
Photoelectrocatalytic organic synthesis: a versatile method for the green production of building-block chemicals
Si-Yang
Li
 abc,
Ke-Fu
Huang
c,
Zhi-Yong
Tang
ac and
Jing-Hao
Wang
*ac
abc,
Ke-Fu
Huang
c,
Zhi-Yong
Tang
ac and
Jing-Hao
Wang
*ac
aChinese Academy of Science (CAS) Key Laboratory of Nanosystem and Hierarchy Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, P. R. China
bSino-Danish College, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 13th January 2023
Abstract
The rising energy crisis and environmental problems are urging the development of more sustainable organic synthetic methods. Photoelectrocatalytic organic synthesis (PECOS) is emerging as an attractive strategy because it utilizes solar power to drive the organic synthesis and can greatly reduce the dependency on fossil fuels. Recently, a variety of organic reactions have been successfully realized using this strategy by rationally designing the photoelectrode and carefully tuning the reaction conditions. In this review, we introduce and categorize the recent advances in PECOS based on their specific reaction types, ranging from photoanode-mediated alcohol oxidation, C–H functionalization, furan oxidation, and sulfide oxidation, to (photo) cathode-mediated in situ H2O2 generation, cofactor regeneration, and the functionalization of aryl halides. The material of the photoelectrode, the reaction conditions of PECOS, and the proposed mechanism will be highlighted during the introduction of each example. Finally, we conclude by providing some perspectives on the future direction of PECOS.
1. Introduction
As a sustainable, cost-free, environment-friendly energy, solar power has been continuously gaining the scientific community's attention.1,2 Fixing solar energy in value-added compounds, like fuels and building-block chemicals, is a promising way to realize the storage of solar energy and has received considerable attention from the chemical community.3–6 Developing green and efficient methods of converting solar energy into chemical energy is becoming the future trend of energy and synthetic chemistry.Inheriting the advantages of photocatalysis and electrocatalysis, photoelectrocatalysis is a potential candidate for the green and efficient conversion of solar power. A typical setup for photoelectrocatalysis consists of a semiconductor photoelectrode and a counter electrode. Upon light irradiation, an electron and hole are separated at the photoelectrode. One of them migrates to the surface of the photoelectrode and motivates the redox reactions; with the help of an external circuit, another would transfer to the counter electrode (in the case of he photoanode) or be neutralized by the electron transported from the counter electrode (in the case of the photocathode).7 Compared with photocatalysis, the separation efficiency of photogenerated carriers could be tuned by the external potential in photoelectrocatalysis, enabling the careful control of reaction selectivity.8,9 Besides, the existence of an external circuit allows the reduction and oxidation half reactions to take place in different electrodes, making it possible to conduct the reductive and oxidative synthesis in a spatially separated manner.10 The spatial separation of reduction and oxidation half reactions can prevent the undesired side reactions of the products and overcome the product separation problems in photo-redox organic synthesis.11,12 In the meantime, the introduction of light energy reduces the applied bias so that photoelectrocatalysis could be carried out in a more energy-saving manner than electrocatalysis. It should be noticed that there is another kind of organic synthesis which is conducted using the combination of inert electrodes and photocatalyst: upon light irradiation, the photocatalyst were first transformed into their excited state, which then participate in the redox reaction of the organic substrates; with the help of inert electrodes, the photocatalysts are finally regenerated. In fact, this kind of catalysis could be classified as “electrophotocatalysis” and König, Wu, and Lambert et al. have already presented some excellent studies on this topic.13–15
Since Fujishima and Honda reported the feasibility of photoelectrocatalytic water splitting,16 for the first time, much effort has been put into the development of photoelectrocatalysis. Nowadays, the application of photoelectrochemical cells (PECs) is no longer limited to the decomposition of water. The capability of PECs in fuel production,17–23 carbon dioxide reduction,24–29 and biomass conversion30–36 has been widely investigated. However, the research on photoelectrocatalytic organic synthesis (PECOS) is still in an early stage.
In the past decade, many attempts on promoting the development of PECOS have been made. The photoelectrode material, the reaction type, and the substrate scope of PECOS have been greatly diversified. Now, PECs equipped with a classical metal oxide photoanode can do much more than oxidizing simple alcohols.37 More complex oxidations such as the functionalization of a C–H bond and the selective oxidation of polyhydric alcohol, furan, and sulfide can also be realized. Besides, by properly introducing enzymes or heterogenous catalysts, the place where redox reactions take place is no longer limited to the photoanode, which makes the cathode-mediated PECOS possible. Moreover, the successful application of photocathodes in PECs further broadens the reaction types of PECOS. This progress shows the compatibility of PECs with organic synthesis.
In this article, we provide a concise review of the recent development of PECOS. Based on the exact electrode which mediates the organic synthesis, we classify the literature into two main categories: photoanode-mediated organic synthesis and (photo) cathode-mediated organic synthesis. Each category is further divided into subcategories according to the specific reaction types. Finally, we offer some perspectives on the future direction of PECOS.
2. Photoanode-mediated organic synthesis
The most common type of PECOS is carried out in a photoanode-mediated manner, in which photogenerated holes directly or indirectly oxidize the organic substrates. In direct oxidation, the organic substrates are oxidized on the surface of a photoanode (Fig. 1A). Generally speaking, the careful control of the reaction selectivity is relatively difficult to obtain in direct oxidation because of the high reactivity of photogenerated holes.38–40 In indirect oxidation, however, the photogenerated holes first oxidize the redox mediator to form a reactive intermediate, which then oxidizes the organic substrates and return to its original state (Fig. 1B). With these two kinds of reaction pathways, the photoanode-mediated organic synthesis reported thus far exhibits diverse reaction types, which are roughly classified into alcohol oxidation, C–H functionalization, and the oxidation of other organic compounds.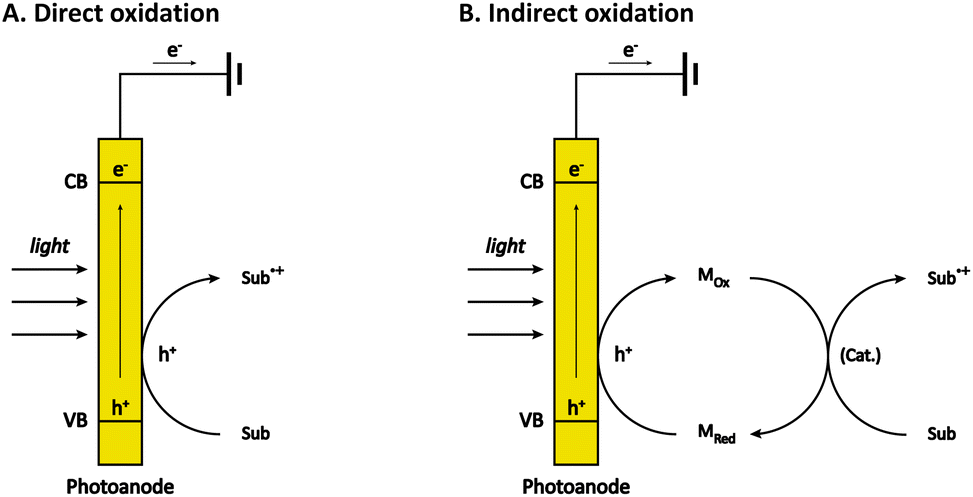 | ||
| Fig. 1 Schematics for direct and indirect oxidation on a photoanode. | ||
2.1 Alcohol oxidation
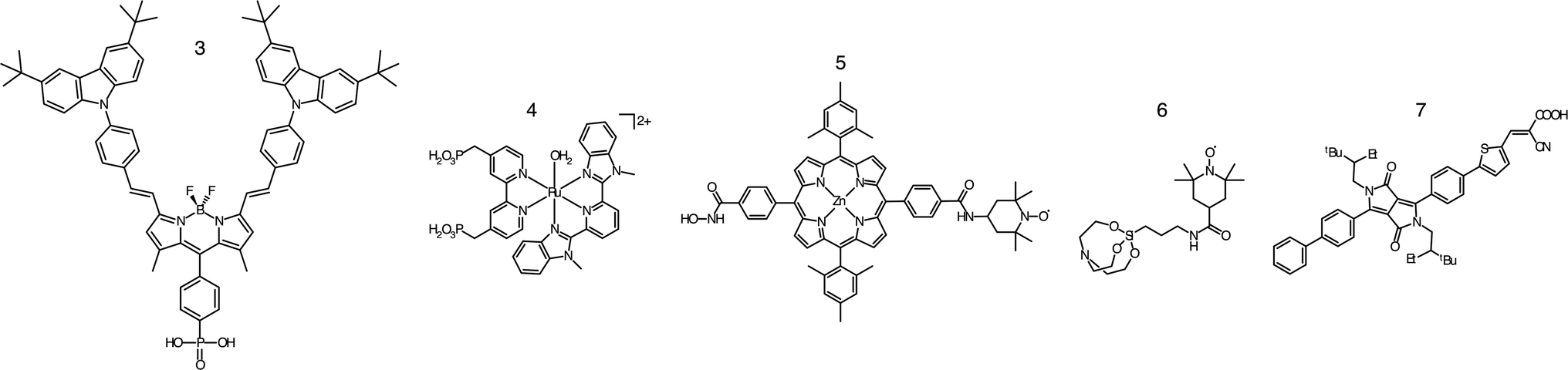
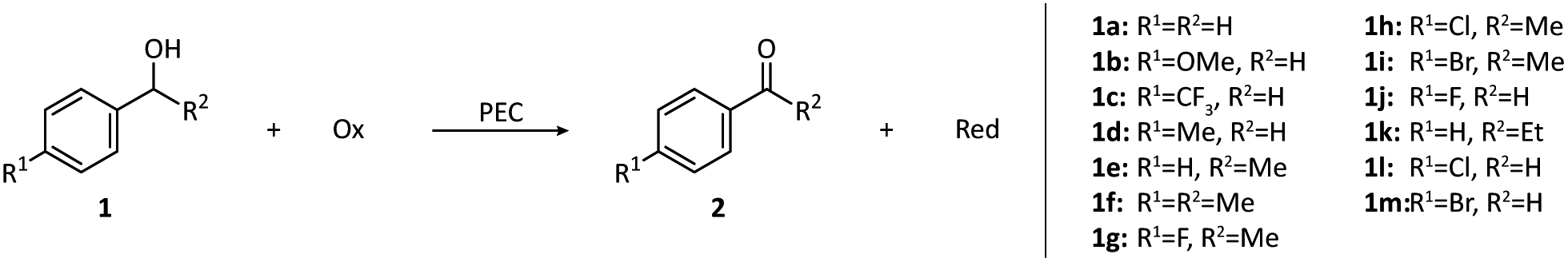
According to the structural complexity of alcohol molecules, we divide the alcohol oxidation that happens in PECs into the oxidation of simple alcohols and the oxidation of polyhydric alcohols. Since simple alcohols have only one hydroxyl group, their oxidation has no regioselectivity problem. In contrast, the regioselectivity must be taken into account if one wants to achieve the selective oxidation of polyhydric alcohols with hydroxyl groups within different environments.In 2020, Meyer and co-workers introduced organic photosensitizers to the surface of a SnO2/TiO2 photoanode.46 The phosphonate-derivatized carbazole and boron dipyrromethene (BODIPY(CBZ)2PO3H2) chromophore (3) has a high molar extinction coefficient and good visible light response. Together with a ruthenium-based catalyst (4), the photoanode could sustain the oxidation of benzyl alcohol (1a) in a pH 4.5 acetic buffer under visible light irradiation. At an applied bias of 0.2 V vs. Ag/AgCl, the density of photocurrent was maintained at around 35 μA cm−2. The low current level may be caused by the back electron transfer after electron injection.49
Even though being frequently used,41–46,50 ruthenium-based dye molecules are not the only choice for the photosensitizer used in a DSPEC. The noble metal ruthenium could be replaced by earth-abundant elements as well.51,52 For example, Nikoloudakis et al. presented efficient and selective conversion from para-substituted phenyl alcohols (1a–1c) into the corresponding aldehydes (2a–2c) with a zinc porphyrin (ZnP) sensitizer (5).47 The photosensitizer was anchored on the surface of a TiO2 photoanode by a robust hydroxamic group and hence was able to work both in aqueous solutions and organic solvents. ZnP was also covalently linked with 2,2,6,6-tetramethyl-1-piperidine N-oxyl (TEMPO), which served as the catalyst for alcohol oxidation. Upon light illumination, the holes generated by ZnP efficiently transferred to TEMPO, thus realizing the selective production of para-methoxy benzaldehyde with an average photocurrent density of 200 μA cm−2 at 0 V vs. SCE in a pH 8.0 borate buffer.
Recently, Reisner's group established a DSPEC where both the dye molecule and alcohol oxidation catalyst were metal-free.48 The photoanode was prepared by co-immobilizing silatrane-functionalized TEMPO (STEMPO, 6) and diketopyrrolopyrrole-based dye (DPP-CA, 7) on mesoporous TiO2 (mTiO2). Under the irradiation of ultraviolet-filtered simulated solar light, 4-methylbenzylalcohol (1d) can be selectively oxidized to its aldehyde form on this photoanode with a photocurrent density of around 90 μA cm−2 at +0.4 V vs. RHE in pH 8.0 sodium borate buffer. The author also coupled the photoanode with a CO2-reducing cathode. The as-constructed PEC could achieve a bias-free, simultaneous alcohol oxidation and CO2 reduction with a photocurrent density up to 30 μA cm−2.
The oxidation of simple alcohol can also be realized in PECs without dye molecules. Earlier in 2017, the oxidation of 1-phenylethanol derivatives on a BiVO4/WO3 photoanode was reported by Sayama's group.53 1-phenylethanol derivatives (1e–1i) were directly oxidized by the photogenerated holes at the surface of the photoanode. The applied potential required for oxidation in this system (approximately +0.7 V vs. SHE) was much lower than that in an electrocatalytic system taking Pt as the anode (>1.9 V vs. SHE). Meanwhile, the para-substituent effect observed in an electrocatalytic system was negligible in this system, suggesting that the external bias did not influence the oxidation ability of photogenerated holes. Under visible light irradiation (>420 nm), 1-phenylethanol can be oxidized with excellent yield (97%) and faradaic efficiency (>99%).
In the same year, Duan and co-workers developed a TiO2/C composite photoanode and tested its catalytic performance on the oxidation of benzyl alcohol derivatives (1a, 1d, 1j, & 1k).54 The photoanode was prepared through a three-step method: first, TiO2 nanowire arrays, with an average diameter of around 200 nm, were hydrothermally grown on a fluorine-doped tin oxide (FTO) substrate; then, polypyrrole was coated on the surface of TiO2via a photo-assisted deposition method; finally, calcination in a N2 atmosphere finally led to the formation of a graphite coating layer. The existence of a graphite layer maximized the separation efficiency of photogenerated carriers and accelerated the oxidation of H2O to generate O2˙−, which plays an important role in alcohol oxidation. With the help of light and external bias, the TiO2/C photoanode was capable of oxidizing benzyl alcohol and its derivatives to their corresponding aldehydes at a selectivity of 100%.
In 2020, Ye, Yan, and Xiang et al. conceived a radical relay strategy and successfully coupled water oxidation with the selective oxidation of benzyl alcohol derivatives (1a, 1d, 1j, 1l, & 1m) in a PEC.55 The photoanode they adopted was a ternary composite consisting of an ultrathin Co-based layered double hydroxide, graphene, and a layer of BiVO4 particles (G@U-LDH@BVO). In the preparation of the photoanode, BiVO4 particles were first deposited on an FTO through electrodeposition; after a hydrothermal process, the as-prepared BiVO4 photoanode was finally modified by graphene and U-LDH. The conducting characteristics of graphene facilitate the transfer of photoexcited electrons from U-LDH to BiVO4. Meanwhile, U-LDH can accept the photogenerated holes and catalyze water oxidation. The relayed ˙OH adsorbed on the surface of U-LDH was key to the selective oxidation of benzyl alcohol derivatives. This radical relay strategy enables the efficient coupling between water splitting and the selective oxidation of organic molecules under mild conditions.56 Under a neutral condition (8 mL phosphate-buffered saline (PBS) solution + 2 mL MeCN), G@U-LDG@BVO could realize the photoelectrocatalytic oxidation of substituted benzyl alcohols into the corresponding aldehyde products with excellent selectivity (>96%) and production rate (125–188 μmol h−1). For comparison, the results of the photoanode-mediated benzyl alcohol oxidation are summarized in Table 1.
|
|
|||||
|---|---|---|---|---|---|
| Substrate | Photoanode | Conditions | Ox/red | Performance | Ref. |
| 1a | SnO2|TiO2|-3,4 | 0.2 V vs. Ag/AgCl, 100 mW cm−2 visible light, 0.4 M LiClO4 pH 4.5 acetic buffer | H+/H2 | J ≈ 35 μA cm−2 | 46 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| 1a | TiO2|5 | 0 V vs. SCE, 100 mW cm−2 visible light, 0.1 M NaClO4 pH 8.0 borate buffer | H+/H2 | J ≈ 0.19 mA cm−2 | 47 |
| 1b | J ≈ 0.20 mA cm−2 | ||||
| 1c | J ≈ 0.50 mA cm−2 | ||||
|
|||||
| 1d | mTiO2|6,7 | Bias-free, 100 mW cm−2 visible light, pH 8.0 borate buffer | CO2/formate | J ≈ 30 μA cm−2 | 48 |
|
|||||
| 1e | BiVO4/WO3 | 0.2 mA cm−2, 100 mW cm−2 visible light, 0.1 M Bu4NBF4, MeCN | NA | Yield 97% | 53 |
| 1f | Yield 63% | ||||
| 1g | Yield 89% | ||||
| 1h | Yield 92% | ||||
| 1i | Yield 91% | ||||
|
|||||
| 1a | TiO2/C | 0.6 V vs. SCE, 120 mW cm−2 Xe lamp, 0.5 M Na2SO4 | H+/H2 | 144 μmol h−1 | 54 |
| 1d | 180 μmol h−1 | ||||
| 1j | 161 μmol h−1 | ||||
| 1k | 45 μmol h−1 | ||||
|
|||||
| 1a | G@U-LDH@BVO | 1.2 V vs. RHE, 100 mW cm−2 visible light, 8 mL pH 7.0 PBS solution, 2 mL MeCN | H+/H2 | 150 μmol h−1 | 55 |
| 1d | 188 μmol h−1 | ||||
| 1j | 144 μmol h−1 | ||||
| 1l | 125 μmol h−1 | ||||
| 1m | 151 μmol h−1 | ||||
|
|||||
Another example of simple alcohol oxidation was provided by Cha, Goodwin, and Yehezkeli et al. using BiVO4 as the photoanode.57 The oxidation of n-butanol (8) was catalyzed by alcohol oxidase (AOx), which reduced O2 to H2O2 at the same time. BiVO4 not only afforded the regeneration of oxidant O2 but also maintained the stability of AOx by timely scavenging the H2O2 in the solution. It is worth mentioning that n-butyraldehyde (9) produced from alcohol oxidation can then undergo an aldol condensation catalyzed by β-alanine and finally be upgraded to 2-ethylhexenal (10) within a PEC (Fig. 2), which vividly demonstrated that PECs can be compatible with an enzymatic and homogenous catalyst.
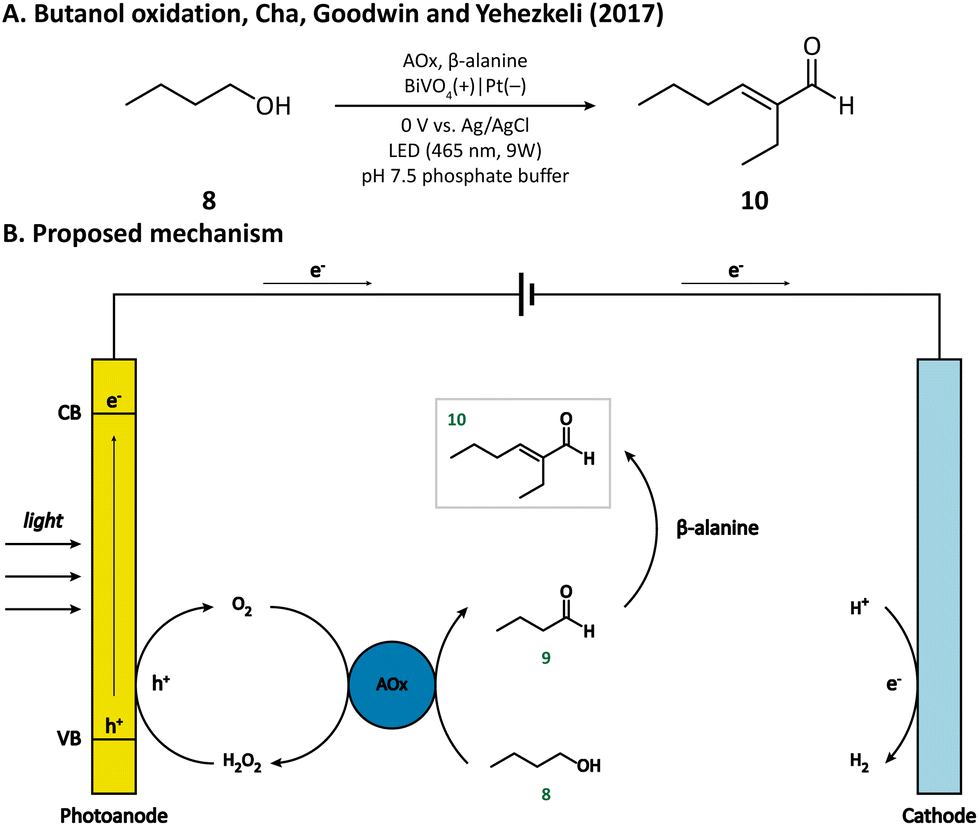 | ||
| Fig. 2 Photoanode-mediated simple alcohol oxidation coupled with aldol condensation. | ||
Subsequently, many attempts have been made to investigate the performance of a PEC in glycerol conversion.59–64 For instance, in early 2022, Duan, Li, and co-workers reported the same reaction with a higher selectivity towards DHA (75.4%) by modifying Bi2O3 on TiO2 (Bi2O3/TiO2).62 The cocatalyst Bi2O3, prepared by an electrodeposition-electrooxidation process, could not only improve the light absorption ability of TiO2 but also facilitate the charge transfer by forming a p–n junction with TiO2, which greatly enhances the photocurrent density. Unlike the BiVO4 system, the glycerol oxidation on Bi2O3/TiO2 was considered as an *OH radical-mediated process. It was demonstrated by in situ Fourier transform infrared spectroscopy and density functional theory (DFT) calculations that glycerol prefers to adsorb over Bi2O3 with its middle hydroxyl group and DHA can poorly adsorb on Bi2O3, which explained the relatively high selectivity of DHA production. At an external bias of 1.0 V vs. RHE, the Bi2O3/TiO2 photoanode sustained glycerol conversion at a rate of 228 mmol m−2 h−1 when being exposed to light irradiation in an acidic environment (pH = 2.0).
Only two months later, Xiang and Yan et al. successfully conducted glycerol oxidation in a neutral medium.63 They established a ternary photoanode by depositing silver nanoparticles on layered double hydroxide nanosheets grown on TiO2 arrays (denoted as Ag@LDH@TiO2). In this ternary photoanode, TiO2 played the role of harvesting light energy, the LDH nanosheets selectively adsorbed and activated the middle hydroxyl group in glycerol, and the loaded Ag nanoparticles facilitated glycerol oxidation by transferring electrons with LDH nanosheets. Using isotopic labelling experiments, the author proposed a mechanism where glycerol first undergoes a dehydration and dehydrogenation process on LDH and then reacts with an *OH radical to form DHA, which was different from the gem-diol mechanism reported in a previous PEC glycerol oxidation system.58,62 Under neutral conditions (0.5 M Na2SO4, pH = 7), glycerol can be converted at a considerable rate (315 mmol m−2 h−1) with a DHA selectivity of 72.1%.
More recently, a tantalum-doped BiVO4 (Ta:BiVO4) photoanode was fabricated and applied in glycerol oxidation by Sayama's group.64 In the fabrication of the Ta:BiVO4 photoanode, BiVO4 was first deposited on a WO3-coated FTO substrate via a spin-coating method and a following calcination; then, a solution containing bismuth oxide, vanadium, and tantalum oxides was spin-coated and calcinated on the as-prepared BiVO4/WO3/FTO photoanode. Ta atoms were found to be concentrated on the surface of the photoanode by EDS mapping and they were considered to exist in the form of Ta5+. The existence of a Ta-rich overlayer improved the light-harvesting efficiency of BiVO4, which resulted in an enhanced glycerol oxidation photocurrent density compared with that of the bare BiVO4 photoanode. When Ta:BiVO4 was placed under an acidic condition (100 mM H2SO4 in acetone/H2O), the faradaic efficiency of the DHA reached 96% and the selectivity towards DHA was impressively close to 100%, which was significantly higher than those in previous studies (Table 2).
|
|
|||
|---|---|---|---|
| Photoanode | Conditions | Performance | Ref. |
| BiVO4 | 1.2 V vs. RHE, 0.5 M Na2SO4, pH 2.0 H2SO4 solution | Photocurrent density: 3.7 mA cm−2 | 58 |
| DHA selectivity: 51% | |||
| DHA production: 200 mmol−1 m−2 | |||
| FE for DHA production:≈30% | |||
|
|||
| BiVO4 | 0.7 V vs. RHE, 0.1 M Na2B4O7 (pH = 2) | DHA selectivity: 15% | 59 |
|
|||
| {010}-BiVO4 | 1.1 V vs. RHE, 0.1 M Na2B4O7, pH 2.0 H2SO4 solution | Photocurrent density:≈1.4 mA cm−2 | 60 |
| DHA selectivity:≈60% | |||
|
|||
| NiOx(OH)y/W:BiVO4 | 1.2 V vs. RHE, pH 9.3 borate buffer | Photocurrent density: 3.5 mA cm−2 | 61 |
| DHA selectivity:≈35% | |||
| DHA production: 138 mmol−1 m−2 | |||
| FE for DHA production: 19% | |||
|
|||
| Bi2O3/TiO2 | 1.0 V vs. RHE, 0.5 M Na2SO4, pH 2.0 H2SO4 solution | Photocurrent density: 0.27 mA cm−2 | 62 |
| DHA selectivity: 75.4% | |||
| DHA production:≈172 mmol−1 m−2 | |||
| FE for DHA production: 62.2% | |||
|
|||
| Ag@LDH@TiO2 | 1.2 V vs. RHE, 0.5 M Na2SO4 | Photocurrent density: 2.12 mA cm−2 | 63 |
| DHA selectivity: 72.1% | |||
| DHA production:≈227 mmol−1 m−2 | |||
| FE for DHA production:≈55% | |||
|
|||
| Ta:BiVO4 | 1.0 V vs. RHE, 100 mM H2SO4 acetone/H2O | Photocurrent density: 2.5 mA cm−2 | 64 |
| DHA selectivity:≈100% | |||
| FE for DHA production: 96% | |||
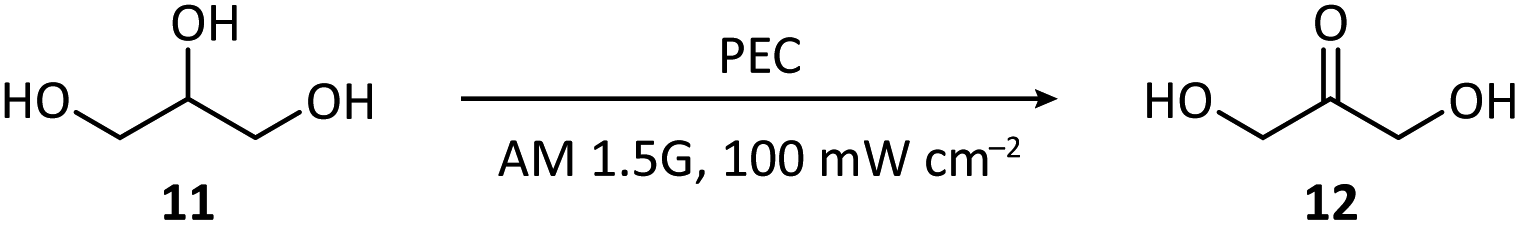
2.2 C–H functionalization
The direct functionalization of a C–H bond can achieve the construction of a carbon–carbon or carbon-heteroatom bond without the independent preparation of reactive functional groups, which is of high atomic and redox efficiency.65–67 However, the ubiquity and stability of C–H bonds make it challenging to achieve high catalytic activity while maintaining high selectivity.68 PECOS, harvesting light energy to provide Gibbs energy for C–H activation and using electricity to control the reaction rate, is a promising method to resolve this dilemma. We offer here an overview of the hitherto developed PEC-mediated C–H functionalization methods, including examples of C–H halogenation, oxygenation, amination, phosphorylation, and C–C coupling.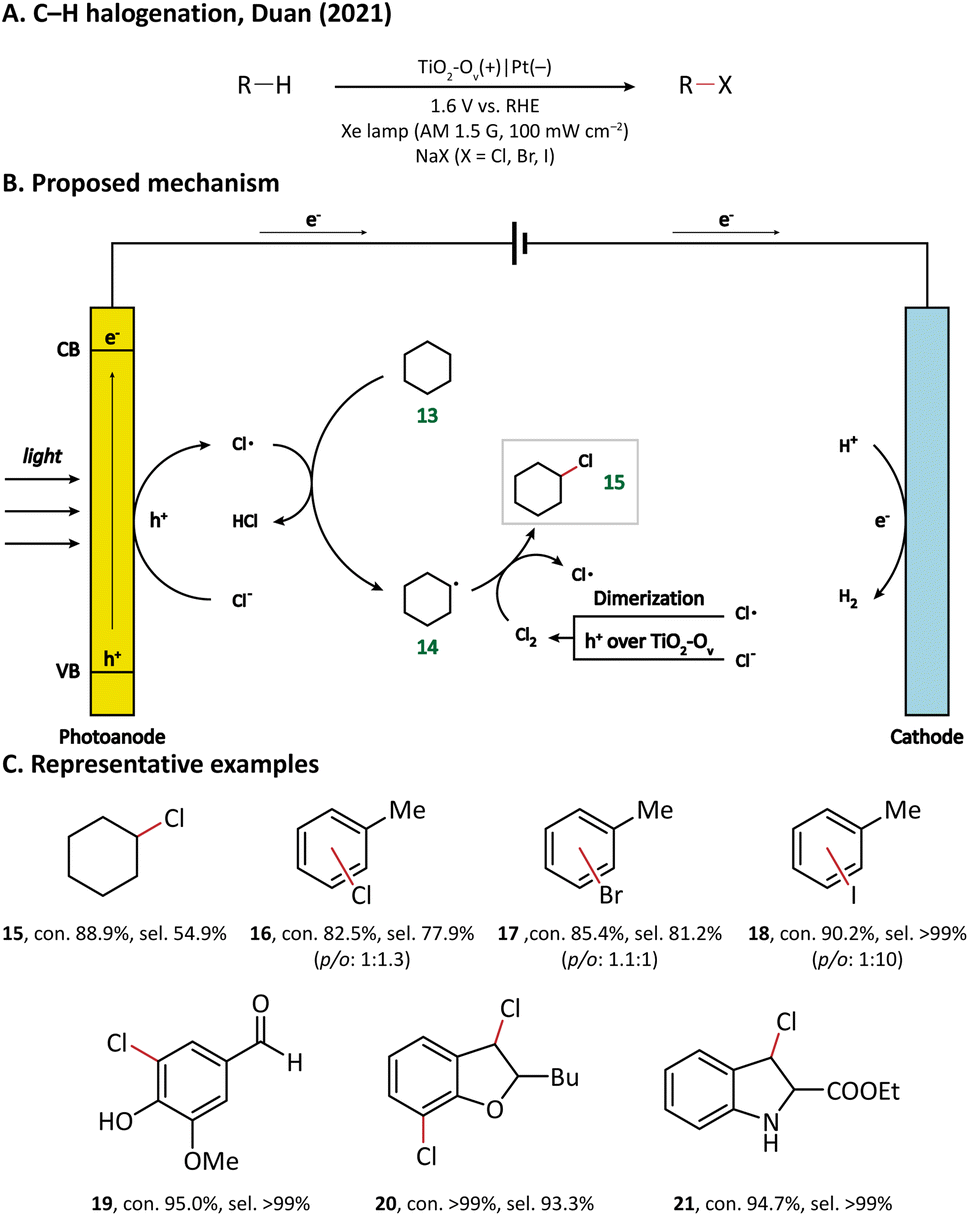 | ||
| Fig. 3 Photoanode-mediated cyclohexane halogenation. | ||
In 2017, Berlinguette and Sammis et al. carried out C–H oxygenation at the α-position of the alkene and arene in a PEC taking BiVO4 as the photoanode.76 The oxygenation of substrates takes place in an indirect oxidation pathway: photogenerated holes first oxidize an N-hydroxysuccinimide anion (NHS−) into NHS˙, which then abstracts the α-H from the substrate (22); the as-formed carbon-centered active species (23 & 24) subsequently react with tert-butyl hydroperoxide (tBuOOH) and finally yield oxygenated products (Fig. 4). Notably, the introduction of light energy enabled the reduction of applied bias from 1.8 V in an electrochemical cell to 0.8 V in a PEC, leading to a 60% decrease in energy consumption.
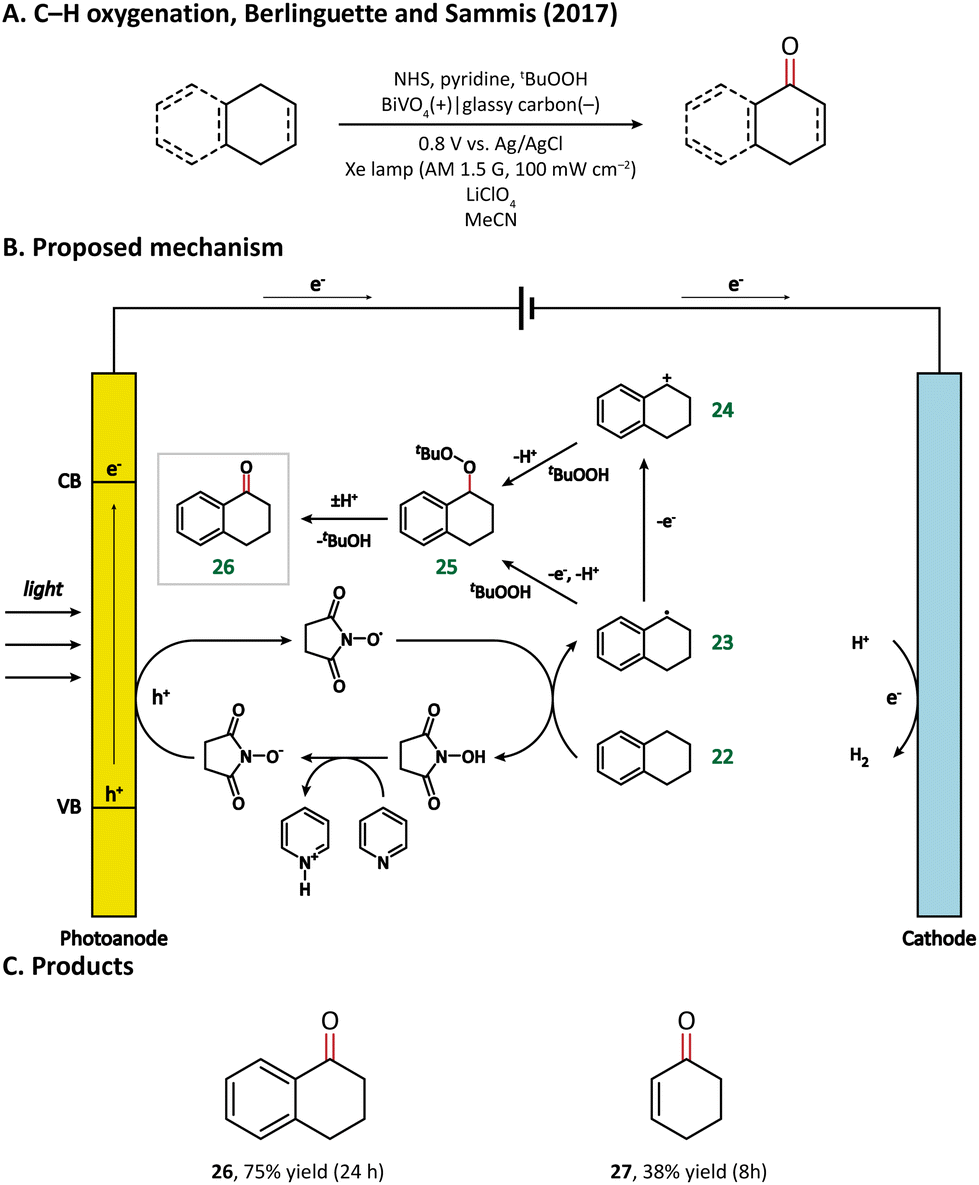 | ||
| Fig. 4 Photoanode-mediated C–H oxygenation at the α-position of the alkane and arene. | ||
Using porous WO3 as the photoanode, Sayama's group succeeded in C–H oxygenation of cyclohexane at room temperature and atmospheric pressure.77 As shown in Fig. 5, the C–H bond in cyclohexane (13) is directly activated by the photogenerated holes on the photoanode and the generated cyclohexyl radical (14) immediately captures a molecule O2 in air to form a cyclohexylperoxyl radical (28), whose disproportionated reaction yields cyclohexanol (29) and cyclohexanone (30). Cyclohexanol may also undergo a two-electron oxidation into cyclohexanone. Under simulated-solar light irradiation, cyclohexane could be converted efficiently with a high partial oxidation selectivity of 99%.
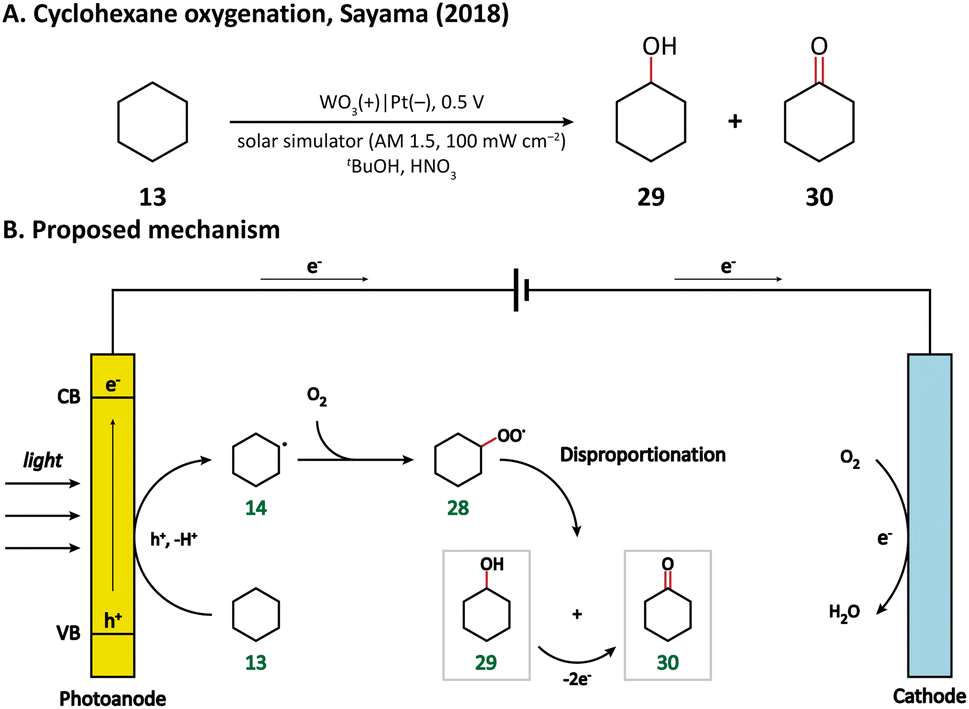 | ||
| Fig. 5 Photoanode-mediated cyclohexane oxygenation. | ||
Compared with cyclohexane, methane (31) has a stronger C–H bond (bond dissociation energy, BDE = 105 kcal mol−1),80 and hence, is more difficult to activate. The challenging methane oxygenation towards ethylene glycol (EG, 35) has been accomplished by Xiong and co-workers in a PEC equipped with a monoclinic WO3 photoanode.79 Through the hydrothermal method, they successfully grew WO3 nanobar, nanoplate, and nanoflake arrays on FTO substrates. The facet-dependent performance of WO3 was studied. It was found that WO3 nanobars, exhibiting the highest {010} facet ratio, has the best EG productivity and selectivity upon light irradiation in an acidic medium (pH 2.0 H2SO4 solution), which may be due to the high reactivity of ˙OH bound on {010} facets. A plausible mechanism proposed by the author is illustrated in Fig. 6. First, the H2O molecule is oxidized by the photoanode to generate the surface-bound ˙OH. Methane (31) is then activated by ˙OH and ˙CH3 (32) is formed. The combination of ˙OH and ˙CH3 leads to the generation of CH3OH (33), which subsequently undergoes a similar activation process to form ˙CH2OH (34). The coupling between two ˙CH2OH yields the final product EG (35). The byproduct ethane (36) may result from the coupling of two ˙CH3 radicals.
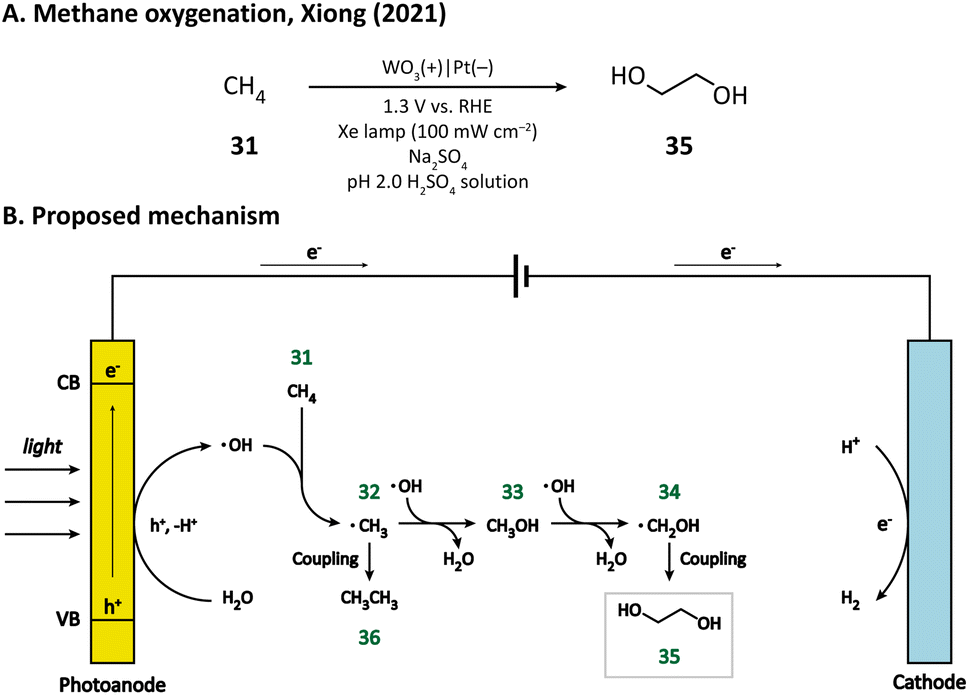 | ||
| Fig. 6 Photoanode-mediated methane oxygenation. | ||
Surprisingly, an unusual ortho selectivity could be obtained in this hematite-mediated PEC, which was speculated to be originated from the interactions between the radical cations and the solvent HFIP. Considering these experimental results, the author proposed a mechanism for C–H amination (Fig. 7). To begin with, the electron-rich arene (37) is directly oxidized by the photogenerated holes to form a radical cation (38). With the help of the HFIP molecule, the nucleophilic azole (39) attacks the ortho position of arene and generates an adduct 40, which then loses one electron and two protons to form the final product 42.
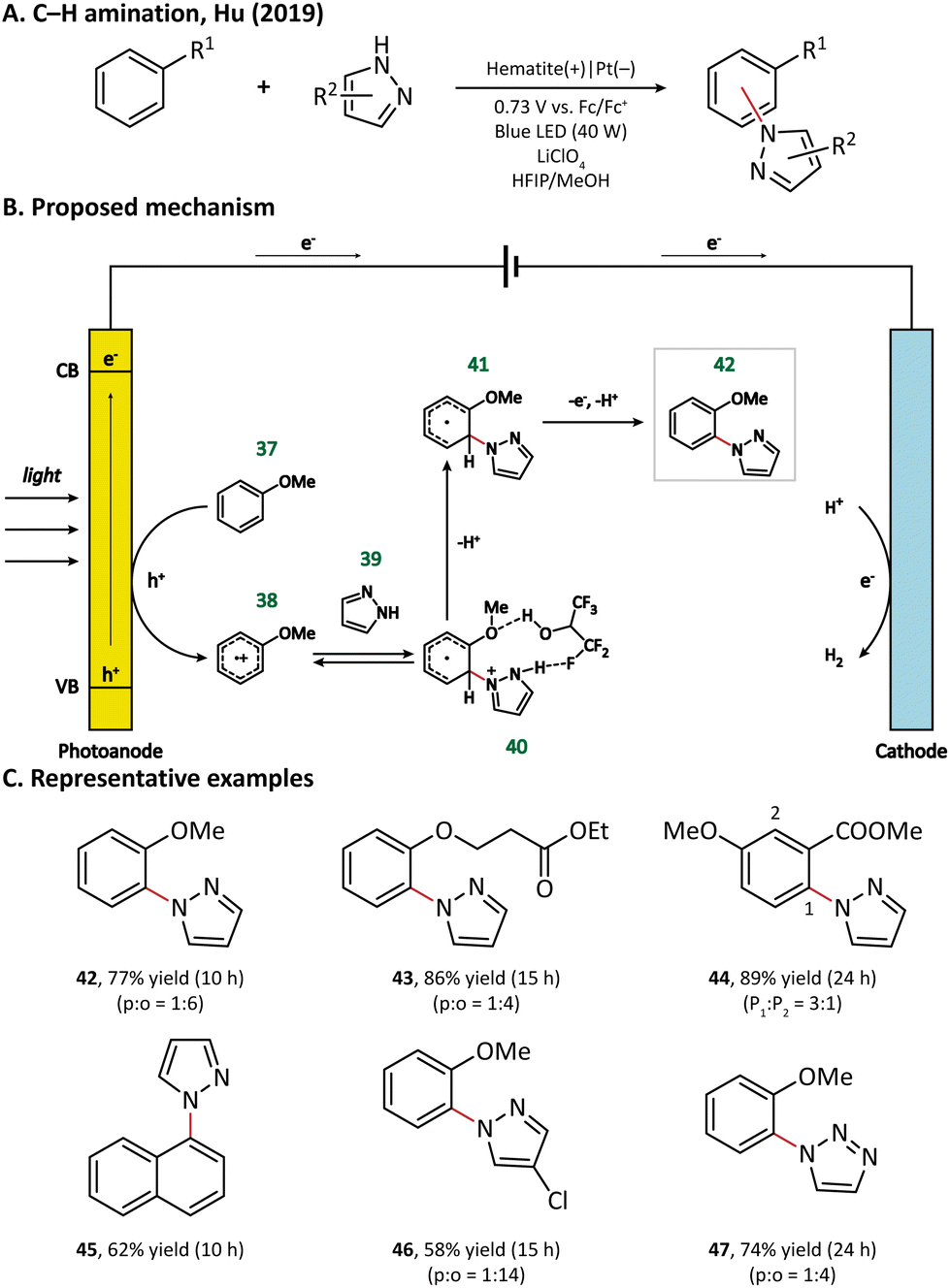 | ||
| Fig. 7 Photoanode-mediated arene amination. | ||
A plausible mechanism for photoelectrochemical P–H/C–H cross-coupling is outlined in Fig. 8. The holes reach the surface of BiVO4 to oxidize 48 into a radical cation intermediate (49). Then, the N-hydroxyphthalimide (NHPI) deprotonated by 2,6-lutidine goes through an anodic oxidation to give the corresponding PINO radicals. Next, the PINO radical extracts a hydrogen atom from 49 to regenerate NHPI, and 49 is further oxidized to afford an iminium ion intermediate (50), which reacts with diphenyl-phosphine oxide (51) via a nucleophilic attack to enable the formation of the final product with a C–P bond (52).
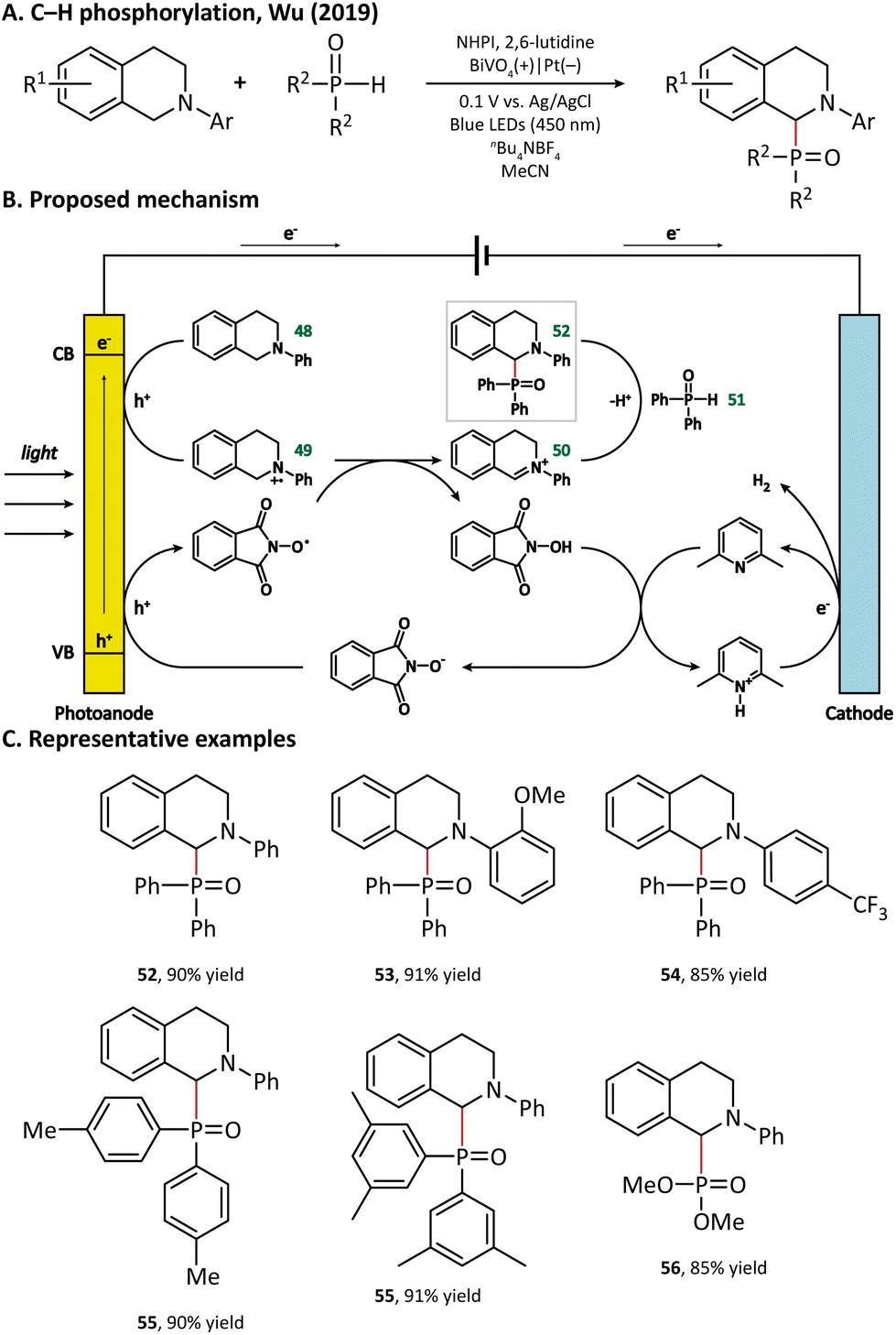 | ||
| Fig. 8 Photoanode-mediated C–H phosphorylation. | ||
The possible reaction pathways are illustrated in Fig. 9. Generally speaking, 48 loses its electron(s) at the photoanode and forms either a radical (57) or cation (58) intermediate. In the meantime, 63 undergoes a deprotonation process at the cathode to generate a malononitrile anion 65. 65 could be oxidized by the photoanode to give the malononitrile radical 66. The intermediate product 58 is generated either from a radical process (57 + 66) or from an ion process (50 + 65). After complex oxidation, deprotonation, and radical cyclization processes, 58 is finally converted into N-containing fused ring compound 67. The generation of byproduct 66 originated from the coupling of 50 and cyanide, which is produced via the oxidation of 63 or 66 by a superoxide anion reduced from the O2 molecule at the cathode.
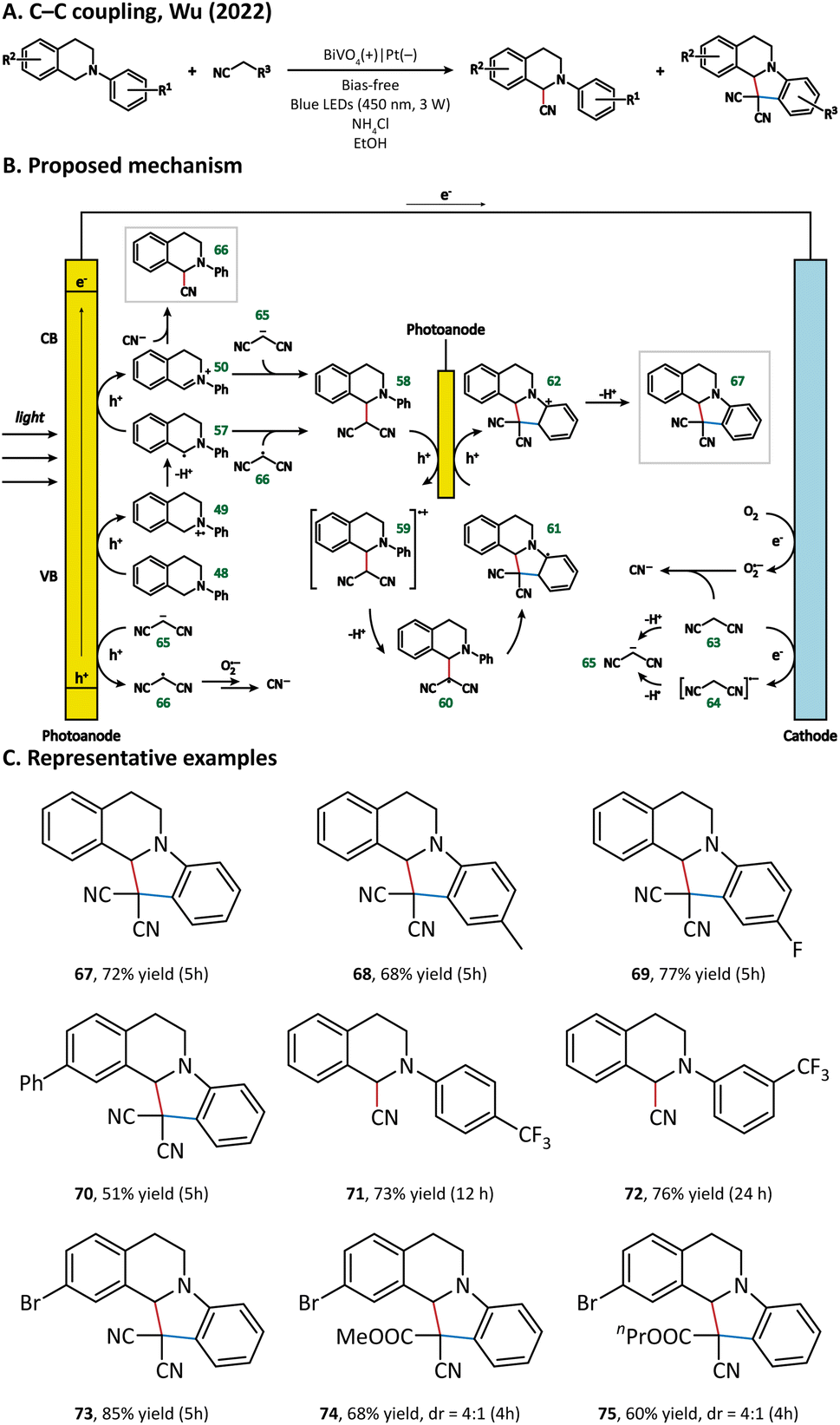 | ||
| Fig. 9 Photoanode-mediated C–C coupling. | ||
2.3 Oxidation of other organic compounds
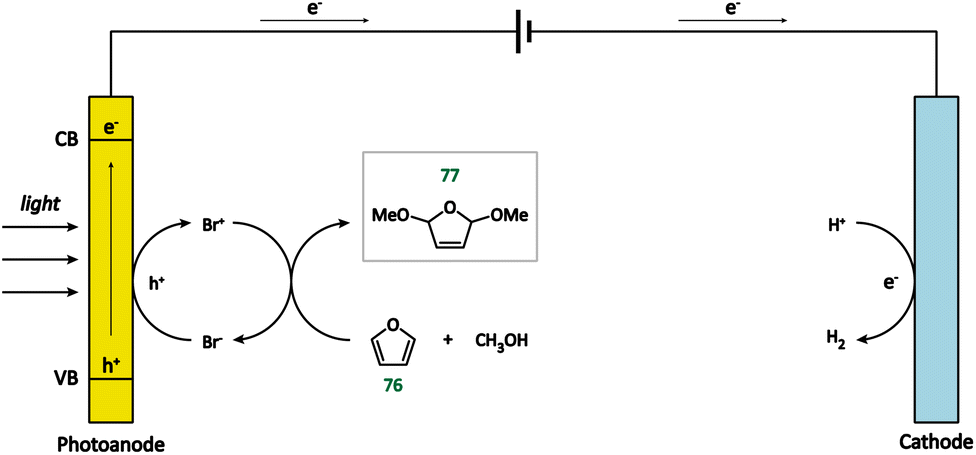 | ||
| Fig. 10 Photoanode-mediated furan oxidation. | ||
In 2021, Kuang's group improved this reaction by using a perovskite single-crystal thin film (SCTF) as the photoanode.106 A space-limited crystallization method was applied to obtain the in situ growth of a methylammonium lead bromide (MAPbBr3) SCTF on an FTO/TiO2 substrate.107,108 Later, the surface defects of the MAPbBr3 SCTF were greatly passivated by the deposition of an ultrathin Al2O3 layer, which effectively enhanced the stability of the photoanode. After depositing a Ti3+-rich titanium layer, the stability and PEC performance of the photoanode were further advanced. The maximum photocurrent density of the as-prepared MAPbBr3 SCTF/Al2O3/Ti photoanode was up to 7.8 mA cm−2 at 0.8 V vs. Ag/AgCl, which was significantly higher than that reported by Sayama's group (0.55 mA cm−2 at 0.5 V vs. SHE). Besides, this photoanode was capable of maintaining its reactivity after 6 h of continual DMDF production at an applied potential of 0.2 V vs. AgCl, indicating that it possessed eminent stability towards PECOS. DFT simulations unveiled the advantageous Br adsorption and significant charge transfer on the Ti3+-titanium layer, which explained the excellent PEC performance of the MAPbBr3 SCTF/Al2O3/Ti photoanode.
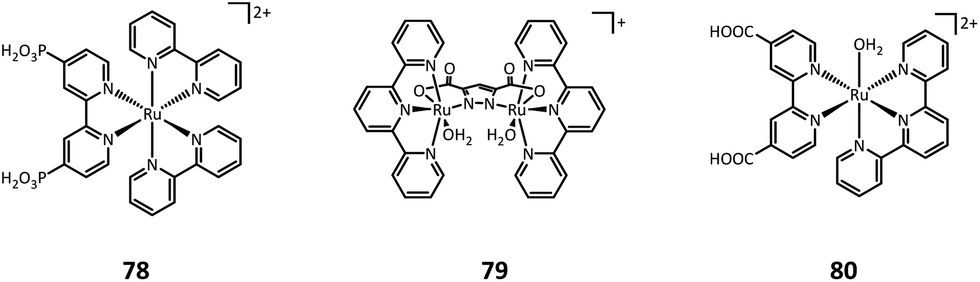 | ||
| Fig. 11 Dyes and catalysts used in photoanode-mediated sulfide oxidation. | ||
One year later, Sun's group developed a molecular ruthenium catalyst (80) modified hematite photoanode and applied it to the PEC oxidation of thioanisole in aqueous solutions (pH = 3 potassium hydrogen phthalate buffer).44 At an applied bias of 1 V vs. RHE, the photocurrent density for thioanisole oxidation reached around 300 μA cm−2. Long-term photoelectrolysis exhibited a high faradaic efficiency of 93% and benzyl methyl sulfoxide was found to be the only organic product.
The performance of the hematite photoanode was further improved by Zhao, Chen, and Zhang et al. in 2021, where MeCN rather than water was used as the solvent.110 In the preparation of the photoanode, FeOOH was first hydrothermally grown on a clean FTO substrate; then, a temperature-programmed annealing treatment transformed FeOOH into hematite with a nanowire morphology. It was found that a bare hematite photoanode is able to produce methyl phenyl sulfoxide (81) from thioanisole in an efficient manner. The author also investigated the sulfoxidation reaction of para-substituted thioanisole derivatives (82–85) and discovered a negative correlation in the Hammett plot, which suggested that the rate-limiting step of hematite-mediated sulfoxidation has a positively charged transition state.111,112 Based on the experimental results and theoretical calculations, an oxygen atom transfer (OAT) mechanism was established (Fig. 12). Upon light illumination, holes are generated and trapped on the surface of hematite to form two adjacent FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O sites. After that, the nucleophilic substrate attacks the oxygen atom of one FeIVO site. The following step is the rate-determining step, where an oxygen atom is transferred from the hematite surface to the substrate with a concerted two-hole transfer after a positively charged transition state. Finally, the oxygen vacancy left on the surface is replenished by the water molecules in the solvent and the catalytic cycle is completed. Notably, with such an OAT mechanism, the hematite photoanode can also be applied to other organic transformations such as PPh3 oxygenation and CC epoxidation, but we will not introduce them in detail in this review.
O sites. After that, the nucleophilic substrate attacks the oxygen atom of one FeIVO site. The following step is the rate-determining step, where an oxygen atom is transferred from the hematite surface to the substrate with a concerted two-hole transfer after a positively charged transition state. Finally, the oxygen vacancy left on the surface is replenished by the water molecules in the solvent and the catalytic cycle is completed. Notably, with such an OAT mechanism, the hematite photoanode can also be applied to other organic transformations such as PPh3 oxygenation and CC epoxidation, but we will not introduce them in detail in this review.
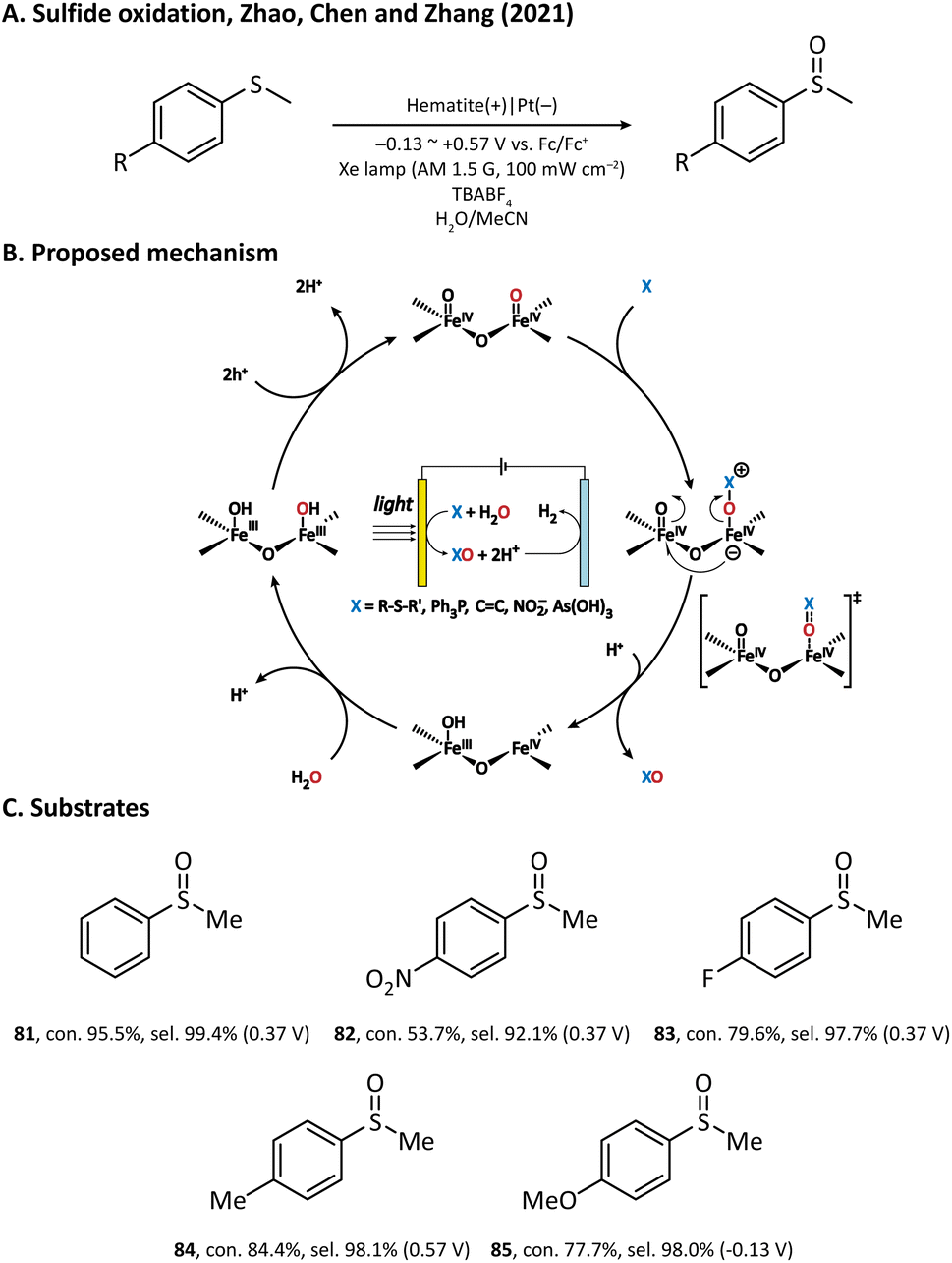 | ||
| Fig. 12 Hematite-mediated oxygen atom transfer reaction. | ||
3. (Photo)cathode-mediated organic synthesis
Compared with the widely researched photoanode-mediated organic synthesis, the potential of a PEC cathode in organic synthesis is nearly undeveloped. But there are, indeed, a few published studies trying to cultivate (photo)cathode-mediated organic synthesis. Some of them utilize the electron transported from the photoanode for in situ H2O2 generation113–115 or cofactor regeneration48,115,116 to accomplish the oxidation or reduction of organic substrates, while others adopt a photocathode as an electron source.117,1183.1 Cathode-mediated in situ H2O2 generation
H2O2 is a powerful, environmentally benign oxidizing agent because it has considerable oxidation potential (H2O2/H2O: +1.76 V vs. NHE)119,120 and does not generate toxic reduction products. It has been reported that heterogenous catalyst titanium silicalite-1 (TS-1) has the capacity of using H2O2 to selectively epoxidize propylene (86).121,122 The current method for H2O2 production, however, is the inefficient anthraquinone process which meanwhile produces organic waste.123 In 2021, Kwak, Jang, and Joo et al. presented a bias-free PEC system where in situ photoelectrocatalytic H2O2 generation was coupled with propylene epoxidation mediated by TS-1.114 In this system, the photoanode consisted of a BiVO4 film for light harvesting and cobalt phosphate (CoPi) deposition as an oxygen evolution catalyst; the cathode was made of carbon nanotubes with atomically dispersed Co–Nx sites (Co–N/CNT), which enabled the selective reduction of O2 to H2O2 (Fig. 13). When TS-1 was dispersed in the cathodic electrolyte (pH 6.0 NaPi buffer), the PEC managed to continuously produce propylene oxide (87) at a rate of 11.8 μmol h−1 with a selectivity higher than 99%. This work demonstrated that it is practical to carry out propylene epoxidation in a green and sustainable manner by coupling PEC-mediated in situ H2O2 generation with heterogeneous catalysis.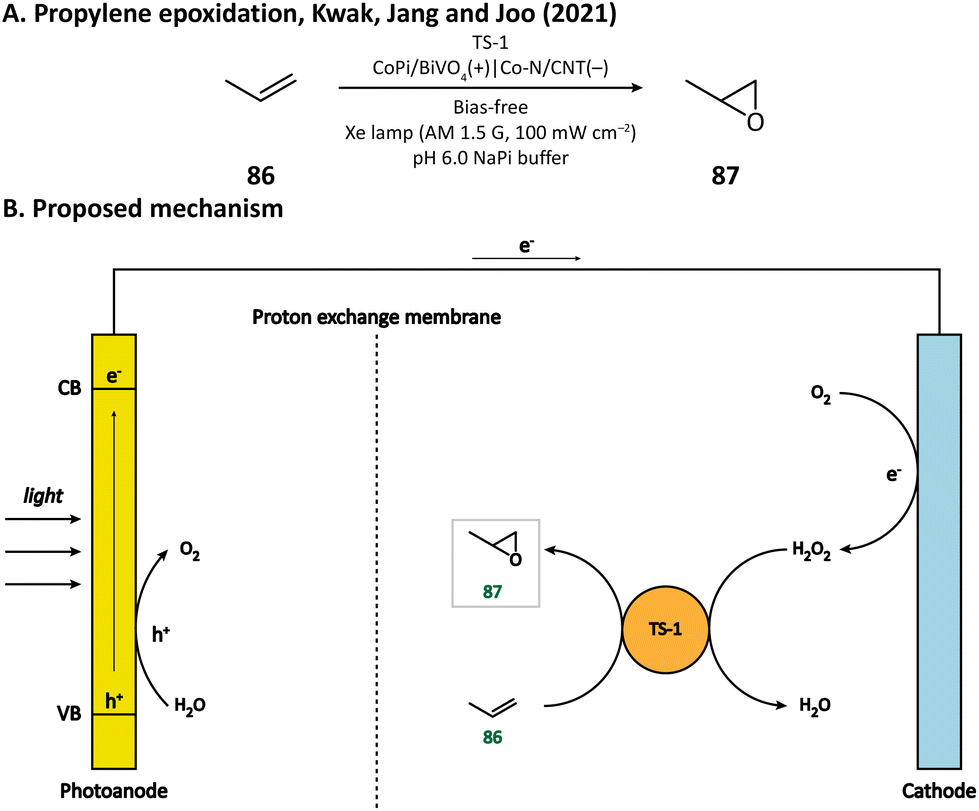 | ||
| Fig. 13 Cathode-mediated in situ H2O2 generation for propylene epoxidation. | ||
Besides heterogeneous catalysts, heme-dependent enzymes, especially unspecific peroxygenases (UPOs), also have the ability to transfer the oxygen atom in H2O2 to organic substrates.124 Therefore, UPOs should be able to cooperate well with a PEC in the oxidation of organic compounds. In fact, a recombinant peroxygenase from Agrocybe aegerita (rAaeUPO) has been used in a PEC for the C–H oxygenation of ethylbenzene (85) by Park's group in 2019.113 In their PEC system, the cathode was a graphitic carbon nitride (CN)/reduced graphene oxide (rGO) hybrid film. The cathode-mediated in situ H2O2 generation was powered by a FeOOH/BiVO4–Cu(In, Ga)Se2 photoanode-photovoltaic tandem structure which provided sufficient photovoltage. By carefully tuning the loading amount of rGO, the production rate of H2O2 was optimized, which maximized the conversion rate of ethylbenzene and maintains the activity of rAaeUPO. Upon illumination, (R)-1-phenylethanol (89) was produced at a rate of 0.89 mM h−1 with an enantiomeric excess (ee) value higher than 99% in a pH 7.0 KPi buffer, indicating the successful cooperation between the PEC and the enzyme (Fig. 14).
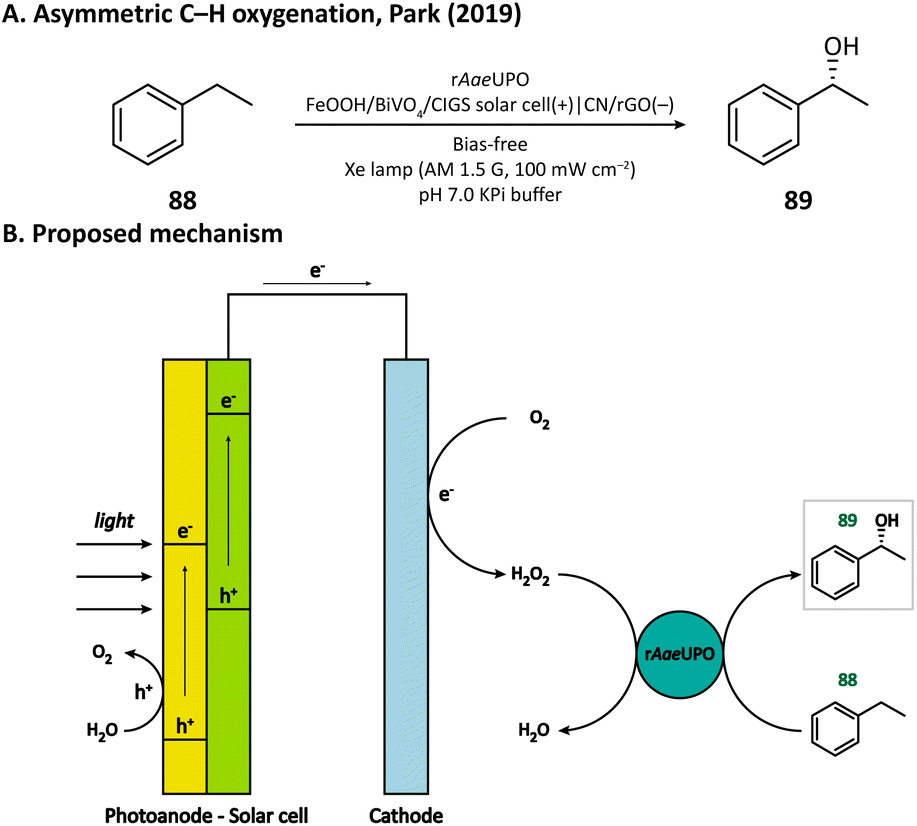 | ||
| Fig. 14 Cathode-mediated in situ H2O2 generation for asymmetric C–H oxygenation. | ||
Recently, Park's group provided another example by using anthraquinone-2-carboxylic acid (AQC) anchored carbon fiber paper (CFP) as the cathode.115 They adopted Zr-doped hematite as the photoanode, which had the capability of converting poly(ethylene terephthalate) (PET) microplastics into chemical fuels in an alkaline medium (5 M NaOH). The electron extracted from PET is transferred to the cathode and reduces the anchored AQC into AQCH2, which then reacts with O2 and realizes the in situ H2O2 generation in a pH 6.0 KPi buffer (Fig. 16). With the help of rAaeUPO, the oxygen atom in H2O2 could be transferred into a variety of organic substrates such as ethylbenzene, cyclohexane, tetralin, and cis-β-methylstyrene (92).
3.2 Cathode-mediated cofactor regeneration
Having extraordinary catalytic activity and selectivity, redox enzymes are ideal catalysts for redox reactions in organic synthesis.125,126 Nevertheless, many redox enzymes can only reduce organic substrates in the presence of expensive cofactors, which limits their applications in industrial production.127,128 Regenerating cofactors from their oxidized forms during the reaction can not only sustain the reduction of organic substrates but also minimize the amount of cofactor addition.In 2018, Park's group employed this strategy for the production of L-glutamate (91) in a PEC system.116 A tandem structure of light-absorbing layers (FeOOH/BiVO4 photoanode and perovskite solar cell) was constructed to generate the bias for the cathode reaction. On the surface of the carbon nanotube (CNT) film cathode, the electron is transferred to an Rh-based electron mediator M ([Cp*Rh(bpy)H2O]2+, Cp* = C5Me5, bpy = 2,2′-bipyridine), which then realizes the regeneration of nicotinamide adenine dinucleotide (NADH) by reducing NAD+. The rapid rate of NADH regeneration enabled glutamate dehydrogenase (GDH) to efficiently convert α-ketoglutarate (90) into L-glutamate at an initial rate of 2.4 mM h−1 in a pH 7.5 phosphate buffer, which was comparable to the production rate obtained in the enzymatic NADH recycling system (Fig. 15).
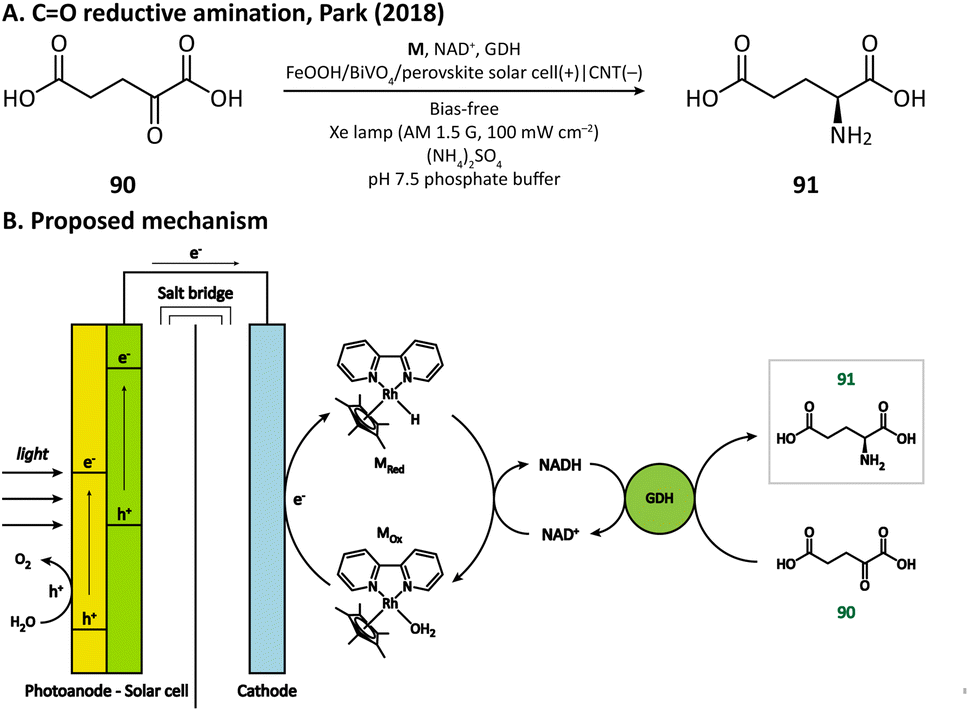 | ||
| Fig. 15 Cathode mediated cofactor regeneration for CO reductive amination. | ||
Cathode-mediated cofactor regeneration has also been coupled with the aforementioned PET conversion by Park's group.115M was also used as the mediator for the electron transfer between the bare CFP cathode and NAD+. The regenerated NADH can participate in either the reductive amination of CO bonds catalyzed by GDH or the hydrogenation of CC bonds driven by the old yellow enzyme (OYE) (Fig. 16).
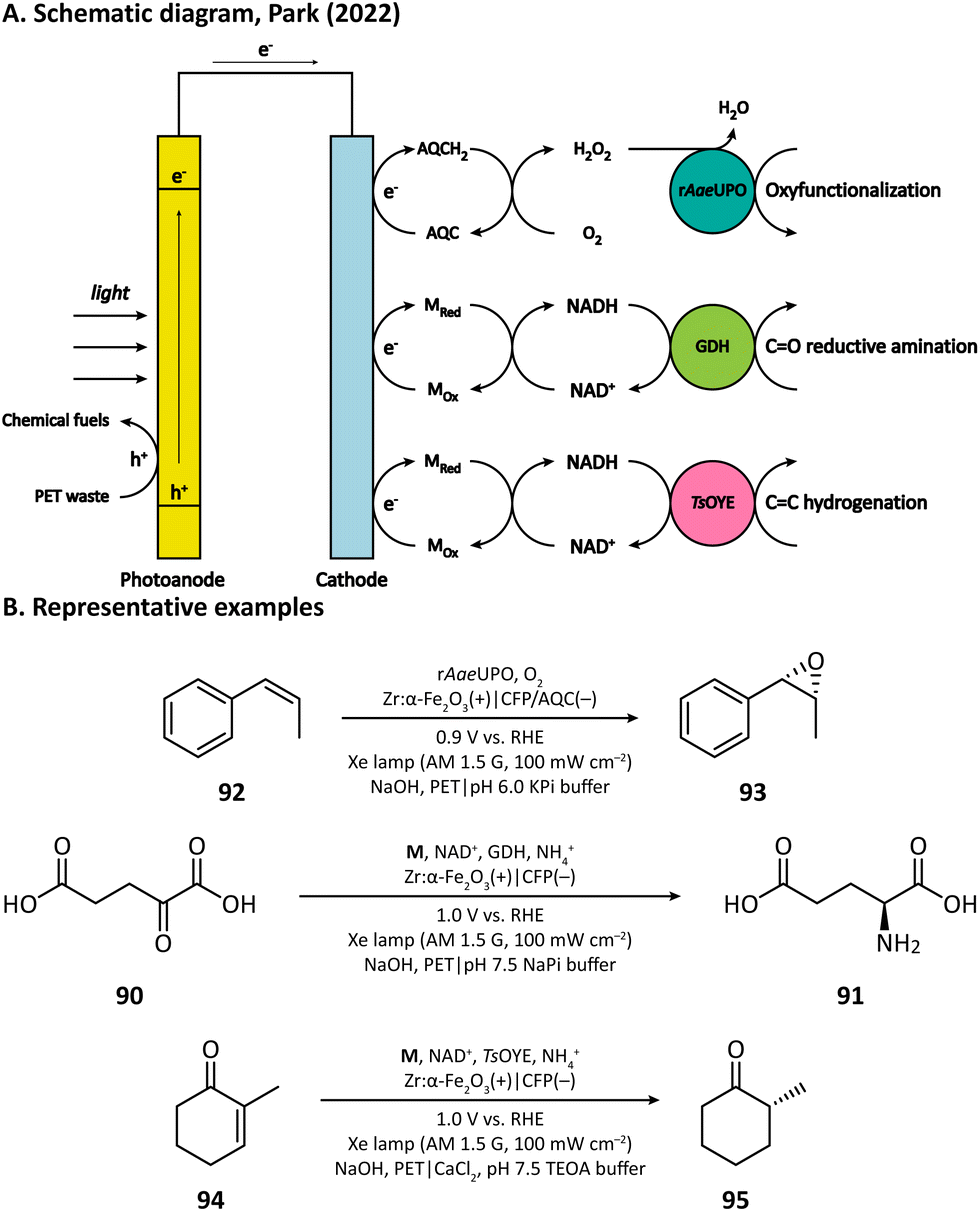 | ||
| Fig. 16 PET conversion coupled with cathode-mediated biosynthesis. | ||
3.3 Photocathode-mediated organic synthesis
The first photocathode-mediated PECOS was reported by Park's group in 2017.117 In this work, the photocathode was prepared by anchoring flavin molecules on the surface of a single-walled carbon nanotube (SWNT) cathode. Under illumination, photosensitive flavin molecules are excited and then converted to their reduced form by receiving electrons from an SWNT cathode. Reduced flavins facilitate the charge transfer between SWNTs and O2 thereby decreasing the overpotential of O2 reduction. When luminchrome (LC), a kind of flavin derivative, was immobilized on the surface of SWNTs, O2 was reduced to H2O2 at −0.32 V vs. Ag/AgCl with a photocurrent density of −1.42 mA cm−2, which was better than the performance of pristine SWNTs (−0.49 V vs. Ag/AgCl and −0.87 mA cm−2). By coupling photocathode-mediated H2O2 generation with proper peroxygenase-catalyzed reactions, the oxidation of alkane (88), arene (96), and indole (98) in phosphate buffer was respectively achieved (Fig. 17).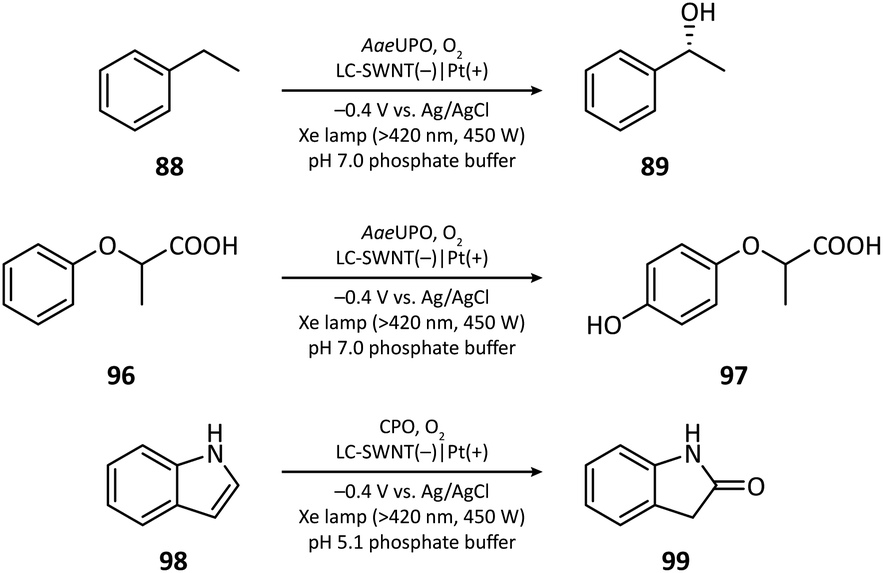 | ||
| Fig. 17 Photocathode-mediated C–H oxygenation. | ||
In 2021, Wu's group developed an Sb2(S, Se)3 photocathode for the selective functionalization of aryl halide.118 The Sb2(S, Se)3 photocathode had an ultra-narrow band gap (1.20 eV) and a rod-like structure with high surface area, which were beneficial for the light absorption performance in the visible and near-infrared region and for the electron and mass transportation ability, respectively. An organic photocatalyst N,N-bis(2,6-ddiisopropylphenyl)perylene-3,4,9,10-bis(dicarboximide) (PDI) was introduced in the electrolyte to promote the electron transfer between the photocathode and aryl halides. It was found that light and PDI played crucial roles in the catalytic activity, and nBu4NAcO-containing DMSO was the best reaction medium. Under optimized reaction conditions, 4-bromoacetophenone could be activated and coupled with N-methyl pyrrole with an excellent yield of 89% (Fig. 18, 104), which was much higher than that in photocatalytic and electrophotocatalytic C–X activation mediated by PDI, demonstrating the superiority of this coupled PEC-photocatalysis (c-PEC/PC) system. The substrate scope was also explored. It turned out that such c-PEC/PC was tolerant to a broad scope of aryl halides and trapping agents (105–111).
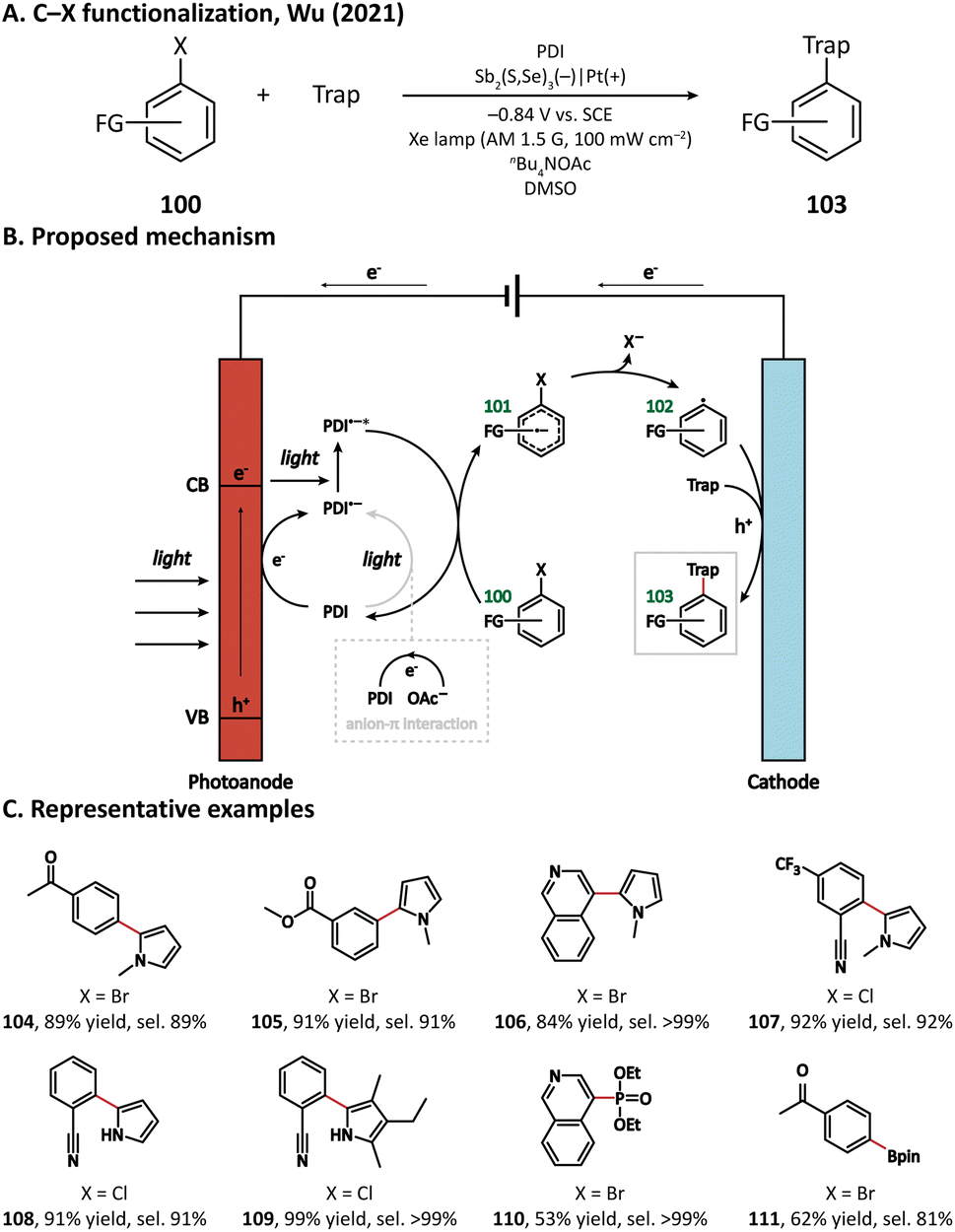 | ||
| Fig. 18 Photocathode-mediated C–X activation. | ||
A plausible mechanism for this photocathode-mediated C–X functionalization is displayed in Fig. 18. Upon light illumination, electrons in Sb2(S, Se)3 are excited from the valence band to the conduction band. The anion-π interaction between the electrolyte anion OAc− and photocatalyst PDI makes PDI easier to be oxidized. PDI can either be reduced by the photogenerated electron at the surface of the photocathode or accept an electron from OAc− to form PDI˙–, which can be further excited by a photon into the more reductive PDI˙–*. PDI˙–* then transfers one electron to the aryl halide (100), leading to the formation of a radical anion intermediate (101). After that, the C–X bond breaks heterolytically, forming an aryl radical (102). The aryl radical subsequently reacts with the trapping reagent and loses one electron at the anode to yield the coupling product (103).
4. Conclusions and perspectives
In this review, we summarize recent advances in photoelectrochemical alcohol oxidation, C–H functionalization, and (photo) cathode-mediated organic synthesis. Compared with electrochemical systems, PEC systems are still in their infancy but offer prospects for the evolution of solar-induced organic transformations towards better, cheaper, higher efficiency, and sustainable alternative reaction methodologies. Following on from here are four major aspects that will enable photoelectrochemical organic transformations to become a broad subject of interest in the future:(1) In conventional photoelectrochemical setups, typically, electrode distances range from millimeters to centimeters. The PEC reaction efficiency could be affected by the inefficient mixing of the persistent radical and transient radical. Therefore, how to enhance substrate diffusion will be the key problem of PECOS. (I) The extremely thin interelectrode gap would prevent active intermediate decomposition, selectively producing the target product. Considering this aspect, new PEC devices should be developed to minimize electrode spacing and improve reaction efficiency. (II) In heterogeneous catalysis, the porous nanocatalyst formed on a photoanode can increase the exposure of active sites. The active intermediates produced on the catalyst surface can accelerate the diffusion of thesubstrate during photoelectrolysis.
(2) Heterogeneous catalysis has attracted more attention from researchers due to the tunability of the catalyst structure, morphology and size. From the perspective of economic resources, heterogeneous catalysis avoids the separation process, reduces the cost of catalysts, and realizes recyclable applications. In the future, various catalysts can be modified on the surface of the photoelectrode to generate high-value-added chemicals. For example, high surface area nanopores can be deposited on the photoanode by atomic layer deposition, chemical synthesis, and chemical vapor deposition, which can play a vital role in various organic transformations with enhanced efficiency.
(3) Transition metal catalysis plays an important role in biochemistry and medicinal chemistry, but its large-scale production is limited by harsh reaction conditions. At present, great progress has been made in electrochemical transition metal catalysis, but not in photoelectrochemical transition metal catalysis. Transition metal catalyst deposition on photoanodes can potentially add more value to this process as cocatalysts can offer additional oxidation or reduction active sites. In the future, photoelectrochemical metal catalysis is a new technology for organic synthesis.
(4) Asymmetric catalysis has always been the most challenging research area and the focus of organic chemists. At present, many factors are hindering the development of electrochemical asymmetric catalysis. For example, chiral catalysts are easy to inactivate during electrolysis. The bottleneck of this field is to develop appropriate catalysts that can withstand electrochemical oxidation and provide stereoselective control. In contrast, photoelectrochemical asymmetric catalysis has unique advantages due to its lower bias voltage, which can effectively prevent peroxidation, ligand deactivation, and other problems. In the future, the development of photoelectrochemical asymmetric catalysis will add new vitality to organic synthesis methodology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Key R&G Program of China (2021YFA1200302, Z. Y. T.), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB36000000, Z. Y. T.), and National Natural Science Foundation of China (92056204, 21890381 and 21721002, Z. Y. T.).Notes and references
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- H. Mai, D. Chen, Y. Tachibana, H. Suzuki, R. Abe and R. A. Caruso, Chem. Soc. Rev., 2021, 50, 13692–13729 RSC.
- Z. Bi, R. Guo, X. Hu, J. Wang, X. Chen and W. Pan, Nanoscale, 2022, 14, 3367–3386 RSC.
- X. Yang and D. Wang, ACS Appl. Energy Mater., 2018, 1, 6657–6693 CrossRef CAS.
- J. Li, X. Han, D. Wang, L. Zhu, M. Ha-Thi, T. Pino, J. Arbiol, L. Wu and M. N. Ghazzal, Angew. Chem., Int. Ed., 2022, 61, e202210242 CAS.
- J. Li, X. Gao, L. Zhu, M. N. Ghazzal, J. Zhang, C. Tung and L. Wu, Energy Environ. Sci., 2020, 13, 1326–1346 RSC.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- J. Li and N. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
- P. Chen, Y. Zhang, Y. Zhou and F. Dong, Nano Mater. Sci., 2021, 3, 344–367 CrossRef CAS.
- G. Hilt, ChemElectroChem, 2020, 7, 395–405 CrossRef CAS.
- A. Landman, H. Dotan, G. E. Shter, M. Wullenkord, A. Houaijia, A. Maljusch, G. S. Grader and A. Rothschild, Nat. Mater., 2017, 16, 646–651 CrossRef CAS PubMed.
- C. Ros, T. Andreu and J. R. Morante, J. Mater. Chem. A, 2020, 8, 10625–10669 RSC.
- J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed.
- Y. Chen, W. Deng, J. Guo, R. Ci, C. Zhou, B. Chen, X. Li, X. Guo, R. Liao, C. Tung and L. Wu, J. Am. Chem. Soc., 2022, 144, 20975 CrossRef CAS PubMed.
- H. Huang, K. A. Steiniger and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- S. Wang, G. Liu and L. Wang, Chem. Rev., 2019, 119, 5192–5247 CrossRef CAS PubMed.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- M. A. Lumley, A. Radmilovic, Y. J. Jang, A. E. Lindberg and K. S. Choi, J. Am. Chem. Soc., 2019, 141, 18358–18369 CrossRef CAS PubMed.
- V. Kumaravel, J. Bartlett and S. C. Pillai, ACS Energy Lett., 2020, 5, 486–519 CrossRef CAS.
- Y. Liu, S. Bai, F. Wang and Y. Chen, Environ. Chem. Lett., 2022, 20, 1169–1192 CrossRef CAS.
- S. Xu and E. A. Carter, Chem. Rev., 2019, 119, 6631–6669 CrossRef CAS PubMed.
- C. Li, T. Wang, B. Liu, M. Chen, A. Li, G. Zhang, M. Du, H. Wang, S. F. Liu and J. Gong, Energy Environ. Sci., 2019, 12, 923–928 RSC.
- R. Kamata, H. Kumagai, Y. Yamazaki, M. Higashi, R. Abe and O. Ishitani, J. Mater. Chem. A, 2021, 9, 1517–1529 RSC.
- S. Navarro, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem., 2021, 5, 564–579 CrossRef.
- W. J. Dong, I. A. Navid, Y. Xiao, T. H. Lee, J. W. Lim, D. Lee, H. W. Jang, J. L. Lee and Z. Mi, J. Mater. Chem. A, 2022, 10, 7869–7877 RSC.
- I. Roh, S. Yu, C. Lin, S. Louisia, S. Cestellos and P. Yang, J. Am. Chem. Soc., 2022, 144, 8002–8006 CrossRef CAS PubMed.
- X. Lu, S. Xie, H. Yang, Y. Tong and H. Ji, Chem. Soc. Rev., 2014, 43, 7581–7593 RSC.
- Z. Zhang and H. Wu, RSC Adv., 2014, 4, 37395–37399 RSC.
- H. G. Cha and K. S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- J. J. Roylance, T. W. Kim and K. S. Choi, ACS Catal., 2016, 6, 1840–1847 CrossRef CAS.
- C. R. Lhermitte and K. Sivula, ACS Catal., 2019, 9, 2007–2017 CrossRef CAS.
- S. Li, Z. Li, H. Yu, M. R. Sytu, Y. Wang, D. Beeri, W. Zheng, B. D. Sherman, C. G. Yoo and G. Leem, ACS Energy Lett., 2020, 5, 777–784 CrossRef CAS.
- C. R. Lhermitte, N. Plainpan, P. Canjura, F. Boudoirea and K. Sivula, RSC Adv., 2021, 11, 198–202 RSC.
- Y. Wu, R. Song and J. Li, Org. Chem. Front., 2020, 7, 1895–1902 RSC.
- M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730–9733 CrossRef CAS PubMed.
- H. Lu, J. Zhao, L. Li, L. Gong, J. Zheng, L. Zhang, Z. Wang, J. Zhang and Z. Zhu, Energy Environ. Sci., 2011, 4, 3384–3388 RSC.
- S. Meng, X. Ye, J. Zhang, X. Fu and S. Chen, J. Catal., 2018, 367, 159–170 CrossRef CAS.
- J. A. Treadway, J. A. Moss and T. J. Meyer, Inorg. Chem., 1999, 38, 4386–4387 CrossRef CAS PubMed.
- W. Song, A. K. Vannucci, B. H. Farnum, A. M. Lapides, M. K. Brennaman, B. Kalanyan, L. Alibabaei, J. J. Concepcion, M. D. Losego, G. N. Parsons and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 9773–9779 CrossRef CAS PubMed.
- T. V. Pho, M. V. Sheridan, Z. A. Morseth, B. D. Sherman, T. J. Meyer, J. M. Papanikolas, K. S. Schanze and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2016, 8, 9125–9133 CrossRef CAS PubMed.
- L. Bai, F. Li, Y. Wang, H. Li, X. Jiang and L. Sun, Chem. Commun., 2016, 52, 9711–9714 RSC.
- J. Jiang, B. D. Sherman, Y. Zhao, R. He, I. Ghiviriga, L. Alibabaei, T. J. Meyer, G. Leem and K. S. Schanze, ACS Appl. Mater. Interfaces, 2017, 9, 19529–19534 CrossRef CAS PubMed.
- D. Badgurjar, B. Shan, A. Nayak, L. Wu, R. Chitta and T. J. Meyer, ACS Appl. Mater. Interfaces, 2020, 12, 7768–7776 CrossRef CAS PubMed.
- E. Nikoloudakis, P. B. Pati, G. Charalambidis, D. S. Budkina, S. Diring, A. Planchat, D. Jacquemin, E. Vauthey, A. G. Coutsolelos and F. Odobel, ACS Catal., 2021, 11, 12075–12086 CrossRef CAS.
- D. Antón, E. E. Moore, M. A. Bajada, A. Eisenschmidt, A. R. Oliveira, I. A. C. Pereira, J. Warnan and E. Reisner, Nat. Synth., 2022, 1, 77–86 CrossRef.
- J. J. Concepcion, J. W. Jurss, M. R. Norris, Z. Chen, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2010, 49, 1277–1279 CrossRef CAS PubMed.
- S. Yun, N. Vlachopoulos, A. Qurashi, S. Ahmad and A. Hagfeldt, Chem. Soc. Rev., 2019, 48, 3705–3722 RSC.
- B. Bozic, E. C. Constable and C. E. Housecroft, Coord. Chem. Rev., 2013, 257, 3089–3106 CrossRef.
- P. Subramanyam, M. Deepa, S. S. K. Raavi, H. Misawa, V. Biju and C. Subrahmanyam, Nanoscale Adv., 2020, 2, 5591–5599 RSC.
- H. Tateno, Y. Miseki and K. Sayama, ChemElectroChem, 2017, 4, 3283–3287 CrossRef CAS.
- R. Zhang, M. Shao, Z. Li, F. Ning, M. Wei, D. G. Evans and X. Duan, Chem. Eur. J., 2017, 23, 8142–8147 CrossRef CAS PubMed.
- L. Luo, Z. Wang, X. Xiang, D. Yan and J. Ye, ACS Catal., 2020, 10, 4906–4913 CrossRef CAS.
- L. Luo, T. Zhang, M. Wang, R. Yun and X. Xiang, ChemSusChem, 2020, 13, 5173–5184 CrossRef CAS PubMed.
- A. W. Harris, O. Yehezkeli, G. R. Hafenstine, A. P. Goodwin and J. N. Cha, ACS Sustainable Chem. Eng., 2017, 5, 8199–8204 CrossRef CAS PubMed.
- D. Liu, J. Liu, W. Cai, J. Ma, H. B. Yang, H. Xiao, J. Li, Y. Xiong, Y. Huang and B. Liu, Nat. Commun., 2019, 1779 CrossRef PubMed.
- L. Huang, T. Vo and C. Chiang, Electrochim. Acta, 2019, 322, 134725 CrossRef CAS.
- T. Vo, C. Kao, J. Kuo, C. Chiu and C. Chiang, Appl. Catal., B, 2020, 278, 119303 CrossRef CAS.
- Y. Wu, D. A. Kuznetsov, N. C. Pflug and C. R. Müller, J. Mater. Chem. A, 2021, 9, 6252–6260 RSC.
- L. Luo, W. Chen, S. Xu, J. Yang, M. Li, H. Zhou, M. Xu, M. Shao, X. Kong, Z. Li and H. Duan, J. Am. Chem. Soc., 2022, 144, 7720–7730 CrossRef CAS PubMed.
- Y. Liu, M. Wang, B. Zhang, D. Yan and X. Xiang, ACS Catal., 2022, 12, 6946–6957 CrossRef CAS.
- H. Tateno, S. Chen, Y. Miseki, T. Nakajima, T. Mochizuki and K. Sayama, ACS Sustainable Chem. Eng., 2022, 10, 7586–7594 CrossRef CAS.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachalb and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- W. R. Gutekunsta and P. S. Bara, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- M. Zhao and W. Lu, Org. Lett., 2017, 19, 4560–4563 CrossRef CAS PubMed.
- D. G. Yu, T. Gensch, F. de Azambuja, S. Vasquez and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722–17725 CrossRef CAS PubMed.
- X. Yang, Q. Yang, X. Wang, H. Xu, T. Mei, Y. Huang and P. Fang, J. Org. Chem., 2020, 85, 3497–3507 CrossRef CAS PubMed.
- A. John and K. M. Nicholas, Organometallics, 2012, 31, 7914–7920 CrossRef CAS.
- Z. Li, L. Luo, M. Li, W. Chen, Y. Liu, J. Yang, S. Xu, H. Zhou, L. Ma, M. Xu, X. Kong and H. Duan, Nat. Commun., 2021, 12, 6698 CrossRef CAS PubMed.
- K. Ohkubo and K. Hirose, Angew. Chem., Int. Ed., 2018, 57, 2126–2129 CrossRef CAS PubMed.
- Y. Wang, Q. Yang, P. J. Walsh and E. J. Schelter, Org. Chem. Front., 2022, 9, 2612–2620 RSC.
- T. Li, T. Kasahara, J. He, K. E. Dettelbach, G. M. Sammis and C. P. Berlinguette, Nat. Commun., 2017, 8, 390 CrossRef PubMed.
- H. Tateno, S. Iguchi, Y. Miseki and K. Sayama, Angew. Chem., Int. Ed., 2018, 57, 11238–11241 CrossRef CAS PubMed.
- M. Gong, J. K. Kim, X. Zhao, Y. Li, M. Huang and Y. Wu, Green Chem., 2019, 21, 3615–3620 RSC.
- J. Ma, K. Mao, J. Low, Z. Wang, D. Xi, W. Zhang, H. Ju, Z. Qi, R. Long, X. Wu, L. Song and Y. Xiong, Angew. Chem., Int. Ed., 2021, 60, 9357–9361 CrossRef CAS PubMed.
- Y. Jiang, Y. Fan, S. Li and Z. Tang, CCS Chem., 2022, 1–15 Search PubMed.
- R. Hili and A. Yudin, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef CAS PubMed.
- C. Lamberth, S. Jeanmart and T. Luksch, Science, 2013, 341, 742–746 CrossRef PubMed.
- I. Sorribes, K. Junge and M. Beller, J. Am. Chem. Soc., 2014, 136, 14314–14319 CrossRef CAS PubMed.
- F. Collet, C. Lescota and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926–1936 RSC.
- D. Hazelard, P. Nocquet and P. Compain, Org. Chem. Front., 2017, 4, 2500–2521 RSC.
- H. J. Kim, J. Kim, S. H. Cho and S. Chang, J. Am. Chem. Soc., 2011, 133, 16382–16385 CrossRef CAS PubMed.
- N. Sauermann, R. Mei and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5090 CrossRef CAS PubMed.
- T. Morofuji, A. Shimizu and J. Yoshida, J. Am. Chem. Soc., 2013, 135, 5000–5003 CrossRef CAS PubMed.
- T. Morofuji, A. Shimizu and J. Yoshida, Chem. Eur. J., 2015, 21, 3211–3214 CrossRef CAS PubMed.
- C. Song, H. Yi, B. Dou, Y. Li, A. K. Singha and A. Lei, Chem. Commun., 2017, 53, 3689–3692 RSC.
- S. Das, P. Natarajan and B. König, Chem. Eur. J., 2017, 23, 18161 CrossRef CAS PubMed.
- L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS PubMed.
- H. Fu, P. Tan, R. W. S. Li, H. Liu, Y. Yang and Z. Wu, J. Hazard. Mater., 2022, 424, 127494 CrossRef CAS PubMed.
- D. Joly, P. A. Bouit and M. Hissler, J. Mater. Chem. C, 2016, 4, 3686–3698 RSC.
- J. Wang, X. Li, J. Li, T. Lei, H. Wu, X. Nan, C. Tung and L. Wu, Chem. Commun., 2019, 55, 10376–10379 RSC.
- Y. Liu, C. Wang, D. Xue, M. Xiao, C. Li and J. Xiao, Chem. Eur. J., 2017, 23, 3051 CrossRef CAS PubMed.
- B. Lin, S. Shi, R. Lin, Y. Cui, M. Fang, G. Tang and Y. Zhao, J. Org. Chem., 2018, 83, 6754–6761 CrossRef CAS PubMed.
- M. Ghobrial, K. Harhammer, M. D. Mihovilovica and M. Schnürch, Chem. Commun., 2010, 46, 8836–8838 RSC.
- T. Nobuta, N. Tada, A. Fujiya, A. Kariya, T. Miura and A. Itoh, Org. Lett., 2013, 15, 574–577 CrossRef CAS PubMed.
- Q. Li, X. Zhao, Y. Li, M. Huang, J. K. Kim and Y. Wu, Org. Biomol. Chem., 2017, 15, 9775–9778 RSC.
- X. Su, F. Yang, Y. Wu and Y. Wu, Org. Biomol. Chem., 2018, 16, 2753–2756 RSC.
- D. S. Brandes and J. A. Ellman, Chem. Soc. Rev., 2022, 51, 6738–6756 RSC.
- D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705–1718 RSC.
- M. Gong, M. Huang, Y. Li, J. Zhang, J. K. Kim, J. S. Kim and Y. Wu, Green Chem., 2022, 24, 837–845 RSC.
- H. Tateno, Y. Miseki and K. Sayama, Chem. Commun., 2017, 53, 4378–4381 RSC.
- X. Wang, Y. Huang, J. Liao, Z. Wei, W. Li, Y. Xu, H. Chen and D. Kuang, Nat. Commun., 2021, 12, 1202 CrossRef CAS PubMed.
- H. Rao, W. Li, B. Chen, D. Kuang and C. Su, Adv. Mater., 2017, 29, 1602639 CrossRef PubMed.
- H. Rao, B. Chen, X. Wang, D. Kuang and C. Su, Chem. Commun., 2017, 53, 5163–5166 RSC.
- P. Farràs, C. D. Giovanni, J. N. Clifford, E. Palomares and A. Llobet, Coord. Chem. Rev., 2015, 304–305, 202–208 CrossRef.
- Y. Zhao, C. Deng, D. Tang, L. Ding, Y. Zhang, H. Sheng, H. Ji, W. Song, W. Ma, C. Chen and J. Zhao, Nat. Catal., 2021, 4, 684–691 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS PubMed.
- D. S. Choi, H. Lee, F. Tieves, Y. W. Lee, E. J. Son, W. Zhang, B. Shin, F. Hollmann and C. B. Park, ACS Catal., 2019, 9, 10562–10566 CrossRef CAS.
- M. Ko, Y. Kim, J. Woo, B. Lee, R. Mehrotra, P. Sharma, J. Kim, S. W. Hwang, H. Y. Jeong, H. Lim, S. H. Joo, J. W. Jang and J. H. Kwak, Nat. Catal., 2022, 5, 37–44 CrossRef CAS.
- J. Kim, J. Jang, T. Hilberath, F. Hollmann and C. B. Park, Nat. Synth., 2022, 1, 776–786 CrossRef.
- Y. W. Lee, P. Boonmongkolras, E. J. Son, J. Kim, S. H. Lee, S. K. Kuk, J. W. Ko, B. Shin and C. B. Park, Nat. Commun., 2018, 9, 4208 CrossRef PubMed.
- D. S. Choi, Y. Ni, E. Fernandez, M. Lee, F. Hollmann and C. B. Park, ACS Catal., 2017, 7, 1563–1567 CrossRef CAS.
- Y. Chen, T. Lei, H. Hu, H. Wu, S. Zhou, X. Li, B. Chen, C. Tung and L. Wu, Matter, 2021, 4, 2354–2366 CrossRef CAS.
- C. Ponce de León, Nat. Catal., 2020, 3, 96–97 CrossRef.
- X. Shi, S. Back, T. M. Gill, S. Siahrostami and X. Zheng, Chem, 2021, 7, 38–63 CAS.
- I. Schmidt, A. Krogh, K. Wienberg, A. Carlsson, M. Brorson and C. J. H. Jacobsen, Chem. Commun., 2000, 21, 2157–2158 RSC.
- Z. Song, X. Feng, N. Sheng, D. Lin, Y. Li, Y. Liu, X. Chen, X. Zhou, D. Chen and C. Yang, Catal. Today, 2020, 347, 102–109 CrossRef CAS.
- J. M. Campos, G. Blanco and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef PubMed.
- Y. Wang, D. Lan, R. Durrani and F. Hollmann, Curr. Opin. Chem. Biol., 2017, 37, 1–9 CrossRef PubMed.
- D. Monti, G. Ottolina, G. Carrea and S. Riva, Chem. Rev., 2011, 111, 4111–4140 CrossRef CAS PubMed.
- Y. Liang, J. Wei, X. Qiu and N. Jiao, Chem. Rev., 2018, 118, 4912–4945 CrossRef CAS PubMed.
- W. B. Black, L. Zhang, W. S. Mak, S. Maxel, Y. Cui, E. King, B. Fong, A. S. Martinez, J. B. Siegel and H. Li, Nat. Chem. Biol., 2020, 16, 87–94 CrossRef CAS PubMed.
- I. Zachos, S. Güner, A. Essert, P. Lommes and V. Sieber, Chem. Commun., 2022, 58, 11945–11948 RSC.
| This journal is © The Royal Society of Chemistry 2023 |