DOI:
10.1039/C5RA24985F
(Paper)
RSC Adv., 2015,
5, 105279-105287
Porphyrin-uptake in liposomes and living cells using an exchange method with cyclodextrin†
Received
25th November 2015
, Accepted 27th November 2015
First published on 8th December 2015
Abstract
The water-solubilisation of porphyrin derivatives is very important for biological applications. Although liposomal drug carriers for porphyrin derivatives have shown significant promise in the field of medicinal chemistry (e.g., as sensitisers for photodynamic therapy), it is currently not possible to prepare lipid-membrane-incorporated tetraphenylporphyrin (TPP) with a high concentration of TPP using conventional methods. In this study, we have succeeded in preparing lipid-membrane-incorporated TPP and zinc(II) tetraphenylporphyrin (ZnTPP) from the corresponding TPP or ZnTPP·cyclodextrin complex using the exchange method in lipid-membranes composed of liposomes. Furthermore, the exchange method allowed for the incorporation of TPP or ZnTPP into the plasma membranes of HeLa cells. However, it was not possible to prepare lipid-membrane-incorporated porphyrin derivatives with polar and hydrophilic groups in the meso positions using this exchange reaction.
Introduction
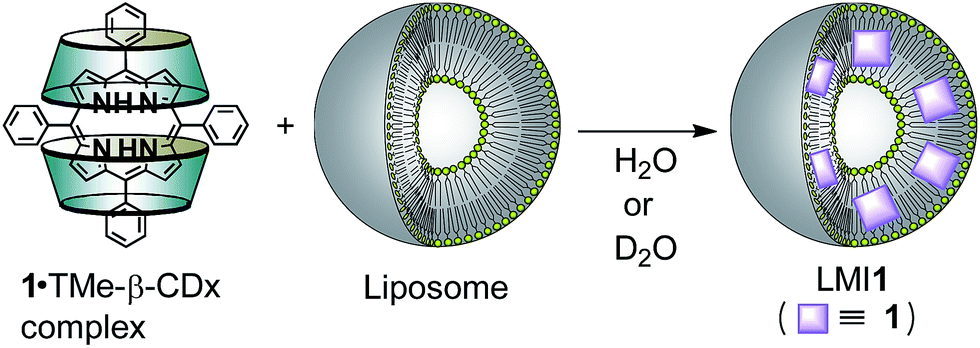
Liposomes have been proven to be of great value as drug carriers because of their ability to encapsulate a variety of different biological or biomedical materials.1–3 Hydrophilic molecules and polymers can be enclosed in the internal aqueous phase of liposomes or adsorbed onto their surfaces, whereas hydrophobic molecules can be incorporated into the lipid-membranes of liposomes. Two major methods have been developed for the design of lipid-membrane-incorporated hydrophobic guest π-molecules (LMIG) such as drugs, including (i) the dissolution of lipids and hydrophobic π-molecules in a suitable organic solvent followed by the drying and subsequent dispersion of the resulting mixture in water (which we previously defined as the ‘premixing method’);4–7 and (ii) the injection of a solution of hydrophobic π-molecules dissolved in a suitable co-solvent, such as ethanol or dimethyl sulfoxide (DMSO), into an aqueous solution of liposomes (‘injection method’).4,8,9 These methods are very useful for the simple preparation of LMIG, but their general uses have been limited by several drawbacks. With regard to the former method, hydrophobic π-molecules tend to form large self-aggregates, making them poorly soluble and therefore enhancing the lability of the LMIG. For the latter method, the use of highly hydrophobic π-molecules can be significantly restricted because of their poor solubility in the co-solvent systems required of this method. We recently proposed a third method for the design of LMIG based on the exchange reaction of a hydrophobic π-molecule (e.g. C60, C70, azobenzene and stilbene) from a hydrophobic π-molecule·cyclodextrin (CDx) complex to a liposome, which we called the ‘exchange method’.10–16 In this context, the porphyrin derivatives are regarded as π-molecules because most of the photosensitizers currently used in clinical practice for the photodynamic treatment of cancer are porphyrin derivatives (e.g., photofrin and laserphyrin).17 To improve the cellular uptake of these materials, several lipid-membrane-incorporated porphyrin derivatives, such as hematoporphyrin and 5-[4-(1-dodecanoylpyridinium)]-10,15,20-triphenylporphyrinyl chloride, have been investigated as potent photosensitisers.18,19 There is therefore an urgent need to develop robust and efficient methods for the preparation of lipid-membrane-incorporated porphyrin derivatives with high stability capable of carrying high concentrations of porphyrin derivatives. In this paper, we have reported the advantages of lipid-membrane-incorporated tetraphenylporphyrin (1) (LMI1), which was prepared by the exchange method (Scheme S1†), as well as the introduction of 1 into the plasma membranes of HeLa cells using the exchange method. We have also investigated the formation of several other lipid-membrane-incorporated systems, including zinc(II) 5,10,15,20-tetraphenyl-21H,23H-porphine (2), 5,10,15,20-tetra(4-pyridyl)porphyrin (3), 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (4) and 5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin (5) (LMI2–5) by the exchange reaction.
Results and discussion
Preparation of LMI1 by the premixing method
LMI1 was initially prepared using the premixing method (Fig. S1A†), but the resulting material gave very weak absorption bands in its UV-vis spectrum (Fig. 1A), which indicated that the liposome could not solubilise 1 in water using the premixing method. The large self-aggregates of 1 that formed in dry thin lipid membranes following the concentration step were removed by the filtration through a polytetrafluoroethylene filter (0.45 μm pore size) after the addition of water.
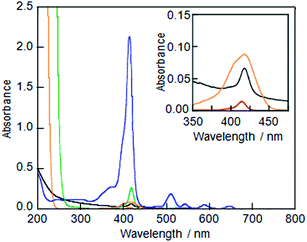 |
| | Fig. 1 UV-vis absorption spectra of (A) LMI1 prepared by the premixing method (black line), (B) an ethanol solution of 1 (red line), (C) a DMSO solution of 1 (green line), (D) LMI1 prepared by the injection method (DMSO) (orange line) and (E) an aqueous solution of the 1·TMe-β-CDx complex (blue line). All of these spectra were measured at 25 °C (1 mm cell). The ethanol and DMSO solutions of 1, LMI1 prepared by the injection method, and the aqueous solutions of the 1·TMe-β-CDx complex were diluted to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20. Inset: expansion of the region 350–475 nm for (A), (B) and (D). :20. Inset: expansion of the region 350–475 nm for (A), (B) and (D). | |
Preparation of LMI1 by the injection method
Although several attempts were then made to solubilise 1 in ethanol or DMSO, the concentrations of 1 achieved in this way were too low (Fig. 1B and C, [1] = ca. 0.1 μM and 0.1 mM, respectively; Fig. S1B†) to allow for the preparation of LMI1 by the injection method. For example, when LMI1 was used with a [1]/[DMPC] value of 10 mol% in a 5% (v/v) mixture of DMSO in water, the concentration of DMPC was lower than 0.05 mM. Furthermore, the LMI1 prepared by the DMSO injection method gave a broad absorption spectrum, which indicated that 1 had aggregated in the lipid membrane (Fig. 1D).
Preparation of LMI1 by the exchange method
To use for the exchange method, the 1·TMe-β-CDx complex was prepared according to a previously described procedure (Scheme 1 and Fig. S1C†),20 and the formation of this material was confirmed by the UV-vis absorption and 1H NMR spectroscopy (Fig. 1E and 4). The UV-vis spectrum of the 1·TMe-β-CDx complex showed an absorption maximum (λmax) at 414 nm in water, corresponding to the Soret band of the porphyrin, as well as several other bands at 511, 543, 588, and 649 nm, which were attributed to the Q bands of the porphyrin (Fig. 1E). The concentration of the 1·TMe-β-CDx complex was evaluated based on the ratio of the 1H NMR peak integrals for the signals of porphyrin 1 and DMSO after the addition of a known amount of DMSO. The value was determined to be 1.33 mM in water. Furthermore, the molar absorption coefficient of the 1·TMe-β-CDx complex was determined based on the absorption spectrum of the same solution prior to the addition of DMSO (ε415 = 3.30 × 104 dm3 mol−1 cm−1). This value therefore indicated that this complex could be used for the exchange method.
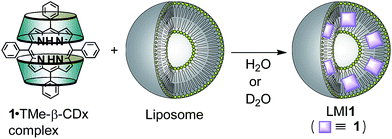 |
| | Scheme 1 Structures of compounds and exchange reaction of 1. | |
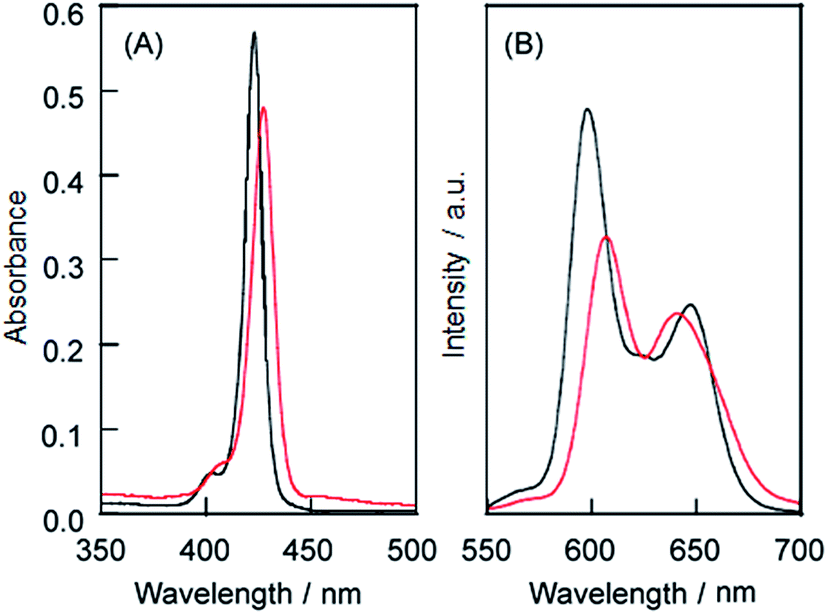
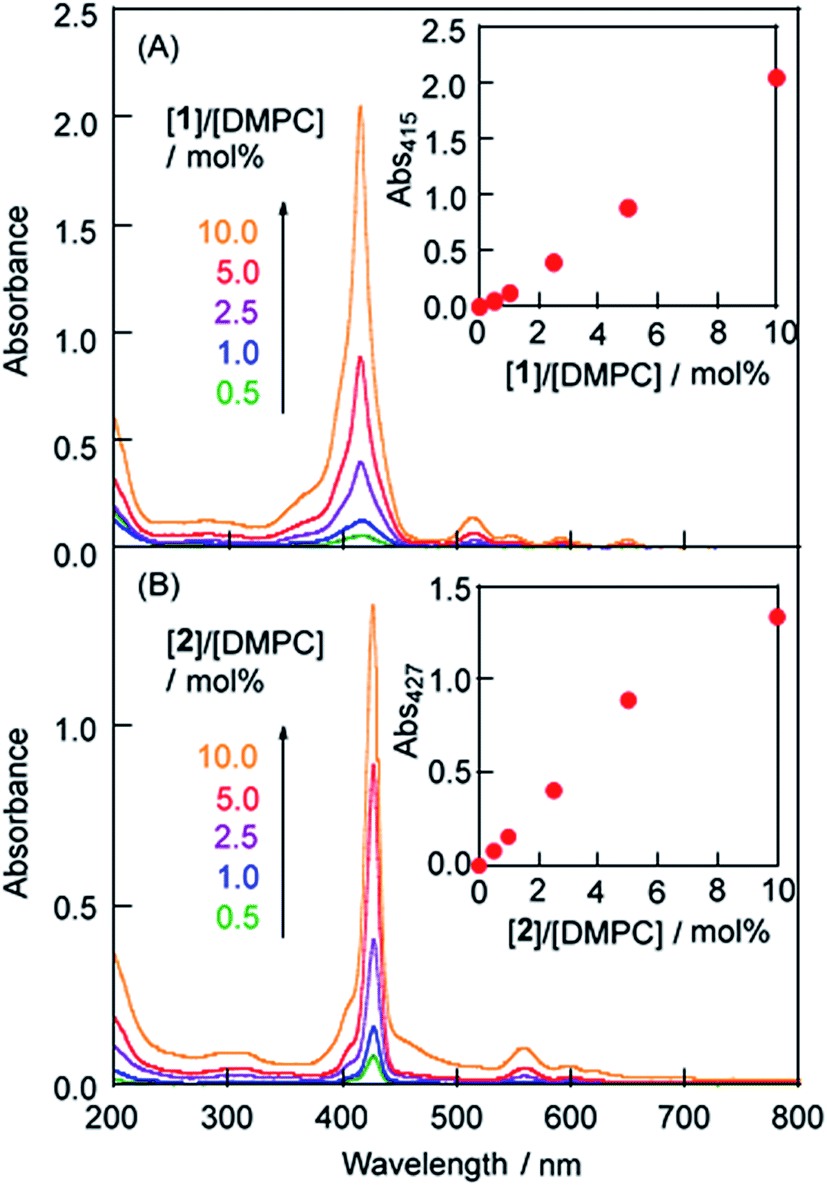
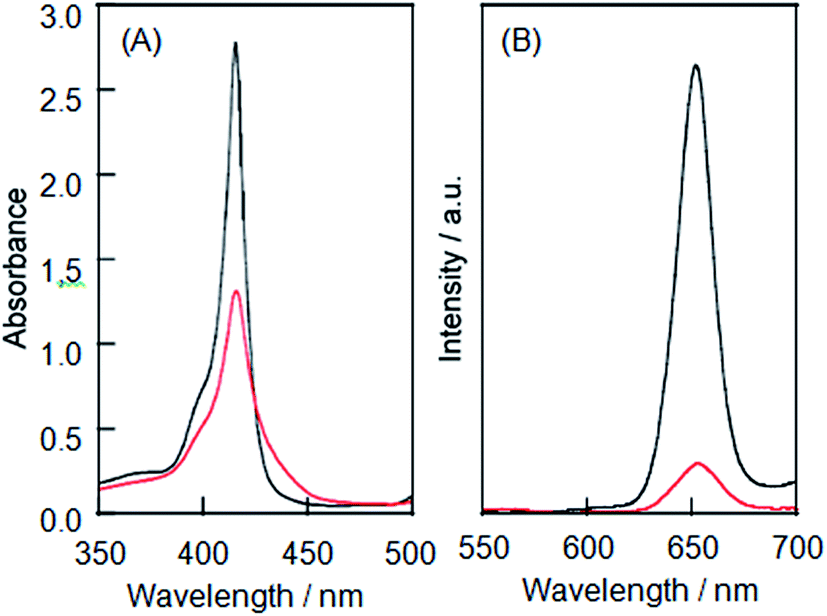
LMI1 was prepared by an exchange reaction between the liposomes and the 1·TMe-β-CDx complex at 30 °C for only 1 min. The UV-vis absorption spectra were recorded for different ratios (0.5–10.0 mol%) of [1]/[DMPC] in D2O following the exchange method (Fig. 2A). Compared with the 1·TMe-β-CDx complex, there was a little broadening in the absorption spectrum of LMI1 ([1]/[DMPC] = 10.0 mol%) (Fig. 1E and 2A). These results therefore suggested that 1 existed in a more isolated state in the lipid membranes than 1 in the LMI1 prepared by the DMSO injection method (Fig. 1D). However, compound 1 apparently aggregated in LMI1 because the fluorescence intensity of LMI1 decreased significantly compared with that of the 1·TMe-β-CDx complex at the same concentration (Fig. 3B). The UV-vis spectrum of LMI1 revealed that the shape of the Soret band broadened significantly after 12 h (Fig. S2A†). Precipitation was also observed in this case, with the self-aggregation of 1 having an adverse impact on the stability of LMI1.
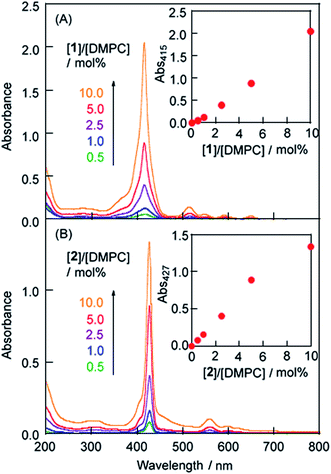 |
| | Fig. 2 UV-vis absorption spectra of (a) LMI1 and (b) LMI2 prepared by the exchange method at 30 °C for 1 h ([DMPC] = 1.0 mM, [1 or 2]/[DMPC] = 0.5, 1.0, 2.5, 5.0 and 10.0 mol%). Inset: the absorbance (Abs) at λmaxversus [1]/[DMPC] in LMI1 or [2]/[DMPC] in LMI2 prepared by the exchange method. | |
 |
| | Fig. 3 (a) UV-vis absorption spectra of the 1·TMe-β-CDx complex (black line) and LMI1 (red line) and (b) the fluorescence spectra of the 1·TMe-β-CDx complex (black line) and LMI1 (red line). Excitation wavelength: 425 nm at the isosbestic point of the Soret band region [1] = 0.05 mM. | |
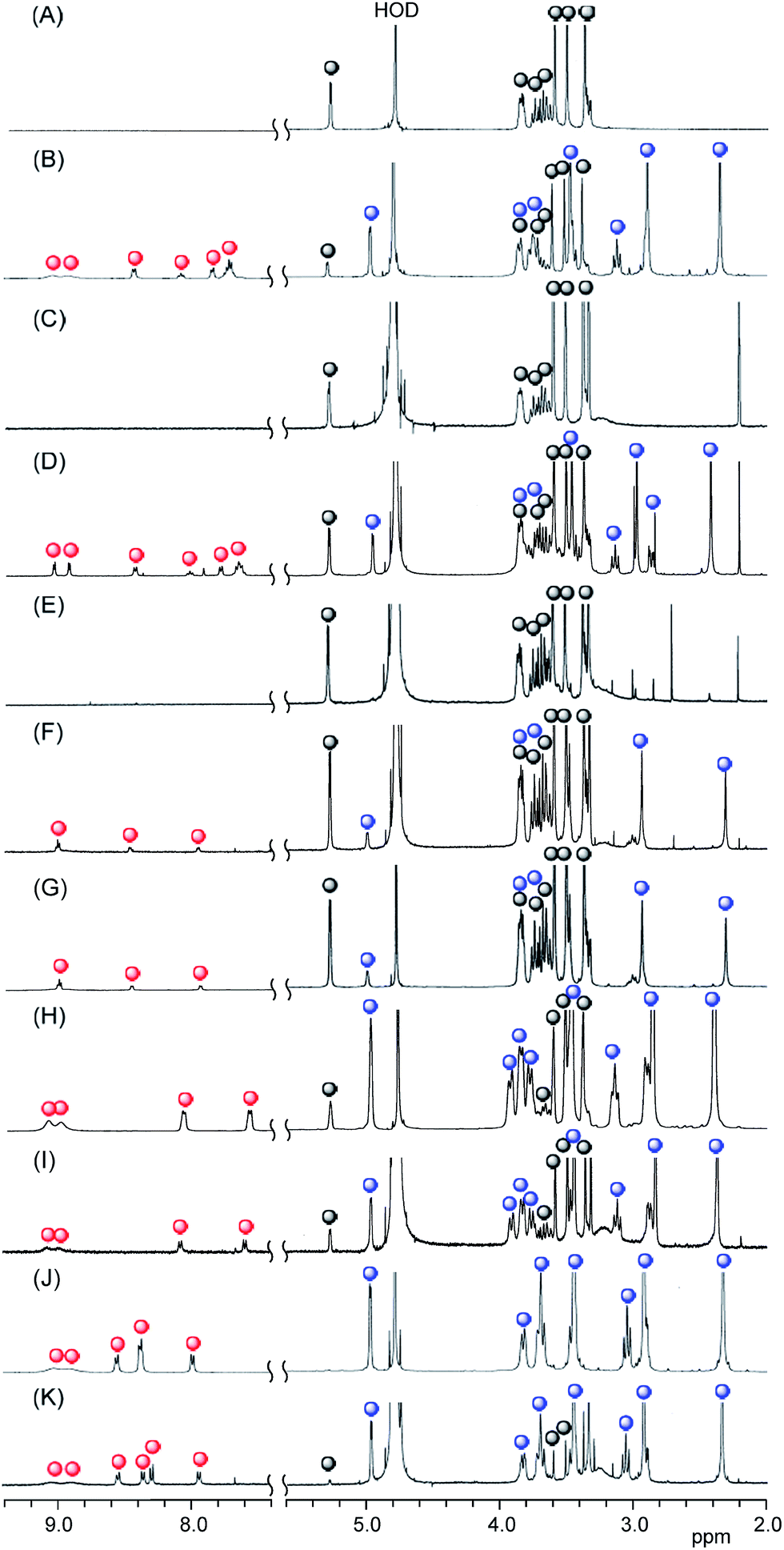
To confirm that all of porphyrins had been released from the two TMe-β-CDx cavities, we measured the 1H NMR spectra (Fig. 4 and S3†) of the aqueous solutions recorded by dissolving 5.0 and 10.0 mol% [1]/[DMPC] mixtures in D2O at 30 °C for 1 h. Fig. 4C shows that there were no peaks assignable to the 1·TMe-β-CDx complex (red and blue circles in Fig. 4B). These results clearly indicated that 1 had been completely released from the TMe-β-CDx cavities. The peaks belonging to 1 disappeared because of broadening of these signals when the material was incorporated into the liposomes or they underwent a self-aggregation process.
 |
| | Fig. 4 Partial 1H NMR spectra of (A) TMe-β-CDx, (B) the 1·TMe-β-CDx complex, (C) a mixture of the 1·TMe-β-CDx complex and the DMPC liposomes, (D) the 2·TMe-β-CDx complex, (E) a mixture of the 2·TMe-β-CDx complex and the DMPC liposomes, (F) the 3·TMe-β-CDx complex, (G) a mixture of the 3·TMe-β-CDx complex and the DMPC liposomes, (H) the 4·TMe-β-CDx complex, (I) a mixture of the 4·TMe-β-CDx complex and the DMPC liposomes (J) the 5·TMe-β-CDx complex and (K) a mixture of the 5·TMe-β-CDx complex and the DMPC liposomes ([1, 2, 3, 4 or 5]/[DMPC] = 10.0 mol%) in D2O at 25 °C ( : free TMe-β-CDx, : free TMe-β-CDx,  : 1, 2, 3, 4 or 5 and : 1, 2, 3, 4 or 5 and  : TMe-β-CDx in the 1, 2, 3, 4 or 5·TMe-β-CDx complex). : TMe-β-CDx in the 1, 2, 3, 4 or 5·TMe-β-CDx complex). | |
Effect of the central metal ion and substituents at the meso positions on the exchange reaction
The effect of a central metal ion on the exchange reaction was evaluated by investigating the transfer of 2 from the TMe-β-CDx cavities to a lipid membrane. The 2·TMe-β-CDx complex was mixed with the DMPC-liposomes at 30 °C for 1 h. Subsequent analysis by the UV-vis adsorption spectroscopy showed that LMI2 gave sharper peaks in its UV-vis absorption spectrum than LMI1 (Fig. 2A and B). Furthermore, the fluorescence intensity of this system decreased slightly after the exchange reaction, as shown in Fig. 5B. These results indicated that 2 existed in a more isolated state in the lipid membranes than 1. As shown in Fig. 4E, the peaks corresponding to complexed TMe-β-CDx (blue circles in Fig. 4D) and 2 (red circles in Fig. 4D) disappeared completely in a similar manner to those of 1 (Fig. 4C). These results clearly indicated that all of the molecules of 2 had been released from the TMe-β-CDx cavities and transferred to the lipid membrane. Although the preparation of LMI1 using an exchange reaction at 30 °C required only 1 min, the preparation of LMI2 under the same conditions required more than 45 min of mixing. This difference in the mixing time can be interpreted in three different ways: (i) the lability of the 1·TMe-β-CDx complex; (ii) the lower affinity of 2 for the lipid membrane; and (iii) the slow penetration of 2 through the surface of the membrane. To test explanation (i), we conducted an experiment to determine the stabilities of the 1·TMe-β-CDx and 2·TMe-β-CDx complexes towards the addition of DMSO. Both complexes decomposed after the addition of DMSO because DMSO effectively lowered the extent of the hydrophobic interaction between the porphyrins and TMe-β-CDx. Fig. 6 shows the resistance properties of the 1:2 complexes towards DMSO that were determined based on the sum of the split peak intensities at ca. 5 ppm versus the peak intensities of the free TMe-β-CDx (5.11 ppm). After the addition of DMSO, the 2·TMe-β-CDx complex was slightly less stable than the 1·TMe-β-CDx complex. This result indicated that 2 was released from the TMe-β-CDx cavity more readily than 1, and therefore suggested that explanation (i) was incorrect. Furthermore, given that LMI2 was determined to be more stable than LMI1 (the details on this are mentioned later), we could also discount explanation (ii). Based on these results, explanation (iii) was determined to be the best explanation of the experimental results, albeit by a process of elimination. However, the reason for the slow penetration of 2 remains unclear at this time.
 |
| | Fig. 5 (A) UV-vis absorption spectra of the 2·TMe-β-CDx complex (black line) and LMI2 (red line) and (B) the fluorescence spectra of the 2·TMe-β-CDx complex (black line) and LMI2 (red line). Excitation wavelength: 425 nm at the isosbestic point of the Soret band region [2] = 0.05 mM. | |
 |
| | Fig. 6 Stability of the 1·TMe-β-CDx complex (black line) and the 2·TMe-β-CDx complex (red line) towards the addition of DMSO ([complex]/[complex]0, where [complex] is the concentration of the 1 or 2·TMe-β-CDx complexes following the addition of DMSO and [complex]0 is the initial concentration). Each value represents the mean ± SD of three experiments. | |
In contrast to porphyrins 1 and 2, porphyrins 3, 4 and 5 have polar and hydrophilic groups at their meso positions. As shown in Fig. 4G and I, the peaks corresponding to complexed TMe-β-CDx (blue circles in Fig. 4F and H) and 3 or 4 (red circles in Fig. 4F and H) remained unchanged following the mixing of the 3·TMe-β-CDx and 4·TMe-β-CDx complexes with the DMPC-liposomes at 30 °C for 1 h. In contrast, although several peaks corresponding to 5 had shifted following the mixing of the 5·TMe-β-CDx complex with the DMPC-liposomes (red circles in Fig. S4C†), the peaks assignable to TMe-β-CDx in the 5·TMe-β-CDx complex remained unchanged in solution (blue circles in Fig. S4C†). This result indicated that the 5·TMe-β-CDx complex did not decompose under these conditions, but interacted with the liposomes. The details of these interactions will be described elsewhere. Notably, no changes were observed in the spectra of any of these mixtures, even after they had heated at 80 °C for 1 h (Fig. S4†). These results indicated that it was not possible to transfer 3, 4 or 5 into the lipid membranes using the exchange reaction. To determine whether the stability of the 3, 4 and 5·TMe-β-CDx complexes or the low affinity of compounds 3, 4 and 5 for the lipid membrane was responsible for the failure of the complexation process, we attempted to prepare LMI3 using the DMSO injection method. However, most of 3 formed had formed a precipitate 1.5 h after the injection. This result clearly showed that 3 could not be incorporated into the liposomes by the injection method. It can therefore be concluded that the formation of lipid-membrane-incorporated porphyrin derivatives is greatly affected by the affinity of the porphyrin derivatives for the alkyl chains in the lipid membranes. These results are supported by a report from the literature, which showed that 5 was not incorporated into the lipid phase at pH 7.3, with the fluorescence spectrum of 5 remaining unchanged at various lipid concentrations.21
Phase transition of LMI1
Differential scanning calorimetry (DSC) was used to observe changes in the phase-transition temperature (Tm) of the large unilamellar vesicles both with and without 1.22 The Tm of LMI1 (24.7 °C, [1]/[DMPC] = 10 mol%) underwent small changes compared with that of the DMPC liposomes (23.2 °C) (Fig. 7). In contrast, the half-height width of the transition peak (ΔT1/2) of LMI1 was nearly the same value as that of the DMPC liposomes [DMPC liposomes: ΔT1/2 = 0.6 °C, LMI1: ΔT1/2 = 0.4 °C]. These results indicated that 1 was perturbing the packing of the alkyl chains in the DMPC liposomes and that the presence of 1 was therefore having an adverse impact on the stability of the gel phase or leading to the instability of the liquid crystal phase.
 |
| | Fig. 7 DSC curves of DMPC (black line) and LMI1 (red line). [1]/[DMPC] = 10 mol%. [DMPC] = 4.0 mM. | |
Liposome diameters before and after the exchange method
The hydrodynamic diameters (Dhy) of the liposomes were measured by dynamic light scattering (DLS) (Fig. S5 and Table S1†). The average Dhy values were determined for all of the LMI1 materials after the exchange method and found to be in the range of 71–76 nm. Furthermore, similar morphologies were observed before and after the exchange method by cryo-TEM measurements (Fig. S6A and B†). Taken together, these results indicated that the introduction of 1 by the exchange method did not lead to the disruption or fusion of the liposomes.
Stabilities of LMI1 and LMI2
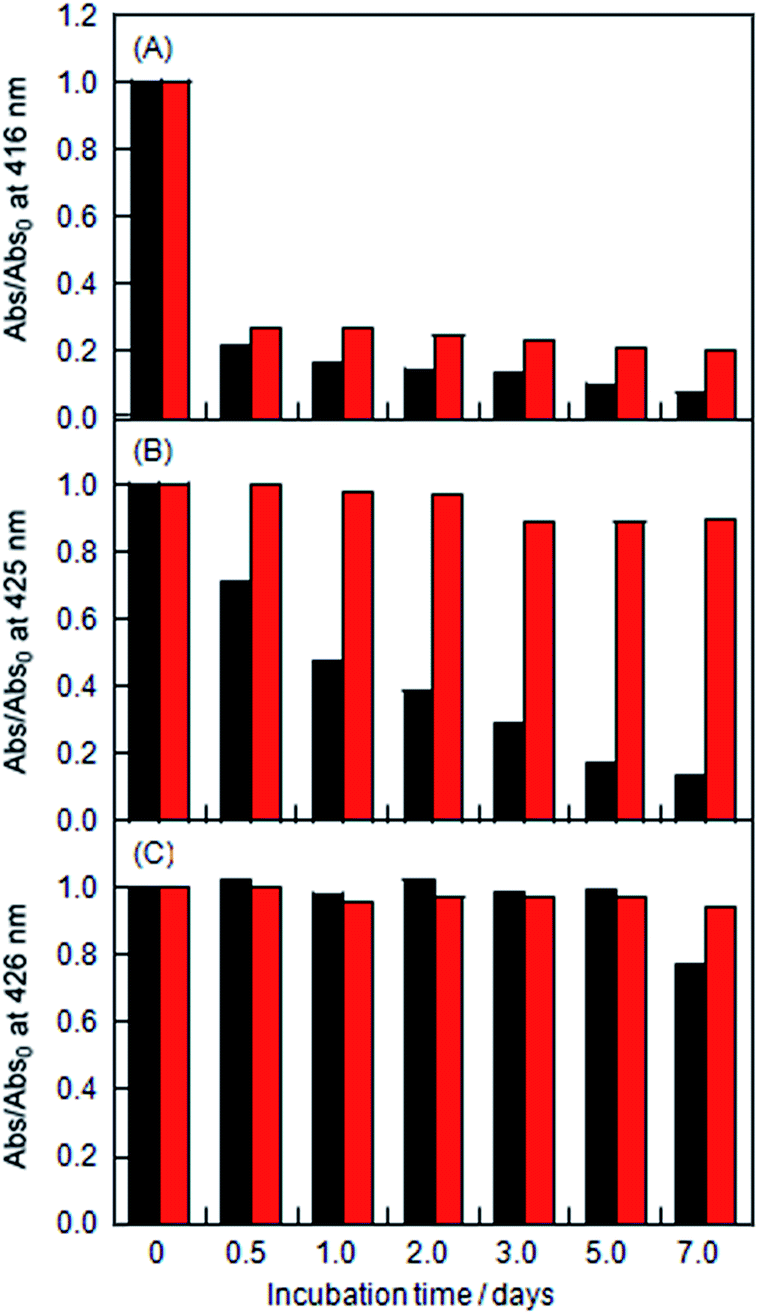
The stabilities of the LMI1 and LMI2 solutions were determined based on changes in the absorbance characteristics of the Soret bands of 1 and 2 (Fig. S2† and 8). The LMI2 solution was more stable than the LMI1 solution at ambient temperature (Fig. 8A and 7B black bars). However, less than 15% of the porphyrins in the liposomes of these solutions were soluble in water, and significant levels of precipitation were observed after 7 days at ambient temperature. The LMI1 and LMI2 solutions were predicted to become unstable because of the formation of large self-aggregates of porphyrins in the lipid membranes, which would result in the fusion and fission of liposomes. To inhibit the movement of the porphyrins in the lipid membranes, the LMI1 and LMI2 solutions were kept for 7 days at a lower temperature than the Tm (DMPC; 23 °C). Below the Tm, the DMPC liposomes would progress from a fluid phase (liquid-crystal) to a gel phase, which is associated with the straightening of the hydrophobic lipid tails. As expected, over 98% of the LMI2 remained in aqueous solution after 7 days and no precipitation was observed for at least 1 week at 4 °C (Fig. 8B red bar). In contrast, although the stability of LMI1 was slightly improved at 4 °C, the LMI1 solutions showed similar stability profiles after 7 days at both of these temperatures (Fig. 8A red bar). These differences in the stabilities between LMI1 and LMI2 could be attributed to the fact that that 1 tends to form the self-aggregations more readily than 2.23
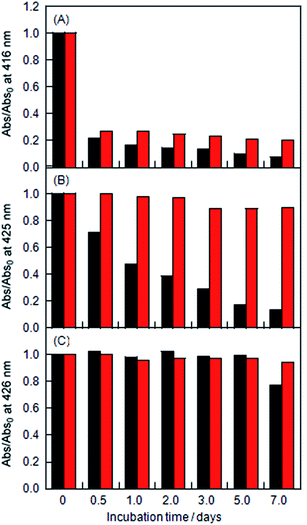 |
| | Fig. 8 Changes in the absorption at λmax of (A) LMI1, (B) LMI2 and (C) LMI2–6 kept at ambient temperature (black bar) and at 4 °C (red bar) with incubation times 0, 0.5, 1, 2, 3, 5, and 7 days (Abs0: initial absorbance). The values of λmax were 416 nm for LMI1, 425 nm for LMI2, and 426 nm for LMI2–6. [1 or 2]/[DMPC] = 5.0 mol% and [6]/[DMPC] = 5.0 mol%. | |
Improvement in the stability of LMI2 by the addition of an axial ligand
To investigate the effect of adding an axial ligand for 2, we prepared a lipid-membrane incorporating 2 and 4-phenylpyridine (6) (LMI2–6) by the transfer of 2 from the cavity of TMe-β-CDx to the lipid membranes of LMI6, which was prepared using the premixing method ([2]/[DMPC] = [6]/[DMPC] = 5 mol%). As shown in Fig. S7,† the Q band of the porphyrin slightly shifted to a long-wavelength in the presence of 6. This result clearly showed that compound 6 was interacting with 2, which was as an axial ligand.24 The LMI2–6 solution was found to be stable at ambient temperature and 4 °C (Fig. 8C and S8†). These results therefore indicated that the axial coordination of 6 stabilised LMI2 by inhibiting the self-aggregation of 2 rather than through the bulkiness of the complex formed between 2 and 6. Overall, this result suggested that the self-aggregation of the porphyrins was responsible for the fusion and fission of the liposomes.
Direct observation for the formation of LMI1
The dynamic behaviour of the giant unilamellar vesicles (GUVs) was monitored by time-lapse microscopy to obtain direct evidence for the transfer of 1 from the TMe-β-CDx cavities to the lipid membranes by the exchange method (Fig. 9 and S9†).25,26 Two minutes after the injection of a solution of the 1·TMe-β-CDx complex, there was no discernible difference in the morphology of the GUVs (Fig. 9A and C). However, fluorescence signals were detected on the surfaces of GUVs during the same time period, which indicated that 1 was being incorporated into lipid membrane of the liposome (Fig. 9D).
 |
| | Fig. 9 Time-lapse images of the GUVs consisting of POPC (A and B) before and (C and D) after the addition of the 1·TMe-β-CDx complex (0.1 mM). (A and C) Phase contrast and (B and C) fluorescence images. The time elapsed following the starting injection of the 1·TMe-β-CDx complex solution through the micropipette has been indicated for each image. All of the scale bars show 50 μm. | |
Direct introduction of 1 and 2 into the plasma membranes by the exchange method
The exchange method developed in the current study was also applied to the direct and short-term introduction of guest molecules into the plasma membranes of living cells.27 The introduction of 1 into HeLa cells was observed by fluorescence microscopy (Fig. 10). After being treated with the 1·TMe-β-CDx complex for 30 min, the plasma membranes of the HeLa cells became slightly stained with 1 (Fig. 10D). This result suggested that the 1·TMe-β-CDx complex was decomposing in the medium to form a precipitate prior to the introduction of 1 into the HeLa cells because of the lability of the 1·TMe-β-CDx complex. Furthermore, the fluorescence intensity of 1 following the exchange method was higher than that of the DMSO injection method (Fig. 10F).
 |
| | Fig. 10 Phase contrast (A, C and E) and fluorescence (B, D and F) images of the HeLa cells both before (A and B) and after (C and D) being treated with the 1·TMe-β-CDx complex for 30 min at 37 °C, and after (E and F) being treated with the DMSO solution of 1 for 30 min at 37 °C. The scale bar represents 100 μm. | |
In contrast, the plasma membranes of the HeLa cells were heavily stained with 2 after being treated with the 2·TMe-β-CDx complex for only 30 min (Fig. 11B). The fluorescence intensity of 2 was much higher than that of 1. To further demonstrate that 2 had been successfully incorporated into the plasma membrane, the HeLa cells were treated sequentially with the 2·TMe-β-CDx and C70·γ-CDx complexes because C70 can also move into the plasma membrane, where it can act as a quencher.28 The results of this experiment revealed that almost all of the signals corresponding to 2 were quenched following the addition of C70 (Fig. 11D), which provided further proof that 2 had been successfully delivered into the plasma membranes of the HeLa cells by the exchange method. To develop a deeper understanding of the mechanism underlying the uptake of 2, we evaluated the effect of a low temperature because it is well known that endocytotic processes can be inhibited at low temperatures. HeLa cells were incubated with the 2·TMe-β-CDx complex for 30 min at 4 °C. The results revealed that although the liposome uptake of 2 was considerably inhibited at 4 °C compared with the uptake at 37 °C (Fig. 11F), the fluorescence of 2 was still observed. This result indicated that 2 had been directly internalised into the HeLa cells by the exchange reaction. Furthermore, the fluorescence intensity of 2 following the DMSO injection method (Fig. 11H) was much lower it was following the exchange method.
 |
| | Fig. 11 Phase contrast (A, C, E and G) and fluorescence (B, D, F and H) images of the HeLa cells after (A and B) being treated with the 2·TMe-β-CDx complex for 30 min at 37 °C, and then after the addition of the C70·γ-CDx complex (C and D). The images (E and F) after being treated with the 2·TMe-β-CDx complex for 1 h at 4 °C. The images (G and H) after being treated with the DMSO solution of 2 for 30 min at 37 °C. The scale bar represents 100 μm. | |
Conclusions
Although tetraphenylporphyrin 1 and zinc(II) tetraphenylporphyrin 2 cannot be incorporated directly into a lipid membrane using conventional methods, such as the premixing and the injection methods, the preparation of LMI1 and LMI2 using an exchange method was achieved. Very few self-aggregates of 1 and 2 formed in the liposomes, as evidenced by the sharp spectral features of LMI1 and LMI2, which were similar to those of isolated 1 and 2. The long-term stability of LMI2 was much higher than that of LMI1 when the two materials were kept at 4 °C. Furthermore, the long-term stability of LMI2 was improved by the addition of the axial ligand 6 even at ambient temperature. In contrast, it was not possible to transfer porphyrins 3, 4 and 5 with four polar and hydrophilic groups at their circumference to lipid membranes using the exchange method. Furthermore, the incorporation of 1 and 2 into the plasma membrane of HeLa cells was observed by the exchange method. It is envisaged that these findings will have a significant impact on a variety of different applications in biological, medicinal and material chemistry because this newly developed exchange method allows for the facile preparation of cells containing porphyrin derivatives. Furthermore, this new exchange method could be used to incorporate porphyrin derivatives into a variety of different biomaterials such as DNA molecules and proteins.
Experimental
Experimental materials
Tetraphenylporphyrin (TPP, 1), 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (4) and 5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin (5) were purchased from Tokyo Chemical Industries Co. Ltd. (Tokyo, Japan). Zinc(II) tetraphenylporphyrin (2) was purchased from Sigma-Aldrich Chemical Co., Inc. (St. Louis, MO, USA). TMe-β-CDx and 5,10,15,20-tetra(4-pyridyl)porphyrin (3) were purchased from Wako Pure Chemical Industries Ltd. (Tokyo, Japan). Dimyristoylphosphatidylcholine (DMPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC) were obtained from NOF Corp. (Tokyo, Japan).
Preparation of LMI1 by the premixing method
A mixture of compound 1 (0.12 mg, 2.0 × 10−7 mol) and DMPC (2.71 mg, 4.0 × 10−6 mol) in chloroform (1 mL) was evaporated to dryness using a rotary evaporator at 40 °C. Water (4.0 mL) was then added, and the resulting mixture was shaken for 5 min on a vortex mixer. To allow for a change from multilamellar to unilamellar vesicles and the formation of a narrow size distribution, the solution was frozen and thawed three times and extruded eleven times (LiposoFast-Basic; Avestin Inc., Ottawa, Canada) with two stacked polycarbonate membranes, pore size 50 nm.
Preparation of LMI1 and LMI2 by the injection method
A DMSO solution of 1 (0.1 mM, 0.05 mL) was dropped into a aqueous solution of unilamellar vesicles of DMPC (0.053 mM, 0.95 mL) at 30 °C, and the resulting mixture was stirred for 1 h. The material was then centrifuged (18000 × g, 25 °C, 20 min) to allow for the removal of non-dispersed 1 from the solution. LMI2 ([2]/[DMPC] = 2.5–20 mol%) was also prepared by mixing an aqueous solution of the DMPC liposomes (2.11 mM, 0.95 mL) and a DMSO solution of 2 (4.0 mM, 0.05 mL) at 30 °C for 1 h in a 5 mL glass vial.
Preparation of the 1–4·TMe-β-CDx complexes
Compound 1 (5.00 mg, 8.13 × 10−6 mol) and TMe-β-CDx (23.3 mg, 1.63 × 10−5 mol) were placed in an agate capsule with two agate-mixing balls, and the resulting mixture was vigorously agitated at 30 Hz for 20 min using a high-speed vibration mill (MM 200; Retsch Co., Ltd., Haan, Germany). The solid mixture was suspended in pure water (1.5 mL) to produce a dark purple emulsion. Subsequent centrifugation (18000 × g, 25 °C, 20 min) allowed for the removal of non-dispersed 1 from the solution. The concentration of 1 in the 1·TMe-β-CDx complex was determined to be 1.33 mM by measuring the absorbance of the solution at 415 nm in water (the molar absorption coefficient for the water-soluble 1·TMe-β-CDx complex was ε415 = 3.30 × 104 dm3 mol−1 cm−1). The TMe-β-CDx complexes of porphyrins 2–5 were also prepared in the same manner.
Preparation of LMI1–4 by the exchange method
A mixture of DMPC (9.76 mg, 1.44 × 10−5 mol) in chloroform (1 mL) was evaporated to dryness using a rotary evaporator at 40 °C. Water (4.0 mL) was then added, and the resulting mixture was shaken for 5 min on a vortex mixer. To allow for a change from multilamellar to unilamellar vesicles and the formation of a narrow size distribution, the solution was frozen and thawed three times and extruded eleven times (LiposoFast-Basic; Avestin Inc., Ottawa, Canada) with two stacked polycarbonate membranes, pore size 50 nm. The solution was diluted with water to a final lipid concentration of 8.0 mM for 1. An aqueous solution of the 1·TMe-β-CDx complex (x mL, x = 0.028, 0.069, 0.139, or 0.278, [1] = 1.44 mM), liposome (1.0 mL, [DMPC] = 4.00 mM) and water (1.0 − x mL) was heated at 30 °C for 1 h in a 5 mL glass vial. The values of the [1]/[DMPC] ratio were changed from 2.5 to 10 mol%. LMI2–5 were also prepared in the same manner.
Cryogenic temperature transmission electron microscopy (Cryo-TEM)
Cryo-TEM samples were prepared using a universal cryofixation and cryopreparation system (Leica EM CPC, Wetzlar, Germany). To eliminate the possibility of water being evaporated from the sample, the isolation chamber was humidified to near saturation prior to the introduction of the sample. Sample droplets (2–3 μL) were placed on a microperforated cryo-TEM grid and soaked on to a filter paper, resulting in the formation of thin liquid films of 10–300 nm in thickness, which freely spanned the micropores in a carbon-coated lace-like polymer layer supported by a metal mesh grid. After a minimum holding time of 30 s, the sample grid assembly was rapidly vitrified with liquid ethane at its melting temperature (−163 to −170 °C). The holding time was adopted to avoid the possibility of any deformations in the flow that may have resulted from the blotting process. The vitreous specimen was held under liquid nitrogen until it was loaded into the cryogenic sample holder (Gatan 626.DH). Imaging was performed on a JEOL JEM-3100 FEF imaging system operating at 300 kV (Tokyo, Japan). The use of a minimal dose system (MDS) was necessitated by the electron radiation sensitivity of the sample being probed. Images were recorded on a Gatan 794 multiscan digital camera (Gatan, Inc., Pleasanton, California) and processed using version 3.8.1 of the Digital Micrographs software (Gatan, Inc.). The optical density gradients in the background, which are normally ramp-shaped were digitally corrected using a custom-made subroutine compatible with Digital Micrographs.
Dynamic light scattering (DLS)
The hydrodynamic diameter of LMI1 was measured on an electrophoretic light scattering instrument with a laser Doppler system (Zetasizer Nano ZS, Malvern Instruments Ltd., Malvern, UK).
Preparation of giant unilamellar vesicles (GUVs)
GUVs were fabricated by the electroformation method on indium tin oxide (ITO) electrodes, which were originally designed by Angelova et al.25,26 A chloroform solution of POPC (1.0 mM) was prepared for the electroformation of the vesicles. These conditions generally provide access to large amounts of GUVs with diameters >10 μm under the appropriate conditions (see below). Ten microliter samples of the lipid solution in chloroform were therefore spread in a snake-like pattern without overlap over a 1.5 × 1.5 cm2 surface area using a 10 μL Hamilton syringe. Following the evaporation of the lipid film deposited on the ITO-coated glass under a stream of nitrogen, the electroformation chamber was assembled. The chamber consisted of two ITO-coated coverslips fitted with a copper wire. The ITO-coverslips were configured in such a way that they were facing each other, with each strip being separated by a 3 mm thick polydimethylsiloxane (PDMS) film, which was used to seal the chamber. The assembled vesicle electroformation chamber was slowly filled with 450 μL of deionised water. A sinusoidal AC electric field of 10 Hz and 2.0 V (rms) was applied for 20 min to form GUVs over a specific phase transition temperature (Tm) using a thermostated observation stage (Linkam 10022/23) (30 °C).
Fluorescence microscopy of GUVs
A solution of the 1·TMe-β-CDx complex (0.1 mM, 10 μL) was injected into the chamber in the presence of the GUVs. A microscopic image was acquired using an Olympus IX71 epifluorescence microscope (Olympus Optical Co. Ltd, Tokyo, Japan) equipped with a 60× objective lens. Fluorescent images were recorded using a Hamamatsu C11440-10C CMOS digital camera (Hamamatsu Photonics K.K., Shizuoka, Japan) under the irradiation of an excitation light beam from a mercury lamp through an optical filter (U-MNIBA2, Olympus). The temperature of the sample was maintained using a thermostated observation stage (Linkam 10022/23).
Cell culture
HeLa cells were maintained in CO2 independent medium (Gibco BRL) supplemented with 10% fetal calf serum at 37 °C in 5% CO2. The cells were seeded on glass-bottomed dishes at a density of 3.2 × 105 cells per plate. The cells were grown overnight under these conditions prior to be used in the experiments. Cells were seeded onto glass coverslips to allow for the uptake of the liposomes to be analysed.
Fluorescence microscopy of HeLa cells
The cells were pre-incubated at 37 °C without CO2 for 30 min before being treated with 1 or 2. Following a 3 min period of incubation with 20 μM of the 1 or 2·TMe-β-CDx complex, or DMSO solution of 1 or 2 at 37 °C, the cells were replaced with fresh medium and the introduction of 1 or 2 was monitored by fluorescence microscopy. The cells were observed using an Olympus IX71 epifluorescence microscope equipped with a 100× or 200× objective lens. Fluorescent images were recorded using a Hamamatsu ImagEM EM-CCD camera (C9100-13) under the irradiation of an excitation light beam from a mercury lamp through an optical filter (U-MNIBA2, Olympus). Furthermore, the observed cells were also incubated for 3 min with 61 μM of the C70·TMe-β-CDx complex at 37 °C. Following the incubation period, the cells were washed with phosphate-buffered saline (PBS) and monitored by fluorescence microscopy.
Acknowledgements
This work was supported by JSPS KAKENHI a Grant-in-Aid for Scientific Research (B) (Grant No. 25288037) and a Grant-in-Aid for Young Scientists (A) (Grant No. 24681028). The authors would like to express their deepest gratitude to Ms S. Fujita, Graduate School of Materials Science, Nara Institute of Science and Technology for providing technical assistance with the cryo-TEM observations.
Notes and references
- A. D. Bangham, Prog. Biophys. Mol. Biol., 1968, 18, 29–36 CrossRef CAS PubMed
 .
.
- Y. Kaneda, Adv. Drug Delivery Rev., 2000, 43, 197–205 CrossRef CAS PubMed .
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed .
-
Liposomes: A Practical Approach, ed. V. P. Torchilin and W. Weissig, Oxford University Press, Oxford, 2nd edn, 2003 Search PubMed .
- H. Hungerbühler, D. M. Guldi and K.-D. Asmus, J. Am. Chem. Soc., 1993, 115, 3386–3387 CrossRef .
- R. V. Bensasson, E. Bienvenue, M. Dellinger, S. Leach and P. Seta, J. Phys. Chem., 1994, 98, 3492–3500 CrossRef CAS .
- A. Ikeda, T. Hida, T. Nakano, S. Hino, K. Nobusawa, M. Akiyama and K. Sugikawa, Chem. Lett., 2014, 43, 1551–1553 CrossRef CAS .
- M. Pons, M. Foradada and J. Estelrich, Int. J. Pharmacol., 1993, 95, 51–56 CrossRef CAS .
- A. Wagner, K. Vorauer-Uhl, G. Kreismayr and H. Katinger, J. Liposome Res., 2002, 12, 259–270 CrossRef CAS PubMed .
- A. Ikeda, T. Sato, K. Kitamura, K. Nishiguchi, Y. Sasaki, J. Kikuchi, T. Ogawa, K. Yogo and T. Takeya, Org. Biomol. Chem., 2005, 3, 2907–2909 CAS .
- A. Ikeda, Y. Doi, K. Nishiguchi, K. Kitamura, M. Hashizume, J. Kikuchi, K. Yogo, T. Ogawa and T. Takeya, Org. Biomol. Chem., 2007, 5, 1158–1160 CAS .
- A. Ikeda, Y. Doi, M. Hashizume, J. Kikuchi and T. Konishi, J. Am. Chem. Soc., 2007, 129, 4140–4141 CrossRef CAS PubMed .
- A. Ikeda, T. Hida, T. Iizuka, M. Tsukamoto, J. Kikuchi and K. Yasuhara, Chem. Commun., 2014, 50, 1288–1291 RSC .
- A. Ikeda, K. Kiguchi, T. Shigematsu, K. Nobusawa, J. Kikuchi and M. Akiyama, Chem. Commun., 2011, 47, 12095–12097 RSC .
- A. Ikeda, M. Mori, K. Kiguchi, K. Yasuhara, J. Kikuchi, K. Nobusawa, M. Akiyama, M. Hashizume, T. Ogawa and T. Takeya, Chem.–Asian J., 2012, 7, 605–613 CrossRef CAS PubMed .
- A. Ikeda, S. Hino, K. Ashizawa, K. Sugikawa, J. Kikuchi, M. Tsukamoto and K. Yasuhara, Org. Biomol. Chem., 2015, 13, 6175–6182 CAS .
- S. Yano, S. Hirohara, M. Obata, Y. Hagiya, S. Ogura, A. Ikeda, H. Kataoka, M. Tanaka and T. Joh, J. Photochem. Photobiol., C, 2011, 12, 46–67 CrossRef CAS .
- S. Ferro, F. Ricchelli, G. Mancini, G. Tognon and G. Jori, J. Photochem. Photobiol., B, 2006, 83, 98–104 CrossRef CAS PubMed .
- H. Ibrahim, A. Kasselouri, C. You, P. Maillard, V. Rosilio, R. Pansu and P. Prognon, J. Photochem. Photobiol., A, 2011, 217, 10–21 CrossRef CAS .
- Y. Tsuchiya, T. Shiraki, T. Matsumoto, K. Sugikawa, K. Sada, A. Yamano and S. Shinkai, Chem.–Eur. J., 2012, 18, 456–465 CrossRef CAS PubMed .
- M. Kępczyński, R. P. Pandian, K. M. Smith and B. Ehrenberg, Photochem. Photobiol., 2002, 76, 127–134 CrossRef .
- J. A. Castillo, A. Pinazo, J. Carilla, M. R. Infante, M. A. Alsina, I. Haro and P. Clapes, Langmuir, 2004, 20, 3379–3387 CrossRef CAS PubMed .
- R. F. Pasternack, L. Francesconi, D. Raff and E. Spiro, Inorg. Chem., 1973, 12, 2606–2611 CrossRef CAS .
- C. H. Kirkesy, P. Hambright and C. B. Storm, Inorg. Chem., 1969, 8, 2141–2144 CrossRef .
- M. Angelova and D. Dimitrov, Faraday Discuss. Chem. Soc., 1986, 81, 303–311 RSC .
- G. Staneva, M. I. Angelova and K. Koumanov, Chem. Phys. Lipids, 2004, 129, 53–62 CrossRef CAS PubMed .
- Kano et al. have reported that a cationic porphirin derivative complexed with TMe-β-CDx can be introduced into the HeLa cells. In this case, endocytosis is probably a dominant pathway in the presence of serum proteins, whereas cationic porphirin derivative in the serum-free medium containing TMe-β-CD can pass directly through the cell membrane and disperse in the cytosol. Therefore, the mechanism is different from that in our complex; H. Kitagishi, S. Hatada, T. Itakura, Y. Maki, Y. Maeda and K. Kano, Org. Biomol. Chem., 2013, 11, 3203–3211 CAS .
- A. Ikeda, M. Matsumoto, M. Akiyama, J. Kikuchi, T. Ogawa and T. Takeya, Chem. Commun., 2009, 1547–1549 RSC .
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H NMR spectra, DLS measurements, cryo-TEM images, phase contrast and fluorescence images. See DOI: 10.1039/c5ra24985f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 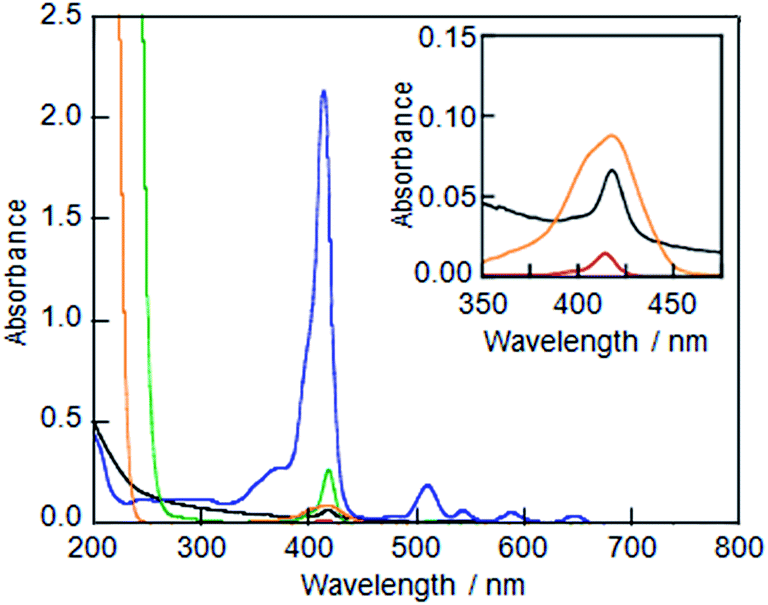
 : free TMe-β-CDx,
: free TMe-β-CDx,  : 1, 2, 3, 4 or 5 and
: 1, 2, 3, 4 or 5 and  : TMe-β-CDx in the 1, 2, 3, 4 or 5·TMe-β-CDx complex).
: TMe-β-CDx in the 1, 2, 3, 4 or 5·TMe-β-CDx complex).