
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA catalyst-free and recycle-reinforcing elastomer vitrimer with exchangeable links†
Jinyun Wang,
Shubin Chen,
Tengfei Lin *,
Jinhuang Ke,
Tianxiang Chen,
Xiao Wu and
Cong Lin
*,
Jinhuang Ke,
Tianxiang Chen,
Xiao Wu and
Cong Lin
College of Materials Science and Engineering, Fuzhou University, Fuzhou 350108, PR China. E-mail: tflin@fzu.edu.cn
First published on 27th October 2020
Abstract
Vitrimers, as intriguing polymers, possess exchangeable links in the crosslinking networks, endowing them with the abilities of recycling and reprocessing. However, most of vitrimers are generally fabricated via complex synthesis and polymerization processes. Toxic and unstable exogenous catalysts are inevitably applied to activate the exchange reaction to rearrange the crosslinking networks. These drawbacks limit the widespread applications of vitrimers. Moreover, most reported vitrimers could only partially maintain or severely deteriorate their mechanical properties after recycling. Herein, to solve the above-mentioned problems, for the first time, a catalyst-free and recycle-reinforcing elastomer vitrimer is revealed. By the reactive blending of commercially available epoxidized natural rubber and carboxylated nitrile rubber, the elastomer vitrimer associated with exchangeable β-hydroxyl ester bonds was obtained. Strikingly, the vitrimer exhibits an exceptional recycle-reinforcing property. This work provides a feasible method to fabricate elastomer vitrimers, which promotes the recycling of crosslinking commercial available elastomers.
Introduction
Traditional vulcanized rubber, with outstanding elasticity, solvent resistance and mechanical properties, are ubiquitously applied in numerous fields.1,2 However, the permanent crosslinks prevent the reprocessing and recycling of end-of-life rubber products, resulting in a huge burden on the environment.3,4 Tremendous efforts have been dedicated to address this issue, including incinerating, grinding, landfilling and devulcanization treatments.5–11 However, these efforts caused inefficient utilization of scraps. Hence, it is highly desirable to employ advanced approaches to promote waste rubber recycling.Vitrimers, a novel class of polymers with dynamic reversible covalent crosslinks, have been receiving considerable attention.12–17 By reshuffling their topology structures via rapid reversible exchange reactions, vitrimers have been noticed to be reprocessable and recyclable.18 The introduction of vitrimer conception into elastomeric networks has been implemented to endow covalently crosslinking elastomers with recyclability.19–24 For instance, Tang et al. reported a covalently crosslinking yet recyclable elastomer vitrimer. With the aid of 1,2-dimethylimidazole (DMI) and Zn(Ac)2 as catalysts, this elastomer vitrimer, which comprised exchangeable β-hydroxyl ester crosslinks between commercial epoxidized natural rubber (ENR) and carbon nanodots, was able to reshuffle its network topology structure.22 Liu et al. fabricated a type of covalently crosslinking yet recyclable and robust elastomer vitrimer composites with the presence of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and DMI as the catalysts.25 Qiu et al. utilized the transesterification reactions of β-hydroxyl ester linkages between acid-modified carbon black and ENR to fabricate robust elastomer vitrimer composites.26 These results indicate that effective strategies have been performed to solve the inherent difficulty referred to the recycling of covalently crosslinking elastomers. It is noticed that the reported elastomer vitrimers were generally fabricated by constructing exchangeable crosslinks between modified fillers and rubbers. Although modified fillers could act as crosslinking and reinforcing agents to construct a three-dimensional reversible crosslinking network, the introduction of modified fillers would lead to the retarding of the topology network rearrangement due to the restriction of the macromolecular chain movement.25,27 Besides, catalysts are inevitably employed to promote the reversible exchange reactions, which could shorten the service lifetime and prevent the recyclability of vitrimers owing to the thermal degradation and leaching of the catalysts.28–30 It is also pointed out that the incorporation of toxic catalysts will potentially limit the widespread applications of these high performance materials. Therefore, catalyst-free and non-filler elastomer vitrimers are required.
Commercially available carboxylated nitrile rubber (XNBR) and ENR possess carboxyl groups and epoxy rings, respectively. It has been reported that the reaction between epoxy groups and carboxyl groups can easily construct crosslinking networks in XNBR/ENR elastomers.31–34 Actually, it is noted that the essence of such linkages is β-hydroxyl ester bonds. Those crosslinks can probably give rise to exchange reactions under appropriate conditions. Inspired by this, we first explored the possibility of this XNBR/ENR composite as an elastomer vitrimer. In the absence of catalysts and modified fillers, the XNBR/ENR composites were found to be recyclable and reprocessable through rearranging topology network via reversible exchange reaction. More particularly, the XNBR/ENR elastomers exhibited surprisingly excellent mechanical performance after multiple recycling, which we term as the recycle-reinforcing phenomenon. To the best of our knowledge, such elastomer vitrimer with excellent mechanical performance has never been reported. The goal of this study is to reveal a practical method for fabricating the above-mentioned elastomer vitrimer and the underlying recycle-reinforcing mechanism.
Experimental section
Materials
XNBR was supplied by NANTEX Industry Co. Ltd., with the carboxyl group content of 7.0 wt% and acrylonitrile content of 27.0 wt% (NANCAR® 1072CG). ENR with an epoxidation degree of 40% (ENR 40) was purchased from the Agricultural Products Processing Research Institute, Chinese Academy of Tropical Agricultural Science (Zhanjiang, PR China). The raw rubbers were used as received without any additional treatments.Preparation of XNBR/ENR elastomers
XNBR and ENR were solely masticated six times on a laboratory two-roll open mill at ambient temperature. Subsequently, after mastication, XNBR and ENR were mixed on the open mill to form XNBR/ENR elastomers. Five different weight ratio mixtures of XNBR/ENR elastomers (10/90, 30/70, 50/50, 70/30 and 90/10) were prepared by the same protocol, respectively. In this study, the sample code of XNBR-χ means that the χ wt% XNBR is related to the total weight of rubber composites. After that, the resultant compounds were stored for 12 h. Finally, they were compression-molded at 160 °C.Sample characterizations
The rheology experiments were conducted on a U-CAN UR-2010SD vulcameter at 160 °C, and the torque–time curves and curing characteristic parameters were determined.Tensile measurements were carried out on a MTS E44 universal testing machine at room temperature. For uniaxial tensile behaviours, the extension rate of dogbone-shaped samples was 500 mm min−1. The values of Young's modulus were obtained by calculating the slope of the stress–strain curve from 0 to 7% strain. Five specimens were tested for each sample to ensure the data reproducibility. For consecutive cyclic tensile performances, all tests were performed at a deformation rate of 100 mm min−1. The sample was loaded to 100% strain and then unloaded. After relaxing for 10 min at ambient temperature, the specimen was reloaded again. After three cycles, the sample went through another loading–unloading cycles with 300% strain and 600% strain, respectively.
Equilibrium swelling experiments were employed to evaluate the crosslinking density of XNBR/ENR elastomers. Three weighed pieces of specimens about 1 g were immersed in toluene at room temperature for 3 days. The swollen samples were gently blotted with filter paper and immediately weighed. Finally, the swollen samples were dried at 80 °C until the weight reached constant values. According to the above weight data, the cross-linking density was determined based on the classical Flory–Rehner equation.35 Fourier transform infrared spectroscopy in an attenuated total reflectance model (ATR-FTIR) was performed on a Nicolet iS50 spectrometer. The tests containing cyclic strain recovery measurements and dynamic mechanical analysis tests (DMA) were operated on a TA Q800 DMA apparatus under the tensile model. Cyclic strain recovery measurements were conducted by stretching alternately between 0.1 MPa for 60 min and 0 MPa for 10 min in every cycle. For DMA investigation, the specimen tests were heated from −50 °C to 100 °C at a rate of 3 °C min−1 as well as at an oscillation frequency of 1 Hz.
To verify the recyclable nature of the XNBR/ENR rubber elastomers, samples were cut into small shards and then masticated on the two-roll open mill for 10 min under an ambient temperature. Subsequently, the masticated rubbers were hot-pressed at 160 °C for 30 min, and then the recycled samples were obtained. Lastly, the reprocessing process was carried out three times. Thermal properties and structural integrity were characterized via thermal gravimetric analysis at a 35–800 °C temperature window and a heating rate of 10 °C min−1 under nitrogen atmosphere. The shore A hardness of samples was measured according to the standard ASTM D2240 with an effective thickness of 6 mm. The cryogenically fractured surfaces of original and recycled XNBR-50 specimens were observed via scanning electron microscopy (SEM, Carl Zeiss, Supra 55). X-ray diffraction (XRD) tests were performed on a Bruker D8 Advance X-ray diffractometer with Cu-Kα radiation (λ = 0.1542 nm). The pre-stretching samples at 100%, 200% and 300% strains were fixed at our home-made clamp.
Results and discussion
The curing behaviour of different compositions of XNBR/ENR elastomers was first examined. All the samples exhibited classic curing profiles and demonstrated the self-crosslinking ability without any additional curing agents or accelerators (Fig. S1, ESI†). It is consistent with the literature reports.31,34 In the present study, the maximum torque (MH) of XNBR-χ first increased and then decreased with the incremental loading of XNBR. Moreover, XNBR-50 exhibited the highest MH, the largest torque difference (ΔM) and the moderate curing time (Tc90) as compared to the others (Table S1, ESI†). Equilibrium swelling experiments of the XNBR/NBR elastomers were also conducted (Fig. S2, ESI†). The cured XNBR-50 was insoluble after immersing in toluene for a week, while the uncured one dissolved thoroughly. This phenomenon further confirmed the self-crosslinking nature of XNBR/ENR elastomers. The crosslinking densities were calculated (Table S2, ESI†). Considering the elastic torque, the crosslinking densities, and mechanical properties of XNBR-χ, XNBR-50 with optimal performance were chosen for the in-depth study.As illustrated in Fig. 1a, the covalent self-crosslinking behaviour occurred via the epoxy-acid reaction between XNBR and ENR. The FTIR spectrum was recorded to verify the esterification reaction between ENR and XNBR and monitor the evolutions of the absorption peaks of epoxy and ester groups during curing at 160 °C (Fig. S3, ESI†). As the curing proceeded, the intensity of epoxy groups at 873 cm−1 gradually declined, while the intensity of ester groups at 1731 cm−1 conversely increased. The peak at 3505 cm−1 attributed to hydroxyl groups appeared in the cured sample. These results supported the formation of β-hydroxyl ester linkages between ENR and XNBR.
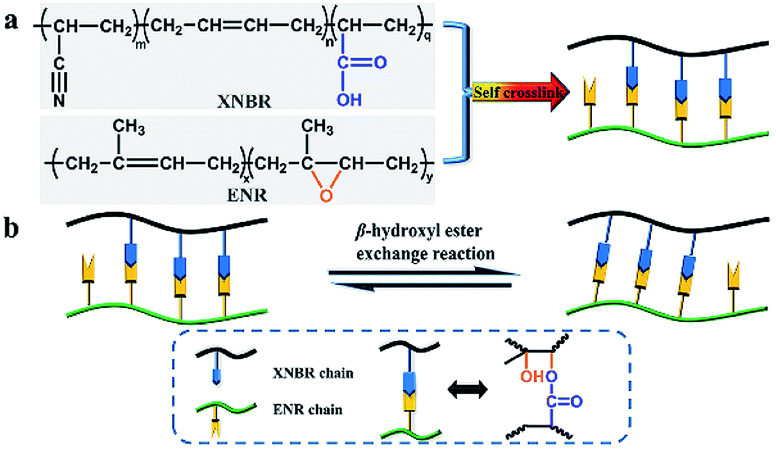 | ||
| Fig. 1 (a) Demonstration of the self-crosslinking behaviour in XNBR/ENR elastomers. (b) Schematic of the topological structure rearrangement based on the β-hydroxyl ester exchange reaction. | ||
It was reported that dynamic ester linkages with reversible exchange reactions are capable of rearranging the topology network structure under appropriate conditions.36,37 Thus, we envisaged the cured XNBR/ENR elastomers, comprising β-hydroxyl ester linkages, are able to rearrange the topological structure via the transesterification reaction (Fig. 1b). The XNBR/ENR elastomers, therefore, could be recycled. To verify this assumption, the cured XNBR-50 was cut into small fragments, subsequently masticated and hot-pressed to fabricate a re-cured sample. As shown in Fig. 2, a recycled XNBR-50 sample with fresh and coherent integrity appearance was obtained after hot-pressing. With the same treatments, the XNBR-50 sample could be recycled over three times, which is not possible in traditional vulcanized rubbers due to the irreversible linkages in crosslinking networks. Such recyclability of XNBR-50 could also be found in other compositions of the XNBR/ENR elastomers. The curing behaviours and crosslinking densities of the original and recycled XNBR-50 samples (XNBR-50s) were examined (Fig. S4 and Table S3, ESI†). All the recycled XNBR-50s exhibited similar typical curing profiles with higher ML (the minimum torque) and MH than the original one. Besides, there were no significant differences in crosslinking densities between the recycled XNBR-50s and the initial one. These results demonstrated that in the absence of any additional catalysts and modified fillers, the as-prepared covalently crosslinking XNBR/ENR elastomers exhibited probable vitrimer-like performance via the transesterification of β-hydroxyl ester linkages in this crosslinking network.
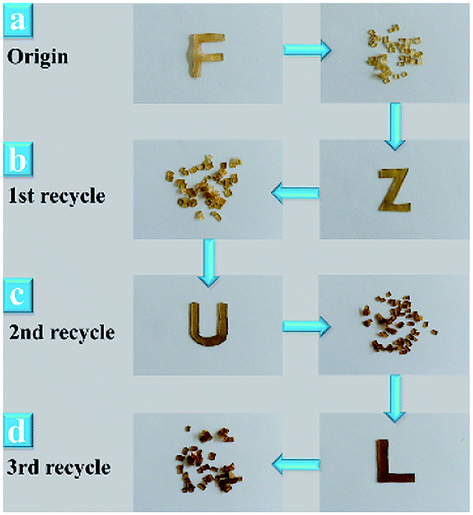 | ||
| Fig. 2 Photographs of XNBR-50s. (a) Original sample. (b) First recycled sample. (c) Second recycled sample. (d) Third recycled sample. | ||
The mechanical performances of the original and recycled XNBR-50s are presented in Fig. 3a and Table S4 (ESI†). The tensile strength and the elongation at break of the original XNBR-50 were 2.8 MPa and 342%, respectively. Strikingly, these values largely increased in the recycled XNBR-50s. For instance, after the first recycle, the tensile strength and the elongation at break of XNBR-50 increased from 2.8 MPa to 5.1 MPa and 342% to 835%, which improved by 182% and 244%, respectively. With the increase in the recycle times, the tensile strength of the recycled XNBR-50 increased. Moreover, the Young's modulus gradually decreased, demonstrating that the recycled samples possessed higher elasticity derived from the easier slippage and alignment of flexible polymer chains under the same strain. Table S5 (ESI†) lists the retention ratio of the tensile strength and elongation at break of the reported vitrimer materials. Partial maintenance or severe deterioration of the mechanical properties of these vitrimers was observed after the recycling process. It is the first reported vitrimer, which exhibits increasing mechanical properties.
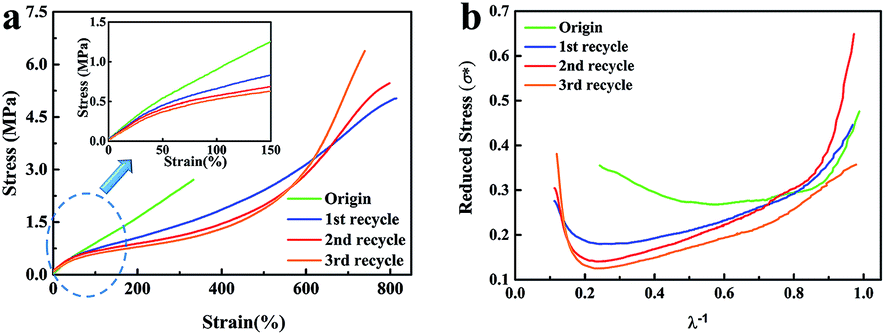 | ||
| Fig. 3 (a) Stress–strain curves of the original and recycled XNBR-50s. (b) Curves of σ* versus λ−1 of the original and recycled XNBR-50s. | ||
The Mooney–Rivlin model was employed to study the recycle-reinforcing phenomenon of XNBR-50. The equation has been listed below, where σ* is the reduced stress, σ is the applied stress, λ is the extension ratio, and C1 and C2 are constants that are independent of λ. By analysing stress–strain curves, the structure–property relationship could be evaluated.38
| σ* = σ/2(λ − λ−2) = C1 + C2 × λ−1 |
As shown in Fig. 3b, the curves of the recycled XNBR-50s were distinctly different with that of the original one. For the original one, at a small extension ratio (λ−1 > 0.85), σ* decreased quickly during stretching, which can be attributed to the Payne effect.38,39 In the region of high extension ratio (λ−1 < 0.50), an upturn of σ* was observed, which could be ascribed to the finite extensibility of polymer chains.40,41 For the recycled XNBR-50s, the upturn point appeared at much higher extension ration (λ−1 < 0.25), indicating that the polymer chains were more easily extended under smaller extension. In the other hand, easier extension could lead to more significant strain-induced crystallization (SIC) effect of the ENR chains.42 Therefore, the tensile strength of the recycled XNBR-50s was remarkably improved.
Two factors may be responsible for the improved mechanical properties of the recycled XNBR-50s. As shown in Fig. 4, during the recycling process, the crosslinking network of XNBR-50 broke due to the large shear stress. The elastomer samples were fragmented into small vulcanized rubber particles.43 According to the classical random coil model,44 it was assumed that the polymer chains in the core region of the rubber particles were highly entangled and the polymer segments motion was difficult. When hot-pressed, the broken crosslinking networks could be reconstructed via the transesterification of the polymer chains. It was supposed that this partial transesterification of the polymer chains could only undergo inside the core region of the particles due to the high entanglement. Conversely, the polymer chains above the shell region of the rubber particles were loose and provided enough movement space for the motion of segments. Thus, this partial transesterification could occur among or inside the rubber particles. Due to the presence of abundant free volume above the shell region of rubber particles, the regenerated crosslinking networks possessed higher motion ability, which therefore greatly improved the elongation of the recycled XNBR-50s. With the large improvement of the elongation of the recycled XNBR-50s, ENR could exhibit a more significant SIC effect. In the other hand, the rubber particles could play roles as reinforced particles, which improved the capability of the stress transferring and absorbing. Considering the SIC effect and particle reinforcement, the mechanical properties of the recycled XNBR-50s were demonstrated to be greatly improved.
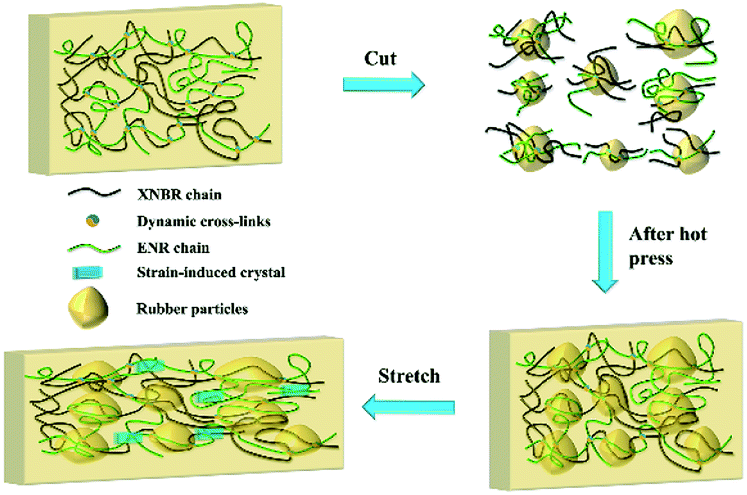 | ||
| Fig. 4 Schematic illustrating of the recycle-reinforcing phenomenon of XNBR-50. | ||
Corresponding characterizations were conducted to study the vitrimer-like performance of XNBR-50. The cyclic strain/recovery tests for the original XNBR-50 at 25 °C and 160 °C are shown in Fig. 5a. The XNBR-50 was first deformed under a constant external force at 25 °C. Similar to other covalently cross-linked elastomers, the deformed XNBR-50 almost recovered to its original length when unloading the stress. In the other hand, the deformed sample cannot recover to its original length when heating to 160 °C. The heated XNBR-50 exhibited a typical viscoelastic behaviour. It was attributed to the topological network rearrangement of the XNBR-50, which was triggered by the activation of the β-hydroxyl ester transesterification reaction at high temperatures. These results indeed confirmed the progress of the transesterification reaction in the XNBR/ENR elastomer vitrimer. Similar results have been reported in other elastomer vitrimers.45–48
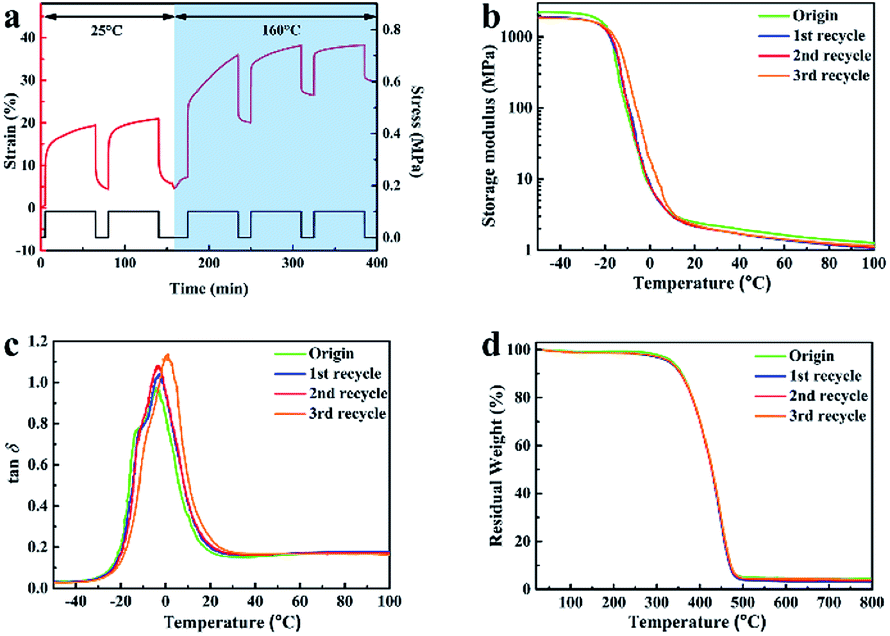 | ||
Fig. 5 (a) Cyclic strain/recovery curves of XNBR-50 at 25 °C and 160 °C. (b) Temperature dependence of storage modulus curves, and (c) tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ curves of XNBR-50s. (d) TGA curves of XNBR-50s. δ curves of XNBR-50s. (d) TGA curves of XNBR-50s. | ||
Fig. 5b shows the storage modulus of the original and recycled of XNBR-50s as a function of temperature. The storage modulus of the recycled XNBR-50s slightly decreased yet similar when compared with that of the original one. Besides, the recycled XNBR-50s almost overlapped with each other, indicating that the recycled XNBR-50s still maintained an integral and stable crosslinking network even after being recycled for several times. The tanδ peak intensities of the XNBR-50s consistently increased with the multiple reprocessing times (Fig. 5c). This suggested a continuous increase in chain mobility, which was associated with the decrease in the crosslinking density and Young's modulus of the recycled XNBR-50s as compared to the origin (Table S4, ESI†). The Tg values of the XNBR-50s increased from −3.7 °C to 1.1 °C with the incremental recycling times. The slight increase in the Tg values may be attributed to the confinement of molecular chains between the ENR and the XNBR matrix. The TGA curves of the recycled XNBR-50s (Fig. 5d) were similar and overlapped with that of the original one. The TGA results also indicated that the recycled XNBR-50s maintained an integral and stable cross-linked network, which is consistent with the DMA results. Fig. S5 (ESI†) shows the FTIR curves of the origin and recycled XNBR-50s. As compared to the original sample, the peak intensity and position of the recycled XNBR-50s exhibits almost no change, which further confirmed the integral linkage structure of the recycled XNBR-50s and the progress of the transesterification.
Wide-angle XRD (WAXD) patterns of the original and the recycled XNBR-50s at different strain are shown in Fig. S6 (ESI†). There were no diffraction peaks displayed in the WAXD spectrum of the original sample under different elongation. As compared to the original XNBR-50, the recycled XNBR-50s exhibited two diffraction peaks upon stretching in the region of 42–45° and 73–77°. This was ascribed to the SIC effect of ENR, corresponding with previously reported work.49 The results of WAXD demonstrated that the recycled XNBR-50s were more prone to show SIC behaviour. Fig. S7 (ESI†) shows SEM images of the cryogenically fractured surfaces of the original and recycled XNBR-50s. The SEM results exhibit that the fractured surface of the original XNBR-50 is rather smooth, indicating a brittle fracture. Conversely, the surfaces of the recycled XNBR-50s distinctly become rough, exhibiting a brittle–ductile transition behaviour. Such behaviour could be ascribed to the improvement in the interfacial interaction of the ENR and XNBR matrix in the recycled XNBR-50s. According to the loading–unloading curves and corresponding integrated value of different samples (Fig. S8, ESI† and Fig. 6), the recycled XNBR-50s showed an integral crosslinking network and good elastic resilience. With the increase in the recycle times, the difference between the dissipating energy of the first loading–unloading curve at 300% strain (W1) and the dissipating energy of second cycle (W2) decreased. Meanwhile, the W2/W1 value increased with the increase in the recycle times. It could be concluded that the value of W1 gradually decreased. This result demonstrated that upon the loading and unloading process, energy dissipation used for macromolecular conformation and configuration changes (particularly alignment and entropy increase of polymer chains) was reduced among the recycled XNBR-50s. It proved that more flexible and elastic polymer chains existed in the recycled XNBR-50s than in the origin one, which provided the possibility for polymer chains to show SIC effect upon stretching. These results are consistent with the WAXD spectrum.
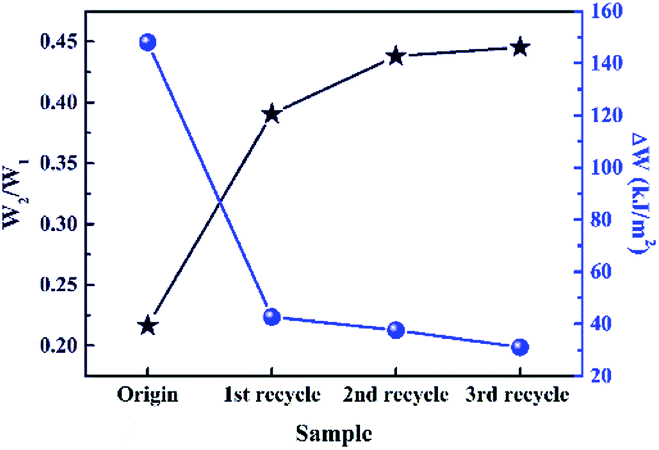 | ||
| Fig. 6 Dissipating energy dependence of samples before and after recycling. ΔW = W1 − W2. | ||
The recycled XNBR-50s with self-healing properties were further demonstrated. The first recycled XNBR-50 sample was cut and then kept at room temperature for 24 h without external stimuli. The damaged rubber sample achieved the self-repairing (Fig. S9a, ESI†). Analogously, when the cut-off rubber strip recompressed at 160 °C for 30 min, the welding sample possessed about 85% recovery of the tensile strength (Fig. S9b, ESI†). Moreover, the XNBR-50 sample was fixed on the home-made shape and could recover (Fig. S9c, ESI†), exhibiting a shape memory performance. These results demonstrate that XNBR-50s could act as a macroscopic flow-like viscoelastic liquid at an elevated temperature, which exhibits their intrinsic vitrimer property.
Conclusions
In this study, a catalyst-free elastomer vitrimer was prepared through the reactive blending of commercially available XNBR and ENR. Carboxyl groups in XNBR and epoxy groups in ENR could react to form β-hydroxyl ester bonds during vulcanization. It was found that these crosslinks in the XNBR/ENR elastomers were capable of undergoing reversible breaking and reformation to rearrange the crosslinking network without any additives, which endowed the elastomers with recyclability. More strikingly, during multiple generations of recycling, the vitrimer elastomers exhibited climbing mechanical properties. A further study showed that such unexpected recycle-reinforcing phenomenon could be attributed to the strain-induced crystallization behaviour of ENR and the particle reinforcement. Taking advantage of the reactive groups in the commercial rubbers to construct dynamic crosslinking linkages, we envision that this work extends the realm of the vitrimers and provides a feasible method to fabricate elastomer vitrimers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (No. 51803030), China Postdoctoral Science Foundation (2016M590590), Natural Science Foundation of Fujian Province, China (No. 2017J05075), Science Foundation of Education Department of Fujian Province, China (No. JAT160062).References
- M. Akiba and A. S. Hashim, Prog. Polym. Sci., 1997, 22, 475 CrossRef CAS.
- H. Xiang, H. Qian, Z. Lu, M. Rong and M. Zhang, Green Chem., 2015, 17, 4315–4325 RSC.
- L. Imbernon and S. Norvez, Eur. Polym. J., 2016, 82, 347–376 CrossRef CAS.
- B. S. Thomas and R. C. Gupta, Renewable Sustainable Energy Rev., 2016, 54, 1323–1333 CrossRef CAS.
- B. Adhikari, D. De and S. Maiti, Prog. Polym. Sci., 2000, 25, 909–948 CrossRef CAS.
- M. Myhre and D. A. MacKillop, Rubber Chem. Technol., 2002, 75, 429–474 CrossRef CAS.
- J. E. Morin, D. E. Williams and R. J. Farris, Rubber Chem. Technol., 2002, 75, 955–968 CrossRef CAS.
- E. Bilgili, A. Dybek, H. Arastoopour and B. Bernstein, J. Elastomers Plast., 2016, 35, 235–256 CrossRef.
- K. Formela, M. Cysewska, J. Haponiuk and A. Stasiek, Polimery, 2013, 58, 906–912 CrossRef CAS.
- R. F. Smith, S. C. Boothroyd, R. L. Thompson and E. Khosravi, Green Chem., 2016, 18, 3448–3455 RSC.
- A. Mouawia, A. Nourry, A.-C. Gaumont, J.-F. Pilard and I. Dez, ACS Sustainable Chem. Eng., 2016, 5, 696–700 CrossRef.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- M. Röttger, T. Domenech, R. van der Weegen, A. B. R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS.
- C. He, S. Shi, D. Wang, B. A. Helms and T. P. Russell, J. Am. Chem. Soc., 2019, 141, 13753–13757 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- L. Imbernon, E. K. Oikonomou, S. Norvez and L. Leibler, Polym. Chem., 2015, 6, 4271–4278 RSC.
- J. P. Brutman, G. X. De Hoe, D. K. Schneiderman, T. N. Le and M. A. Hillmyer, Ind. Eng. Chem. Res., 2016, 55, 11097–11106 CrossRef CAS.
- W. Denissen, M. Droesbeke, R. Nicolay, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef CAS.
- Z. Tang, Y. Liu, B. Guo and L. Zhang, Macromolecules, 2017, 50, 7584–7592 CrossRef CAS.
- Y. Chen, Z. Tang, Y. Liu, S. Wu and B. Guo, Macromolecules, 2019, 52, 3805–3812 CrossRef CAS.
- G. Zhang, H. Feng, K. Liang, Z. Wang, X. Li, X. Zhou, B. Guo and L. Zhang, Sci. Bull., 2020, 65, 889–898 CrossRef CAS.
- Y. Liu, Z. Tang, Y. Chen, C. Zhang and B. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 2992–3001 CrossRef CAS.
- M. Qiu, S. Wu, S. Fang, Z. Tang and B. Guo, J. Mater. Chem. A, 2018, 6, 13607–13612 RSC.
- Y. Liu, Z. Tang, S. Wu and B. Guo, ACS Macro Lett., 2019, 8, 193–199 CrossRef CAS.
- H. Liu, H. Zhang, H. Wang, X. Huang, G. Huang and J. Wu, Chem. Eng. J., 2019, 368, 61–70 CrossRef CAS.
- C. Xu, R. Cui, L. Fu and B. Lin, Compos. Sci. Technol., 2018, 167, 421–430 CrossRef CAS.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- R. Alex, P. P. De and S. K. De, J. Polym. Sci., Part C: Polym. Lett., 1989, 27, 361–367 CrossRef CAS.
- R. Alex, P. P. De and S. K. De, Polym. Commun., 1990, 31, 118–120 CAS.
- R. Alex, P. P. De, N. M. Mathew and S. K. De, Plast. Rubber Process. Appl., 1990, 14, 223–234 CAS.
- R. Alex and P. P. De, Kautsch. Gummi Kunstst., 1990, 43, 1002–1005 CAS.
- P. J. Flory, J. Chem. Phys., 1950, 18, 108–111 CrossRef CAS.
- L. Cao, J. Fan, J. Huang and Y. Chen, J. Mater. Chem. A, 2019, 7, 4922–4933 RSC.
- T. Liu, B. Zhao and J. Zhang, Polymer, 2020, 194, 122392 CrossRef CAS.
- L. Bokobza and B. Erman, Macromolecules, 2000, 33, 8858–8864 CrossRef CAS.
- A. H. Muhr, Rubber Chem. Technol., 2005, 78, 391–425 CrossRef CAS.
- S. Schlögl, M.-L. Trutschel, W. Chassé, G. Riess and K. Saalwächter, Macromolecules, 2014, 47, 2759–2773 CrossRef.
- J. Shen, X. Lin, J. Liu and X. Li, Macromolecules, 2018, 52, 121–134 CrossRef.
- X. Zhang, K. Niu, W. Song, S. Yan, X. Zhao, Y. Lu and L. Zhang, Macromol. Rapid Commun., 2019, 40, e1900042 CrossRef.
- C. Wang, J. X. Su, J. Li, H. Yang, Q. Zhang, R. N. Du and Q. Fu, Polymer, 2006, 47, 3197–3206 CrossRef CAS.
- J. S. Crespo, S. Lecommandoux, R. Borsali, H.-A. Klok and V. Soldi, Macromolecules, 2003, 36, 1253–1256 CrossRef CAS.
- A. Legrand and C. Soulié-Ziakovic, Macromolecules, 2016, 49, 5893–5902 CrossRef CAS.
- S. Wu, Z. Yang, S. Fang, Z. Tang, F. Liu and B. Guo, J. Mater. Chem. A, 2019, 7, 1459–1467 RSC.
- Y. Spiesschaert, M. Guerre, L. Imbernon, J. M. Winne and F. Du Prez, Polymer, 2019, 172, 239–246 CrossRef CAS.
- Y. Li, T. Liu, S. Zhang, L. Shao, M. Fei, H. Yu and J. Zhang, Green Chem., 2020, 22, 870–881 RSC.
- L. Imbernon, R. Pauchet, M. Pire, P. A. Albouy, S. Tencé-Girault and S. Norvez, Polymer, 2016, 93, 189–197 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07728c |
| This journal is © The Royal Society of Chemistry 2020 |