular ion [M + H]+.
 | ||
| Fig. 2 Characterization of 1e using NMR spectroscopy. | ||
Pharmacology
| Organism | Compound | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 1d | 1e | 1f | 1g | 1h | 1i | 1j | 1k | 1l | 1m | 1n | C | |
| a C: streptomycin – standard antibacterial agent; C: ketoconazole – standard antifungal agent. | |||||||||||||||
| Gram positive bacteria | |||||||||||||||
| Bacillus subtilis | 16 | 18 | — | 14 | 12 | 14 | 18 | 20 | 19 | 19 | 14 | — | 14 | 18 | 24 |
| Micrococcus luteus | 15 | 12 | — | 14 | 20 | 12 | 17 | 15 | 21 | 16 | 12 | — | 14 | 18 | 26 |
| Staphylococcus aureus | 13 | — | — | 12 | 13 | 13 | 12 | 15 | 11 | 14 | 10 | — | 17 | 16 | 14 |
| Staphylococcus epidermidis | 14 | 16 | — | 10 | 14 | 14 | 22 | 21 | 24 | 19 | 14 | — | 15 | 20 | 26 |
|
|||||||||||||||
| Gram negative bacteria | |||||||||||||||
| Klebsiella pneumoniae | 13 | 14 | — | 15 | 12 | 17 | 21 | 18 | 19 | 18 | 10 | — | 13 | 16 | 20 |
| Salmonella typhimurium | 12 | 13 | — | — | 16 | 16 | 14 | — | — | — | — | — | — | 14 | 24 |
| Proteus vulgaris | 13 | 16 | — | 14 | 18 | 13 | 17 | 20 | 15 | 20 | 10 | — | 16 | 18 | 30 |
| Shigella flexneri | 15 | — | — | 15 | 19 | 12 | 18 | 25 | 21 | 20 | 12 | — | 18 | 19 | 30 |
| Enterobacter aerogenes | 16 | 15 | — | 14 | 15 | 16 | 20 | 18 | 18 | 17 | 10 | — | 16 | 17 | 22 |
| Pseudomonas aeruginosa | 14 | 18 | — | 15 | 16 | 13 | 20 | 22 | 21 | 18 | 13 | — | 16 | 18 | 30 |
| Staphylococcus aureus MRSA | 15 | 12 | — | 13 | 18 | 10 | 23 | 22 | 19 | 17 | 16 | — | 14 | 16 | 30 |
|
|||||||||||||||
| Fungi | |||||||||||||||
| Candida albicans | 16 | 15 | — | — | — | — | 15 | — | 14 | 16 | — | — | 10 | 14 | 28 |
| Malassezia pachydermatis | — | 13 | — | — | — | — | — | — | — | 12 | — | — | 13 | — | 26 |
| Organism | Compound | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1d | 1e | 1f | 1g | 1h | 1i | 1j | 1k | 1m | 1n | C-1 | C-2 | |
| a C-1: streptomycin and C-2: ciprofloxacin – standard antibacterial agents. | ||||||||||||||
| Gram positive bacteria | ||||||||||||||
| Bacillus subtilis | 62.5 | 62.5 | 125 | 62.5 | 125 | 62.5 | 31.25 | 31.25 | 62.5 | 125 | 250 | 125 | 25 | <0.78 |
| Micrococcus luteus | 250 | 500 | 250 | 62.5 | 125 | 500 | 62.5 | 62.5 | 125 | 500 | 250 | 125 | 6.25 | >100 |
| Staphylococcus aureus | 250 | — | 500 | 250 | 250 | 250 | 250 | 125 | 250 | 500 | 125 | 125 | 6.25 | <0.78 |
| Staphylococcus epidermidis | 250 | 125 | 500 | 250 | 62.5 | 250 | 62.5 | 31.2 | 62.5 | 250 | 250 | 62.5 | 25 | 6.25 |
|
||||||||||||||
| Gram negative bacteria | ||||||||||||||
| Klebsiella pneumonia | 250 | 250 | 250 | 125 | 62.5 | 125 | 62.5 | 125 | 62.5 | 500 | 250 | 125 | 6.25 | 6.25 |
| Salmonella typhimurium | 500 | 250 | — | 125 | 250 | 125 | 125 | — | — | — | — | 250 | 30 | >100 |
| Proteus vulgaris | 250 | 125 | 250 | 125 | 125 | 250 | 125 | 250 | 62.5 | 500 | 125 | 125 | 6.25 | 6.25 |
| Shigella flexneri | 250 | — | 250 | 125 | 125 | 500 | 125 | 62.5 | 62.5 | 500 | 125 | 125 | 6.25 | <0.78 |
| Enterobacter aerogenes | 125 | 250 | 250 | 250 | 125 | 125 | 62.5 | 62.5 | 125 | 500 | 125 | 125 | 25 | <0.78 |
| Pseudomonas aeruginosa | 125 | 62.5 | 125 | 62.5 | 125 | 31.25 | 31.25 | 31.25 | 62.5 | 250 | 125 | 125 | 25 | 6.25 |
| Staphylococcus aureus MRSA | 250 | 500 | 500 | 125 | 31.2 | 500 | 31.2 | 125 | 125 | 250 | 250 | 125 | 6.25 | <0.78 |
The obtained results disclosed good antimicrobial activity of the synthesized compounds 1a–n when compared with the standard antimicrobial drugs used in this study. P. aeruginosa has emerged as one of the most problematic Gram-negative pathogens, with an alarmingly high antibiotic resistance rate. Even with the most effective antibiotics against this pathogen, namely carbapenems (imipenem and meropenem), the resistance rate was found to be 15–20.4% amongst aeruginosa strains.54 Our current study showed that the synthesized compounds were active against P. aeruginosa. Compounds, 1e,f & h–j show better activity when compared with others against tested bacteria and fungi. The best MIC values were observed for compounds 1h–j. The grounds for this better activity in 1h and 1j may be due to the presence of methoxy substitutions on the benzoic acid which can increase the in vitro activity against Gram positive bacteria when compared to other compounds.55,56 The reason for the best activity of 1i may be due to the presence of para-nitro substitution on the benzoic acid.
| Concentration (μg mL−1) | Compound | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1b | 1e | 1f | 1g | 1h | 1i | 1j | 1k | 1l | 1m | 1n | ||
| 250 | % | 31.3 | 89.2 | 91.5 | 96.3 | 94.7 | 96.2 | 94.2 | 47.2 | 95.6 | 19 | 93.7 |
| Mean ± S.D. | 1.345 ± 0.00325 | 0.211 ± 0.00839 | 0.166 ± 0.00712 | 0.072 ± 0.00337 | 0.103 ± 0.00441 | 0.074 ± 0.00118 | 0.114 ± 0.00890 | 1.033 ± 0.00881 | 0.087 ± 0.00879 | 1.586 ± 0.00557 | 0.123 ± 0.00997 | |
| 125 | % | 27 | 84.9 | 62.5 | 95.8 | 87 | 95.5 | 90.9 | 35.6 | 90.8 | 16.7 | 80.9 |
| Mean ± S.D. | 1.429 ± 0.00412 | 0.295 ± 0.00559 | 0.735 ± 0.00660 | 0.082 ± 0.00559 | 0.255 ± 0.00578 | 0.088 ± 0.00229 | 0.179 ± 0.00773 | 1.261 ± 0.00836 | 0.18 ± 0.00706 | 1.631 ± 0.00967 | 0.123 ± 0.00997 | |
| 62.5 | % | 24.9 | 51.2 | 18.2 | 82.7 | 81.5 | 95.3 | 86.3 | 25.6 | 80.6 | 14.9 | 56.1 |
| Mean ± S.D. | 1.47 ± 0.00522 | 0.955 ± 0.00436 | 1.602 ± 0.00578 | 0.338 ± 0.00631 | 0.363 ± 0.00669 | 0.091 ± 0.00397 | 0.268 ± 0.00559 | 1.456 ± 0.00771 | 0.38 ± 0.00756 | 1.666 ± 0.00805 | 0.859 ± 0.00361 | |
| 31.25 | % | 16.4 | 42.9 | 13.5 | 71.9 | 60.6 | 79.9 | 75 | 14.5 | 77.9 | 10.2 | 18.6 |
| Mean ± S.D. | 1.636 ± 0.00632 | 1.118 ± 0.00947 | 1.694 ± 0.00309 | 0.55 ± 0.00779 | 0.771 ± 0.00330 | 0.393 ± 0.00433 | 0.489 ± 0.00397 | 1.675 ± 0.00743 | 0.432 ± 0.00801 | 1.759 ± 0.00598 | 1.594 ± 0.00772 | |
| 15.63 | % | 13.1 | 30.9 | 11.9 | 68.1 | 51.2 | 75 | 71.6 | 11.1 | 66.1 | 8.2 | 15.5 |
| Mean ± S.D. | 1.701 ± 0.00203 | 1.352 ± 0.00871 | 1.725 ± 0.00399 | 0.625 ± 0.00597 | 0.956 ± 0.00449 | 0.489 ± 0.00507 | 0.556 ± 0.00227 | 1.741 ± 0.00491 | 0.663 ± 0.00699 | 1.797 ± 0.00671 | 1.654 ± 0.00589 | |
| 7.81 | % | 5.2 | 26.7 | 8.6 | 62 | 30.5 | 68.2 | 57.8 | 8.1 | 54.1 | 6.7 | 9.6 |
| Mean ± S.D. | 1.856 ± 0.00654 | 1.436 ± 0.00856 | 1.789 ± 0.00580 | 0.745 ± 0.00888 | 1.361 ± 0.00379 | 0.623 ± 0.00409 | 0.826 ± 0.01025 | 1.799 ± 0.00513 | 0.899 ± 0.00712 | 1.826 ± 0.00334 | 1.77 ± 0.00665 | |
| Concentration (μg mL−1) | Compound | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1b | 1e | 1f | 1g | 1h | 1i | 1j | 1lk | 1l | 1m | 1n | ||
| 250 | % | 48.9 | 71.2 | 81.2 | 94.3 | 94 | 94.7 | 92.6 | 39.9 | 95.3 | 84.7 | 91.6 |
| Mean ± S.D. | 0.653 ± 0.00338 | 0.368 ± 0.00443 | 0.24 ± 0.00556 | 0.073 ± 0.00702 | 0.077 ± 0.00507 | 0.068 ± 0.00560 | 0.095 ± 0.00363 | 0.768 ± 0.00338 | 0.06 ± 0.00447 | 0.195 ± 0.00337 | 0.107 ± 0.00447 | |
| 125 | % | 40.5 | 54 | 64.9 | 87.2 | 92.2 | 93.7 | 83.6 | 34.9 | 93.9 | 77.7 | 81 |
| Mean ± S.D. | 0.76 ± 0.00561 | 0.588 ± 0.00881 | 0.448 ± 0.00773 | 0.164 ± 0.00564 | 0.1 ± 0.00812 | 0.08 ± 0.00609 | 0.21 ± 0.00569 | 0.831 ± 0.00501 | 0.078 ± 0.00510 | 0.285 ± 0.00429 | 0.242 ± 0.00366 | |
| 62.5 | % | 34.1 | 42 | 40.4 | 80.3 | 89.8 | 91.6 | 81.8 | 23.4 | 63.8 | 65.3 | 86.3 |
| Mean ± S.D. | 0.841 ± 0.00201 | 0.741 ± 0.00779 | 0.761 ± 0.00501 | 0.252 ± 0.00328 | 0.13 ± 0.00328 | 0.107 ± 0.00544 | 0.232 ± 0.00438 | 0.978 ± 0.00588 | 0.462 ± 0.00553 | 0.443 ± 0.00577 | 0.175 ± 0.00228 | |
| 31.25 | % | 31.6 | 40.1 | 30.1 | 69.5 | 65.8 | 85.2 | 77.6 | 16.2 | 54.9 | 50.1 | 80.7 |
| Mean ± S.D. | 0.874 ± 0.00198 | 0.765 ± 0.00278 | 0.893 ± 0.00337 | 0.389 ± 0.00209 | 0.437 ± 0.00449 | 0.189 ± 0.00224 | 0.286 ± 0.00371 | 1.07 ± 0.00579 | 0.576 ± 0.00438 | 0.637 ± 0.00804 | 0.246 ± 0.00509 | |
| 15.63 | % | 24.6 | 28.6 | 22 | 63.6 | 57.1 | 81.9 | 72.1 | 9.2 | 51.1 | 31.2 | 75.3 |
| Mean ± S.D. | 0.963 ± 0.00854 | 0.912 ± 0.00900 | 0.996 ± 0.00886 | 0.465 ± 0.00667 | 0.548 ± 0.00579 | 0.231 ± 0.00697 | 0.356 ± 0.00399 | 1.159 ± 0.00416 | 0.625 ± 0.00688 | 0.878 ± 0.00331 | 0.315 ± 0.00708 | |
| 7.81 | % | 6 | 22.7 | 9.5 | 60.1 | 50.3 | 76.4 | 63.4 | 3.2 | 33.7 | 22.2 | 63.3 |
| Mean ± S.D. | 1.201 ± 0.00337 | 0.987 ± 0.00458 | 1.156 ± 0.00927 | 0.51 ± 0.00345 | 0.635 ± 0.00670 | 0.301 ± 0.00555 | 0.468 ± 0.00608 | 1.236 ± 0.00399 | 0.847 ± 0.00701 | 0.993 ± 0.00209 | 0.469 ± 0.00884 | |
 | ||
| Fig. 3 Comparison of anticancer activity of synthesized compounds against A 549 cancer cell line (note: no toxicity was observed up to 2 mg mL−1 in Vero cells). | ||
 | ||
| Fig. 4 Comparison of anticancer activity of synthesized compounds against SKOV-3 cancer cell line. | ||
| Compound | Free energy of bindinga (kcal mol−1) | |
|---|---|---|
| DNA topoisomerase IV (4EMV) | Anaplastic lymphoma kinase (2XP2) | |
| a Calculated by AutoDock, NC – not calculated, inhibitor: co-crystallized inhibitor with protein. | ||
| 1a | −6.95 | NC |
| 1b | −5.23 | −8.54 |
| 1c | −6.58 | NC |
| 1d | −5.53 | NC |
| 1e | −9.29 | −8.66 |
| 1f | −7.97 | −8.01 |
| 1g | −8.40 | −8.32 |
| 1h | −6.71 | −9.55 |
| 1i | −7.09 | −7.94 |
| 1j | −8.13 | −6.70 |
| 1k | −7.04 | −8.70 |
| 1l | −6.08 | −8.42 |
| 1m | −7.44 | −8.78 |
| 1n | −7.28 | −8.28 |
| Inhibitor | −9.80 | −8.42 |
The molecular docking outcomes of synthesized compounds 1a–n with the 4EMV receptor established that all the docked compounds 1a–n bind efficiently with the receptor and exhibit free energy of binding values from −5.23 to −9.29 kcal mol−1. Interestingly, among all the compounds docked, compound 1e exhibits very high binding with the 4EMV receptor and forms five polar interactions with three amino acids, namely ASP-78, GLY-82 and ARG-140, resulting in a binding energy of −9.29 kcal mol−1. In compound 1e, the N–H attached to the triazole interacts with ASP-78 and forms two polar interactions with bond lengths of 1.7 and 3.3 Å respectively. Likewise, the two nitrogens of the triazole interact with GLY-82 and form two polar interactions with bond lengths of 3.2 and 3.5 Å respectively. Aside from this, the pyrimidine nitrogen forms a polar interaction with the ARG-140 with a bond length of 2.9 Å (Fig. 5).
 | ||
| Fig. 5 Docking with the 4EMV receptor (A) method validation using crystallised and docked ligand; (B) docking mode of all the compounds; (C) docking mode of the highest binding energy compound 1e. | ||
Docking of the synthesized compounds with 2XP2 revealed that compounds efficiently bind with the active site of the 2XP2 receptor and exhibit free energies of binding from −6.70 to −9.55 kcal mol−1. Compounds interact with the active site amino acids of 2XP2 namely ARG-1120, LEU-1122, GLY-1123, VAL-1130, GLU-1132, ALA-1148, LYS-1150, LEU-1196, GLU-1197, LEU-1198, MET-1199, ALA-1200, GLY-1201, GLY-1202, ASP-1203, SER-1206, PHE-1207, GLU-1210, ARG-1253, ASN-1254, CYS-1255, LEU-1256, GLY-1269 and ASP-1270. Among all the compounds docked, compound 1h exhibited very high binding with the 2XP2 receptor and formed seven polar interactions with five amino acids and resulted in a binding energy of −9.55 kcal mol−1. In compound 1h, the nitrogens of the two pyrimidines interact with CO of LEU-1122 and ASP-1203 to form three polar interactions with bond lengths of 2.7, 3.2 and 3.4 Å. In addition, the nitrogen and oxygen of the triazole and phenoxy groups interact with the CO and N–H of MET-1199 and LYS-1150 to form two polar interactions of 2.0 and 3.4 Å respectively. Also, the N–H and SO interact with the CO of GLY-1123 to form two polar interactions of 2.0 and 3.4 Å respectively. The binding interaction with the 2XP2 receptor is shown in Fig. 6.
 | ||
| Fig. 6 Docking with 2XP2 receptor (A) method validation using crystallised and docked ligand; (B) docking mode of all the compounds; (C) docking mode of the highest binding energy compound 1h. | ||
Conclusions
In summary, we have described the facile one pot synthesis of novel bis-pyrimidinyl linked 1,4-disubstituted 1,2,3-triazoles 1a–n (triazolo-bosentans) with good yields from bosentan 2 as potent hybrid drugs with dual biological activity (antimicrobial and anticancer) and rationalized the activity with the respective proteins using a docking study. During this work good antimicrobial activities were observed for compounds 1e,f & 1h–j against tested bacteria and fungi, amongst which, 1h and 1j exhibited very good activity. In vitro cytotoxicity (lung and ovarian) studies against A549 and SKOV-3 cell lines revealed prominent cytotoxic activity of compounds 1e–n. Among all triazolo-bosentans screened, (1g, i, j) and (1l, e–j, l–n) showed very significant in vitro cytotoxicity against A549 and SKOV-3 cell lines respectively with an IC50 value at 7.81 μg mL−1. Active compounds 1e–j, 1l and 1n were tested against Vero cells and no toxicity was observed up to 2 mg mL−1. When the structure and activity were correlated, it was concluded that antimicrobial and anticancer activities were enhanced by methoxy substitutions on the benzoic acid ring. Among the tested compounds, the best anticancer activity was observed for p-nitro substituted triazolo-bosentan 1i which also exhibited good antimicrobial activity. Free energies of binding were calculated for the synthesized compounds against 4EMV and 2XP2 receptors. Among them, 1e shows a better binding energy (−9.29 kcal mol−1) against the 4EMV receptor with five polar interactions by three amino acids and 1h shows a better binding energy (−9.55 kcal mol−1) against the 2PX2 receptor with seven polar interactions by five amino acids. Based on the significant anticancer results, this class of compounds can be considered as a good starting point for the development of bosentan 2 based dual endothelin receptor (ET) antagonists for efficient mono therapy for cancer.Experimental
Chemistry
Melting points were measured using a Veego melting point apparatus model VMP-PM. Thin layer chromatography (TLC) was carried out using pre-coated Merck TLC Silica gel 60 F254 and spots were detected using Ultra-Violet light. IR spectra were recorded (KBr pellet) on a Shimadzu Prestige 21 FTIR instrument in the range of 4000 to 400 cm−1. 1H NMR and 13C NMR spectra were recorded using a Bruker-Avance 300 MHz FT-NMR spectrometer (300 and 75 MHz, respectively) using DMSO-d6 as solvent and TMS as internal standard. Chemical shifts (δ) are given in parts per million (ppm). The following abbreviations are used: s-singlet, d-doublet, t-triplet, q-quintet and m-multiplet. Low resolution mass spectra were recorded on an Agilent 6110LC/MS mass spectrophotometer using the ESI mode. The elemental analysis was done (sample thoroughly dried under vacuum) using a Thermo Fischer Flash 1112 Series elemental analyzer. For antimicrobial activity, the reference cultures were obtained from the Institute of Microbial Technology (IMTECH), Chandigarh, India-160 036 and the remaining cultures were obtained from the Department of Microbiology, Christian Medical College, Vellore, Tamil Nadu, India.4-tert-Butyl-N-(6-(2-chloroethoxy)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl)benzene-sulfonamide (3). Yellow solid. Yield 85%. Mp: 198–200 °C; IR: 3252, 2954, 2904, 2835, 1684, 1618, 1570, 1564, 1554, 1499, 1455, 1438, 1397, 1384, 1284, 1253, 1213, 1173, 1162, 1155, 1111, 1083, 1026, 993, 874, 833, 827, 741, 698, 679, 625, 572, and 545 cm−1. 1H NMR (400 MHz, DMSO-d6): δH: 1.36 (s, 9H), 3.88 (s, 3H), 4.51 (t, 2H, J = 8.0 Hz), 5.18 (t, 2H, J = 8.0 Hz), 6.76–10.02 (m, 11H, aromatic) and 11.62 (bs, 1H). 13C NMR (100 MHz, DMSO-d6): δC: 31.33, 35.41, 37.73, 52.59, 56.27, 113.48, 115.93, 120.76, 123.86, 124.58, 126.04, 127.33, 129.15, 138.24, 141.61, 145.71, 147.01, 149.26, 149.52, 153.73, 155.09, 156.59 and 165.98. ESI-MS: [M + H]+ at m/z 570 (Cl35) & 572 (Cl37) in 3
:1 ratio. Anal. calcd for C27H28ClN5O5S: C, 56.89; H, 4.95; N, 12.29. Found C, 57.12; H, 4.99; N, 12.35.
N-(6-(2-Azidoethoxy)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl)-4-tert-butylbenzenesulfonamide (azide 4). Pale brown solid. Yield 93%. Mp: 172–175 °C. IR: 3245, 3066, 2959, 2903, 2835, 2795, 2090, 1681, 1617, 1563, 1550, 1499, 1455, 1397, 1386, 1333, 1254, 1215, 1179, 1162, 1111, 1084, 926, 873, 833, 824, 768, 749, 625, 574 and 545 cm−1. 1H NMR (400 MHz, DMSO-d6): δH: 1.26 (s, 9H), 3.47 (t, 2H, J = 6.2 Hz), 3.83 (s, 3H), 4.12 (t, 2H, J = 18.3 Hz), 6.67–9.11 (m, 11H, aromatic), 11.30 (bs, 1H). 13C NMR (100 MHz, DMSO-d6): δC: 31.23, 35.24, 44.34, 49.06, 56.18, 113.35, 115.33, 120.85, 122.84, 123.29, 124.33, 125.60, 128.53, 138.48, 146.14, 146.92, 149.33, 151.31, 156.26, 157.27, 158.38 and 159.58. ESI-MS: [M + H]+ at m/z 577. Anal. calcd for C27H28N8O5S: C, 56.24; H, 4.89; N, 19.43. Found C, 56.55; H, 4.93; N, 19.52.
4-tert-Butyl-N-(6-(2-(4-((((1S,2R)-1-hydroxy-1-phenyl-2-propyl)(methyl)amino)methyl)-1H-1,2,3-triazol-1-yl)-ethoxy)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl)-benzenesulfonamide (1a). Pale brown amorphous solid. Yield 6.7 g (88%). Mp: 122–125 °C. IR: 3373, 2961, 2924, 2853, 1658, 1651, 1587, 1562, 1520, 1499, 1454, 1395, 1250, 1217, 1177, 1138, 1103, 1078, 851, 750, 702, 629 and 577 cm−1. 1H NMR (400 MHz, DMSO-d6): δH 0.90 (d, 3H, J = 14.4 Hz), 1.01 (s, 9H), 2.71 (t, 2H, J = 12.2 Hz), 3.29 (s, 2H), 3.81 (s, 3H), 4.19 (s, 3H), 4.64 (t, 2H), 4.98 (m, 1H), 5.79 (bs, 1H), 6.78–7.72 (m, 17H, aromatic) and 8.97 (bs, 1H). 13C NMR (100 MHz, DMSO-d6): δC 21.59, 26.92, 27.17, 29.75, 30.03, 31.59, 34.73, 48.15, 56.15, 63.48, 113.18, 113.29, 113.55, 113.77, 120.84, 120.86, 124.41, 124.55, 125.18, 125.25, 126.29, 126.32, 126.69, 126.76, 126.79, 127.03, 127.10, 127.89, 128.15, 128.67 and 157.98. ESI-MS m/z 780 [M + H]+. Anal. calcd for C40H45N9O6S: C, 61.60; H, 5.82; N, 16.16. Found C, 62.20; H, 5.87; N, 16.10.
4-tert-Butyl-N-(6-(2-(4-(((1S,2R)-1-hydroxy-1-phenyl-2-propyl-amino)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)-5-(2-methoxy-phenoxy)-2,2′-bipyrimidin-4-yl)benzenesulfonamide (1b). Brown amorphous solid. Yield 6.0 g (78%). Mp: 140–143 °C. IR: 3356, 2959, 2924, 2853, 1668, 1603, 1562, 1499, 1456, 1393, 1341, 1252, 1219, 1175, 1140, 1103, 1080, 1022, 908, 852, 750, 702, 627, 575 and 548 cm−1. 1H NMR (400 MHz, DMSO-d6): δH 0.94 (d, 3H, J = 10.5 Hz), 1.24 (s, 9H), 3.18 (s, 2H), 3.83 (s, 3H), 4.11 (t, 2H, J = 12.2 Hz), 4.59 (t, 2H, J = 12.2 Hz), 5.10 (d of d, 1H, J = 56 Hz & J = 20.2 Hz), 5.92 (m, 1H), 6.56–7.59 (m, 17H), 8.94 (bs, 1H) and 9.00 (bs, 1H). 13C NMR (100 MHz, DMSO-d6): δC 14.33, 22.62, 29.46, 29.90, 31.40, 31.76, 32.02, 34.86, 56.14, 113.23, 114.51, 115.11, 116.05, 120.35, 120.84, 123.19, 123.64, 125.15, 126.31, 127.84, 128.19, 128.21, 128.24, 128.32, 128.66, 128.99, 129.92, 131.86, 133.48, 139.58, 153.83 and 158.22. ESI-MS m/z 766 [M + H]+. Anal. calcd for C39H43N9O6S: C, 61.16; H, 5.66; N, 16.46. Found C, 61.30; H, 5.69; N, 16.50.
4-tert-Butyl-N-(5-(2-methoxyphenoxy)-6-(2-(4-((methyl(1-phenylpropan-2-yl)-amino)methyl)-1H-1,2,3-triazol-1-yl)-ethoxy)-2,2′-bipyrimidin-4-yl)benzenesulfonamide (1c). Brown amorphous solid. Yield 6.9 g (90%). Mp: 99–102 °C. IR: 3447, 3063, 2961, 2924, 2853, 1653, 1595, 1562, 1499, 1458, 1393, 1250, 1221, 1175, 1140, 1107, 1076, 849, 746, 702, 631, 577 and 546 cm−1. 1H NMR (400 MHz, DMSO-d6): δH1.04 (s, 3H), 1.24 (s, 9H), 2.62 (t, 2H, J = 11.3 Hz), 3.14 (t, 2H, J = 15.3 Hz), 3.48 (s, 3H), 3.83 (s, 3H), 4.18 (1H, m), 4.24 (d of d, 2H), 4.65 (t, 2H), 6.60–7.73 (m, 17H, aromatic) and 9.01 (bs, 1H). 13C NMR (100 MHz, DMSO-d6): δC 29.47, 29.90, 30.34, 31.39, 31.76, 32.09, 34.99, 36.07, 45.11, 56.11, 113.13, 114.37, 120.84, 122.46, 122.54, 122.80, 124.79, 124.95, 126.92, 127.50, 127.78, 128.08, 128.17, 128.91, 129.08, 129.72, 130.12, 133.33, 153.45 and 158.15. ESI-MS m/z 764 [M + H]+. Anal. calcd for C40H45N9O5S: C, 62.89; H, 5.94; N, 16.50. Found C, 62.85; H, 5.98; N, 16.55.
4-tert-Butyl-N-(6-(2-(4-(((2-hydroxy-2-(3-hydroxyphenyl)-ethyl)(methyl)amino)-methyl)-1H-1,2,3-triazol-1-yl)ethoxy)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl)-benzenesulfonamide (1d). Brown amorphous solid. Yield 7.1 g (90%). Mp 148–150 °C. IR: 3443, 3067, 2963, 2868, 1653, 1693, 1593, 1564, 1499, 1456, 1396, 1252, 1217, 1180, 1138, 1103, 1078, 1047, 906, 837, 752, 702, 630 and 577 cm−1. ESI-MS m/z 782 [M + H]+. Anal. calcd for C39H43N9O7S: C, 59.91; H, 5.54; N, 16.12. Found C, 60.05; H, 5.57; N, 16.24.
4-tert-Butyl-N-(6-(2-(4-((2,3-dihydro-1H-inden-1-ylamino)-methyl)-1H-1,2,3-triazol-1-yl)ethoxy)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl)benzenesulfonamide (1e). Yellowish brown solid. Yield 6.6 g (88%). Mp: 102–105 °C. IR: 3422, 2963, 2868, 1674, 1605, 1564, 1499, 1458, 1393, 1340, 1252, 1219, 1173, 1111, 1084, 1020, 864, 837, 762, 625 and 575 cm−1. 1H NMR (400 MHz, DMSO-d6): δH 1.25 (s, 9H), 2.24 &2.41 (m, 2H, J = 7.5 Hz), 2.87 (m, 2H), 3.14 (m, 2H), 3.82 (s, 3H), 4.22 (s, 2H), 4.25 (t, 2H, J = 8.0 Hz), 4.73 (t, 2H, J = 12 Hz), 6.67–8.24 (m, 16H, aromatic) and 9.75 (bs, 2H). 13C NMR (100 MHz, DMSO-d6): δC 28.80, 28.90, 30.28, 31.28, 35.21, 36.61, 45.35, 56.14, 61.13, 97.74, 104.52, 108.11, 113.21, 115.20, 120.80, 125.45, 125.50, 125.53, 125.57, 126.47, 126.71, 127.05, 127.16, 128.45, 129.91, 137.73, 138.20, 138.79, 145.41, 149.17, 157.42, 158.36 and 159.24. ESI-MS m/z 748 [M + H]+. Anal. calcd for C39H41N9O5S: C, 62.63; H, 5.53; N, 16.86. Found C, 62.53; H, 5.59; N, 16.94.
N-(1-(2-(1-((6-(4-tert-Butylphenylsulfonamido)-5-(2-methoxy-phenoxy)-2,2′-bipyrimidin-4-yl)methyl)-1H-1,2,3-triazol-4-yl-oxy)ethyl)-4-(methoxymethyl)piperidin-4-yl)-N-phenylpropionamide (1f). Pale brown solid. Yield 8.1 g (91%). Mp 180–181 °C. IR: 3474, 2965, 2833, 1651, 1589, 1562, 1518, 1499, 1456, 1396, 1377, 1323, 1250, 1229, 1178, 1140, 1107, 1078, 1002, 849, 750, 706, 633, 579 and 552 cm−1. 1H NMR (400 MHz, DMSO-d6): δH 0.79 (t, 3H, J = 7.3 Hz), 1.23 (s, 9H), 1.69, (t, 4H), 1.98 (t, 4H), 3.32 (s, 3H), 3.52 (q, 2H), 3.85 (t, 2H), 3.93 (s, 2H), 4.12 (s, 2H), 4.56 (t, 2H), 6.51–7.74 (m, 17H) and 8.95 (bs, 1H). 13C NMR (100 MHz, DMSO-d6): δC 9.97, 23.30, 28.29, 28.70, 30.47, 31.53, 31.59, 34.80, 59.28, 76.57, 79.46, 89.08, 113.80, 120.86, 122.40, 124.47, 129.13, 131.52, 133.47, 134.01, 135.10, 136.29, 139.18, 140.51, 141.35, 146.09, 149.15, 150.32, 152.92 and 168.34. ESI-MS m/z 891 [M + H]+. Anal. calcd for C46H54N10O7S: C, 62.00; H, 6.11; N, 15.72. Found C, 62.39; H, 6.15; N, 15.89.
(1-(2-(6-(4-tert-Butylphenylsulfonamido)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl-oxy)ethyl)-1H-1,2,3-triazol-4-yl)-methyl benzoate (1g). Yellowish brown solid. Yield 6.8 g (92%). Mp: 95–98 °C. IR: 3144, 3067, 2963, 2926, 2868, 2853, 1719, 1676, 1605, 1564, 1499, 1452, 1393, 1340, 1271, 1254, 1219, 1173, 1111, 1084, 1024, 864, 835, 752, 714, 692, 625, 573 and 552 cm−1. 1H NMR (400 MHz, DMSO-d6): δH 1.25 (s, 9H), 3.84 (s, 3H), 4.34 (t, 2H, J = 9.5 Hz), 4.66 (t, 2H, J = 8.0 Hz), 5.32 (s, 2H), 6.70–9.03 (m, 17H, aromatic) and 11.30 (bs, 1H). 13C NMR (100 MHz, DMSO-d6): δC 31.23, 35.25, 51.34, 56.16, 58.33, 59.58, 113.24 to 165.90. ESI-MS m/z 737 [M + H]+. Anal. calcd for C37H36N8O7S: C, 60.31; H, 4.92; N, 15.21. Found C, 60.25; H, 4.96; N, 15.20.
(1-(2-(6-(4-tert-Butylphenylsulfonamido)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl-oxy)ethyl)-1H-1,2,3-triazol-4-yl)-methyl 2-bromo-5-methoxybenzoate (1h). Pale brown solid. Yield 7.2 g (84%). Mp: 160–164 °C. IR: 3068, 2965, 2905, 1709, 1674, 1602, 1565, 1515, 1500, 1464, 1456, 1419, 1394, 1343, 1290, 1271, 1253, 1220, 1176, 1140, 1111, 1084, 1022, 869, 835, 763, 753, 696, 627, 575 and 547 cm−1. 1H NMR (400 MHz, DMSO-d6): δH 1.26 (s, 9H), 3.77 (s, 3H), 3.83 (s, 3H), 4.32 (t, 2H), 4.67 (t, 2H), 5.32 (s, 2H), 6.69–9.03 (m, 15H, aromatic) and 11.30 (bs, 1H). 13C NMR (100 MHz, DMSO-d6): δC 31.24, 35.24, 45.69, 45.76, 47.91, 56.20, 58.98, 110.74, 113.29, 115.52, 116.56, 119.56, 120.85, 122.75, 123.23, 124.44, 125.65, 125.91, 128.52, 133.26, 135.26, 138.38, 146.09, 146.69, 149.27, 151.17, 156.31, 157.38, 158.26, 158.85, 159.10 and 165.60. ESI-MS m/z 847 [M + H]+. Anal. calcd for C38H37BrN8O8S; C, 59.60; H, 4.87; N, 14.63. Found C, 59.69; H, 4.93; N, 14.70.
(1-(2-(6-(4-tert-Butylphenylsulfonamido)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl-oxy)ethyl)-1H-1,2,3-triazol-4-yl)-methyl 4-nitrobenzoate (1i). Brown solid. Yield 7.9 g (88%). Mp: 168–171 °C. IR: 3111, 3078, 2965, 2869, 2838, 1726, 1675, 1607, 1565, 1528, 1499, 1456, 1393, 1342, 1267, 1253, 1219, 1172, 1112, 1102, 1084, 1014, 872, 858, 762, 752, 720, 625, 575 and 546 cm−1. 1H NMR (400 MHz, DMSO-d6): δH 1.25 (s, 9H), 3.83 (s, 3H), 4.34 (t, 2H, J = 9.5 Hz), 4.67 (t, 2H, J = 8.0 Hz), 5.38 (s, 2H), 6.69–9.03 (m, 16H, aromatic) and 11.31 (bs, 1H). 13C NMR (100 MHz, DMSO-d6): δC 31.58, 35.24, 47.82, 56.13, 59.05, 59.26, 124.39, 124.75, 125.46, 125.78, 127.62, 128.51, 131.22, 131.86, 132.12, 135.23, 142.50, 142.81, 146.92, 148.26, 150.77, 158.25, 160.23, 161.22, 161.58, 162.12, 164.45 and 164.56; ESI-MS m/z 891 [M + H]+. Anal. calcd for C37H35N9O9S: C, 56.84; H, 4.51; N, 16.12. Found C, 56.98; H, 4.55; N, 16.19.
(1-(2-(6-(4-tert-Butylphenylsulfonamido)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl-oxy)ethyl)-1H-1,2,3-triazol-4-yl)-methyl 3,4-dimethoxybenzoate (1j). Brown solid. Mp 128–132 °C. Yield 7.2 g (90%). IR: 3605, 3150, 3076, 2964, 2905, 2837, 1707, 1675, 1601, 1565, 1514, 1500, 1457, 1418, 1392, 1343, 1289, 1271, 1253, 1219, 1174, 1111, 1022, 868, 834, 763, 694, 626, 575 and 546 cm−1. 1H NMR (400 MHz, DMSO-d6): δH1.26 (s, 9H), 3.80 (s, 6H), 4.32 (t, 2H), 4.66 (t, 2H), 5.30 (s, 2H), 6.71–9.02 (m, 17H, aromatic) and 11.30 (bs, 1H). ESI-MS: [M + H]+ at m/z 797; anal. calcd for C39H40N8O9S: C, 58.78; H, 5.06; N, 14.06. Found C, 58.69; H, 5.12; N, 14.04.
(1-(2-(6-(4-tert-Butylphenylsulfonamido)-5-(2-methoxyphen-oxy)-2,2′-bipyrimidin-4-yl-oxy)ethyl)-1H-1,2,3-triazol-4-yl)-methyl 3-methylbenzoate (1k). Brown solid. Mp 140–144 °C. Yield 6.6 g (87%). IR: 3455, 3139, 3067, 2964, 2904, 2869, 1718, 1653, 1562, 1500, 1457, 1397, 1278, 1251, 1228, 1214, 1202, 1179, 1141, 1105, 1081, 1021, 851, 835, 748, 668, 633, 580 and 551 cm−1. 1H NMR (400 MHz, DMSO-d6): δH 1.23 (s, 9H), 2.36 (s, 3H), 3.83 (s, 3H), 4.16 (t, 2H), 4.62 (t, 2H), 5.32 (s, 2H), 6.57–8.07 (m, 15H, aromatic) and 8.95 (bs, 1H). ESI-MS: [M + H]+ at m/z 751; anal. calcd for C38H38N8O7S: C, 60.79; H, 5.10; N, 14.92. Found C, 60.95; H, 5.14; N, 14.96.
4-tert-Butyl-N-(6-(2-(4-(hydroxymethyl)-1H-1,2,3-triazol-1-yl)-ethoxy)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl)-benzenesulfonamide (1l). Yellowish brown solid. Mp 168–170 °C. Yield 6.0 g (95%). IR: 3447, 3134, 2965, 2870, 2835, 1678, 1661, 1609, 1568, 1499, 1427, 1394, 1348, 1250, 1219, 1196, 1167, 1113, 1084, 1026, 856, 833, 756, 692, 625, 573 and 546 cm−1. 1H NMR (400 MHz, DMSO-d6): δH 1.27 (s, 9H), 3.84 (s, 3H), 4.30 (t, 2H, J = 9.5 Hz), 4.45 (t, 2H, J = 9.5 Hz), 4.61 (s, 2H), 5.13 (bs, 1H), 6.70–9.05 (m, 12H, aromatic) and 11.27 (bs, 1H). 13C NMR (100 MHz, DMSO-d6): δC 22.41, 28.69, 47.77, 55.46, 69.75, 71.11, 111.40, 113.65, 115.24, 118.88, 123.45, 126.56, 128.54, 128.68, 129.04, 134.68, 136.43, 143.13, 143.91, 147.00, 148.41, 150.75, 157.66 and 161.20. ESI-MS: [M + H]+ at m/z 633; anal. calcd for C30H32N8O6S: C, 56.95; H, 5.10; N, 17.71. Found C, 57.15; H, 5.14; N, 17.85.
4-tert-Butyl-N-(6-(2-(4-butyl-1H-1,2,3-triazol-1-yl)ethoxy)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl)-benzenesulfonamide (1m). Brown solid. Mp 101–103 °C. Yield 5.9 g (89%). IR: 3136, 3069, 2959, 2870, 1676, 1605, 1564, 1499, 1456, 1393, 1340, 1252, 1219, 1173, 1111, 1084, 1045, 1022, 864, 837, 752, 694, 625, 575 and 546 cm−1. 1H NMR (400 MHz, DMSO-d6): δH 0.88 (t, 3H, J = 6.8 Hz), 1.27 (m, 13H, aromatic), 1.51 (t, 2H, J = 7.5 Hz), 3.83 (s, 3H), 4.31 (t, 2H, J = 9.5 Hz), 4.57 (t, 2H, J = 9.5 Hz), 6.71–9.04 (m, 12H, aromatic) and 11.27 (bs, 1H). ESI-MS: [M + H]+ at m/z 659. Anal. calcd for C33H38N8O5S: C, 60.17; H, 5.81; N, 17.01. Found C, 60.85; H, 5.86; N, 17.15.
4-tert-Butyl-N-(6-(2-(4-(1-hydroxycyclohexyl)-1H-1,2,3-triazol-1-yl)ethoxy)-5-(2-methoxyphenoxy)-2,2′-bipyrimidin-4-yl)-benzenesulfonamide (1n). Brown solid. Mp 105–107 °C. Yield 5.9 g (87%). IR: 3418, 3140, 3069, 2936, 2860, 1674, 1607, 1566, 1499, 1456, 1393, 1340, 1285, 1252, 1219, 1171, 1111, 1084, 1047, 1020, 866, 835, 752, 702, 625, 575 & 546 cm−1. 1H NMR (400 MHz, DMSO-d6): δH 1.27 (m, 11H), 1.41 (m, 2H), 1.64 (m, 4H, J = 5.2 Hz), 1.78 (m, 2H), 3.85 (s, 3H), 4.29 (t, 2H, J = 5.6 Hz), 4.60 (m, 2H, J = 6.4 Hz), 4.77 (bs, 1H), 6.74–9.06 (m, 12H, aromatic), 11.27 (bs, 1H); 13C NMR (100 MHz, DMSO-d6): δC 22.06, 25.72, 31.24, 35.24, 38.19, 45.89, 47.42, 56.21, 68.41, 113.30, 115.49, 120.90, 121.89, 122.84, 123.22, 124.42, 125.62, 128.51, 138.43, 146.15, 146.80, 147.52, 149.29, 151.25, 156.25, 157.41, 158.32 & 159.15; ESI-MS: [M + H]+ at m/z 683; anal. calcd for C35H40N8O6S: C, 59.98; H, 5.75; N, 15.99. Found C, 60.12; H, 5.79; N, 16.11.
Biological assays
Microbes. The following bacteria and fungi were used for the experiments. Bacteria: Bacillus subtilis MTCC 44.1, Klebsiella pneumoniae MTCC 109, Staphylococcus epidermidis MTCC 3615, Micrococcus luteus MTCC 106, Salmonella typhimurium MTCC 1251, Proteus vulgaris MTCC 1771, Shigella flexneri MTCC 1457, Enterobacter aerogenes MTCC 111, Staphylococcus aureus MTCC 96, Pseudomonas aeruginosa MTCC 741 and Staphylococcus aureus (MRSA-methicillin resistant). The reference cultures were obtained from the Institute of Microbial Technology (IMTECH), Chandigarh-160036, India; fungi: Candida albicans MTCC 227 and Malassezia pachydermatis. All the remaining cultures were obtained from the Department of Microbiology, Christian Medical College, Vellore, Tamil Nadu, India.
In vitro well method. Petri plates were prepared with 20 mL of sterile Mueller Hinton agar (MHA) (Hi-media, Mumbai). The test cultures were swabbed on top of a solidified media and were allowed to dry for 10 min and a specific amount of synthesised compound at 1 mg per disc was added to each well separately. The negative control was prepared using the respective solvents. Streptomycin was used as positive control against bacteria. Ketoconazole was used as positive control for fungi. The plates were incubated for 24 h at 37 °C for bacteria and for 48 h at 28 °C for fungi. Zones of inhibition were recorded in mm and the experiment was repeated twice. Bacterial inoculums were prepared by growing cells in Mueller Hinton broth (MHB) (Himedia) for 24 h at 37 °C. The filamentous fungi were grown on Sabouraud dextrose agar (SDA) slants at 28 °C for 10 days and the spores were collected using sterile doubled distilled water and were homogenized. Yeast was grown on a Sabouraud dextrose broth (SDB) at 28 °C for 48 h.
Minimum inhibition concentration study. The required concentrations (1000 μg mL−1, 500 μg mL−1, 250 μg mL−1, 125 μg mL−1, 62.5 μg mL−1, 31.25 μg mL−1, 15.62 μg mL−1 and 7.81 μg mL−1) of the compound were dissolved in DMSO (2%), and were diluted to give serial two-fold dilutions that were added to each medium in 96 well plates. An inoculum of 100 μL from each well was inoculated. The antifungal agents, ketoconazole for fungi and streptomycin for bacteria were included in the assays as positive controls. For fungi, the plates were incubated for 48 to 72 h at 28 °C and for bacteria the plates were incubated for 24 h at 37 °C. The MIC for fungi was defined as the lowest extract concentration, showing no visible fungal growth after incubation time. 5 μL of the tested broth was placed on the sterile MHA plates for bacteria and incubated at the respective temperature. The MIC for bacteria was determined as the lowest concentration of the compound inhibiting the visual growth of the test cultures on the agar plate.
Study of cytotoxicity. A549 (lung), SKOV-3 (ovarian) and Vero cells were obtained from ATCC, USA. A549, SKOV-3 and Vero cells were maintained in complete tissue culture medium DMEM with 10% fetal bovine serum and 2 mM L-glutamine, along with antibiotics (about 100 IU mL−1 of penicillin, 100 μg mL−1 of streptomycin) with the pH adjusted to 7.2. The cytotoxicity was determined according to the published method57 with some changes. Cells (5 × 105) were seeded in 96 well plates containing medium with different concentrations such as 250, 125, 62.5, 31.25, 15.63 and 7.81 μg mL−1. The cells were cultivated at 37 °C with 5% CO2 and 95% air in 100% relative humidity. After various durations of cultivation, the solution in the medium was removed. An aliquot of 100 μL of medium containing 1 mg mL−1 of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) was loaded on to the plate. The cells were cultured for 4 h and then the solution in the medium was removed. An aliquot of 100 μL of DMSO was added to the plate, which was shaken until the crystals were dissolved. The cytotoxicity against cancer cells was determined by measuring the absorbance of the converted dye at 570 nm in an ELISA reader. The cytotoxicity of each sample was expressed as IC50 value. The IC50 value is the concentration of test sample that causes 50% inhibition of cell growth, averaged from three replicate experiments. The percentage of growth inhibition was calculated using the following formula; inhibition (%) = A − B/A × 100 (A – control group and B – treated group).
Acknowledgements
Sincere thanks to the management of Malladi Drugs & Pharmaceuticals Ltd., Chennai, India for their support. This project was supported by King Saud University, Deanship Scientific Research, College of Science, Research Center.Notes and references
- WHO, Antimicrobial resistance fact sheet no. 194, World Health Organization, Geneva, 2014, http://www.who.int/mediacentre/factsheets/fs194/en/ Search PubMed.
- A. P. Magirokaos, A. Srinivasan, R. B. Carey, Y. Carmeli, M. E. Falagas, C. G. Giske, S. Harbarth, J. F. Hindler, G. Kahlmeter, B. Olsson-Liljequist, D. L. Paterson, L. B. Rice, J. Stelling, M. J. Struelenes, A. Vatopoulos, J. T. Weber and D. L. Monnet, Clin. Microbiol. Infect., 2012, 18, 268 CrossRef PubMed.
- B. K. Benjamin and T. H. Deborah, Mol. Cell, 2010, 37, 297 CrossRef PubMed.
- I. Pastan and M. G. Michael, Annu. Rev. Med., 1991, 42, 277 CrossRef CAS PubMed.
- P. M. Bennett, Br. J. Pharmacol., 2008, 153, S347 CrossRef CAS PubMed.
- R. Siegel, D. Naishadham and A. Jemal, Ca-Cancer J. Clin., 2013, 63, 11 CrossRef PubMed.
- A. Bagnato, M. Loizidou, B. R. Pflug, J. Curwen and J. Growcott, Br. J. Pharmacol., 2011, 163, 220 CrossRef CAS PubMed.
- D. Tamkus, A. Al-Janadi, G. Fink, G. Krishnan and N. V. Dimitrov, J. Cancer Mol., 2009, 4, 163 CAS.
- N. Cook, R. Brais, W. Qian, C. Chan Wah Hak and P. G. Corrie, J. Clin. Pathol., 2015, 68, 309 CrossRef CAS.
- G. Remuzzi, N. Perico and A. Benigni, Nat. Rev. Drug Discovery, 2002, 1, 986 CrossRef CAS PubMed.
- J. R. Wu-Wong and R. Padley, Curr. Opin. Cardiovasc., Pulm. Renal Invest. Drugs, 2000, 2, 339 CAS.
- C. Wu, Expert Opin. Ther. Pat., 2000, 10, 1653 CrossRef CAS.
- C. Boss, M. Bolli and T. Weller, Curr. Med. Chem., 2002, 9, 349 CrossRef CAS.
- H. Cohen, C. Chahine, A. Hui and R. Mukherji, Am. J. Health-Syst. Pharm., 2004, 61, 1107 CAS.
- J. O’Neil Maryadele, The Merck Index: Am Encyclopedia of Chemicals, Drugs and Biologicals, Merck Research Laboratories, 14th edn, 2006, monograph 1353 Search PubMed.
- C. Funk, M. Farr, B. Werner, S. Dittmann, K. Uberla, C. Piper, K. Niehaus and D. Horstkotte, J. Gen. Virol., 2010, 91, 1959 CrossRef PubMed.
- Z. Jokar, M. Nematbakhsh, M. Moeini and A. Talebi, Adv. Biomed. Res., 2015, 4, 83 CrossRef PubMed.
- M. Clozel and U. Regenass, WO Patent 2009/104150 A1, 2009.
- C. Eriksson, A. Gustavasson, T. Kronvall and C. Tysk, Journal of Gastrointestinal and Liver Diseases, 2011, 20, 77 Search PubMed.
- N. Dwyer, G. Jones and K. D. Kilpatric, Journal of Clinical Rheumatology, 2009, 15, 88 CrossRef.
- K. Nepali, S. Sharma, M. Sharma, P. M. Bedi and K. L. Dhar, Eur. J. Med. Chem., 2014, 77, 422 CrossRef CAS PubMed.
- K. Nepali, S. Sharma, D. Kumar, A. Budhiraja and K. L. Dhar, Recent Pat. Anti-Cancer Drug Discovery, 2014, 9, 303 CrossRef CAS PubMed.
- L. K. Gediya and V. C. Njar, Expert Opin. Drug Discovery, 2009, 4, 1099 CrossRef CAS PubMed.
- F. Sebastien and B. Gervais, Expert Opin. Drug Discovery, 2013, 8, 1029 CrossRef.
- A. Celia Henry, Chem. Eng. News, 2007, 85, 46 Search PubMed.
- P. G. Baraldi, A. N. Zaid, D. Preti, F. Fruttarolo, M. A. Tabrizi, A. Laconinoto, M. G. Pavani, M. D. Carrion, C. L. Cara and R. Romagnoli, ARKIVOC, 2006, vii, 20 Search PubMed.
- H. A. Saadeh, M. M. Ibrahim and M. S. Mubarak, Molecules, 2009, 14, 1483 CrossRef CAS.
- M. Chattopadhyay, R. Kodela, K. R. Olson and K. Kashfi, Biochem. Biophys. Res. Commun., 2012, 419, 523 CrossRef CAS PubMed.
- L. H. Miller, H. C. Auckerman, S. Xin-zhuan and T. E. Wellems, Nat. Med., 2013, 19, 156 CrossRef CAS.
- S. J. Burgess, J. Xu-Kelly, K. Liebmann, B. Gunsaru, W. Morrill and D. H. Peyton, Malar. J., 2010, 9, 6 CrossRef.
- M. Li-Yung, P. Lu-Ping, B. Wang, M. Zhang, B. Hu, X. Deng-Qi, S. Kun-Peng, Z. Bao-Le, L. Ying, Z. En and L. Hong-Min, Eur. J. Med. Chem., 2014, 86, 368 CrossRef PubMed.
- K. S. Jain, T. S. Chitre, P. B. Miniyar, M. K. Kathiravan, V. S. Bendre, V. S. Veer, S. R. Shahane and C. J. Shishoo, Curr. Sci., 2006, 90, 793 CAS.
- K. A. Jacobson, in Comprehensive Medicinal Chemistry, ed. J. C. Ernet, Pergamon Press, Oxford, 1990, vol. 3, p. 623 Search PubMed.
- I. M. Lagoza, Chem. Biodiversity, 2005, 2, 1 Search PubMed.
- E. A. Tanifum, A. Y. Kots, B. K. Choi, F. Murad and S. R. Gilbertson, Bioorg. Med. Chem. Lett., 2009, 19, 3067 CrossRef CAS.
- H. Mitsuya, R. Yarchoan and S. Broder, Science, 1990, 249, 1533 CAS.
- A. S. Jones, J. R. Sayers, R. T. Walker and E. D. Clercq, J. Med. Chem., 1988, 31, 268 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- A. Evans, Aust. J. Chem., 2007, 60, 384 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- J. Hou, X. Liu, J. Shen, G. Zhao and P. G. Wang, Expert Opin. Drug Discovery, 2012, 7, 489 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC.
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278 CrossRef CAS PubMed.
- M. Ravinesh, R. Kumar, S. Kumar, J. Majeed, M. Rashid and S. Sharma, J. Chil. Chem. Soc., 2010, 55, 359 CrossRef.
- A. Lutz and P. H. Kumar, Org. Biomol. Chem., 2010, 8, 4503 Search PubMed.
- A. Lauria, R. Delisi, F. Mingoia, A. Terenzi, A. Martorana, G. Barone and A. M. Almerico, Eur. J. Org. Chem., 2014, 16, 3289 CrossRef.
- R. Kumar, M. Shaharyar, C. Saurabh and S. Atul, Int. J. PharmTech Res., 2013, 5, 1844 CAS.
- C. Sumitra, B. Yogesh and B. Shipra, Arch. Appl. Sci. Res., 2010, 2, 117 Search PubMed.
- G. A. Sandip, R. M. Suleman and S. P. Vandana, Chem.–Asian J., 2011, 6, 2696 CrossRef PubMed.
- C. Balachandran, Y. Arun, V. Duraipandiyan, S. Ignacimuthy, K. Balakrishna and N. A. Al-Dhabi, Appl. Biochem. Biotechnol., 2014, 172, 3513 CrossRef CAS PubMed.
- C. Balachandran, V. Duraipandiyan, R. L. S. Balakrishna, A. Vijayakumar, S. Ignacimuthu and N. A. Al-Dhabi, Environ. Chem. Lett., 2013, 11, 303 CrossRef CAS.
- NCCLS, 2002, M27-A2. Int. Committee for Clinical Laboratory Standards, Reference method for broth dilution antifungal susceptibility testing of yeasts: proposed standard.
- Clinical and Laboratory Standards Institute (CLSI) 2008, Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard second edition, CLSI document M38-A2 (ISBN 1-56238-668-9), Clinical and Laboratory Standards Institute, 940, West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898, USA.
- L. Savafi, N. Duran, N. Savafi, Y. Onlem and S. Ocak, J. Med. Sci., 2005, 35, 317 Search PubMed.
- R. L. Peterson, Clin. Infect. Dis., 2001, 33, S180 CrossRef PubMed.
- M. Takej, H. Fukuda, R. Kishii, Y. Kadowaki, Y. Atobe and M. Hosaka, Antimicrob. Agents Chemother., 2002, 46, 3337 CrossRef.
- C. Balachandran, N. Emi, Y. Arun, Y. Yamamoto, B. Ahilan, B. Sangeetha, V. Duraipandiyan, Y. Inaguma, A. Okamoto, S. Ignacimuthu, N. A. Al-Dhabi and P. T. Perumal, Chem.-Biol. Interact., 2015, 242, 81 CrossRef CAS PubMed.
- B. K. Shoichet, S. L. McGovern, B. Wai and J. J. Irwin, Curr. Opin. Chem. Biol., 2002, 6, 439 CrossRef CAS PubMed.
- M. F. Sanner, J. Mol. Graphics Modell., 1999, 17, 55 CrossRef.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785 CrossRef CAS PubMed.
- K. C. Rao, Y. Arun, K. Easwaramoorthi, C. Balachandran, T. Prakasam, T. E. Yuvaraj and P. T. Perumal, Bioorg. Med. Chem. Lett., 2014, 24, 3057 CrossRef PubMed.
- Y. Arun, K. Saranraj, C. Balachandran and P. T. Perumal, Eur. J. Med. Chem., 2014, 74, 50 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR, NMR and mass spectra and docking. See DOI: 10.1039/c5ra18618h |
| This journal is © The Royal Society of Chemistry 2015 |