DOI:
10.1039/D0RA08257K
(Paper)
RSC Adv., 2020,
10, 39277-39283
Synthesis of poly(N-isopropylacrylamide) polymer crosslinked with an AIE-active azonaphthol for thermoreversible fluorescence†
Received
27th September 2020
, Accepted 20th October 2020
First published on 27th October 2020
Abstract
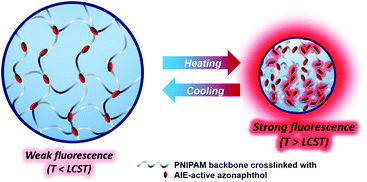
A fluorescent polymer was synthesized using N-isopropylacrylamide (NIPAM) crosslinked with a divinylazonaphthol monomer via radical emulsion polymerization. Because the crosslinked polymer contained an aggregation-induced emissive (AIE) azonaphthol-based crosslinker, a thermoreversible sol–gel transformation and gelation-induced reversible fluorescence alteration were successfully attained in an aqueous medium. Like typical PNIPAM, the size and transmittance of the polymer dramatically decreased near the lower critical solution temperature (LCST, 36 °C). Such gelation facilitated aggregation of the polymer chains, resulting in the close contact between azonaphthol groups producing fluorescence. The crosslinked polymer exhibited changes in dual properties: one is related to PNIPAM structural alteration, which corresponds to conventional swelling/shrinkage behavior; and the other is involved in the reversible fluorescence change in response to the swelling/shrinkage. Because the major backbone of the polymer was composed of NIPAM with an LCST at 36 °C, the resultant polymer is expected to have potential applications in biologically related fields.
Introduction
Recently, stimuli-responsive polymers have attracted much attention because their swelling behavior can be altered in response to stimuli such as pH, temperature, and salt concentration,1–5 and are useful in a variety of applications, including drug delivery, water treatment, scaffolds, molecular sensing, and energy storage.6–13 Poly(N-isopropylacrylamide) (PNIPAM)-based polymers typically exhibit a sol–gel transition at a lower critical solution temperature (LCST) of 32 °C to 33 °C in aqueous solution, depending on the type of copolymer used, in which they become hydrophobic and aggregated above the LCST, and hydrophilic and swollen below the LCST.14–19 PNIPAM polymers have been synthesized as copolymers to demonstrate a versatile thermoresponsive property.20–27 The thermoresponsiveness from PNIPAM has been developed as an on–off switch for various functions such as controlled release and drug delivery.28–31 To adjust the LCST, hydrophobic or hydrophilic comonomers or crosslinkers have been used, which enable controlling the hydrophilicity/hydrophobicity of the PNIPAM backbone. The introduction of comonomers or crosslinkers has led to versatile PNIPAM-based polymers having additional functions such as self-healing,32,33 fluorescence,34,35 photoresponsive properties,36,37 and aggregation-induced emission (AIE).38,39
Smart polymers that exhibit fluorescence change in response to external stimuli have been extensively investigated in biologically relevant fields such as imaging and new drug delivery systems.40–42 Many investigations combined fluorescence with the responsiveness of PNIPAM-based polymers. For practical applications, the choice of fluorophores is becoming important and organic fluorophores are excellent candidates because they are less toxic and easier to design than inorganic materials.43–46 However, most organic fluorophores suffer from aggregation-caused quenching (ACQ), in which fluorescence is decreased in their aggregated solid state or at high concentrations. This limits their versatile application in the solid state. Meanwhile, an AIE effect was discovered, which is the opposite of ACQ.47–50 AIE is a phenomenon by which fluorophores show negligible or weak fluorescence emission in solution, but whose fluorescence is greatly increased in the solid or aggregated state. Recently, the synthesis of PNIPAM-based smart polymers with AIE properties was attempted using tetraphenylethylene (TPE), which is the most representative example.27,51–53 Because the TPE group is easy to synthesize and tether to the polymer backbone, most AIE-active smart polymers are focused on the use of TPE, which might hinder various molecular design for versatile and useful applications.
Unusual fluorescence from azonaphthol compounds that is related to AIE has been reported by us, to our knowledge, for the first time.54 Based on previous results, we expected fluorescence switching via the sol–gel transition of crosslinked PNIPAM containing an AIE-active azonaphthol crosslinker. In this manner, a fluorescent polymer was designed and synthesized comprising PNIPAM as a hydrophilic matrix and a new divinyl crosslinker with AIE-active azonaphthol. As a result, the synthesized fluorescent polymers showed not only the conventional LCST transition of PNIPAM but also AIE characteristics in aqueous media. For instance, both the size and transmittance of the polymers decreased dramatically beyond the LCST (36 °C). In conjunction with this, the fluorescence was enhanced by AIE above the LCST, which could be explained by the aggregation at the LCST. Although various fluorescence manipulation in the PNIPAM was reported earlier,55–57 the fluorescence switching from PNIPAM containing the AIE-active crosslinker is reported for the first time. The AIE-active PNIPAM showed two changes: (1) swelling/shrinkage behavior; (2) fluorescence intensity in response to temperature. Because the LCST of the polymer was determined at 36 °C, similar to body temperature, potential applications are expected in biologically relevant fields, in which thermoresponsive fluorescent PNIPAM with dual signals would be used in bioimaging and controlled drug delivery.
Experimental
Instrumentation and materials
1H-NMR and 13C-NMR spectra were obtained on a Bruker Fourier-300 spectrometer and solid-state NMR spectrometer (AVANCE HD, Bruker), respectively. FT-IR spectra were obtained on a Bruker Tensor 27 spectrometer. A scanning electron microscopic (SEM) image was obtained using a Hitachi S-4800 instrument. UV-vis absorption and transmittance spectra were obtained using a PerkinElmer Lambda 35 spectrophotometer. Photoluminescence spectra were obtained using a Varian Cary Eclipse spectrophotometer equipped with a Xe-flash-lamp excitation source. Size distributions were obtained with dynamic light scattering (DLS, Zetasizer Nano ZS, Malvern). 4-Bromoaniline, sodium nitrite, 6-bromo-2-naphthol, anhydrous toluene, tetrakis(triphenylphosphine)palladium, sodium dodecyl sulfate (SDS), anhydrous N,N-dimethylformamide (DMF), and dialysis tubes (MWCO 12400) were purchased from Sigma-Aldrich (U.S.A.). Di-tert-butyl dicarbonate, N-isopropylacrylamide, and 4-vinylphenylboronic acid were purchased from Tokyo Chemical Industries (Japan). Hydrochloric acid (HCl), potassium carbonate, dichloromethane, magnesium sulfate, potassium persulfate (KPS), and sodium hydroxide were purchased from Samchun Chemicals (Korea). Pyridine, ethanol, n-hexane, and chloroform were purchased from Daejung (Korea). MilliQ deionized water (18.2 MΩ cm−1 at 25 °C) was obtained from a water purification system (Direct-Q3 UV). Flash chromatography was carried out on silica gel (70–230 mesh). All chemicals and solvents were used without further purification.
Synthesis of 1-(p-bromophenylazo)-6-bromo-2-naphthol (Azo-OH)
We synthesized Azo-OH according to a method reported previously.54 For the diazotization of p-bromoaniline, p-bromoaniline (3 g, 17.4 mmol) was added to an HCl solution (4 mL) in water (11 mL). The solution was cooled using an ice-water bath with vigorous stirring. A cold aqueous solution (10 mL) of sodium nitrite (1.20 g, 17.4 mmol) was rapidly added to the solution under stirring at 0 °C. The stirring was continued until the diazotization was completed to obtain p-bromobenzenediazonium chloride. For the coupling reaction, 6-bromo-2-naphthol (3.9 g, 17.4 mmol) and sodium hydroxide (1.40 g) were dissolved in water (300 mL) and the solution was cooled to 0 °C. After obtaining a clear solution, the p-bromobenzenediazonium salt solution was added to the 6-bromo-2-naphthol solution. Subsequently, an HCl solution (10 mL) was poured into the mixture and warmed to room temperature. After stirring for 40 min, the product was isolated by filtration and washed three times with water. The solid was finally dried in a vacuum oven and the product was obtained as a red powder (yield 6.26 g, 89%). 1H NMR (300 MHz, CDCl3): 15.91 (s, 0.38H), 8.44 (d, 1H), 7.78 (d, 1H), 7.67–7.62 (m, 6H), 6.92 (d, 1H).
Synthesis of 1-(p-bromophenylazo)-6-bromo-2-naphthalene-2-yl tert-butyl carbonate (azo-t-boc)
Azo-t-boc was synthesized according to previously reported method.58 Azo-OH (3 g, 7.39 mmol) was dissolved in pyridine (180 mL) under stirring at 40 °C. Then, di-tert-butyl dicarbonate (6.4 g, 29.78 mmol) was added to the Azo-OH solution and stirred at 40 °C for 40 h. The mixture was allowed to cool to room temperature and poured into water (1400 mL). The precipitates were isolated by filtration and the solid was obtained by recrystallization from ethanol. Orange products were dried under vacuum (yield 2.82 g, 76%). 1H NMR (300 MHz, CDCl3): 8.54–8.52 (d, 1H), 8.06 (d, 1H), 7.87–7.66 (m, 6H), 7.42–7.39 (d, 1H).
Synthesis of 1-(p-vinylbiphenylazo)-6-vinylphenyl-2-naphthol (Azo-divinyl)
4-Vinylphenylboronic acid (1.22 g, 8.25 mmol) and Azo-t-boc (1.9 g, 3.75 mmol) were added to 250 mL flask under an atmosphere of Ar. Anhydrous toluene (36 mL) was added to the flask using a syringe and the mixture was stirred at 90 °C. When the reactants were completely dissolved, Pd(0) was added to the solution and the solution was stirred at 100 °C. After 40 min, aqueous K2CO3 solution (2 M, 10 mL) and a few drops of Aliquat 336 were added slowly. The mixture was stirred overnight at 100 °C and cooled to room temperature. The mixture was extracted with dichloromethane. The organic layer was washed with brine and dried over MgSO4. The organic solvent was evaporated, and the crude product was purified by column chromatography using silica gel (eluent: hexane/dichloromethane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2)). A red solid was obtained by recrystallization from ethanol (yield 0.2 g, 12%). 1H NMR (300 MHz, CDCl3): 16.32 (s, 0.3H), 8.65 (d, 1H), 7.83–7.50 (m, 15H), 6.93–6.91 (d, 1H), 6.73–6.72 (dd, 2H), 5.85–5.79 (d, 2H), 5.32–5.28 ppm (d, 2H). 13C NMR (CDCl3): 144.15, 140.06, 139.93, 139.81, 139.81, 139.41, 138.07, 137.02, 136.82, 136.36, 136.31, 132.56, 130.23, 130.03, 128.45, 128.05, 127.75, 127.14, 127.14, 127.02, 126.81, 126.59, 125.16, 122.42, 119.17, 114.22, 114.08 ppm. FT-IR (KBr pellet, cm−1): 3439, 3032, 1625, 1602, 1496, 1398, 1265, 1232, 1155, 989, 908, 829. Anal. calcd for C32H24N2O: C, 84.93; H, 5.35; N, 6.19. Found: C, 85.73; H, 5.33; N, 6.07.
:2)). A red solid was obtained by recrystallization from ethanol (yield 0.2 g, 12%). 1H NMR (300 MHz, CDCl3): 16.32 (s, 0.3H), 8.65 (d, 1H), 7.83–7.50 (m, 15H), 6.93–6.91 (d, 1H), 6.73–6.72 (dd, 2H), 5.85–5.79 (d, 2H), 5.32–5.28 ppm (d, 2H). 13C NMR (CDCl3): 144.15, 140.06, 139.93, 139.81, 139.81, 139.41, 138.07, 137.02, 136.82, 136.36, 136.31, 132.56, 130.23, 130.03, 128.45, 128.05, 127.75, 127.14, 127.14, 127.02, 126.81, 126.59, 125.16, 122.42, 119.17, 114.22, 114.08 ppm. FT-IR (KBr pellet, cm−1): 3439, 3032, 1625, 1602, 1496, 1398, 1265, 1232, 1155, 989, 908, 829. Anal. calcd for C32H24N2O: C, 84.93; H, 5.35; N, 6.19. Found: C, 85.73; H, 5.33; N, 6.07.
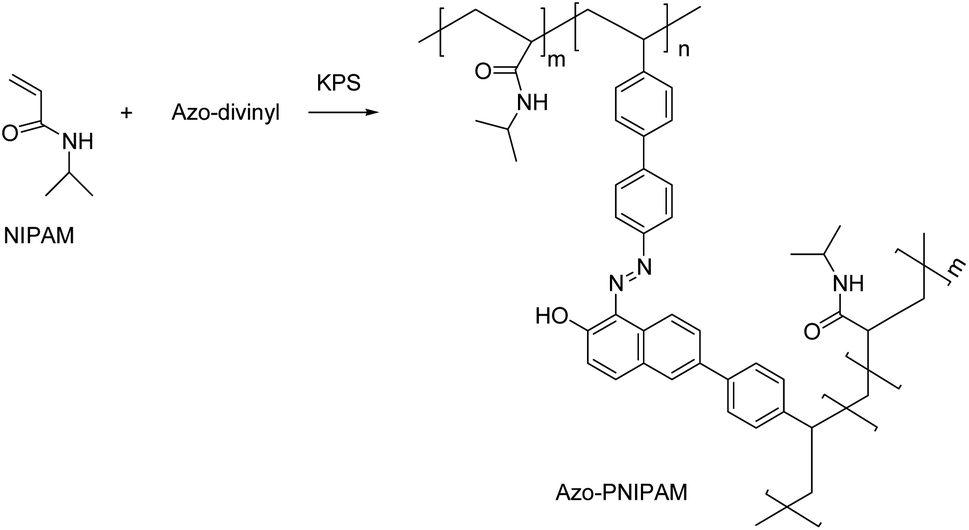
Synthesis of Azo-PNIPAM
Emulsion polymerization was used to obtain crosslinked PNIPAM with Azo-divinyl. A 100 mL round bottom flask was charged with NIPAM (900 mg, 8 mmol), Azo-divinyl (0.036 g, 0.08 mmol), KPS (0.23 g, 0.85 mmol), and SDS (0.33 g, 1.14 mmol) under a N2 purge. DMF (25 mL) was added into the flask and N2 bubbling for 40 min was used to remove O2. Subsequently, deionized water was added slowly to the flask with a syringe. N2 bubbling was continued for a further 40 min. The resulting emulsion was stirred at 80 °C overnight under N2 and cooled to room temperature. The polymer was isolated by centrifugation (14000 rpm) and washed repeatedly with water. The polymer was washed with dichloromethane to remove unreacted monomers. After washing, the solution was dialyzed for 3 days. A pink solid was obtained after freeze-drying (yield 128 mg, 14%). FT-IR (KBr pellet, cm−1): 1649 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1496 (CC), 1265 (C–N, aromatic amine). 13C solid state NMR: 175 (CO in amide), 41 (CH2 in isopropyl group), 22 ppm (CH3 in isopropyl group).
O), 1496 (CC), 1265 (C–N, aromatic amine). 13C solid state NMR: 175 (CO in amide), 41 (CH2 in isopropyl group), 22 ppm (CH3 in isopropyl group).
Results and discussion
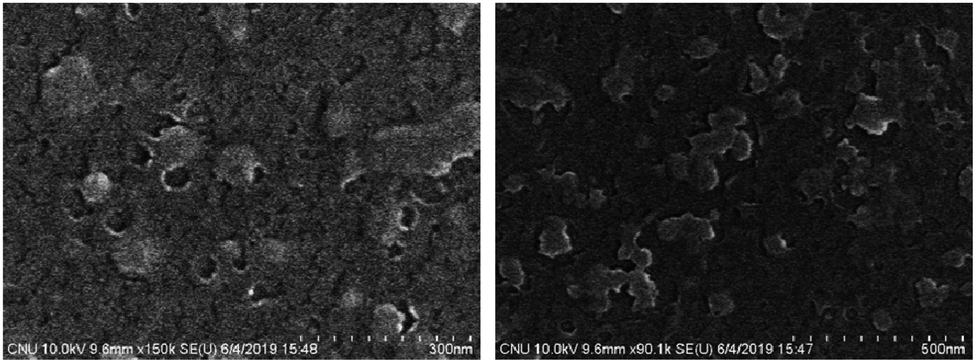
The unusual fluorescence of Azo-OH was found to exhibit AIE behavior, in which an intramolecular hydrogen bond was essential, as we have reported previously.54 A conventional diazotization-coupling reaction easily synthesized Azo-OH (Scheme S1†). Divinyl groups were introduced at the end of the Azo-OH to synthesize a new AIE-active crosslinker (Azo-divinyl) for further use in radical polymerization with NIPAM. We synthesized Azo-divinyl according to the synthetic route shown in Scheme 1. Initially, we attempted to attach vinyl groups at the end group of Azo-OH via a Suzuki coupling reaction between Azo-OH and 4-vinylphenylboronic acid but found that it was not possible.59 It is possible that free hydroxyl group interfered with the Suzuki coupling reaction. Thus, a t-boc group was introduced to protect the hydroxyl groups, and subsequently, vinyl groups were successfully introduced using 4-vinylphenylboronic acid (Scheme S1†). Because the basic K2CO3 solution was used for the Suzuki coupling reaction, the attached t-boc group was spontaneously hydrolyzed and converted to a hydroxyl group during the reaction, which circumvented additional deprotection procedure. The chemical structures of Azo-OH and Azo-t-boc were confirmed with 1H NMR spectra, in which an OH peak was observed at 16 ppm for Azo-OH and was not present for Azo-t-boc. In addition, a new t-butoxy peak was observed at 1.5 ppm for Azo-t-boc (Fig. S1a, b†). The chemical structure of Azo-divinyl was confirmed with spectroscopic methods, including 1H and 13C NMR and FT-IR. The t-butoxy peak in the 1H NMR spectrum of Azo-t-boc disappeared, and the OH peak at 16 ppm and vinyl group peaks at 5.2, 5.7, and 6.7 ppm newly appeared (Fig. S1c†). In addition, FT-IR spectra confirmed the structures of Azo-divinyl and Azo-PNIPAM (Fig. S2†). To obtain PNIPAM-based fluorescent polymer, exhibiting both LCST and AIE, Azo-PNIPAM was synthesized by conventional emulsion polymerization using SDS as the surfactant and KPS as the radical initiator (Scheme 1). The vinyl groups at both ends of the Azo-divinyl monomer facilitated the cross-linking of NIPAM monomer during the polymerization. The Azo-PNIPAM was washed with water and dichloromethane repeatedly until unreacted monomers were completely removed. After washing, dialysis was performed for 3 days to remove small molecules using a membrane with an MWCO 12400, in which the molecular weight of the Azo-PNIPAM was estimated to be above 12400. The chemical structure of crosslinked Azo-PNIPAM was confirmed with NMR (Fig. S1d†) and FT-IR spectra (Fig. S2†). In the 13C solid-state NMR, peaks corresponding to CO of the amide bond, CH2 of the isopropyl group, and CH3 of the isopropyl group were observed at 175, 41, and 22 ppm, respectively. The peak from azobenzene should be observed around 130 ppm, but was not clearly seen in the spectrum, presumably because of the relatively small amount of Azo-divinyl (the feed mole ratio of NIPAM to Azo-divinyl: 100 to 1).60,61 The characteristic band of the amide from PNIPAM was observed at 1649 cm−1 in the IR spectrum of Azo-PNIPAM. The characteristic bands of the azo group and C–N (aromatic amine) from Azo-divinyl were found at 1496 and 1265 cm−1, respectively. The surface morphology of Azo-PNIPAM was observed with SEM, showing the presence of nonuniform and porous microstructure (Fig. 1).27,62
 |
| | Scheme 1 Synthetic routes to Azo-divinyl (AIE-active crosslinker) and polymerization of Azo-PNIPAM. | |
 |
| | Fig. 1 SEM images of Azo-PNIPAM. | |
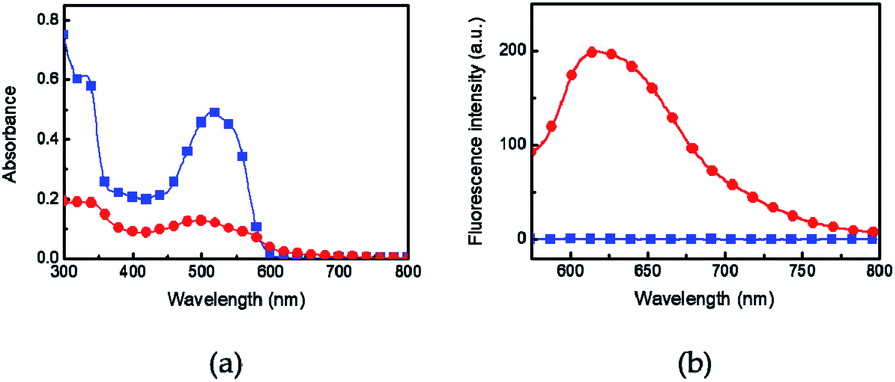
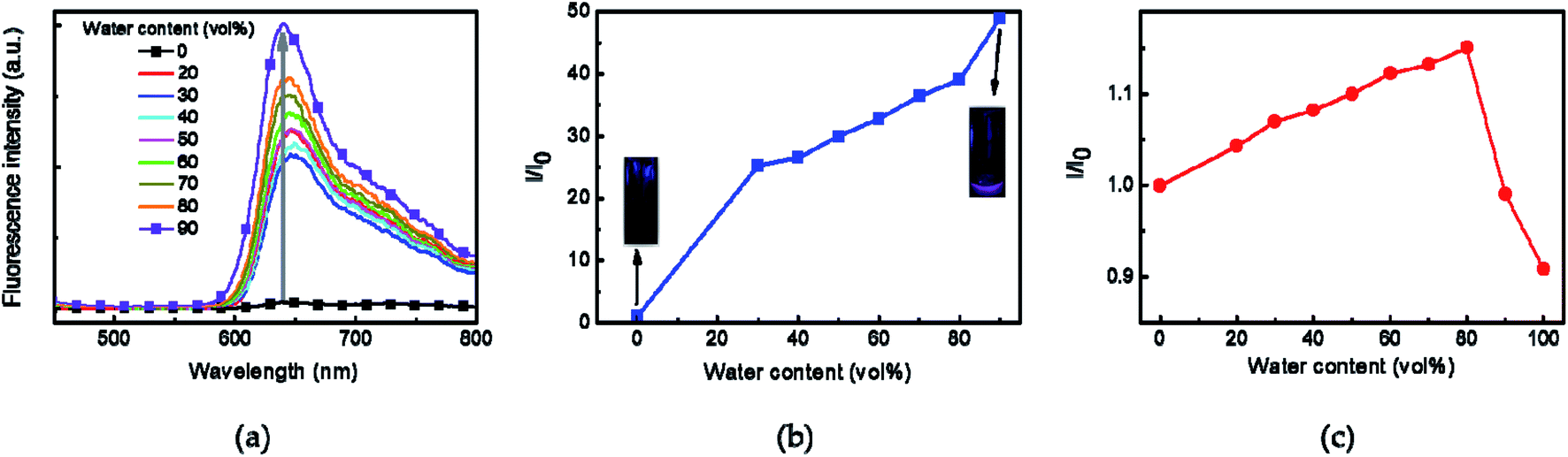
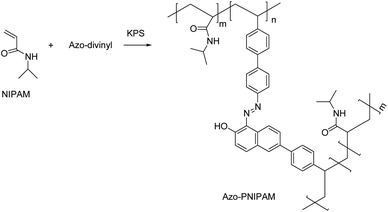
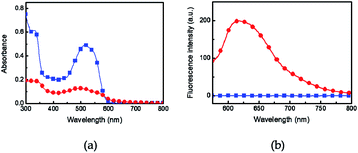
The UV-vis absorption and fluorescence of Azo-divinyl were investigated in solution and in the spin-cast film. The UV-vis absorption of Azo-divinyl in chloroform solution was observed at 515 nm, resulting from an o-azonaphthol moiety, while blue-shifted emission at 490 nm was observed in the film (Fig. 2a). Fluorescence was not observed for the Azo-divinyl solution, while obvious fluorescence was found in the solid state with an emission wavelength at 617 nm (Fig. 2b). Such AIE behavior originating from Azo-OH was maintained, regardless of the conversion of Azo-OH to Azo-divinyl. Because the Azo-divinyl interconnects NIPAM units acting as a crosslinker, AIE is expected during PNIPAM gel shrinkage by temperature increase. To elucidate the AIE of Azo-divinyl, related experimental changes in the fluorescence were investigated upon the addition of water (Fig. 3a). As the water volume fractions (fw = 0–90%) to THF solution of Azo-divinyl increase, the fluorescence intensity at 640 nm increased. This indicates that Azo-divinyl exhibited bright fluorescence only in a solid or aggregated state, but fluorescence was hardly observed in its THF solution. The changes in the fluorescence intensity of Azo-divinyl in the THF/water system at 640 nm is plotted in Fig. 3b. The AIE can be clearly observed by the naked eye in inset photographs of Azo-divinyl under 365 nm UV light in its solution and in the aggregated state, which corresponds to 0 and 90% water content, respectively. As a result, it was confirmed that the AIE of Azo-divinyl could be used as an AIE-active crosslinker. Like the Azo-divinyl, the AIE of Azo-PNIPAM was investigated using fluorescence spectroscopy (Fig. S3†). The effect of water content in THF solution of Azo-PNIPAM was elucidated. The fluorescence intensity of Azo-PNIPAM solution increased linearly with increasing water content up to 80 vol% (Fig. 3c). When water was added in the THF solution, the polymer became partially aggregated and its fluorescence increased. Unexpectedly, a decrease in the fluorescence intensity was observed after 90 vol% of water was added to the THF solution. It is unusual and even decreased further than the initial fluorescence intensity. It is presumed that upon addition of a large amount of water to the THF solution of Azo-PNIPAM, the major solvent changed to water, and then the main chain of Azo-PNIPAM became swollen, resulting in the decrease in the fluorescence of Azo-PNIPAM.
 |
| | Fig. 2 (a) UV-vis and (b) fluorescence spectra of Azo-divinyl in chloroform solution (■; 2.5 × 10−5 M) and in the film (●) prepared by spin coating from chloroform solution. Excitation wavelength 515 nm. | |
 |
| | Fig. 3 (a) Changes in the fluorescence spectra of Azo-divinyl in THF solution with various water contents ([Azo-divinyl] = 10 mg mL−1, 2.5 × 10−2 M). (b) Relative fluorescence intensity of Azo-divinyl in the THF/water system at 640 nm. Excitation wavelength 515 nm. I0 and I correspond to the fluorescence intensity of Azo-divinyl in THF solution at 640 nm in the absence and presence of water, respectively. Inset photographs show Azo-divinyl in THF solution under UV light (365 nm) containing 0 (left) and 90 vol% (right) water. (c) Relative fluorescence intensity of Azo-PNIPAM in the THF/water system at 601 nm. I0 and I correspond to the fluorescence intensity of Azo-PNIPAM in THF solution at 601 nm in the absence and presence of water, respectively. [Azo-PNIPAM] = 3 mg mL−1. | |
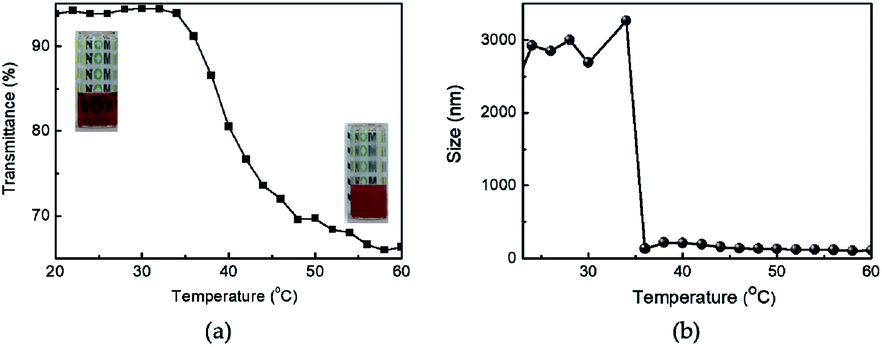
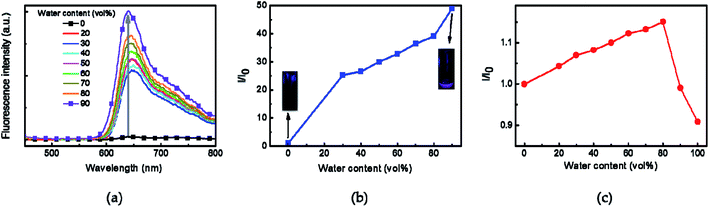
The transmittance changes of Azo-PNIPAM were investigated with increasing temperature to determine its LCST. The phase transition temperature of Azo-PNIPAM was determined at 600 nm using UV-vis spectroscopy (Fig. 4a). The changes in transmittance were investigated at different temperatures in a range from 20 to 60 °C. The transmittance of Azo-PNIPAM abruptly decreased from around 35 °C (called LCST), showing thermally reversible solubility changes in response to temperature. The transmittance of Azo-PNIPAM decreased from 94% to 65% with increasing temperature to 60 °C. Though transmittance much lower than 65% was expected at a higher temperature, it was not possible to obtain a transmittance lower than 65%, presumably because highly hydrophobic phenylene-linked azonaphthol derivatives were introduced to the polymer, leading to narrowing the difference between hydrophilicity and hydrophobicity in aqueous solution.63
 |
| | Fig. 4 Changes in the (a) transmittance at 600 nm and (b) DLS particle size of aqueous Azo-PNIPAM solutions. Inset photographs were taken below (left) and above the LCST (right). [Azo-PNIPAM] = 1.25 mg mL−1 for transmittance; 1.1 mg mL−1 for DLS. | |
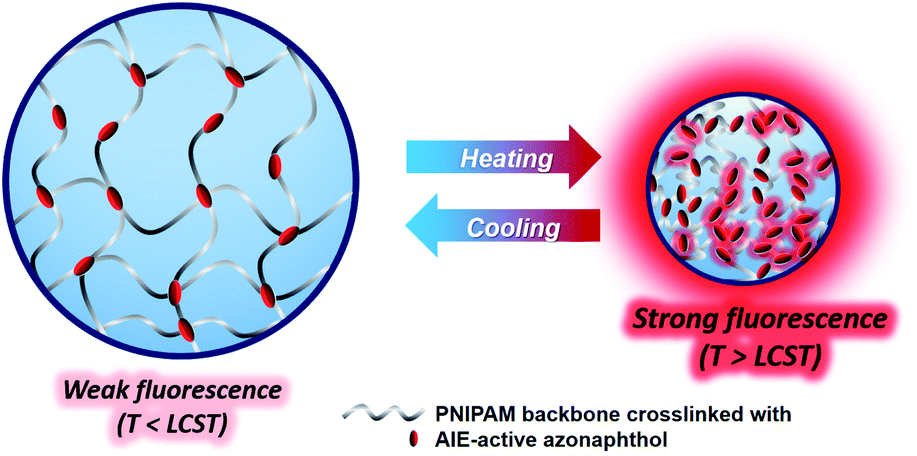
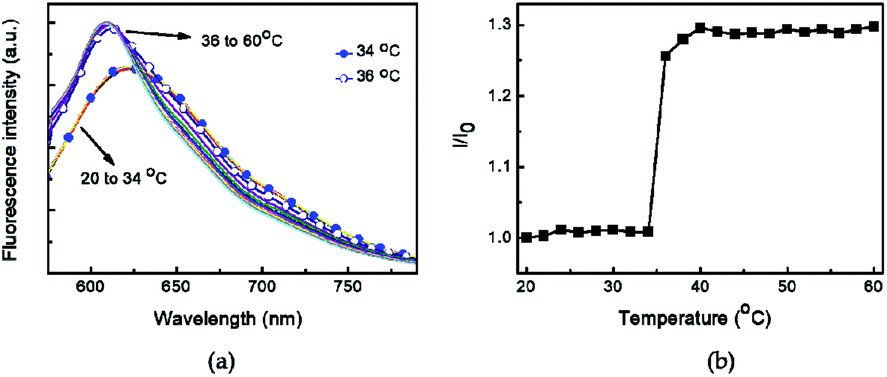
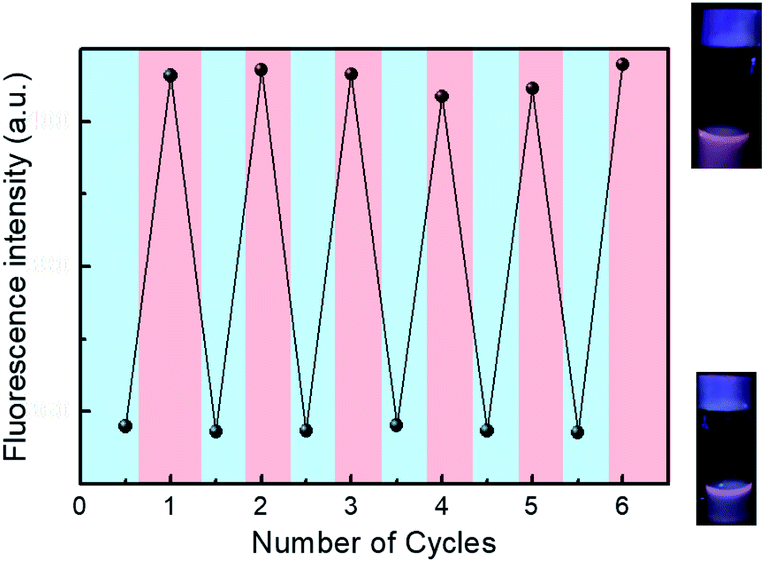
The hydrodynamic diameter and size distribution of Azo-PNIPAM were obtained in aqueous solution using DLS and they were investigated by increasing the temperature from 20 to 60 °C (Fig. 4b). In accordance with the transmittance changes, the aggregation of Azo-PNIAPM was observed above the LCST of 36 °C. The diameters of Azo-PNIPAM were determined to be 3100 and 144 nm below and above the LCST, respectively, indicating that disaggregation and aggregation occurred in response to temperature. In addition, the polydispersity index values were found to be 0.074 at 24 °C (below the LCST) and 0.041 at 42 °C (above the LCST), indicative of Azo-PNIPAM of uniform size with a narrow distribution, regardless of the temperature (Fig. S4a†). The aggregation of Azo-PNIPAM can be visualized using laser light, in which the beam path can be observed only above the LCST, indicating the formation of aggregates (Fig. S4b†). The effect of temperature on the fluorescence of Azo-PNIPAM was investigated to elucidate whether the AIE would be affected by the aggregation of the polymer gel. Upon aggregation above the LCST, the AIE-active crosslinking points in Azo-NIPAM will associate with each other to be in close proximity, resulting in AIE (Scheme 2). As the temperature increased, a dramatic increase in the fluorescence intensity of Azo-PNIPAM was observed above 36 °C, which corresponds to the LCST of Azo-PNIPAM (Fig. 5). The fluorescence increment was estimated to be 30% compared with the initial intensity below the LCST. The fluorescence increase was saturated above the LCST, indicating that such dramatic fluorescence changes coincide with size changes. The Azo-PNIPAM was swollen up to 34 °C and became aggregated above 36 °C. At the LCST, Azo-PNIPAM became aggregated because of the increase in hydrophobicity of the PNIPAM, resulting in association of AIE-active azonaphthol-based crosslinkers and, ultimately, fluorescence intensity was increased. In addition, a reversible fluorescence switching of Azo-PNIPAM was found in response to temperature (Fig. 6). The fluorescence intensity of Azo-PNIPAM was investigated by changing the temperature between 30 and 50 °C repeatedly. The reversible changes in the temperature were repeated six times and we found that fluorescence was highly dependent on the temperature reversibly without any fatigue. Therefore, we confirmed that the fluorescence of Azo-PNIPAM depended on the swelling and contraction of Azo-PNIPAM in response to temperature. More detailed observation of the fluorescence of Azo-PNIPAM enabled us to realize that Azo-PNIPAM showed weak fluorescence even at a lower temperature (inset photographs in Fig. 6). Although the fluorescence intensity of Azo-PNIPAM was increased above the LCST, weak emission was also observed below the LCST. It is presumed that because the azonaphthol-based crosslinkers are connected by NIPAM molecules, the surrounding environment restricts free rotation of the azo unit in the excited state.64,65 This led to the weak fluorescence of Azo-PNIPAM in solution. In addition, the changes in the fluorescence of Azo-OH were investigated to elucidate the effect of temperature on intramolecular hydrogen bonding (Fig. S5†). In previous study, we suggested that the fluorescence of Azo-OH resulted from intramolecular hydrogen bonding.54 As a result, it was found that the fluorescence of Azo-OH decreased with increasing temperature. This was originated from the weakening of intramolecular hydrogen bonding by the temperature increase. In conjunction with this, the fluorescence of Azo-PNIPAM increased upon heating. Though the increment was small (1.3 times), AIE was obviously exhibited in Azo-PNIPAM. It was not high compared with other PNIPAM polymers containing AIE units (Table S1†).66–70 However, thermoreversible fluorescence was elucidated using a newly synthesized Azo-PNIPAM.
 |
| | Scheme 2 Schematic illustration of AIE above LCST of Azo-PNIPAM. | |
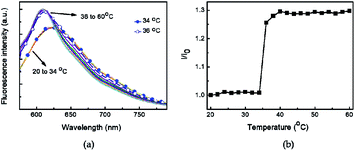 |
| | Fig. 5 (a) Changes in the fluorescence spectra of aqueous Azo-PNIPAM solution with increasing temperature from 20 °C to 60 °C. Excitation wavelength 450 nm. (b) Relative fluorescence intensity of Azo-PNIPAM at various temperatures. I0 and I correspond to the fluorescence intensity at 608 nm at 20 °C and at each temperature, respectively. | |
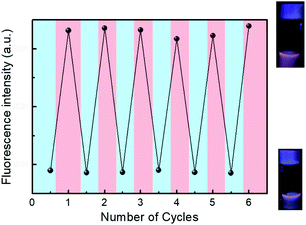 |
| | Fig. 6 Reversible fluorescence switching of Azo-PNIPAM in aqueous solution (1.5 mg mL−1) at 30 and 50 °C. Excitation wavelength 450 nm. Inset photographs indicate Azo-PNIPAM in aqueous solution under UV light (365 nm) at 30 °C (bottom) and 50 °C (upper). Blue and red shaded areas correspond to cooling (30 °C) and heating (50 °C), respectively. | |
Conclusions
AIE-active, PNIPAM-based thermoresponsive polymer was successfully synthesized using an AIE-active crosslinker composed of an azonaphthol derivative, in which the PNIPAM showed reversible sol–gel transition and thermoresponsive fluorescence. Although the AIE-active azonaphthol-based crosslinker was used to only 0.01 mol% compared with the NIPAM monomer during the radical polymerization, the crosslinked polymer demonstrated two kinds of responsiveness to a thermal stimulus. In addition to the conventional sol–gel transition of PNIPAM upon cooling below or heating above the LCST, thermally induced fluorescence variation was newly observed. The newly synthesized Azo-PNIPAM showed an LCST at 36 °C and exhibited noticeable size change with sol–gel transition, as well as notable AIE. In particular, a reversible response to temperature with a combination of sol–gel transition and AIE behavior indicated good solubility and stability of the polymer in aqueous media. These features indicate potential uses in bioimaging, environmental remediation, and drug delivery.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the National Research Foundation (NRF) of Korean government through Nuclear R&D Project (2016M2B2B1945085) and Regional Leading Research Center (2020R1A5A8017671) is gratefully acknowledged.
Notes and references
- Q. Shi, H. Liu, D. Tang, Y. Li and F. Xu, NPG Asia Mater., 2019, 11, 64 CrossRef.
- M. Vazquez-Gonzalez and I. Willner, Angew. Chem., Int. Ed., 2020, 59, 2–38 CrossRef.
- Y. Guo, J. Bae, Z. Fang, P. Li, F. Zhao and G. Yu, Chem. Rev., 2020, 120, 7642–7707 CrossRef CAS.
- M. Ding, L. Jing, H. Yang, C. E. Machnicki, X. Fu, K. Li, I. Y. Wong and P.-Y. Chen, Mater. Today Adv., 2020, 8, 100088 CrossRef.
- S. Chatterjee, P. C.-L. Hui, C.-W. Kan and W. Wang, Sci. Rep., 2019, 9, 11658 CrossRef.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2012, 64, 154–162 CrossRef.
- L. Hu, Q. Zhang, X. Li and M. J. Serpe, Mater. Horiz., 2019, 6, 1774–1793 RSC.
- J. Y. C. Lim, S. S. Goh, S. S. Liow, K. Xue and X. J. Loh, J. Mater. Chem. A, 2019, 7, 18759–18791 RSC.
- C. D. Spicer, Polym. Chem., 2020, 11, 184–219 RSC.
- A. Kawamura, Polym. J., 2017, 49, 751–757 CrossRef CAS.
- Z. Wang, H. Li, Z. Tang, Z. Liu, Z. Ruan, L. Ma, Q. Yang, D. Wang and C. Zhi, Adv. Funct. Mater., 2018, 28, 1804560 CrossRef.
- X. Wang, K. Xu, H. Yao, L. Chang, Y. Wang, W. Li, Y. Zhao and J. Qin, Polym. Chem., 2018, 9, 5002–5013 RSC.
- Y. Wang, A. G. Penas, S. G. Ruiz and F. J. Stadler, Macromol. Chem. Phys., 2020, 221, 2000035 CrossRef CAS.
- D. Zehm, A. Lieske and A. Stoll, Macromol. Chem. Phys., 2020, 221, 2000018 CrossRef CAS.
- M. Khimani, H. Patel, V. Patel, P. Parekh and R. L. Vekariya, Polym. Bull., 2020, 77, 5783–5810 CrossRef CAS.
- S. Gupta, A. Singh and N. Matsumi, ACS Omega, 2019, 4, 20923–20930 CrossRef CAS.
- S. Qian, R. Liu, G. Han, K. Shi and W. Zhang, Polym. Chem., 2020, 11, 2532–2541 RSC.
- Y. Zhang, C. Fu, Y. Li, K. Wang, X. Wang, Y. Wei and L. Tao, Polym. Chem., 2017, 8, 537–544 RSC.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC.
- G. Fundueanu, M. Onstantin, S. Ucatariu and P. Ascenzi, Polymer, 2017, 110, 177–186 CrossRef CAS.
- C. Ma, W. Lu, X. Yang, J. He, X. Le, L. Wang, J. Zhang, M. J. Serpe, Y. Huang and T. Chen, Adv. Funct. Mater., 2018, 28, 1704568 CrossRef.
- R. Chang, X. Wang, X. Li, H. An and J. Qin, ACS Appl. Mater. Interfaces, 2016, 8, 25544–25551 CrossRef CAS.
- I.-N. Lee, O. Dorbre, D. Richards, C. Ballestrem, J. M. Curran, J. A. Hunt, S. M. Richardson, J. Swift and L. S. Wong, ACS Appl. Mater. Interfaces, 2018, 10, 7765–7776 CrossRef CAS.
- S. Tang, M. Floy, R. Bhandari, M. Sunkara, A. J. Morris, T. D. Dziubla and J. Z. Hilt, ACS Omega, 2017, 2, 8723–8729 CrossRef CAS.
- H. Ma, C. Qi, C. Cheng, Z. Yang, H. Cao, Z. Yang, J. Tong, X. Yao and Z. Lei, ACS Appl. Mater. Interfaces, 2016, 8, 8341–8348 CrossRef CAS.
- S. Hajebi, A. Abdollahi, H. R. Mamaqani and M. S. Kalajahi, Langmuir, 2020, 36, 2683–2694 CrossRef CAS.
- A. Karmakar, P. G. M. Mileo, I. Bok, S. B. Peh, J. Zhang, H. Yuan, G. Maurin and D. Zhao, Angew. Chem., Int. Ed., 2020, 132, 11096–11102 CrossRef.
- M. Cao, Y. Wang, X. Hu, H. Gong, R. Li, H. Cox, J. Zhang, T. A. Waigh, H. Xu and J. R. Lu, Biomacromolecules, 2019, 20, 3601–3610 CrossRef CAS.
- Z. Lu, Z. Zhang and Y. Tang, ACS Appl. Bio Mater., 2019, 2, 4485–4492 CrossRef CAS.
- C. Sun, H. Jia, K. Lei, D. Zhu, Y. Gao, Z. Zheng and X. Wang, Polymer, 2019, 160, 246–253 CrossRef CAS.
- A. R. Uribe and L. Albanil, ACS Appl. Mater. Interfaces, 2019, 11, 24447–24458 CrossRef.
- C. J. Christopherson, D. M. Mayder, J. Poisson, N. R. Paisley, C. M. Tonge and Z. M. Hudson, ACS Appl. Mater. Interfaces, 2020, 12, 20000–20011 CrossRef CAS.
- S. Uchiyama, J. Synth. Org. Chem., Jpn., 2019, 77, 1116–1127 CrossRef CAS.
- A. Abdollahi, H. R. Mamagani, B. Razavi and M. S. Kalajahi, Polym. Chem., 2019, 10, 5686–5720 RSC.
- S. Qian, S. Li, W. Xiong, H. Khan, J. Huang and W. Zhang, Polym. Chem., 2019, 10, 5001–5009 RSC.
- J. Yang, K. Gu, C. Shi, M. Li, P. Zhao and W.-H. Zhu, Mater. Chem. Front., 2019, 3, 1503–1509 RSC.
- H. Guo, X. Cheng, H. Li, J. Li, J. Wei and C. Feng, RSC Adv., 2020, 10, 23532–23542 RSC.
- K. Lei, Q. Ma, L. Yu and J. Ding, J. Mater. Chem. B, 2016, 4, 7793–7812 RSC.
- C. Wei, X. Dong, Y. Zhang, J. Liang, A. Yang, D. Zhu, T. Liu, D. Kong and F. Lv, Sens. Actuators, B, 2018, 273, 264–275 CrossRef CAS.
- G. K. Park, S.-H. Kim, K. Kim, P. Das, B.-G. Kim, S. Kashiwagi, H. S. Choi and N. S. Hwang, Theranostics, 2019, 9, 4255–4264 CrossRef.
- K. Khalid, X. Tan, H. F. M. Zaid, Y. Tao, C. L. Chew, D.-T. Chu, M. K. Lam, Y.-C. Ho, J. W. Lim and L. C. Wei, Bioengineered, 2020, 11, 328–355 CrossRef CAS.
- D. Broadwater, M. Bates, M. Jayaram, M. Young, J. He, A. L. Raithel, T. W. Hamann, W. Zhang, B. Borhan, R. R. Lunt and S. Y. Lunt, Sci. Rep., 2019, 9, 15288 CrossRef.
- V. K. Sharma, J. Filip, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 8410–8423 RSC.
- Y. Li, Z. Cai, S. Liu, H. Zhang, S. T. H. Wong, J. W. Y. Lam, R. T. K. Kwok, J. Qian and B. Z. Tang, Nat. Commun., 2020, 11, 1255 CrossRef CAS.
- J. Qian and B. Z. Tang, Chem, 2017, 3, 56–91 CAS.
- H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365–377 CrossRef CAS.
- S. Gan, J. Zhou, T. A. Smith, H. Su, W. Luo, Y. Hong, Z. Zhao and B. Z. Tang, Mater. Chem. Front., 2017, 1, 2554–2558 RSC.
- E. Zhao, Y. Chen, H. Wang, S. Chen, J. W. Y. Lam, C. W. T. Leung, Y. Hong and B. Z. Tang, ACS Appl. Mater. Interfaces, 2015, 7, 7180–7188 CrossRef CAS.
- C. Zhang, X. Yao, J. Wang and X. Ma, Polym. Chem., 2017, 8, 4835–4841 RSC.
- Y. Zhao, Y. Wu, S. Chen, H. Deng and X. Zhu, Macromolecules, 2018, 51, 5234–5244 CrossRef CAS.
- P. Q. Nhien, W.-L. Chou, T. T. K. Cuc, T. M. Khang, C.-H. Wu, N. Thirumalaivasan, B. T. B. Hue, J. I. Wu, S.-P. Wu and H. C. Lin, ACS Appl. Mater. Interfaces, 2020, 12, 10959–10972 CrossRef.
- Y. Yoon, S. Jo, S. J. Park, H. M. Kim, D. Kim and T. S. Lee, Chem. Commun., 2019, 55, 6747–6750 RSC.
- J.-H. Kim, Y. Jung, D. Lee and W.-D. Jang, Adv. Mater., 2016, 28, 3499–3503 CrossRef CAS.
- W. Wu, F. Huang, S. Pan, W. Mu, X. Meng, H. Yang, Z. Xu, A. J. Ragauskas and Y. Deng, J. Mater. Chem. A, 2015, 3, 1995–2005 RSC.
- G. Liang, J. Wu, H. Gao, Q. Wu, J. Lu, F. Zhu and B. Z. Tang, ACS Macro Lett., 2016, 5, 909–914 CrossRef CAS.
- M. S. Choi, J. H. Lee, D. W. Kim and T. S. Lee, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4430–4440 CrossRef CAS.
- T. H. Kim, H. J. Kim, C. G. Kwak, W. H. Park and T. S. Lee, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2059–2068 CrossRef CAS.
- R. Yi, G. Ye, D. Lv and J. Chen, RSC Adv., 2015, 5, 55277–55284 RSC.
- Y. Xu, Z. Li, F. Zhang, X. Zhuang, Z. Zeng and J. Wei, RSC Adv., 2016, 6, 30048–30055 RSC.
- X.-J. Ju, L.-Y. Chu, X.-L. Zhu, L. Hu, H. Song and W.-M. Chen, Smart Mater. Struct., 2006, 15, 1767–1774 CrossRef CAS.
- D. Zhang, Y. Fan, H. Chen, S. Trepout and M.-H. Li, Angew. Chem., Int. Ed., 2019, 58, 10260–10265 CrossRef CAS.
- Y. Li, Y. Dong, L. Cheng, C. Qin, H. Nian, H. Zhang, Y. Yu and L. Cao, J. Am. Chem. Soc., 2019, 141, 8412–8415 CrossRef CAS.
- J.-B. Xiong, Y.-X. Yuan, L. Wang, J.-P. Sun, W.-G. Qiao, H.-C. Zhang, M. Duan, H. Han, S. Zhang and Y.-S. Zheng, Org. Lett., 2018, 20, 373–376 CrossRef CAS.
- X. Guan, D. Zhang, L. Meng, Y. Zhang, T. Jia, Q. Jin, Q. Wei, D. Lu and H. Ma, Ind. Eng. Chem. Res., 2017, 56, 680–686 CrossRef CAS.
- T. Li, S. He, J. Qu, H. Wu, S. Wu, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 2964–2970 RSC.
- H. Zhou, F. Liu, X. Wang, H. Yan, J. Song, Q. Ye, B. Z. Tang and J. Xu, J. Mater. Chem. C, 2015, 3, 5490–5498 RSC.
- S. Sasaki and G.-I. Konishi, RSC Adv., 2017, 7, 17403–17416 RSC.
- C.-M. Yang, Y.-W. Lai, S.-W. Kuo and J.-L. Hong, Langmuir, 2012, 28, 15725–15735 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of monomers and polymer, LCST behaviour of polymer. See DOI: 10.1039/d0ra08257k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*