DOI:
10.1039/D1SC02819G
(Edge Article)
Chem. Sci., 2021,
12, 12174-12180
Mesoionic N-heterocyclic olefin catalysed reductive functionalization of CO2 for consecutive N-methylation of amines†‡
Received
24th May 2021
, Accepted 2nd August 2021
First published on 3rd August 2021
Abstract
A mesoionic N-heterocyclic olefin (mNHO) was introduced as a metal-free catalyst for the reductive functionalization of CO2 leading to consecutive double N-methylation of primary amines in the presence of 9-borabicyclo[3.3.1]nonane (9-BBN). A wide range of secondary amines and primary amines were successfully methylated under mild conditions. The catalyst sustained over six successive cycles of N-methylation of secondary amines without compromising its activity, which encouraged us to check its efficacy towards double N-methylation of primary amines. Moreover, this method was utilized for the synthesis of two commercially available drug molecules. A detailed mechanistic cycle was proposed by performing a series of control reactions along with the successful characterisation of active catalytic intermediates either by single-crystal X-ray study or by NMR spectroscopic studies in association with DFT calculations.
Introduction
Carbon dioxide (CO2), the major nontoxic and increasingly abundant greenhouse gas, has potential to be one of the most useful reagents as a sustainable C1 building block with an enormous industrial prospect for the synthesis of value-added fine chemicals.1,2 In recent years, the catalytic activation of CO2 to form new C–O, C–N, and C–C bonds has become more useful in synthetic chemistry through the reductive functionalization pathway.3–5 Towards reductive functionalization of CO2, N-methylation for the construction of new C–N bonds in the presence of a hydride source such as hydroboranes or hydrosilanes is considered to be one of the promising and efficient routes.6,7 In particular, N-methylation of amines can give rise to products which are broadly used in the synthesis of medicines, dyes, agrochemicals, perfumes and drugs.8,9 Traditionally, N-methylamines are synthesized using stoichiometric amounts of toxic reagents such as formaldehyde, methyl iodide, dimethyl sulphate etc.10,11 Thus, the use of CO2 as a methylating reagent under a reductive atmosphere is an attractive alternative to produce N-methyl amines through direct functionalization of amines. In the last decade, several metal catalysts12–21 and a few metal-free catalysts22–25 were reported for this catalytic transformation in the presence of hydrosilanes as a reducing agent and all of these studies were carried out at an elevated temperature. However, these methods have very limited potential towards consecutive N-methylation involving primary amines.12,13,22,24 In contrast to N-methylation of secondary amines, double N-methylation of primary amines has been considered challenging due to their much lower reactivity.21 Moreover, hydroboranes as reducing agents were less studied than hydrosilanes for the reductive functionalization of CO2 leading to N-methylation.26,27
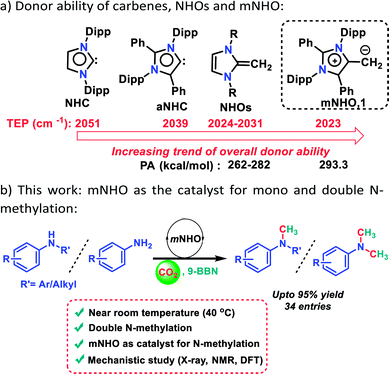
In this context, metal-free nucleophiles such as proazaphosphatrane super bases26 and carbodicarbenes27 have displayed moderate reactivity towards N-methylation using CO2 which works only at a high temperature (100 °C) in the presence of 9-BBN. A closer look at the mechanism described earlier26,27 reveals that it passes through the following key steps: (I) formation of boron formate, (II) formyl group transfer and (III) the reduction of the formyl group to the methyl group. Among these steps, steps II and III can be achieved only at high temperature. In this work, we attempted to lower the activation barrier of steps II and III so that such steps could be achieved at a lower temperature by increasing the nucleophilicity of the catalyst. In this regard, a highly nucleophilic mesoionic N-heterocyclic olefin (mNHO) 1 was explored as a catalyst (Scheme 1a).
 |
| | Scheme 1 (a) NHC, aNHC, NHOs, mNHO, their Tolman electronic parameter (TEP) and proton affinity (PE) values. Dipp = 2,6-diisopropylphenyl;, R = aryl. (b) This work showing mono and double N-methylation of amines catalysed by mNHO under mild conditions. | |
It may be recalled that N-heterocyclic olefins28 (NHOs), alkylidene derivatives of N-heterocyclic carbenes29–31 (NHCs), have been emerging as metal-free catalysts for various organic transformations including CO2 activation, polymerization, and hydrosilylation reactions.32–35 Besides these NHOs, Hansmann and co-workers recently introduced a super nucleophilic mesoionic N-heterocyclic olefin (mNHO) 1 containing a polarised exocyclic double bond.36,37 The Tolman electronic parameter (TEP) of 1 (2023 cm−1)−suggests that mNHO 1 has the strongest donor property compared to NHC (2051 cm−1), NHOs (2024–2031 cm−1) and aNHC (2039 cm−1) (Scheme 1a).36 Moreover, theoretical calculations predicted that mNHO (1) possessing the highest proton affinity value of 293.3 kcal mol−1 is likely to be the strongest nucleophile among earlier reported N-heterocyclic olefins (NHOs) whose proton affinity values lie in the range of 262–282 kcal mol−1 (Scheme 1a).36 Such a high proton affinity of mNHO was attributed to the superior ylidic character of mNHO 1 compared to that of the previously reported NHOs.36 Due to such a high nucleophilic property, mNHO (1) was explored in this work for borane assisted N-methylation of amines using CO2. It was anticipated that the superior nucleophilicity of mNHO will enable the reaction under milder conditions leading to a more efficient catalytic conversion. In this direction, we report the first catalytic application of mNHO 1 for the methylation of secondary and primary amines in the presence of CO2 under mild conditions using 9-BBN as a hydride source (Scheme 1b). The catalytic process unravelled that the mNHO can efficiently convert two N–H bonds of amines consecutively leading to double N-methylation of a range of primary amines nearly at room temperature (40 °C) with very good to excellent yields. Thereafter, we focused on the mechanistic understanding with the combined approach of various experimental and theoretical studies. From a stoichiometric control reaction, the mNHO–9-BBN adduct was isolated and characterized using the X-ray structure, and was found to be the active reaction intermediate.
Results and discussion
The present investigation was started by optimizing the N-methylation of amines using CO2 in the presence of a catalytic amount of mNHO (1), diphenylamine (2a) as a model substrate and borane as a reducing agent (see the ESI; Table S1‡). To our delight, after several trial runs, it was observed that the mNHO works as an efficient catalyst for the N-methylation of secondary amines under mild conditions. Furthermore, such an optimization study unravelled that 9-BBN is the best hydride source delivering the highest isolated yield of 95% using 5 mol% mNHO (1) under 1 atmospheric pressure of CO2 at 40 °C. Having the optimal catalytic conditions in hand, the scope of the reaction was further evaluated for the N-methylation of various secondary amines with 9-BBN as a hydride donor and mNHO (1) as a catalyst under 1 atm pressure of CO2 (Scheme 2).
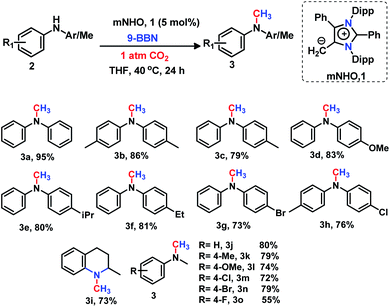 |
| | Scheme 2 mNHO catalysed N-methylation of secondary amines. Reaction conditions: amines (0.2 mmol), CO2 (1 atm), 9-BBN (0.8 mmol), and dry THF (0.5 mL). Isolated yields (average yield of two catalytic runs) are reported. | |
The secondary amines containing N-alkyl or N-aryl groups displayed an excellent activity towards N-methylation in the presence of catalyst 1. Diphenylamine derivatives (2) afforded the corresponding methylated products (3a–3h) in good to excellent isolated yields (73–95%). The diphenylamines containing electron donating groups have been found to be highly reactive under the optimized reaction conditions providing very good yields (79–86%) of N-methyl amines 3b–3f (Scheme 2). The bromo and chloro-substituted diphenylamines underwent N-methylation with 73% (3g) and 76% (3h) yields, respectively (Scheme 2). This catalytic protocol was also applied for the N-methylation of an N-heterocyclic amine, affording the N-methylated product 3i in a good yield (73%, Scheme 2). The substituted N-methylamines resulted in the formation of the N,N-dimethyl aniline derivatives 3j–3o in moderate to excellent isolated yields (55–80%) under the same reaction protocol (Scheme 2).
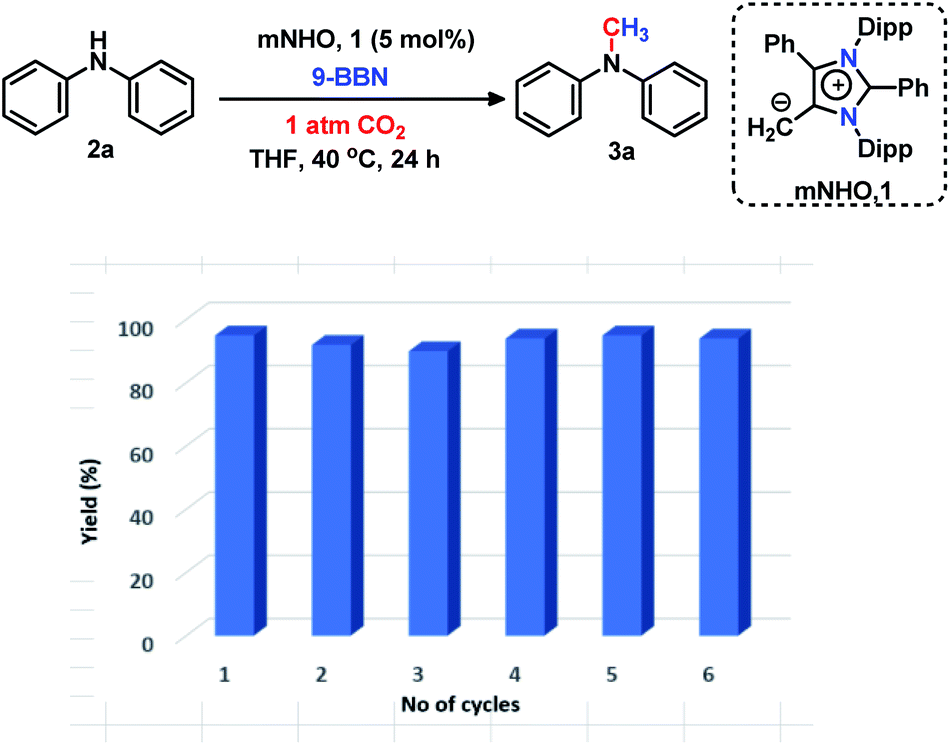
The 6e reduction of CO2 leading to mono-N-methylated products in excellent yields made us curious if the same catalyst can be used for double N-methylation by performing consecutive 12e reduction. To succeed with such an objective, we argued in favor of a catalyst which would be highly sustainable under the reaction conditions to carry out the further methylation process. To this purpose, we checked the longevity of the catalyst by taking 2a as the model substrate under the optimized reaction protocol (Scheme 3), where the catalyst was added once in the beginning and its activity was checked over six successive catalytic runs. After each cycle of catalytic run as per the optimized protocol, a fresh batch of substrate (2a) and 9-BBN were added to the reaction mixture under 1 atmospheric pressure of CO2 without adding any additional catalyst (see the ESI, Scheme S17‡). To our delight, it was noted that the yield for each cycle remains within the range of 90% to 95% (Scheme 3). The result indicates that the catalyst remains active at least up to six successive catalytic cycles without compromising its efficacy. This observation establishes the sustained activity of the catalyst, which prompted us to further investigate consecutive double N-methylation using primary amines. It may be noted that the primary aromatic and aliphatic amines are considered to be less reactive and thus challenging for such double N-methylation by CO2 under mild conditions.23–26 In this direction, a model substrate 4-iodo aniline 4a was considered for the double N-methylation using our standard reaction protocol. To our delight, 4a resulted in the formation of the corresponding product in an excellent yield (88%) showing the efficacy of our catalytic system for double N-methylation of primary amines using CO2 under very mild conditions (Scheme 4). In total 8.0 equivalents of 9-BBN are needed for the double methylation of primary amines. The six hydrogens of two –CH3 groups come from the six equivalents of 9-BBN and another two equivalents of 9-BBN are required for the activation of two N–H bonds leading to the N-borylation process. Following this success, we focused on a variety of primary amines containing electron donating substituents. The electron donating substituents at the para position of primary aryl amines 4b–4f afforded the N,N-dimethylaniline derivatives 5b–5f in good to excellent isolated yields (66–84%, Scheme 4). Under these reaction conditions, 4-halo anilines were observed to be relatively less reactive affording the N,N-dimethylaniline derivatives 5g–5i in moderate to very good yields (52–78%, Scheme 4). The ortho substituted aniline derivatives 4j–4m also exhibited moderate to very good reactivity and afforded the N,N-dimethylaniline derivatives 5j–5m (59–77%, Scheme 4). The 1-napthylamine 4n showed good reactivity and was converted to N,N-dimethyl-1-napthylamine 5n in 73% yield (Scheme 4). Aliphatic primary amines 4o–4p also responded well to our optimized protocol by delivering moderate to good yields (47–69%). The compound 4q containing both the ‘NH2’ group and ‘NH-Ph’ group in the same molecule was successfully methylated to form 5q resulting in 61% yield to achieve consecutive triple N-methylation counting to a total of 18e reduction process (Scheme 4). We have tried selective N-monomethylation of primary amines by lowering the amount of 9-BBN and altering the reaction conditions; however, these attempts were not successful (see the ESI, Scheme S19‡). Furthermore, the current catalytic protocol was applied to synthesize two commercially available drug molecules such as angustureine38,39 (7), an anti-malarial drug (Scheme 5a), and pempidine17 (9), a ganglion-blocking drug (Scheme 5b), with 84% and 72% yields, respectively. Although compound 9 was synthesized earlier17 by reductive functionalization of CO2, this is the first report of the synthesis of 7 using CO2 as a reagent. These examples highlight the synthetic applicability of the current method.
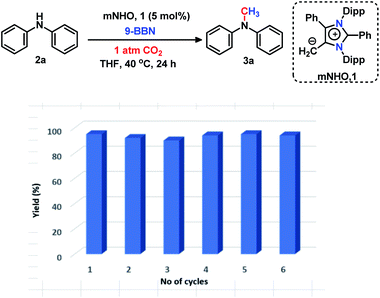 |
| | Scheme 3 Longevity of the catalyst. Reaction conditions: amines (0.2 mmol), CO2 (1 atm), 9-BBN (0.8 mmol), and dry THF (0.5 mL). A bar diagram showing yield (average yield of two experiments) versus number of catalytic cycles, emphasizing that the catalyst remains equally efficient over six successive catalytic cycles. | |
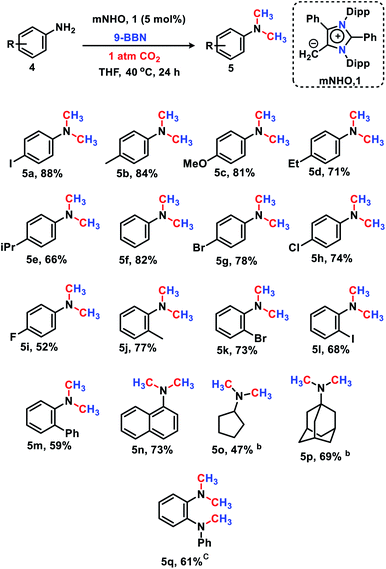 |
| | Scheme 4 mNHO catalysed N,N-dimethylation of primary amines.a aReaction conditions: amines (0.2 mmol), CO2 (1 atm), 9-BBN (1.6 mmol), and dry THF (1 mL). Isolated yields (average yield of two experiments) are reported. bYields were determined by GC using p-xylene as an internal standard. c2.4 mmol of 9-BBN is used. | |
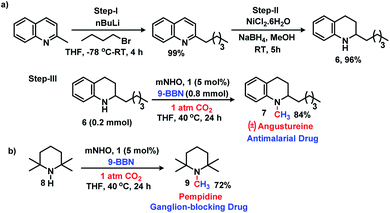 |
| | Scheme 5 Synthetic application. Reaction conditions: amines (0.2 mmol), CO2 (1 atm), 9-BBN (0.8 mmol), and dry THF (0.5 mL). Isolated yields (average yield of two experiments) are reported. | |
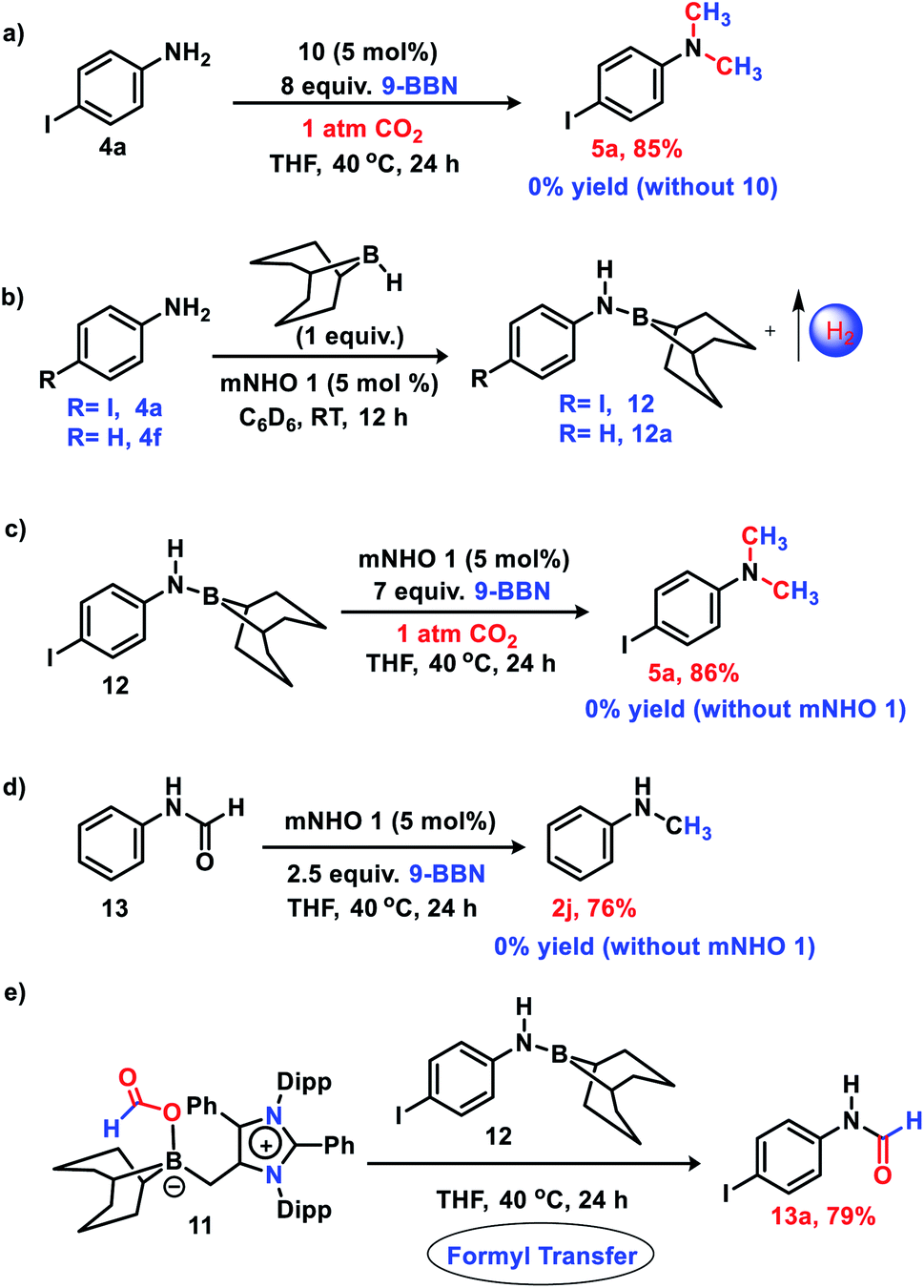
Next, to delve into the mechanistic details of such a catalytic N-methylation process, a series of control reactions were carried out. At first, a stoichiometric reaction between mNHO 1 and 9-BBN in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio was performed in toluene (Fig. 1a). After the completion of the reaction, an off-white solid was isolated from the reaction mixture. The 11B NMR spectrum of this off-white solid in C6D6 revealed a singlet at δ = −11.2 ppm (see the ESI; Fig. S3‡), which is indicative of the formation of a mNHO–9BBN adduct (10).40 In another control reaction between mNHO and CO2, the formation of the mNHO–CO2 adduct was revealed. However, in our reaction protocol, we first mixed mNHO and 9-BBN (excess) and a mNHO–9BBN adduct (10) was formed. This observation discards the possibility of the involvement of the mNHO–CO2 adduct in the reaction considering the sequence of addition of reagents. Furthermore, mNHO–9-BBN (10) was successfully crystallized from toluene under an argon atmosphere at −25 °C with 62% yield (Fig. 1a). The X-ray structure (Fig. 1b) revealed that the C1– B1 bond length {1.689(4) Å} in 10 was slightly elongated compared to that observed for the aNHC–9-BBN adduct {1.636(2) Å}.41,42 The C1–C2 bond length {1.483(4) Å} of 10 was longer than that of mNHO 1{1.432(2) Å},36 which indicates that the π-electron cloud of mNHO has moved towards the boron center and increased the B–H hydride donor ability. To understand the reactivity of 10, a reaction between 10 and CO2 was performed at room temperature for 30 min in THF (Fig. 1c). After completion of the reaction, the solvent was dried and a white solid product was obtained. 1H NMR (δ = 8.67 ppm) and 13C NMR spectroscopy (δ = 166.27 ppm) studies suggest the formation of the mNHO–boron formate intermediate 11 (see the ESI; Fig. S4 and S5‡).26,27,43 To further investigate the reaction mechanism, a series of control reactions were performed (Scheme 6). At first, the isolated 10 was used as a catalyst in the double N-methylation of substrate 4a which afforded the corresponding N,N-dimethylated product 5a in a very good yield (85%) under the optimized conditions (Scheme 6a) and it compares very well with the catalytic outcome (88% yield, see Scheme 4) using free mNHO 1. This experiment suggests that mNHO–9-BBN adduct 10 is a catalytically active intermediate for this N,N-dimethylation reaction. Next, in a screw cap NMR tube, 4-iodo aniline (4a) and 9-BBN were taken in a 1:1 ratio in the presence of 5 mol% 1 in C6D6 at room temperature (Scheme 6b) and the reaction was monitored by 1H NMR spectroscopy. During this reaction, a gas was evolved, which was identified as dihydrogen from the 1H NMR spectroscopy peak at δ = 4.47 ppm and further confirmed by GC-MS analysis (see the ESI; Fig. S7 and S8‡). After completion of this reaction (12 h), 1H and 13C NMR spectroscopy of the reaction mixture confirmed the formation of an N-borylated compound 12 (see the ESI; Fig. S9 and S10‡). Similar to that for 4-iodo aniline, we also observed the dehydrogenative B–N coupling product 12a for the aniline substrate (see the ESI; Fig. S11 and S12‡). Such an N-borylation of amines with hydrogen liberation has been reported earlier.44,45 Furthermore, when 12 was treated with 7 equiv. of 9-BBN and 5 mol% of 1 under 1 atm pressure of CO2 at 40 °C, it afforded 4-iodo N,N-dimethyl aniline 5a in 86% yield, which confirms that the reaction proceeds through the intermediacy of 12 (Scheme 6c). It has been reported earlier that the N-methylation of amines proceeds in a stepwise manner through the N-formylation of amines.26,27 To check this, we performed the reduction of formanilide 13, using 2.5 equiv. of 9-BBN in the presence of catalyst 1 (Scheme 6d). The formation of N-methyl aniline in 76% yield suggests that the N-methylation reaction proceeds via N-formylation. A control reaction between formanilide 13 and 9-BBN in the absence of the mNHO catalyst did not produce N-methyl aniline which indicates the involvement of the catalyst during this step. We performed a stoichiometric reaction between mNHO–boron formate 11 and 12 in a 1:1 ratio which afforded 4-iodo N-formylamine 13a with 79% yield (Scheme 6e), characterized by 1H and 13C{1H} NMR spectroscopies (see the ESI, Fig. S18 and S19‡). Formation of 13a suggests that the N-methylation reaction proceeds through N-formylation.
:1 ratio was performed in toluene (Fig. 1a). After the completion of the reaction, an off-white solid was isolated from the reaction mixture. The 11B NMR spectrum of this off-white solid in C6D6 revealed a singlet at δ = −11.2 ppm (see the ESI; Fig. S3‡), which is indicative of the formation of a mNHO–9BBN adduct (10).40 In another control reaction between mNHO and CO2, the formation of the mNHO–CO2 adduct was revealed. However, in our reaction protocol, we first mixed mNHO and 9-BBN (excess) and a mNHO–9BBN adduct (10) was formed. This observation discards the possibility of the involvement of the mNHO–CO2 adduct in the reaction considering the sequence of addition of reagents. Furthermore, mNHO–9-BBN (10) was successfully crystallized from toluene under an argon atmosphere at −25 °C with 62% yield (Fig. 1a). The X-ray structure (Fig. 1b) revealed that the C1– B1 bond length {1.689(4) Å} in 10 was slightly elongated compared to that observed for the aNHC–9-BBN adduct {1.636(2) Å}.41,42 The C1–C2 bond length {1.483(4) Å} of 10 was longer than that of mNHO 1{1.432(2) Å},36 which indicates that the π-electron cloud of mNHO has moved towards the boron center and increased the B–H hydride donor ability. To understand the reactivity of 10, a reaction between 10 and CO2 was performed at room temperature for 30 min in THF (Fig. 1c). After completion of the reaction, the solvent was dried and a white solid product was obtained. 1H NMR (δ = 8.67 ppm) and 13C NMR spectroscopy (δ = 166.27 ppm) studies suggest the formation of the mNHO–boron formate intermediate 11 (see the ESI; Fig. S4 and S5‡).26,27,43 To further investigate the reaction mechanism, a series of control reactions were performed (Scheme 6). At first, the isolated 10 was used as a catalyst in the double N-methylation of substrate 4a which afforded the corresponding N,N-dimethylated product 5a in a very good yield (85%) under the optimized conditions (Scheme 6a) and it compares very well with the catalytic outcome (88% yield, see Scheme 4) using free mNHO 1. This experiment suggests that mNHO–9-BBN adduct 10 is a catalytically active intermediate for this N,N-dimethylation reaction. Next, in a screw cap NMR tube, 4-iodo aniline (4a) and 9-BBN were taken in a 1:1 ratio in the presence of 5 mol% 1 in C6D6 at room temperature (Scheme 6b) and the reaction was monitored by 1H NMR spectroscopy. During this reaction, a gas was evolved, which was identified as dihydrogen from the 1H NMR spectroscopy peak at δ = 4.47 ppm and further confirmed by GC-MS analysis (see the ESI; Fig. S7 and S8‡). After completion of this reaction (12 h), 1H and 13C NMR spectroscopy of the reaction mixture confirmed the formation of an N-borylated compound 12 (see the ESI; Fig. S9 and S10‡). Similar to that for 4-iodo aniline, we also observed the dehydrogenative B–N coupling product 12a for the aniline substrate (see the ESI; Fig. S11 and S12‡). Such an N-borylation of amines with hydrogen liberation has been reported earlier.44,45 Furthermore, when 12 was treated with 7 equiv. of 9-BBN and 5 mol% of 1 under 1 atm pressure of CO2 at 40 °C, it afforded 4-iodo N,N-dimethyl aniline 5a in 86% yield, which confirms that the reaction proceeds through the intermediacy of 12 (Scheme 6c). It has been reported earlier that the N-methylation of amines proceeds in a stepwise manner through the N-formylation of amines.26,27 To check this, we performed the reduction of formanilide 13, using 2.5 equiv. of 9-BBN in the presence of catalyst 1 (Scheme 6d). The formation of N-methyl aniline in 76% yield suggests that the N-methylation reaction proceeds via N-formylation. A control reaction between formanilide 13 and 9-BBN in the absence of the mNHO catalyst did not produce N-methyl aniline which indicates the involvement of the catalyst during this step. We performed a stoichiometric reaction between mNHO–boron formate 11 and 12 in a 1:1 ratio which afforded 4-iodo N-formylamine 13a with 79% yield (Scheme 6e), characterized by 1H and 13C{1H} NMR spectroscopies (see the ESI, Fig. S18 and S19‡). Formation of 13a suggests that the N-methylation reaction proceeds through N-formylation.
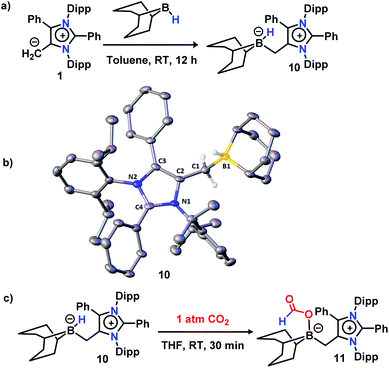 |
| | Fig. 1 (a) Stoichiometric reaction between mNHO 1 and 9-BBN resulting in the formation of 10. (b) The X-ray structure of 10. Ellipsoids are set at the 50% probability level; hydrogen atoms except –CH2 and the B–H hydrogens have been omitted for the sake of clarity. (c) A control reaction between 10 and CO2 reveals the formation of mNHO–boron formate 11via insertion of CO2 into the activated B–H bond. | |
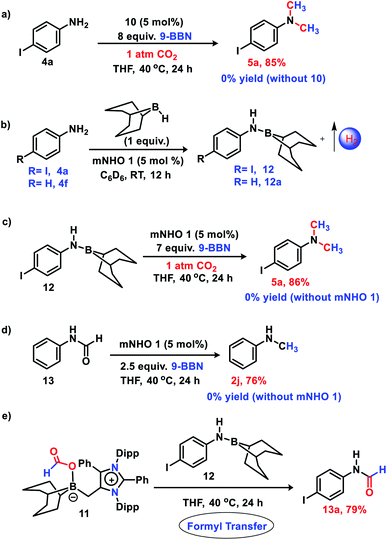 |
| | Scheme 6 Control experiments: (a) reaction of 4a with 9-BBN and CO2 using 10 as a catalyst (b) N-borylation of amines with H2 liberation (c) reaction of 12 with 9-BBN and CO2 in presence of mNHO catalyst (d) reduction of formanilide to N-methylamine (e) formyl transfer reaction from 11 to 12. Average yields of two experiments are reported. | |
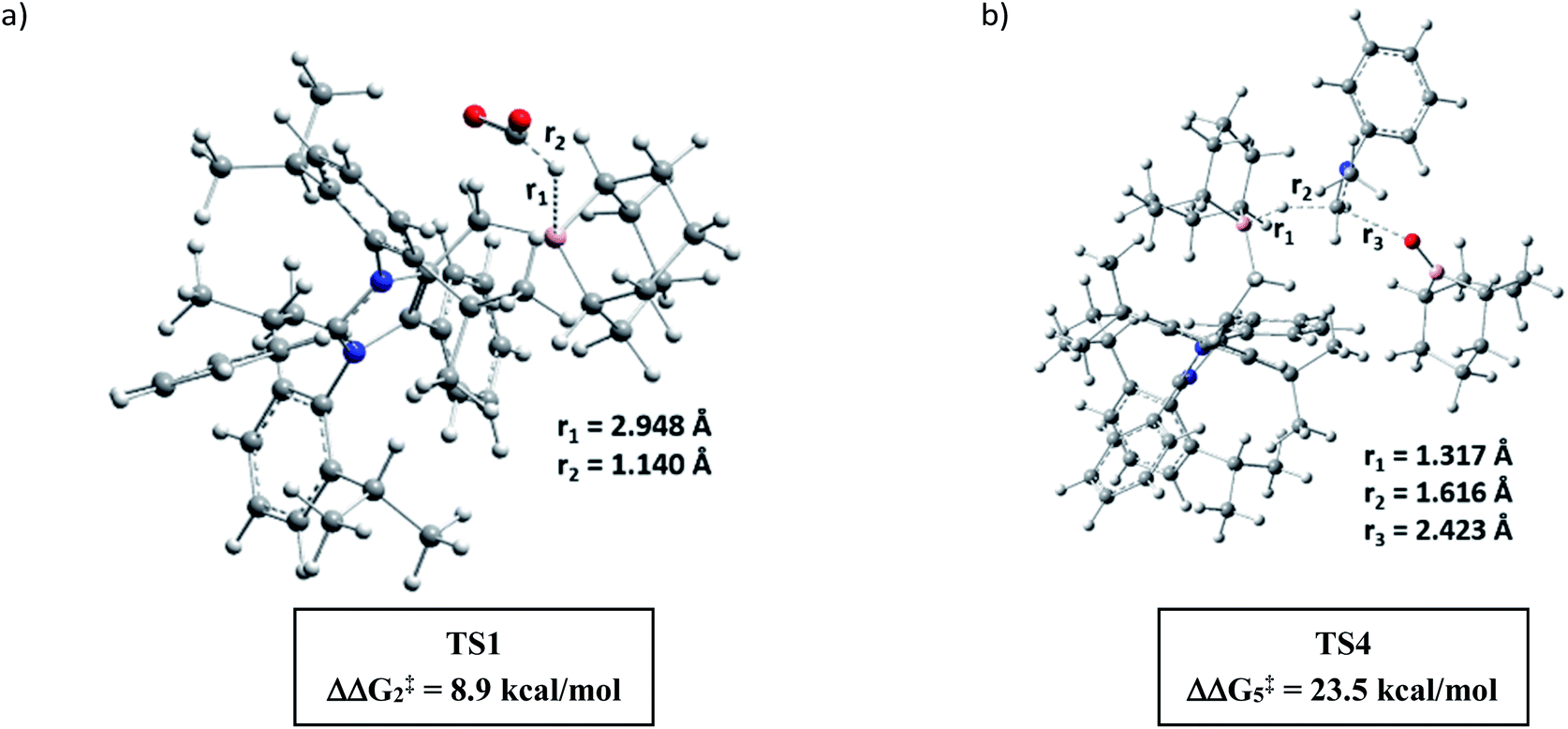
A full reaction path was mapped with the help of DFT calculations using the M06-2X/6-311+G(d)-SMD(THF)//B3LYP/6-31G(d) level of theory (for full energy profile diagrams, see the ESI, Fig. S95 and S96‡). Based on the calculated reaction energetics and the experimental support, a plausible reaction mechanism was sketched for N,N-dimethylation of primary amines in Scheme 7. At first, mNHO 1 reacts with 9-BBN to form mNHO–9-BBN adduct 10, which was crystallographically and NMR spectroscopically characterized. In this step, the B–H bond of the borane gets activated through coordination of a strong nucleophile mNHO. In the next step, the activated B–H bond inserts a CO2 molecule forming the intermediate 11 through TS1 (Fig. 2a) which is 8.9 kcal mol−1 (ΔΔG‡2) lower in energy than that of the catalyst free conditions. Compound 11 was characterized by NMR spectroscopic studies. Simultaneously, the amine is activated by reaction with 9-BBN forming the N-borylated compound 12 with the liberation of hydrogen gas. In the subsequent step, the formyl group was transferred from the intermediate 11 to N-borylated amine 12 leading to the formation of formanilide 13 and (9-BBN)2O as a byproduct, which was characterized by NMR spectroscopy46 (see the ESI; Fig. S24 and S25‡). The theoretical study reveals that the transition state TS2, corresponding to the formyl transfer, is energetically less favorable than that of the uncatalyzed one without the assistance of the mNHO catalyst (ΔΔG‡5 = −22.0 kcal mol−1). However, with the help of a series of control reactions (see Scheme 6e, also see the ESI; Schemes S11–S13†), we indeed found that the involvement of the mNHO catalyst is compulsory for the formyl transfer from mNHO–9-BBN formate 11 to the N-borylamine 12 species. This is in consonance with the fact that the formation of 9-BBN formate is not favorable in the absence of mNHO (indicated from the difference in the transition barrier corresponding to TS1 for catalyzed and uncatalysed reactions). After the formation of N-formylated product 13 as the reaction intermediate, it undergoes further reduction to intermediate 14 involving TS3f. Computational study suggests that this step does not require the involvement of a catalyst. Next, 14 undergoes further reduction with the mNHO–9-BBN adduct through TS4 (Fig. 2b) which is 23.5 kcal mol−1 (ΔΔG‡5) lower than that of the catalyst free conditions. This step afforded the final N,N-dimethyl aniline along with the formation of (9-BBN)2O as a byproduct. For double N-methylation, the mechanistic steps discussed above may repeat to deliver the final N,N-dimethylated product.
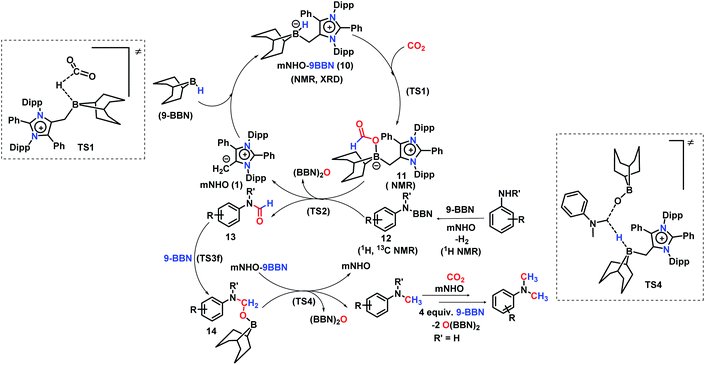 |
| | Scheme 7 Proposed mechanism of the mNHO catalysed N,N-dimethylation of amines. | |
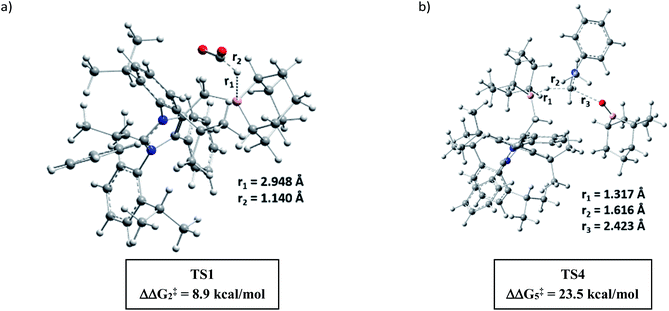 |
| | Fig. 2 DFT optimized key transition states involved in the catalytic cycle (for chemical drawings of TS1 and TS4, see Scheme 7). | |
Conclusions
In the present work, mNHO has been introduced as the catalyst for N,N-dimethylation and N-methylation of primary and secondary amines, under mild conditions, using CO2 as the C1 source and 9-BBN as the hydride source. Moreover, this newly developed methodology was successfully extended towards the preparation of two commercially available drug molecules. This result demonstrates that mNHO can be used as an efficient catalyst for the consecutive 12e reduction process leading to double N-methylation. A series of control reactions and theoretical calculations were performed to understand the underlying mechanistic pathway through the isolation and characterization of various reaction intermediates either by single-crystal X-ray study or by NMR spectroscopy along with the realization of different transition states. Further application for functionalization of CO2 using mNHO as a metal-free catalyst is currently ongoing in our laboratory.
Data availability
All experimental procedures, characterization details, copies of NMR spectra for all compounds, and computational data related to this article have been uploaded as part of the ESI. Crystallographic data for 10 have been deposited at the CCDC under 2065467 and can be obtained from https://www.ccdc.cam.ac.uk/structures/.
Author contributions
SKM and SM conceived the idea of this work. SM carried out all synthetic and catalytic experiments as well as DFT calculations. AD contributed to X-ray structure determination. SKM supervised the overall work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
SKM thanks the STARS Program (Grant No. MoE-STARS/STARS-1/473) of India. SM thanks the CSIR, Delhi (09/921(0233)/2019-EMR-I), for a research fellowship. AD thanks the IISER Kolkata for a research fellowship. SM thanks Jasimuddin Ahmed, IISER-Kolkata, for his help with theoretical calculation. The authors also thank Prof. T. K. Paine, IACS Kolkata, for X-ray data collection.
Note and references
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS.
- Y. Li, X. Cui, K. Dong, K. Junge and M. Beller, ACS Catal., 2017, 7, 1077–1086 CrossRef CAS.
- P. Sreejyothi and S. K. Mandal, Chem. Sci., 2020, 11, 10571–10593 RSC.
- C. D. N. Gomes, O. Jacquet, C. Villiers, P. Thuery, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef.
- R. A. Pramudita and K. Motokura, Green Chem., 2018, 20, 4834–4843 RSC.
- S. Bontemps, Coord. Chem. Rev., 2016, 308, 117–130 CrossRef CAS.
- E. J. Barreiro, A. E. Kümmerle and A. C. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed.
-
M. F. Ali, M. F. Ali and J. G. Speight, Handbook of Industrial Chemistry: Organic Chemicals, McGraw-Hill Education, 2005 Search PubMed.
- T. H. Clarke, B. H. Gillespie and S. Z. Weisshaus, J. Am. Chem. Soc., 1933, 55, 4571–4587 CrossRef.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS.
- O. Jacquet, X. Frogneux, C. D. N. Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS.
- M.-Y. Wang, N. Wang, X.-F. Liu, C. Qiao and L.-N. He, Green Chem., 2018, 20, 1564–1570 RSC.
- R. H. Lam, C. M. A. McQueen, I. Pernik, R. T. McBurney, A. F. Hill and B. A. Messerle, Green Chem., 2019, 21, 538–549 RSC.
- L.-G. Sebastian, M. Flores-Alamo and J. J. García, Organometallics, 2015, 34, 763–769 CrossRef.
- C. Lu, Z. Qiu, Y. Zhu and B.-L. Lin, Sci. Bull., 2019, 64, 723–729 CrossRef CAS.
- X. F. Liu, C. Qiao, X.-Y. Li and L.-N. He, Green Chem., 2017, 19, 1726–1731 RSC.
- Z. Huang, X. Jiang, S. Zhou, P. Yang, C.-X. Du and Y. Li, ChemSusChem, 2019, 12, 3054–3059 CrossRef CAS.
- W.-D. Li, D.-Y. Zhu, G. Li, J. Chen and J.-B. Xia, Adv. Synth. Catal., 2019, 361, 5098–5104 CrossRef CAS.
- C. Fang, C. Lu, M. Liu, Y. Zhu, Y. Fu and B.-L. Lin, ACS Catal., 2016, 6, 7876–7881 CrossRef CAS.
- F. Bobbink, S. Das and P. Dyson, Nat. Protoc., 2017, 12, 417–428 CrossRef CAS PubMed.
- Z. Yang, B. Yu, H. Zhang, Y. Zhao, G. Ji, Z. Ma, X. Gao and Z. Liu, Green Chem., 2015, 17, 4189–4193 RSC.
- X.-F. Liu, X. Y. Li, C. Qiao, H.-C. Fu and L.-N. He, Angew. Chem., Int. Ed., 2017, 56, 7425–7429 CrossRef CAS.
- X.-F. Liu, X.-Y. Li, C. Qiao and L.-N. He, Synlett, 2018, 29, 548–555 CrossRef CAS.
- E. Blondiaux, J. Pouessel and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 12186–12190 CrossRef CAS PubMed.
- W.-C. Chen, J.-S. Shen, T. Jurca, C.-J. Peng, Y.-H. Lin, Y.-P. Wang, W.-C. Shih, G. P. A. Yap and T.-G. Ong, Angew. Chem., Int. Ed., 2015, 54, 15207–15212 CrossRef CAS PubMed.
- N. Kuhn, H. Bohnen, J. Kreutzberg, D. Blaser and R. Boese, J. Chem. Soc., Chem. Commun., 1993, 1136–1137 RSC.
- A. Ghosh and A. T. Biju, Angew. Chem., Int. Ed., 2021, 60, 13712–13724 CrossRef CAS PubMed.
- S. Mondal, S. R. Yetra, S. Mukherjee and A. T. Biju, Acc. Chem. Res., 2019, 52, 425–436 CrossRef CAS.
-
S. Mondal, S. R. Yetra and A. T. Biju, N-Heterocyclic Carbene-Catalyzed Stetter Reaction and Related Chemistry, in N-Heterocyclic Carbenes in Organocatalysis, ed. A. T. Biju, Wiley-VCH, 2019, pp. 59–93, ISBN: 9783527809042 Search PubMed.
- Y. B. Wang, Y. M. Wang, W. Z. Zhang and X. B. Lu, J. Am. Chem. Soc., 2013, 135, 11996–12003 CrossRef CAS.
- V. B. Saptal and B. M. Bhanage, ChemSusChem, 2016, 9, 1980–1985 CrossRef CAS PubMed.
- S. Naumann, A. W. Thomas and A. P. Dove, Angew. Chem., Int. Ed., 2015, 54, 9550–9554 CrossRef CAS.
- U. Kaya, U. P. N. Tran, D. Enders, J. Ho and T. V. Nguyen, Org. Lett., 2017, 19, 1398–1401 CrossRef CAS PubMed.
- M. M. Hansmann, P. W. Antoni and H. Pesch, Angew. Chem., Int. Ed., 2020, 59, 5782–5787 CrossRef CAS PubMed.
- P. W. Antoni, C. Golz, J. J. Holstein, D. A. Pantazis and M. M. Hansmann, Nat. Chem., 2021, 13, 587–593 CrossRef CAS PubMed.
- S. G. Davies, A. M. Fletcher, P. M. Roberts and J. E. Thomson, Eur. J. Org. Chem., 2019, 5093–5119 CrossRef CAS.
- P. Kothandaraman, S. J. Foo and P. W. H. Chan, J. Org. Chem., 2009, 74, 5947–5952 CrossRef CAS PubMed.
- L. Qiuming, H. Kasumi and S. Datong, Organometallics, 2020, 39, 4115–4122 CrossRef.
- S. C. Sau, R. Bhattacharjee, P. K. Hota, P. K. Vardhanapu, G. Vijaykumar, R. Govindarajan, A. Datta and S. K. Mandal, Chem. Sci., 2019, 10, 1879–1884 RSC.
- P. Eisenberger, B. P. Bestvater, E. C. Keske and C. Crudden, Angew. Chem., Int. Ed., 2015, 54, 2467–2471 CrossRef CAS PubMed.
- C. Chauvier, A. Tlili, C. Das Neves Gomes, P. Thuéry and T. Cantat, Chem. Sci., 2015, 6, 2938–2942 RSC.
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2007, 26, 4076–4079 CrossRef CAS.
- D. J. Liptrot, M. S. Hill, M. F. Mahon and A. S. S. Wilson, Angew. Chem., Int. Ed., 2015, 54, 13362–13365 CrossRef CAS.
- A. Das, J. Ahmed, N. M. Rajendran, D. Adhikari and S. K. Mandal, J. Org. Chem., 2021, 86, 1246–1252 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Dr Matthias Drieß on the occasion of his 60th birth anniversary. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2065467. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02819g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Arpan
Das
,
Arpan
Das