
Semiconductor quantum dots: a versatile platform for photoredox organic transformation
Hui-Li
Wu
,
Ming-Yu
Qi
,
Zi-Rong
Tang
 * and
Yi-Jun
Xu
*
* and
Yi-Jun
Xu
*
College of Chemistry, State Key Laboratory of Photocatalysis on Energy and Environment, Fuzhou University, Fuzhou 350116, China. E-mail: zrtang@fzu.edu.cn; yjxu@fzu.edu.cn
First published on 16th January 2023
Abstract
Semiconductor quantum dots (QDs), as a newfashioned light-absorbing material with great promise in artificial photosystems, generally exhibit attractive photoactivity and selectivity in organic photoredox transformation thanks to their tunable redox potential, high-efficiency light harvesting capability, and high extinction coefficient in the visible region. Utilizing QDs as a versatile platform to convert organic compounds into value-added feedstocks provides an effective way to alleviate energy and chemical feedstock supply problems. In this review, we concisely summarize the basic principles of photocatalytic organic conversions over semiconductor QDs and the effects of grain size, surface active sites and ligands on their catalytic performance. Then, we highlight the recent progress of QDs enabling multifarious photocatalytic organic transformations, including nitroaromatic reduction, selective alcohol oxidation, sulfide oxidation, C–H functionalization and so on. In the end, we discuss the current challenges and future prospects in further developing efficient semiconductor QD-based photocatalysts toward photoredox-catalyzed organic conversion.
1. Introduction
Organic synthesis chemistry plays a vital role in the manufacture of bulk commodities and other fine chemicals, which permeates people's daily life from chemical industry.1 Nevertheless, the traditional organic synthesis is typically accompanied by the use of toxic or corrosive chemical reagents, which have caused severe damage to the environment.2,3 For example, traditional industrial production commonly uses strong acids or bases to synthesize important organic chemicals at high temperature and pressure. The complex synthesis routes and harsh operating conditions also limit the sustainable development of traditional organic synthesis methods.3,4 By contrast, the introduction of clean and renewable solar energy into organic synthesis reaction systems promotes the rapid development of photocatalytic organic conversion, which is undoubtedly an environmentally friendly and promising alternative.5,6Among a variety of common photocatalysts, metal complexes (e.g., [Ru], [Rh] and [Ir]) and bulk semiconductors (e.g., TiO2, ZnO and g-C3N4) are not ideal light absorbers for driving organic transformation due to the limited photostability of molecular species or the narrow absorption range of wide-bandgap materials.7–9 In particular, some precious metal-based photocatalysts, such as polypyridyl complexes of Ru(II), Ir(III), and Pt(II), are not well suited for industrial productions on a large scale due to their limited practicability and high-energy consumption.10 In comparison with other traditional semiconductor materials, semiconductor quantum dots (QDs) have become one of the most promising candidate materials in artificial photosystems by virtue of their wide absorption range, high extinction coefficient, effective charge transfer and stable photoactivity, providing a new versatile platform for photocatalytic organic synthesis.10 By adjusting the structure (size or shape) or elemental composition of QDs, the redox potential of QDs, that is the bandgap, can be customized for diverse photocatalytic reactions. Besides, QDs can not only achieve surface functionalization through various organic or inorganic ligands without modifying the spectral absorption range or the redox potentials of the QDs, but also construct multifunctional heterostructure composites by surface assembly with other co-catalysts and/or other semiconductor photocatalysts, thus effectively regulating the photocatalytic performance for organic synthesis.11,12 In addition, The colloidal properties of QD catalysts are conducive to the sufficient contact between catalysts and substrates and promotes the transfer of charge and energy, exhibiting the advantages of both homogeneous and heterogeneous catalysts.10,13,14 The use of QD photocatalysts for organic transformation has been increasingly studied in recent years, because it opens up new and efficient pathways to synthesize high-value-added chemicals. Accordingly, it is highly desirable to afford an overview and detailed classifications of current advances on a QD-based composite system for selective photocatalytic organic synthesis.
Herein, we present a timely review to recap the recent progress of semiconductor QD-based catalysts in various photoredox organic transfoxrmations. First, we clarify the fundamental principles of photocatalytic organic transformation over QD-based catalysts. In this part, the exciton generation, the formation of radical species and the influence of surface-chemistry-modification on selective organic conversion will be summarized. Later, we focus on the application of QD-based catalysts in photoredox organic transformations of various useful chemicals, which include nitroaromatic reduction, selective alcohol oxidation, C–H functionalization, sulfide oxidation, biomass valorization, etc. Finally, we will discuss the perspectives and possible challenges in photoredox-catalyzed organic transformation with QD-based catalysts.
2. Basic principles of photocatalytic organic synthesis of QD-based catalysts
2.1 The generation of charge carriers
The photocatalytic redox process starts with the generation of electron–hole pairs (charge carriers). Generally, after stimulation of QD-based catalysts by solar energy, electrons and holes can be simultaneously generated. On the one hand, the photoexcited electrons can readily be transferred to the conduction band (CB) for reduction reactions. On the other hand, the photogenerated holes left in the initial valence band (VB) can be used for oxidation reactions. Ideally, these photoexcited charge carriers generated in the bulk phase will migrate to the reactive sites on the surface to take part in the redox reactions.15,16Typically, unlike bulk semiconductors (e.g., TiO2, WO3, and g-C3N4) where a single incident photon produces only one exciton pair (electron–hole pair), semiconductor QDs exhibit unique properties in exciton generation due to quantum confinement effects.17,18 For example, as shown in Fig. 1a, semiconductor QDs (especially PbTe, PbSe and PbS) are capable of generating multiple excitons after the absorption of one high-energy photon (hυ > 2Eg).19–21 When excited by an absorbed photon having energy more than twice their bandgap, semiconductor QDs may generate a pair of high-energy excitons, in which the energetic electron in the CB releases partial energy through impact ionization and falls back to the bottom of the CB, thus generating more new excitons and achieving efficient carrier multiplication to improve photocatalytic performance.22–26 However, most single phase QDs have plenty of defects on the surface and a fast recombination rate of charge carriers, which is not favorable for improving the overall photocatalytic efficiency.27 Moreover, the colloidal QDs have a very large specific surface area and many surface dangling bonds, some of which will introduce a large number of trap states, thus strongly localizing the photoexcited carriers and preventing them from further taking part in surface chemical reactions.28 To overcome the above problems, a number of strategies have been explored for constructing efficient QD-based composite photocatalysts, which can better inhibit photogenerated charge carrier recombination and promote more rapid charge separation/transfer (Fig. 1b), such as combination with other matched semiconductors, doping with anions and/or cations or other heteroatoms, hybridization with suitable cocatalysts, and modification with inorganic/organic ligands.29–33 Specifically, composite semiconductors, modified ligands, and loaded ions or atoms can all act as transporters of electrons or holes, and the construction of heterostructures inhibits photogenerated carrier recombination and prolongs the excited-state lifetime, thus achieving efficient selective organic conversion.
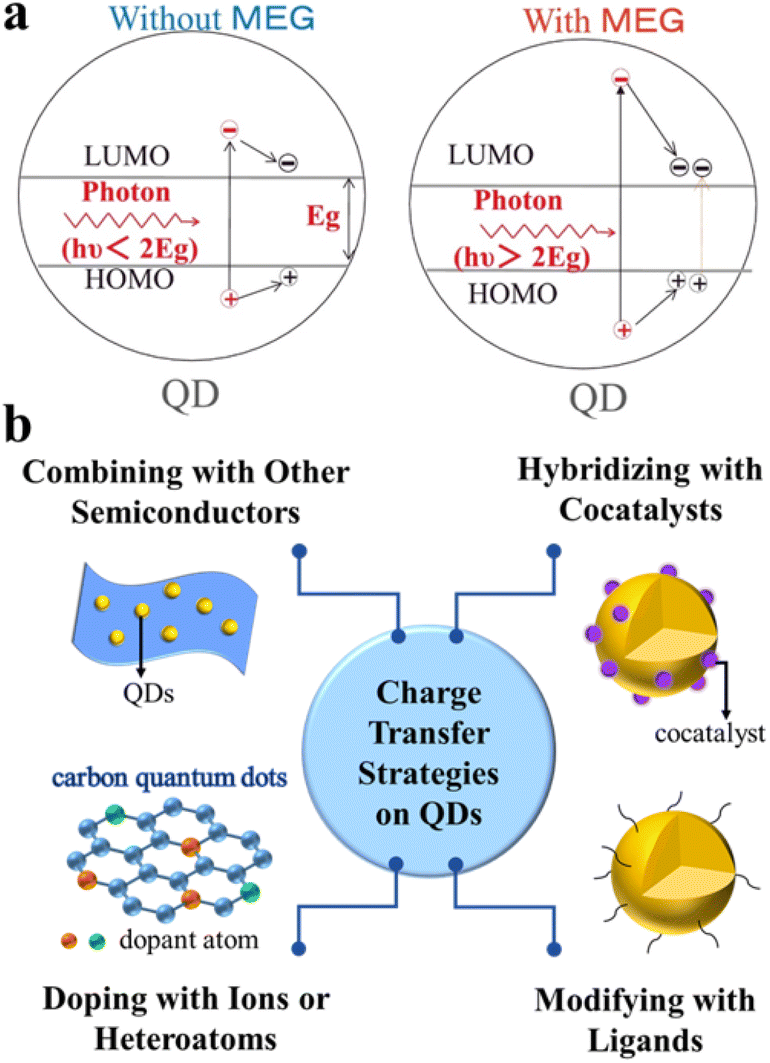 | ||
| Fig. 1 (a) Schematic diagram of multiple exciton generation (MEG). Reprinted with permission from ref. 17. (b) Scheme of typical charge transfer strategies for QD catalysts. | ||
2.2 The formation of radicals
During photocatalytic organic conversion, the highly energetic charge carriers migrating from the catalyst phase to the surface are transferred to organic substrates or the solvent environment to induce the formation of reactive radicals, hence boosting the performance of the photocatalytic system. Fig. 2 summarizes the source and generation pathways of radicals in the organic transformation reaction system. Ideally, organic substrates can be directly activated by photoexcited catalysts via charge or energy transfer to form corresponsive radical intermediates which will further react to obtain target products with high selectivity.34–37 For example, the C–H bond of benzyl alcohol is oxidized to form a ˙CH(OH)Ph radical by the photogenerated holes in the VB of CdS QDs after illumination. The unstable ˙CH(OH)Ph radical which is strongly adsorbed on the bare CdS QD surface can be further oxidized by the holes to produce benzaldehyde. However, over the CdS QDs anchored onto TiO2 nanosheets, such a radical intermediate is directly coupled to form C–C products with high selectivity thanks to the effective transfer and accumulation of multiple electrons in the CdS/TNS heterostructure.38 Additionally, the surface ligands of QDs can also generate radicals, which will come into contact with organic substrates to initiate redox reactions.39 For instance, the thiolate ligand attached to CdSe QDs generates a thiyl radical which extracts the hydrogen atom from the C–H bond of benzyl alcohol to produce a benzylic radical. Then, the benzylic radical induced by the thiyl radical is disproportionated to benzyl alcohol and benzaldehyde.29 Furthermore, some radicals are derived from the gas atmosphere or solution environment, promoting the photocatalytic reaction indirectly as the active species (Fig. 2). On one hand, solvent molecules in the liquid phase can be activated to form radicals, such as water molecules losing electrons to generate hydroxyl radicals (˙OH). On the other hand, O2 can be dissolved in the reaction solvent and is more likely to capture electrons than H+, generating superoxide radicals (˙O2−) readily in the catalytic systems with an O2 atmosphere.40,41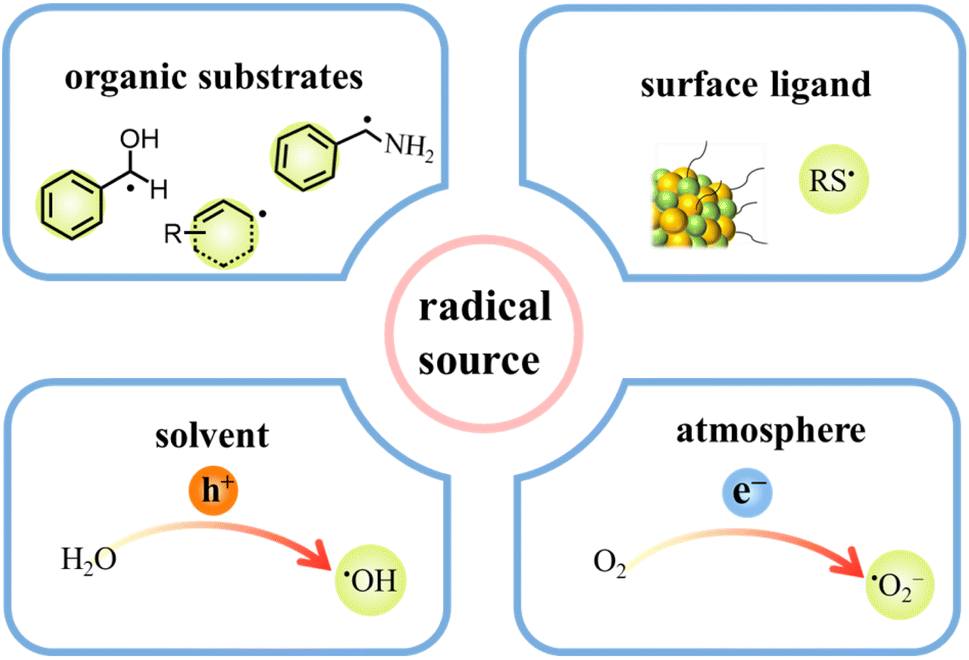 | ||
| Fig. 2 Illustration of radical sources in the photocatalytic reaction system. | ||
Some radical intermediates can be dynamically adsorbed on the surface of QDs, and go through a radical mediated reaction pathway to form a C–C/C–X bond (X = S, N, O).37,42 Heterogeneous photocatalysis based on semiconductor QDs is a promising alternative approach for triggering radical coupling reactions in C–C/C–X bond synthesis, which has emerged as a hot direction in the photocatalysis research field because of its excellent efficiency and convenience in generating a variety of highly reactive radicals by transferring charges between organic molecules and semiconductors.3 However, it is extremely challenging to achieve the synthesis of a C–C/C–X bond on semiconductor surfaces by precisely controlling selective radical bond formation, because semiconductors interact with photogenerated radicals unselectively and radicals are only present in certain and limited circumstances.43 Among various regulatory strategies, combining a semiconductor with a matched cocatalyst, such as a metal cluster or single metal atom, to realize a solar-driven radical controllable reaction is a feasible approach. Studies have shown that atomically dispersed cocatalysts have the merits of both homogeneous catalysts and heterogeneous catalysts, including tunable interactions with ligands, well-defined active sites, high stability and excellent recyclability.44,45 Particularly, these active sites with special electronic structures and unsaturated coordination bonds exert positive effects on the adsorption and activation of reactant molecules.44,46,47 At the reactive sites, atomically dispersed cocatalysts can manipulate radical transformation for divergent synthesis of target products.42
2.3 The impact of surface chemistry in QDs
As shown in Fig. 3a and b, changing the size of QDs can facilely regulate their band gap and the redox potentials of photogenerated electrons and holes,48,49 which means that the substrate distribution can be adjusted by regulating the organic redox path. Fig. 3c depicts the approximate redox potentials of several typical organic substrates. Theoretically, an organic substrate can be oxidized by semiconductors with the VB maximum higher than its redox potential or be reduced by semiconductors with the CB minimum lower than its own redox potential. Virtually, because of the external environment or unfavorable dynamic, certain organic conversion reactions are still challenging to take place under the bandgap-matching semiconductor catalysis. Modulating the size of semiconductor QDs can effectively change the band gap width and redox potential, thus increasing their thermodynamic feasibility. Cd-rich semiconductor QDs based on III–VI elements are known for their tunable band gap, and have a wider band gap than bulk semiconductors (i.e., about 2.43 eV band gap energy for bulk CdS and 2.65 eV for QDs) due to quantum confinement effects.51–53 Additionally, the surface properties of QDs can significantly affect their exciton kinetics for the reason that the surface atoms of QDs make up the majority of total atoms.54,55 In particular, the spatially separated charge carriers can transfer to the semiconductor QD surface independently, and ultimately be captured by molecules adsorbed on their surface or transfer to the reactive sites on the other semiconductors or cocatalysts.56,57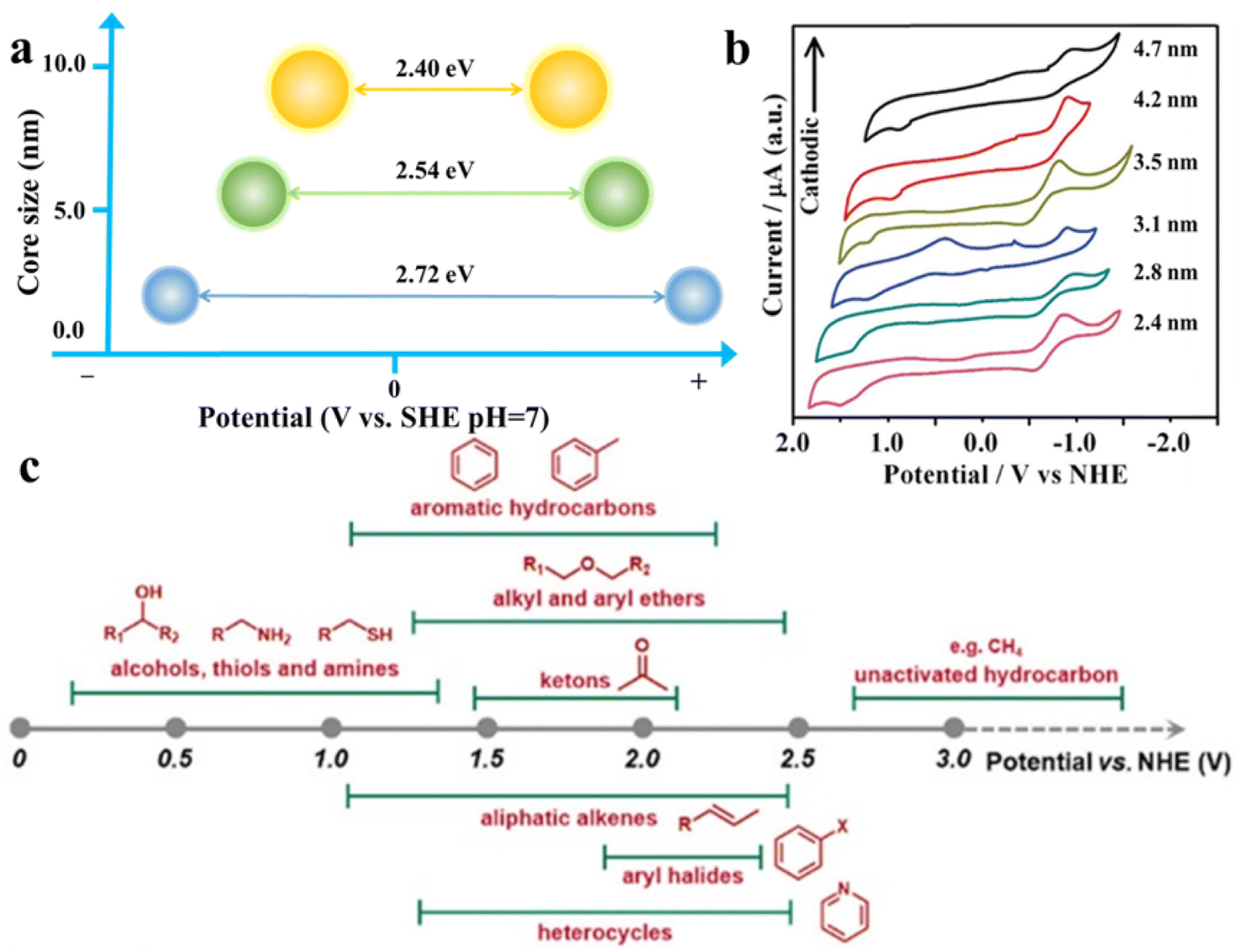 | ||
| Fig. 3 (a) Illustration of the relationship between the core size and band gap of CdS QDs. (b) Cyclic voltammograms (CVs) on CdTe QDs of different sizes in methylene chloride. Scan rates were 100 mV s−1 for all the cases. Reprinted with permission from ref. 50. (c) Approximate redox potential of common chemical molecules. Reprinted with permission from ref. 3. | ||
In addition to QD size, ligand modification, surface reconstruction, and the active sites also contribute significantly to the promotion of photoredox organic transformation. The interaction between QDs and electron donors/acceptors is enhanced by plentiful surface sites, which encourage the interfacial charge transfer rate of photocatalysis.58 Generally, QDs provide abundant catalytic active sites and adsorption centers because of their unique surface structural and adjustable atomic proportions. Furthermore, the quantity of acceptor molecules adsorbed on the QD surface is positively correlated with the efficiency of energy transfer. For example, QD gels, once deprived of the surface ligands, exhibit superior photocatalytic activity due to more exposed active sites which improve the probability of reactant adsorption and promote charge transfer.59 On one hand, constructing a great quantity of surface active sites is of great significance for greatly improving photocatalytic organic conversion, enabling effective reactants and adsorption rapid charge transfer. On the other hand, the application of selective organic conversion can be further expanded by surface structure reconstruction, such as changing the atomic composition and introducing defect vacancies or cocatalysts.
Additionally, organic/inorganic ligands of QDs have a decisive influence on their surface properties, which can functionalize the QD surface and regulate the charge transfer on semiconductor QDs, thus improving the organic conversion rate in photocatalysis.32,39,60–63 The primary functions of ligands include: (i) serving as a stabilizer to prevent quantum dots from gathering, (ii) promoting exciton delocalization and inter-particle charge transmission,64,65 (iii) establishing a stable channel for electron transfer between catalysts and metal ions, metal atoms, or other organic molecules,66,67 and (iv) modulating the properties of catalysts through modification of the surface chemistry.68–70 Particularly, it has been shown that there is efficient charge transfer between organic molecules and surface ligands of QDs.71,72 For example, Weiss et al. constructed hybrid monolayers of oleate and octyl phosphonate to make the ligand shell of CdS QDs disordering. The unique ligand shell is beneficial to facilitate the hole-transfer step which increases the initial rate and enhances the energy efficiency of the C–C coupled reaction.61 Studies have shown that the photocatalytic performance of semiconductor QDs can also be greatly influenced by the length, hydrophilicity/hydrophobicity, intrinsic charge properties, and anchoring groups of surface ligands.30,73,74 For instance, a novel electron tunneling photosystem using surface ligands as the charge transfer medium has been reported. The ligands can not only induce electrostatic self-assembly to construct composite catalysts, but also precisely regulate the directional charge flow of photoredox organic conversion.75 The surface of transition metal chalcogenide quantum dots (TMC QDs) is negatively charged due to the deprotonation of carboxyl groups of mercaptoacetic acid (MAA) ligands, while the surface of metal nanocrystals (NYs) is positively charged because of partial protonation of the exocyclic nitrogen atoms of 4-dimethylaminopyridine (DMAP) ligands.76–78 The interfacial insulating electron tunneling layer, which is composed of layered ligands on the surface of TMC QDs (CdSe and CdS) and NYs (Pd and Au), is beneficial to the unidirectional transfer of electrons from QDs to metal nanocrystals, enhancing the carrier lifetime and promoting charge separation.
3. Applications of QDs for photoredox organic transformation
Under mild reaction conditions, using exhaustless solar energy to drive selective photochemical reactions is an environmentally friendly route that can be potentially applied in various industrial organic transformation processes.79–81 As one of the semiconductor photocatalysts, semiconductor QDs are widely used in photocatalysis such as CO2 photoreduction, water splitting, degradation of organic pollutants, and organic redox reactions due to their inherent strengths in light absorption, exciton generation, and carrier transfer.3,9,36,82–84 In this context, the surface modification of QDs with different ligands30,85 and/or suitable cocatalysts86,87 can effectively adjust the surface composition and charge distribution of catalysts, thus improving the performance of photocatalysts and improving the photocatalytic reaction efficiency.42 In this section, we will focus on the recent advances of QD-based photocatalysts in various photoredox selective organic transformation reactions.3.1 Reduction of nitroaromatics
As a kind of indispensable organic intermediates, amino aromatic compounds are used to prepare industrial products, such as medicines, dyestuff, pesticides, and pigments,88–90 and can be synthesized by photocatalytic reduction of different nitroaromatics. Fig. 4 shows that amino aromatic compounds can be synthesized by a direct method through intermediates of N-phenylhydroxylamine and nitrosobenzene, or by the condensation method through hydrazobenzene, azobenzene, and azoxybenzene.91,92Table 1 summarizes the recent applications for selective photoreduction of nitroaromatics over diverse QD-based photocatalysts. Conventional reduction of nitroaromatics usually involves reducing nitroaromatic compounds to amines by a mild and efficient method with viologens as the electron-transfer catalyst along with the generation of by-products.93 CdS QDs sealed with mercaptoethylamine hydrochloride (MEA) have been reported to be effective in reducing aromatic nitro compounds to the corresponding aromatic amines without generating the by-products from carbonyl and cyano functional group reduction on the aromatic ring.94 To further explore the underlying mechanism of aromatic nitro reduction, Weiss's group reported that benzylamine (NB) can be reduced to aniline (NA) over CdS QD photocatalysts through six sequential photoinduced, proton-coupled electron transfers, which can be depicted as a straightforward kinetic model comprising three sequential processes (Fig. 5a).26 Gas chromatography-mass spectrometry (GC-MS) has detected both four- and six-electron photoproducts of phenylhydroxylamine (PHA) and AN without the two-electron intermediate nitrosobenzene (NSB) (Fig. 5a and b), which is mainly due to the lower reduction potential (+0.29 V) of NSB to PHA, causing the irreversible conversion of the NSB intermediate to the further reduced product PHA. Limited by the production and utilization of a single electron in the QD catalyst under typical photon flux illumination, the photocatalytic reaction mechanism of benzylamine is presented in Fig. 5c.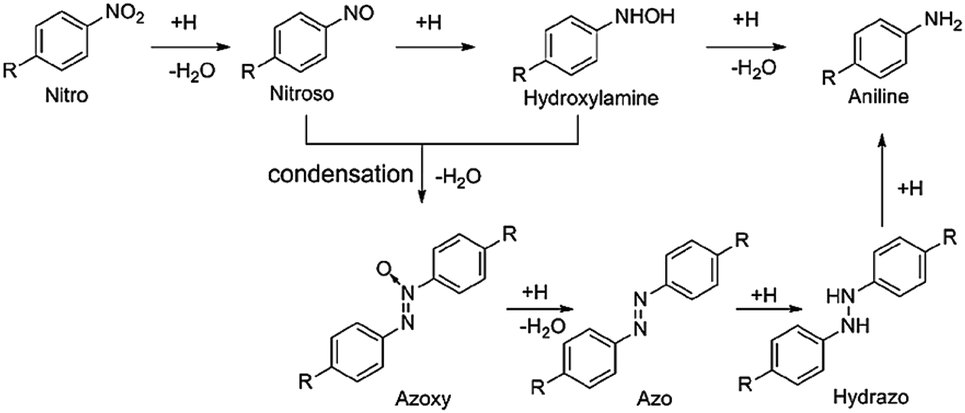 | ||
| Fig. 4 Mechanism of reduction for catalytic nitroaromatic to aniline. Reprinted with permission from ref. 91. | ||
| Entry | Photocatalyst | Atmosphere | Solvent | Scavenger | Light source | Ref. |
|---|---|---|---|---|---|---|
| a MPA refers to 3-mercaptopropionic acid. b T001 refers to anatase TiO2 with exposed facets. c GQDs/TCN stands for graphene quantum dots/Mn–N–TiO2/g-C3N4. d N-GQDs refers to nitrogen-doped graphene quantum dots. e GR refers to graphene. f NYs refers to metal nanocrystals (Au and Pd) and TMCs refers to transition metal chalcogenides (CdSe and CdS). g In metal nanocrystals InOOH. | ||||||
| 1 | C dots/CdS | N2 | H2O | HCOON4 | 3 W LED lamp, λ > 420 nm | 95 |
| 2 | CdS QDs | Ar | H2O/CH3OH | MPAa | 7 mW 405 nm laser | 26 |
| 3 | CdSe QDs-T001b | N2 | H2O | Na2SO3 | 500 W Xe-lamp, 420–800 nm | 77 |
| 4 | GQDs/TCNc | Vacuum | H2O | t-BuOH, K2S2O8, EDTA | 300 W Xe-lamp, 320 nm ≤ λ ≤ 780 nm | 96 |
| 5 | N-GQDsd | Ar | H2O | NaBH4 | NIR light | 97 |
| 6 | CdSe QDs-GRe | N2 | H2O | Na2SO3 | 300 W Xe-lamp, λ > 420 nm | 32 |
| 7 | CdTe QDs/GR | N2 | H2O | Na2SO3 | 300 W Xe-lamp, λ > 420 nm | 27 |
| 8 | CdS QDs | N2 | D2O/CD3OD | Methanol | Blue LED, 450–460 nm | 98 |
| 9 | NYs/TMC QDsf | N2 | H2O | Na2SO3 | 300 W Xe-lamp, λ > 420 nm | 75 |
| 10 | CdS QDs-Ing | N2 | H2O | HCOON4 | 500 W Xe-lamp, 420–800 nm | 99 |
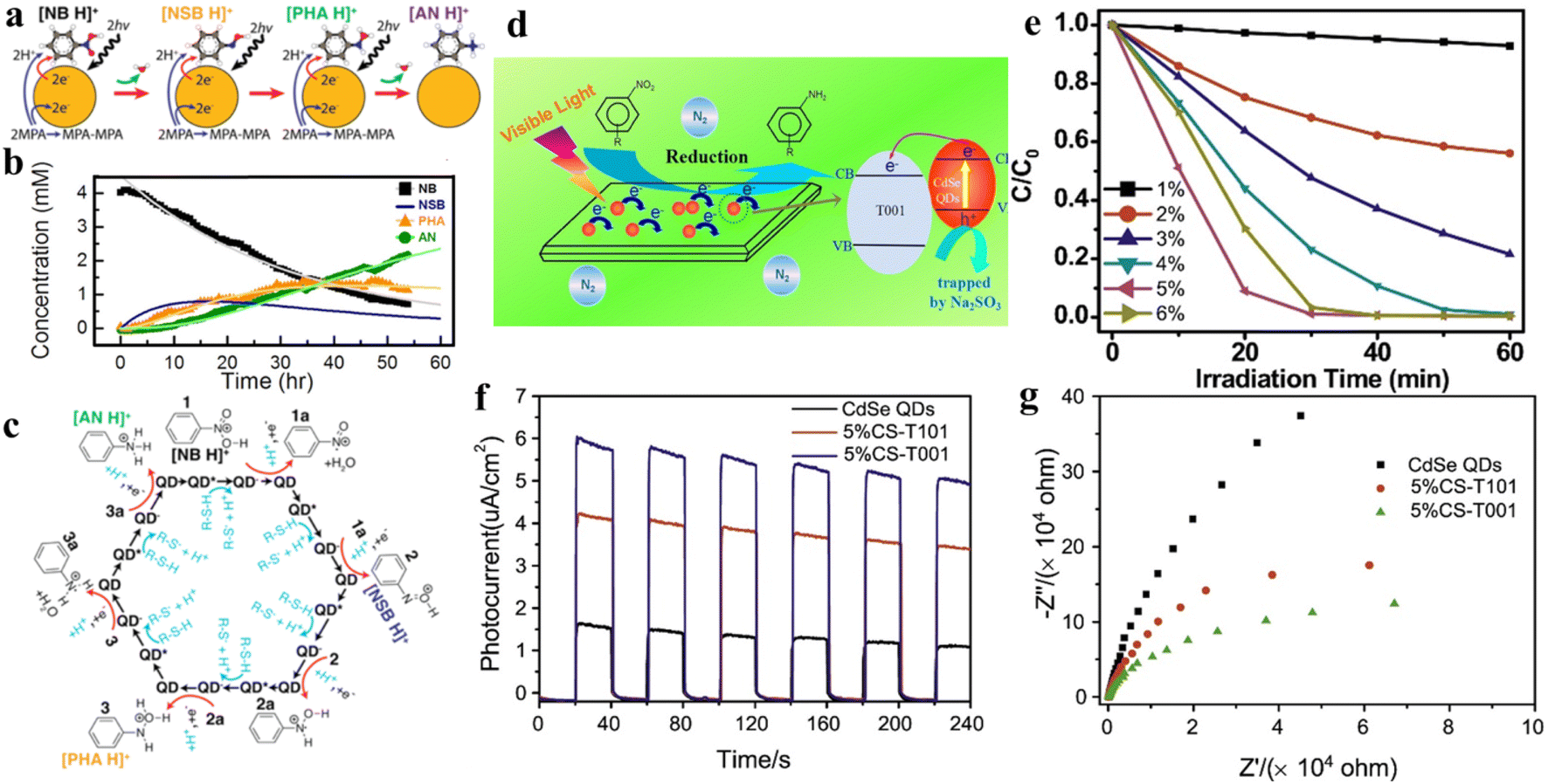 | ||
| Fig. 5 (a) Catalytic steps of conversion of nitrobenzene to aniline with six-electron, six-proton photoreduction. (b) The relationship between the concentration of NB and reduction products and time. (c) Six sequential proton coupled electron transfer steps of photocatalytic reduction of protonated NB to AN over QDs, with intermediates of NSB and PHA. Reprinted with permission from ref. 26. (d) Illustration for nitroaromatic photoreduction over CS-T001. (e) Photocatalytic performance for reduction of 4-NA over CS-T001 with different contents of CdSe QDs. (f) Transient photocurrent spectra of CS-T001. (g) EIS Nyquist curves. Reprinted with permission from ref. 77. | ||
In one instance, an efficient charge-transfer channel for the reduction of nitroaromatics has been constructed on the (3D) CdSe QD–graphene (GR) nanocomposites by tightly bonding CdSe QDs and GR nanosheets through a universal layer-by-layer self-assembly strategy.32 The significantly enhanced photoredox performance of this QD-based composite can be attributed to a conjunction of several factors, including the greatly improved separation of photogenerated excitons and the significantly increased specific surface area caused by seamless GR encapsulation,100–102 thus leading to the superior photocatalytic activity. The obtained results indicated that compared with blank CdSe QDs, the photoactivity of the CdSe QDs-1% GR nanocomposite for selective photoreduction of 4-nitroaniline (4-NA) to 4-phenylenediamine (4-PDA) was markedly enhanced. In addition, it was discovered that the CdSe QDs are universal in nitroaromatic reduction by investigating diverse aromatic nitro compounds possessing various function groups.
Combining QDs with other suitable semiconductor materials (e.g., CdS, ZnSe, and TiO2) to construct QD-based heterostructures has been regarded as a feasible strategy for prolonging the lifetime of photogenerated excitons and promoting the separation/transfer of charge carriers. For instance, He and co-workers successfully constructed CdSe QD-decorated TiO2 with exposed facets (CS-T001) for the first time through a facile method, and proved that it was a stable photocatalyst with high activity for nitroaromatic reduction under visible light.77Fig. 5d displays the mechanism of photocatalytic conversion of nitroaromatics to amino aromatics over CS-T001. The photocatalytic activity of composite catalysts for the reduction of nitroaromatics 4-NA tothe corresponding amino aromatics under ambient conditions has been investigated in the presence of Na2SO3 as a cavity scavenger and N2 purging. Fig. 6e shows that the photocatalytic activity of CS-T001 increases significantly as the content of CdSe QDs increases from 1% to 5%, which indicates that the content of CdSe QDs will affect the composite photocatalytic performance. It is further found that the proper content of CdSe QDs results in uniform particle dispersion on TiO2 and excellent visible light absorption. Transient photocurrent spectroscopy shows that the photocurrent responses of 5%CS-T001 are significantly enhanced, indicating that the transfer and separation efficiency of photogenerated charge carriers is higher in 5%CS-T001 compared with CdSe QDs (Fig. 5f). As can be seen from Fig. 5g, the impedance arc radius of 5%CS-T001 is the smallest in all samples, indicating that 5%CS-T001 has a better electrical conductivity, which is conducive to improving transfer of photogenerated charge carriers and thus boosting the photocatalytic efficiency.
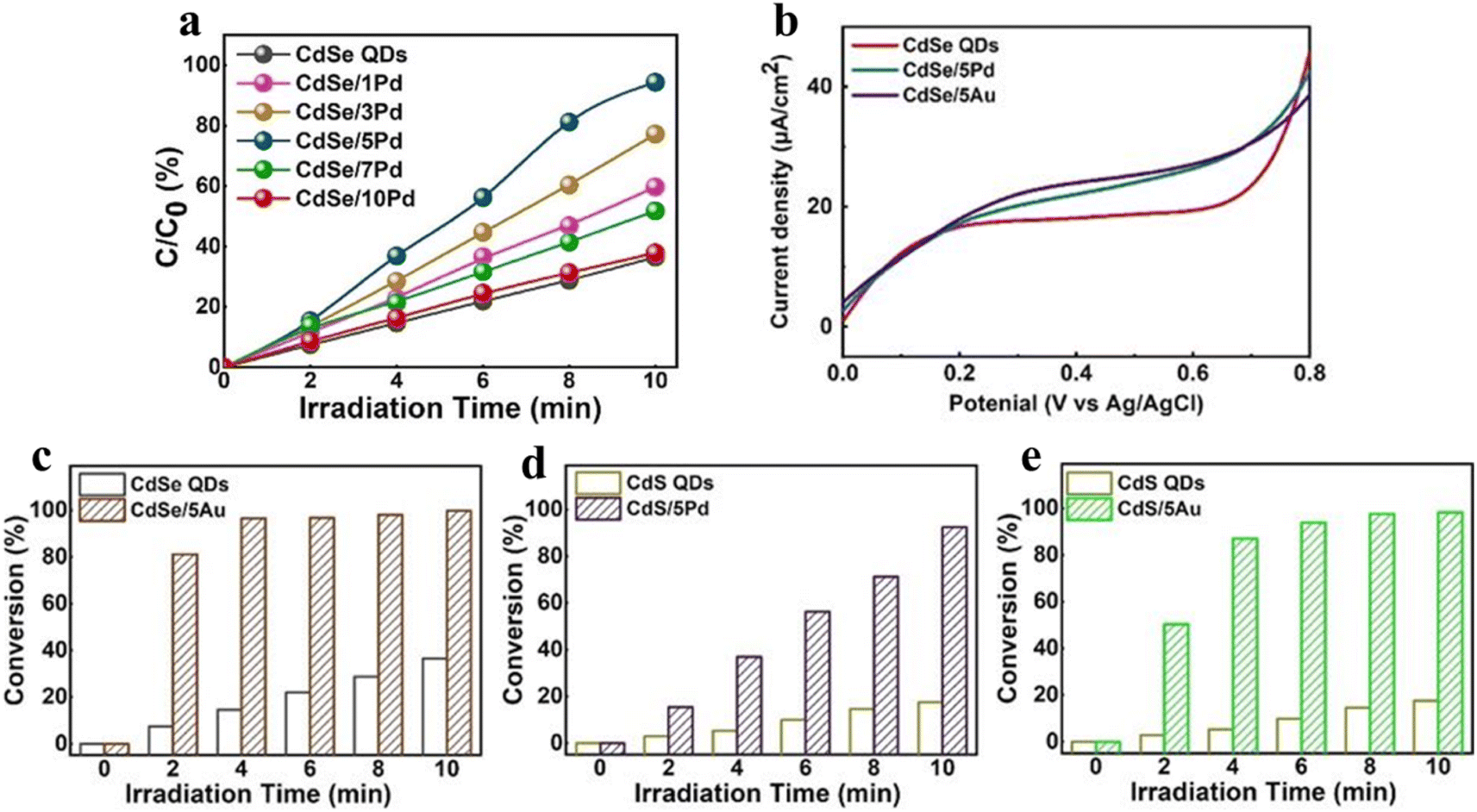 | ||
| Fig. 6 (a) Photoactivity of selective nitroaromatic reduction over CdSe/Pd nanocomposites with different volume of Pd nanocrystals. (b) LSV plots. Nitroaromatic conversion over (c) CdSe/5Au, (d) CdS/5Pd and (e) CdS/5Au compared to their corresponding single QDs. Reprinted with permission from ref. 75. | ||
Additionally, modifying QDs with other cocatalysts has been regarded as a promising strategy, which promotes the photocatalytic stability and activity by inhibiting the recombination of photogenerated charge carriers. Generally, precious metals (e.g., Pt, Au, and Pd) are the most prevalent cocatalysts, which are deposited on the QD surface to form Schottky junctions at the interface, facilitating the separation of charge carriers and ultimately enhancing the photocatalytic performance. For instance, Xiao and co-workers have designed transition metal chalcogenide QD heterojunction catalysts under ambient conditions via a simple ligand-induced electrostatic self-assembly method, exhibiting excellent photocatalytic performance for nitroaromatic reduction due to the improved photogenerated electron transfer and the enhanced response of visible light.75 As can be seen from Fig. 6a, under visible light irradiation, the blank CdSe QDs exhibit lower photoactivity than Pd-decorated CdSe QDs, which is caused by the fast electron–hole recombination rate. The photocatalytic activities of CdSe/5Au, CdS/5Pd, and CdS/5Au exhibit a similar enhancement trend compared with their corresponding single counterparts under the same conditions (Fig. 6b–e). This result implies the universal roles of the positively charged surface ligand capped metal nanocrystals in facilitating the interfacial charge transport. As seen in Fig. 6b, owing to the higher electron–hole pair recombination rate, the blank CdSe QDs exhibit a weak photocurrent response under visible light irradiation, while the photocurrents of CdSe/5Pd and CdSe/5Au are relatively enhanced because of the enhanced charge separation due to unidirectional electron transport of CdSe induced by electron tunneling.
3.2 Oxidation of alcohol
Selective oxidation of alcohols to aldehydes, ketones, carboxylic acids or C–C coupling products has wide and various industrial applications, such as aldehydes and C–C coupled compounds have a wide utilization in confectionary, beverage, fragrance, and pharmaceutical industries.103–106 Nevertheless, traditional conversion of alcohols is not only affected by excessive oxidation and toxic oxidants like chromates and manganese oxides, but also requires harsh operating conditions.107,108 It is expected that QDs with finely tuned redox potentials can be used as photocatalysts to solve the above problems. Table 2 summarizes the recent advances for selective oxidation of alcohols to carbonyl compounds or coupling products over diverse QD-based photocatalysts.| Entry | Photocatalyst | Solvent | Substrate | Product | Light source | Ref. |
|---|---|---|---|---|---|---|
| a MPA refers to 3-mercaptopropionic acid. b BA refers to benzyl alcohol. c BAD stands for benzaldehyde. d BTF denotes benzotrifluoride. e HB refers to hydrobenzoin. f NYs refers to metal nanocrystals (Au and Pd) and TMCs refers to transition metal chalcogenides (CdSe and CdS). g TNS refers to titanium dioxide nanosheets. h CdS-MTA refers to mesoporous CdS-sensitized TiO2 nanoparticle assemblies. i GR refers to graphene. | ||||||
| 1 | MPAa-CdSe QD | H2O | BAb | BADc | LED, λ = 410 nm | 29 |
| 2 | CdTe QDs/5%GR | BTFd | Aromatic alcohols | Aryl aldehydes | 300 W Xe-lamp, λ > 420 nm | 27 |
| 3 | CdS/SiO2 | CH3CN | BA | HBe | 300 W Xe lamp, | 112 |
| 4 | CdS QDs | D2O/CD3OD | BA | BAD/HB | 2.6 mW, λ = 405 nm | 113 |
| 5 | NYs/TMCs QDsf | BTF | Aromatic alcohols | Aryl aldehydes | 300 W Xe-lamp, λ > 420 nm | 75 |
| 6 | CdS/TNSg | CH3CN | BA | HB | 300 W Xe-lamp, λ > 420 nm | 38 |
| 7 | CdS-MTAh | CH3CN | Aromatic alcohols | Aryl aldehydes | 300 W Xe-lamp, λ > 320 nm | 114 |
| 8 | CdSe QDs-GRi | BTF | Aromatic alcohols | Aryl aldehydes | 300 W Xe-lamp, λ > 420 nm | 32 |
| 9 | CdS-TiO2 | BTF | BA | BAD | 300 W Xe-lamp, λ > 420 nm | 40 |
| 10 | CQD | H2O | BA | BAD | 450 W Xe-lamp, λ < 700 nm | 115 |
| 11 | CdSe/CdS QDs | DMF | Phenylethanol | Pinacol | Blue LED lamp, 450 nm | 116 |
| 12 | MPA-CdSe QDs | H2O | Isopropanol | Acetone | 3 W LED lamp, λ = 410 nm | 74 |
| 13 | CdSe QDs-TiO2-Ni(OH)2 | Isopropanol/H2O | Isopropanol | Acetone | High-pressure mercury lamp, λ > 400 nm | 117 |
| 14 | CdS QDs@α-ZrP | CH3CN | BA | BAD | 55 W Xe-lamp | 118 |
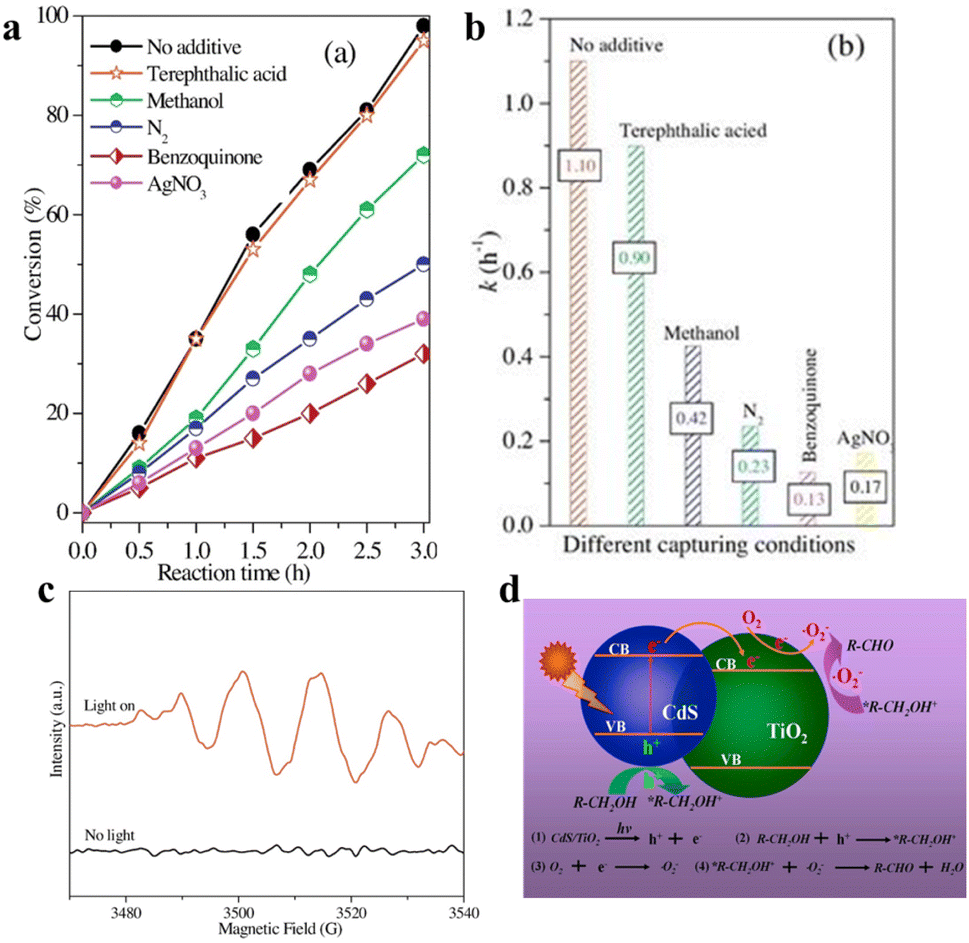 | ||
| Fig. 7 (a) The conversion of BA photooxidation CdS/TiO2 in diverse capturers. (b) Photocatalytic rate constant k of BA oxidation under different capturing conditions. (c) ESR spectra. (d) Schematic for photocatalytic selective oxidation of alcohol to aldehyde. Reprinted with permission from ref. 40. | ||
In addition to the photocatalytic dehydrogenation mechanism of BA above, Wu et al. proposed 3-mercaptopropionic acid-capped CdSe QDs that oxidize alcohols to carbonyl compounds. Electron transfer that facilitates the formation of an active thiyl radical is the most critical initial step. The thiyl radical in turn induces the activation of reactant molecules to generate radical intermediates, which then produce the corresponding carbonyl compounds via a radical relay process. They have also investigated a variety of aromatic alcohols to explore the universality of alcohol oxidation over CdSe QDs under the same reaction conditions.29 The results indicate that the photocatalytic conversion of aromatic alcohols on CdSe QDs is independent of the volume of the ortho-substituted group of the aryl ring, and the benzyl alcohols substituted by electron-withdrawing groups at the para-position has a slightly slower reaction rate than that substituted by electron-donating groups.
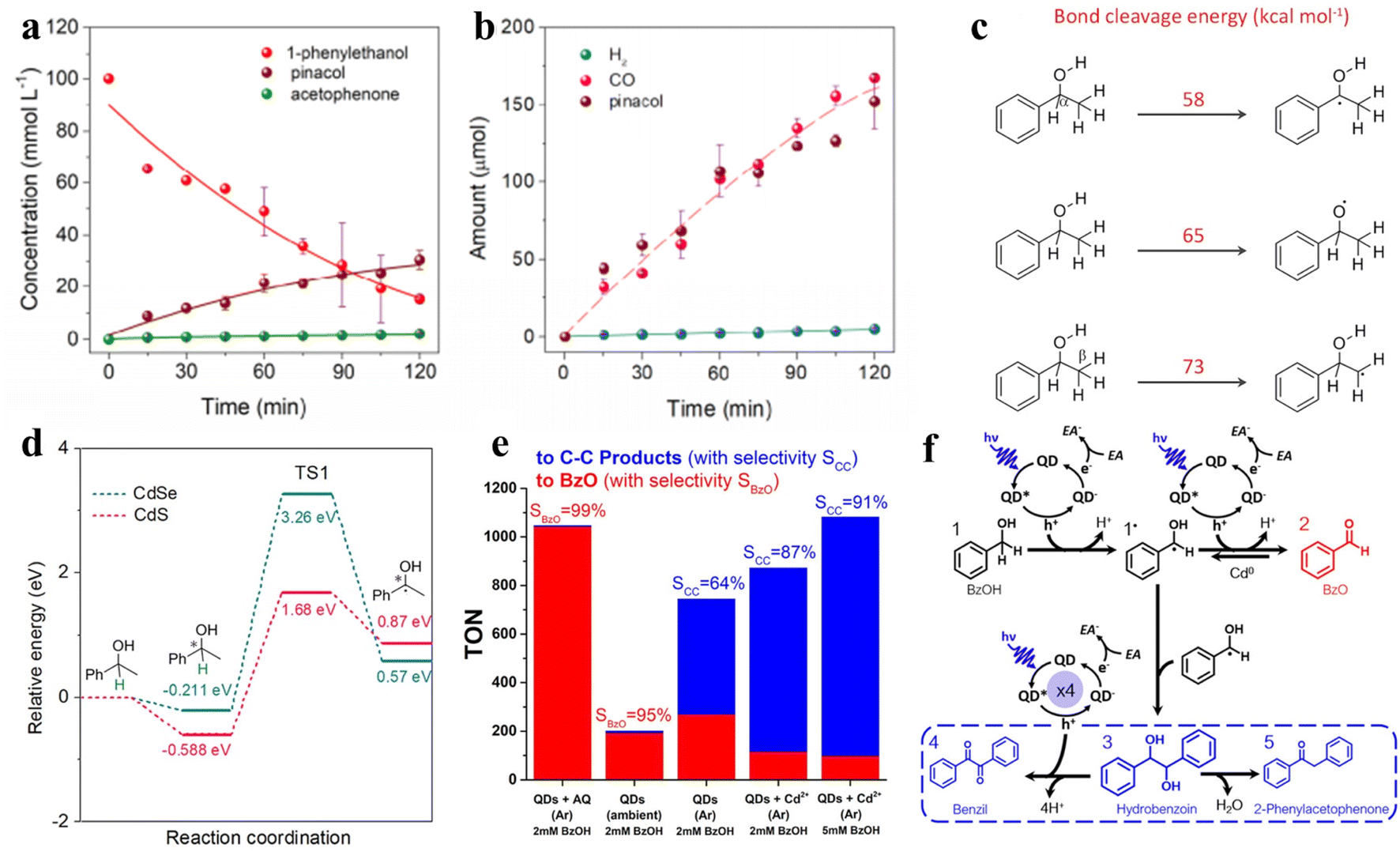 | ||
| Fig. 8 (a) Concentration changes of 1-phenylethanol, pinacol, and acetophenone during the reaction. (b) The generation of gas products and pinacol with increasing irradiation time. (c) Bond cleavage energy. (d) Full potential energy for 1-phenylethanol deprotonation on CdSe/CdS QDs. Reprinted with permission from ref. 116. (e) Production and selectivity of the photocatalyst. “AQ” refers to sodium anthraquinone-2-sulfonate; “BzOH” refers to benzyl alcohol. “BzO” refers to benzaldehyde. (f) The pathway of photocatalytic BzOH oxidation. “EA” refers to an electron acceptor. Reprinted with permission from ref. 113. | ||
In another example, Weiss and co-workers achieved photocatalytic oxidation of BA to BAD or C–C coupling products with the selectivity near 100% by adjusting the quantity of Cd on QD surfaces by in situ deposition (Fig. 8e).113Fig. 8f shows that the photoexcitation of CdS QDs without deposited Cd induces the radical intermediates (1˙) to form BzO (2), while the produced hydrobenzoin (3) sometimes undergoes a four-electron oxidation to benzil (4) over QDs in the presence of metallic cadmium.
Furthermore, our group has constructed a SiO2-surpported CdS QD (CdS/SiO2) material for visible-light-driven dehydrogenative C–C coupling reaction of BA under ambient conditions.112Fig. 9a shows that hydrogen (H2) and hydrobenzoin (HB) production rates are evaluated to be ca. 1.0, indicating a stoichiometric dehydrogenation reaction. Furthermore, the CdS/SiO2 photocatalysts exhibit excellent photoactivity with a yield of 84.6% and an outstanding selectivity of near 100%. Meanwhile, high performance liquid chromatography (HPLC) is used to identify the liquid products produced in the photocatalytic system to explore the evolution kinetics of BA, benzaldehyde (BAD) and HB over the CdS/SiO2 composites. Fig. 9b shows that the BA decreases continuously with the increase in HB and BAD as the reaction proceeds. The mechanism of dehydrocoupling of BA into H2 and HB is proposed in Fig. 9c. Owing to the intrinsic band-gap optical absorption property of semiconductors, CdS QDs can not only absorb direct incident light but also recycle the scattered light in the near field of SiO2. Recently, we designed a bifunctional 0D CdS QDs-2D TiO2 (CdS/TNS) heterostructure for visible-light-driven C–C coupling of BA along with the photoreduction of CO2 to CO.38 The introduction of TNS as the carrier may greatly affect the abstraction of α–H at the interface between QDs and the carrier, thus regulating the selectivity of BA oxidation from BAD to the C–C coupling products. Upon visible-light irradiation, TiO2 cannot be activated, while CdS QDs are excited to produce high-energy electrons and holes. This type-II heterojunction induces the electrons located in the CB of CdS to be transferred to the CB of TNS, thereby driving the dual-function photocatalytic redox processes (Fig. 9d). Photogenerated holes in the VB of CdS can activate the C–H bonds of BA to produce protons and ˙CH(OH)Ph. This produced ˙CH(OH)Ph radicals will then be oxidized by holes for producing BAD or C–C coupling products. At the same time, the CO2 molecules and remaining protons are reduced to CO and H2 by photoexcited electrons.
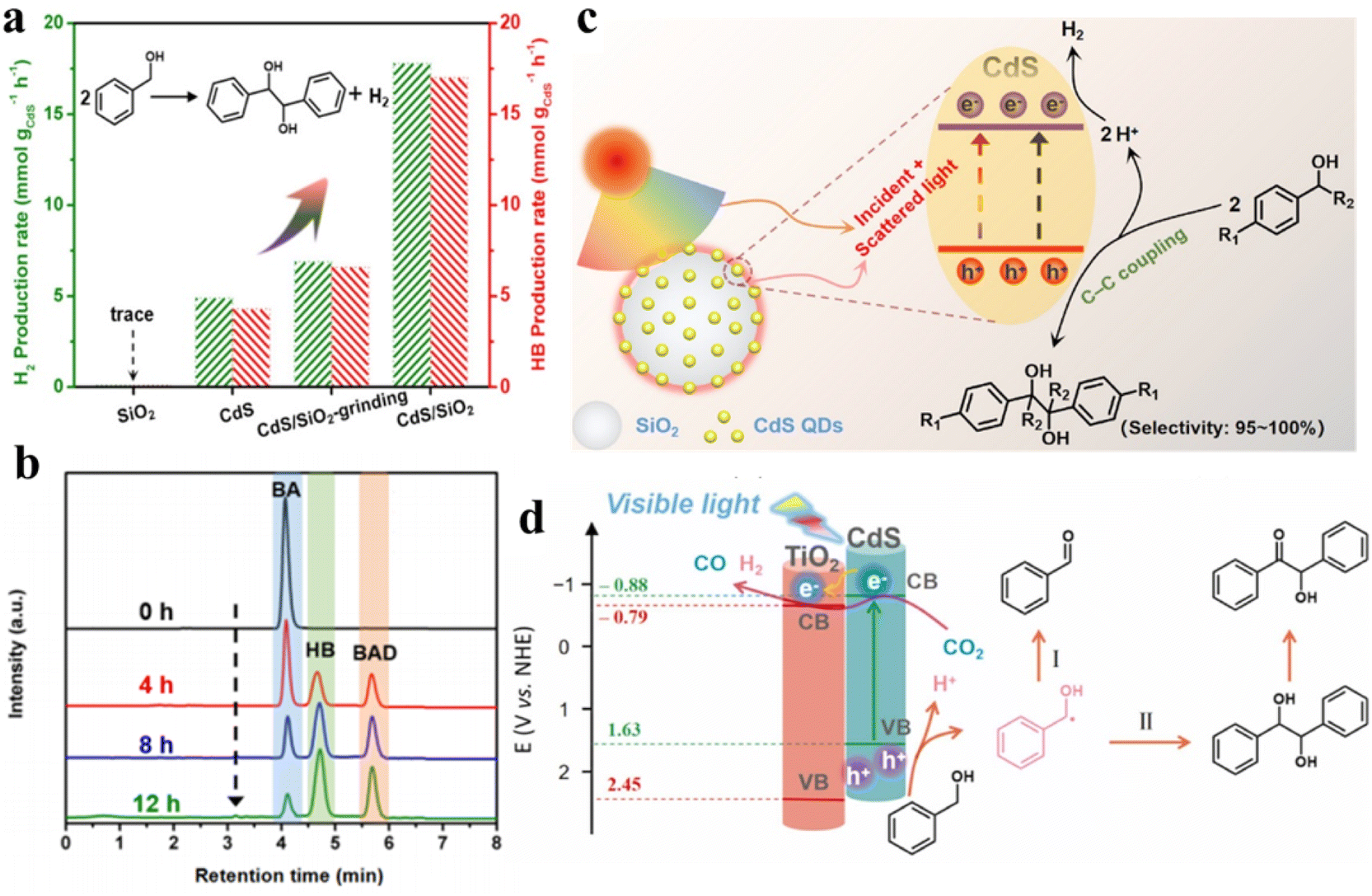 | ||
| Fig. 9 (a) Production of H2 and HB over different samples. (b) HPLC spectra of the liquid products during photocatalytic reaction on CdS/SiO2 composites during photocatalytic reaction. (c) Schematic illustration of CdS QDs/SiO2 for photocatalytic HB synthesis and H2 production. Reprinted with permission from ref. 112. (d) Diagram of the photocatalytic reaction mechanism for CO2 reduction and C–C coupling synthesis over CdS/TNS composites. Reprinted with permission from ref. 38. | ||
3.3 C–H functionalization
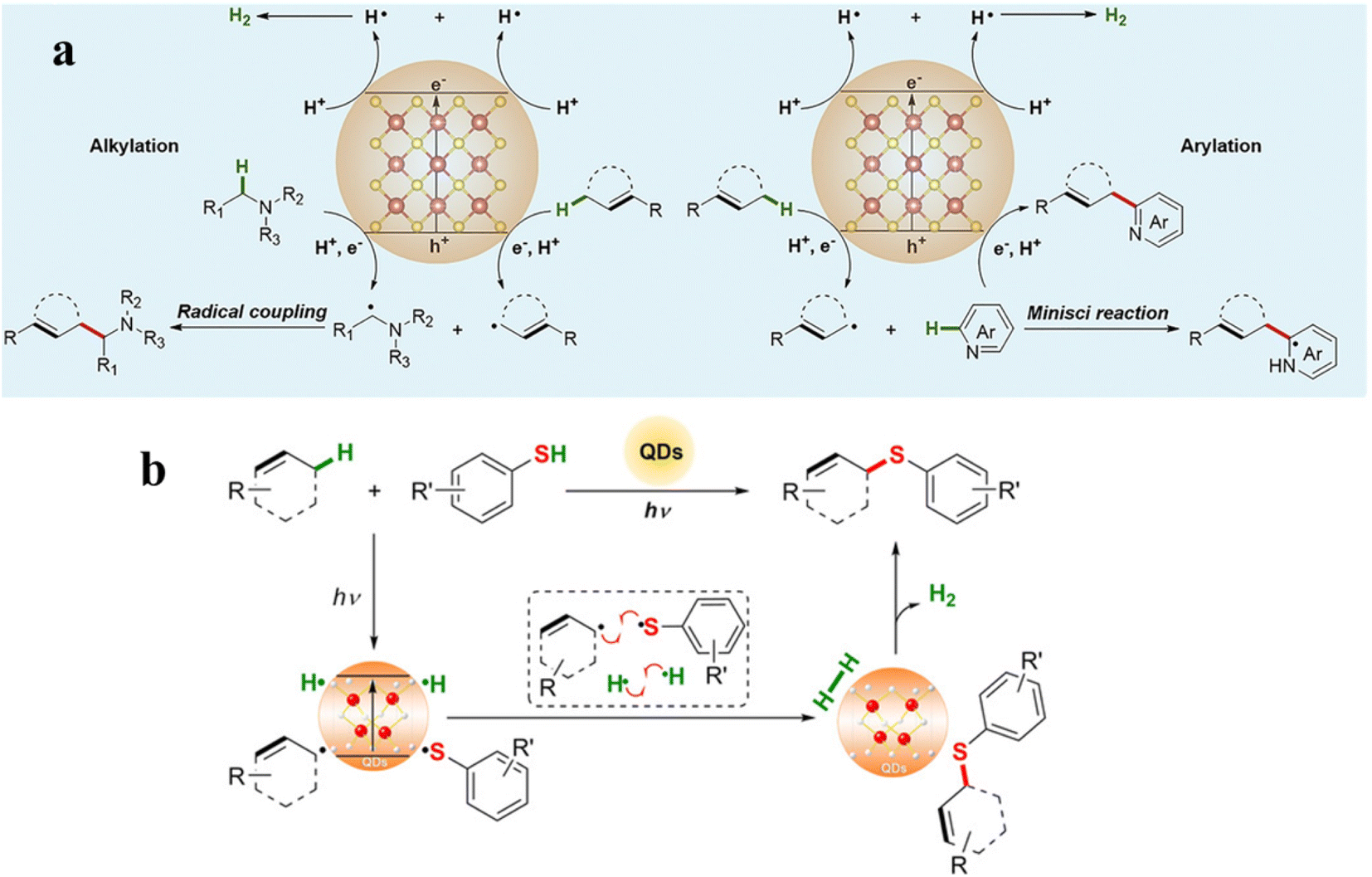
Recent decades have seen a considerable increase interest in functionalizing C–H bonds in the chemistry of organic synthesis. Particularly, using the activation of C–H to synthesize complex organic structures is a highly efficient reaction strategy.119,120 However, oxidative C–H bond cleavage sometimes involves the use of toxic and expensive metal oxidants like Ir(ppy)2(dtbbpy)X and Ru(bpy)3X2, endangering the sustainable study of C–H activation. In this context, single-sized QDs could replace some traditional catalysts and induce C–H bond activation to promote different photoredox reactions (e.g., β-alkylation, β-aminoalkylation, and amine arylation).61,121 Moreover, solar-driven C–H functionalization by using CdSe QDs or CdS QDs as catalysts is currently of increasing interest. Here, we will focus on the photocatalytic C–H bond activation to form C–C and C–S bonds from abundant and easily accessible compounds containing X–H bonds.Recently, using QDs as the photocatalyst, Wu's group reported a direct cross-coupling of allylic C(sp3)–H bonds with α-amino C–H bonds or heteroarenes, respectively (Fig. 10a).122 The formation of an allylic radical can be confirmed by radical-trapping experiments, deuterium experiments, product distribution detection, and electron paramagnetic resonance (EPR) spectroscopy in this catalytic cycle. Under illumination, the allylic C(sp3)–H bonds and α-amino C–H bonds are synchronously activated by photogenerated holes in CdSe QDs to form transient allylic radicals and α-amino alkyl radicals. Later, the intermolecular radical–radical cross coupling promotes the formation of new C–C bonds. Simultaneously, protons generated by C–H bond scission will form H˙ in the CB of CdSe QDs, and the H˙ stably adsorbed on the QD surface will combine to release H2. Additionally, the allylic radicals can also produce arylation products by reacting with heteroarenes through a Minisci-type mechanism125 over CdSe QDs(Fig. 10a).126
3.4 Oxidation of sulfides
Oxidation products of sulfides, such as sulfoxide, sulfone, and disulfide, play an important role in chemistry and biology. However, conventional strategies for sulfide oxidation using toxic oxidants or costly reagents with the need for high temperature and unstable reactive intermediates result in reduced selectivity or excessive oxidation of the products. Accordingly, developing a green route for the oxidation of sulfide compounds is necessary.135–138 Noteworthily, photocatalytic sulfide oxidation over QD-based materials under mild conditions is a promising alternative. For instance, Ford et al. found that the CdSe QDs photocatalyzed the oxidative cleavage of 1,1-dithiooxalate (DTO) to produce carbon disulfide.139 The transfer of photoexcited holes from QDs to surface DTOs triggers this oxidative cleavage reaction.Recently, CdSe QDs have been reported to induce oxidative coupling of various thiols to produce disulfides and H2 in the absence of sacrificial reagents or external oxidants under visible light.142 The photocatalytic conversion of thiols to disulfides and H2 over CdSe QDs is significantly improved after the addition of nickel(II) salt as a cocatalyst. Even under aerobic conditions, the yield of disulfide and H2 could reach 92% and 95%, respectively. To demonstrate the photocatalysis of CdSe QDs, the steady-state emission and typical emission decay profiles of CdSe QDs have been used to study the interaction between organic substrates and catalysts. 3-Mercaptopropionic acid (MPA) significantly reduces the band-edge emission of the QDs and accelerates the rate of emission decay (Fig. 11a and b). Furthermore, EPR spectroscopy confirms the formation of sulfenyl radicals under illumination. According to the control experiment, no spectral signal can be observed before irradiation, and after 10 s of this irradiation (λ > 400 nm), the spectral signals (labeled with *) of a typical sulfenyl radical (RS˙) can be detected (Fig. 11c). Besides, the results of the GC-mass spectrum reveal that the photogenerated sulfenyl radicals are coupled on the CdSe QD surface to generate disulfide products, which then diffuse into the solution. On the basis of the above experimental results, the oxidative coupling mechanism of thiols is proposed in Fig. 11d. Upon illumination, the deprotonated thiols are bound to the surface of CdSe QDs to generate QD/thiolate conjugates containing Cd–S. CdSe QDs are excited by visible light to generate electron–hole pairs and then the adsorbed thiolates reductively quench the excited states of the QDs to produce sulfur-centered radicals (RS˙), which are coupled on the QD surface to form the corresponding disulfides. Meanwhile, the H+ is reduced to H˙ which is stabilized on the QD surfaces to form H2.
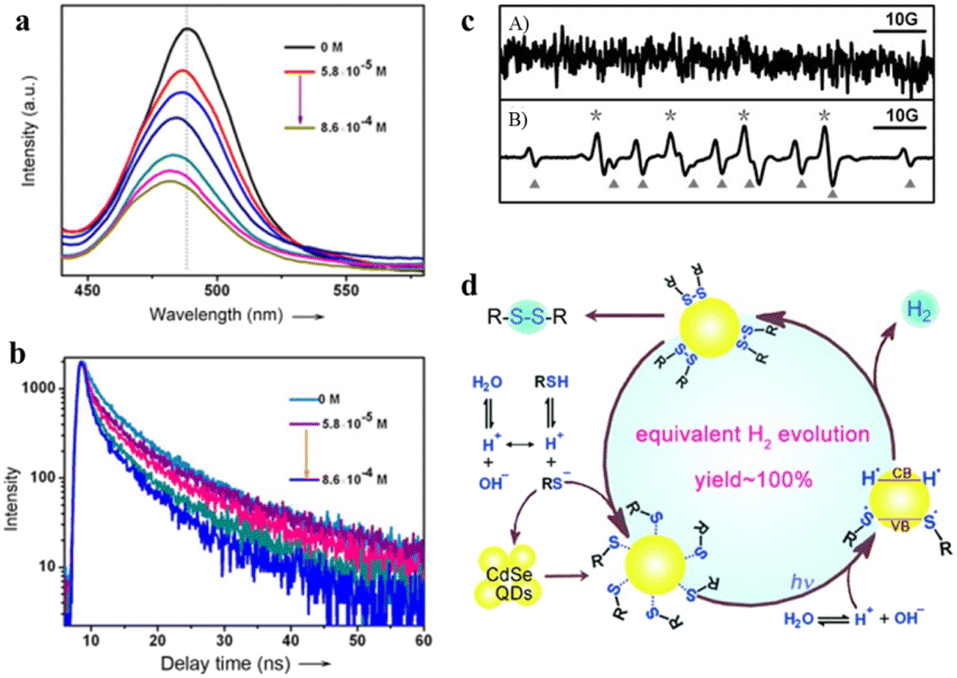 | ||
| Fig. 11 (a) Steady-state emission spectra of CdSe QDs at different MPA concentrations. (b) The emission decay profiles of CdSe QDs. (c) EPR signals before (A) and after (B) irradiation. (d) Mechanism of photocatalytic oxidation of thiols to disulfides and H2. Reprinted with permission from ref. 142. | ||
3.5 Oxidation of amines
Amines are oxidized to synthesize various value-added products including nitriles, oximes, amides, and imines, which constitute the key N-containing building blocks in the contemporary chemical industry. In particular, imines, as important organic synthesis intermediates, play a vital role in the industrial production of fine chemicals like dyestuff and pharmaceuticals. The preparation of imines can be achieved by photocatalytic conversion of amine compounds under aerobic conditions.143,144 For example, Kim and co-workers developed an efficient and facile synthesis strategy to regulate the photocatalytic performance of heteroatom-doped graphene quantum dots (GQDs) by controlling the dopant.140 As shown in Fig. 12a, compared with nitrogen- and oxygen-doped GQDs, nitrogen/sulfur codoped GQDs exhibit much higher activity of photocatalysis for the aerobic oxidative coupling reaction of benzylamine. Besides, Zhu et al. studied in detail the catalytic activity of oxygen-rich carbon quantum dots for aerobic oxidation of amines.146 Li et al., recently, have designed multifunctional graphene QDs decorated with a tertiary amine functional group. Under visible light illumination GQD-DMA exhibits excellent activity and higher photocatalytic efficiency for amine oxidative coupling.147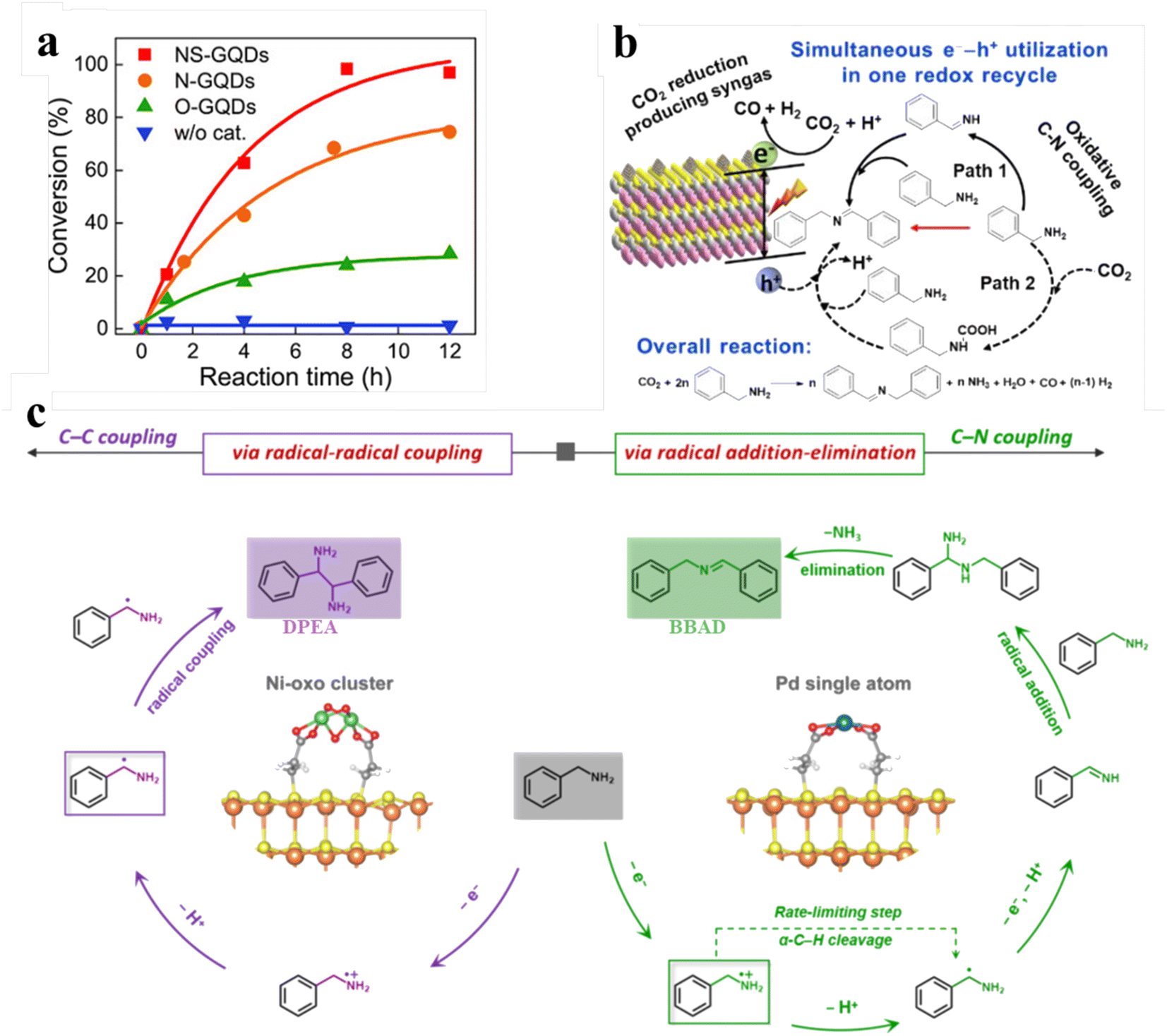 | ||
| Fig. 12 (a) Conversion of benzylamine over NS-GQDs, N-GQDs, and O-GQDs. Reprinted with permission from ref. 140. (b) Illustration of photocatalytic selective benzylamine oxidation integrated with CO2 reduction over (BP/ZIS) heterostructures. Reprinted with permission from ref. 141. (c) Mechanism diagram of photocatalytic selective oxidative coupling of benzylamine over different samples. Reprinted with permission from ref. 42. | ||
Furthermore, our group has reported using a simple electrostatic interaction assembly method to immobilize black phosphorus quantum dots (BP QDs) on the surface of ZnIn2S4 nanosheets (ZIS NSs) to construct BP QDs-ZnIn2S4 (BP/ZIS) heterostructures for oxidative C–N coupling integrated with CO2 reduction (Fig. 12b).141 The experimental results demonstrate that the effective charge transformation in this heterostructure promotes the separation of charge carriers, thereby enhancing the photoactivity of the composite photocatalyst. Very recently, we prepared an atomically dispersed Ni or Pd-decorated CdS/SiO2 composite through in situ photodeposition on the CdS QD surface for the selective photochemical coupling of amines (Fig. 12c).42 Atomically dispersed metal atoms act as reactive sites to manipulate the unselective photogenerated radical conversion to precisely control the dehydrocoupling reaction of benzylamine to C–C or C–N coupling products with excellent activity and selectivity. Specifically, Ph(˙CH)NH2 can produce vicinal diamines through direct C–C coupling over Ni-oxo cluster decorated catalysts, while the Ph(CH)NH produced by the dehydrogenation of Ph(˙CH)NH2 can produce imines by C–N coupling over single Pd atom decorated catalysts. SiO2-supported CdS QDs can catalyze the dehydrocoupling of benzylamine to 1,2-diphenylethylenediamine (DPEA) paired with H2 production under Xe-lamp irradiation. When decorating with Ni, the rate of both H2 evolution and DPEA formation of the Ni-CdS/SiO2 composites improve. Particularly, Ni–CdS/SiO2 can achieve a yield of 90% with the selectivity near 100% after 7 h irradiation. Upon decorating with Pd, Pd–CdS/SiO2 promotes the evolution of H2 and also modulates the selectivity of benzylamine oxidation from DPEA to N-benzylbenzaldimine (BBAD). Their selectivity approaches 100% when it reaches a BBAD production rate of 22.1 mmol gCdS−1 h−1. Moreover, the yield of BBAD on Pd–CdS/SiO2 can reach 99% after 6 h of irradiation.
3.6 Biomass valorization
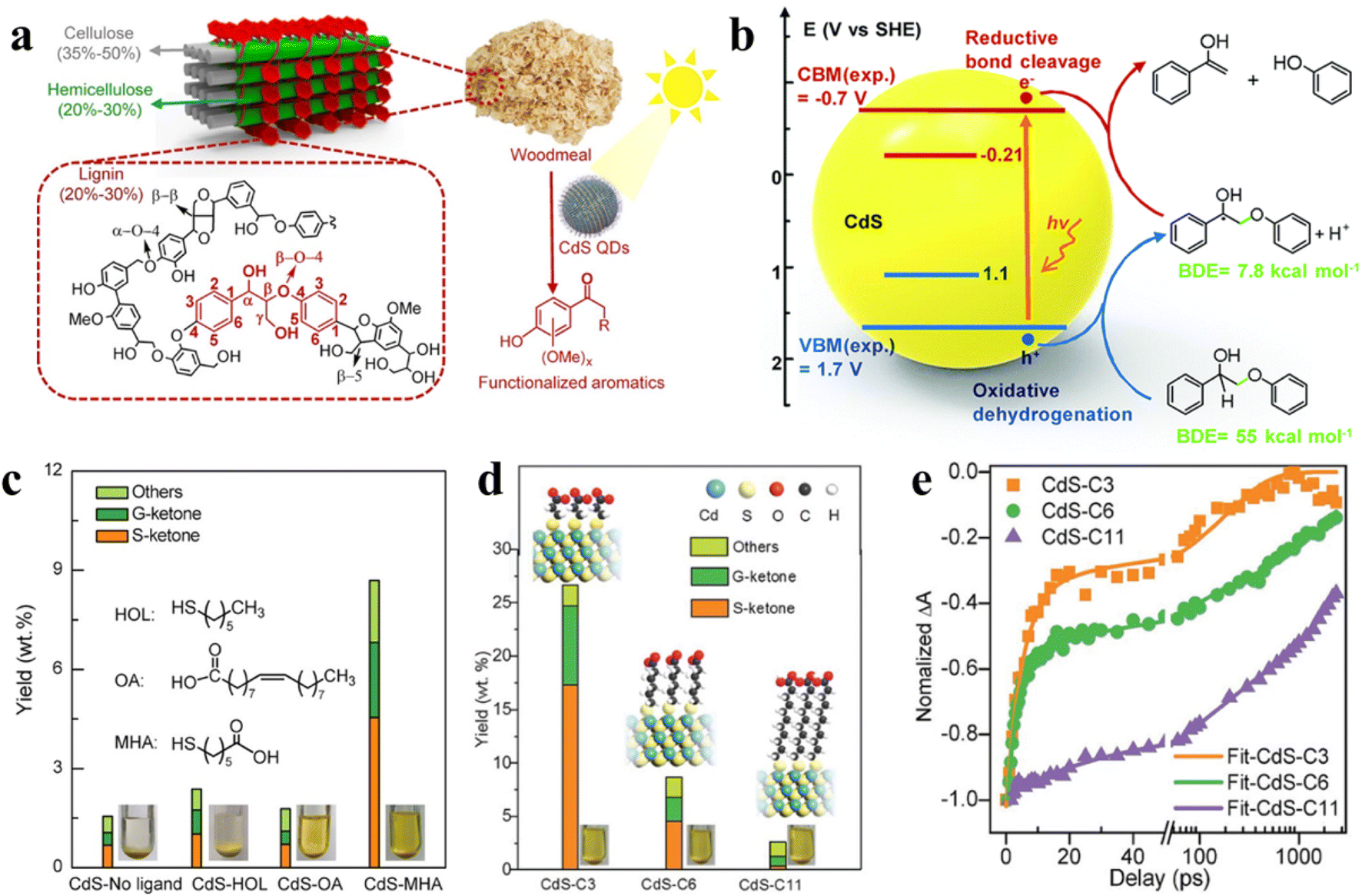 | ||
| Fig. 13 (a) Diagram of the lignocellulose structure. Reprinted with permission from ref. 30. (b) EHCO mechanism for photocatalytic conversion of a dimeric β-O-4 model over CdS QDs. Reprinted with permission from ref. 145. (c) Effect of different surface ligands on photocatalytic valorization of birch woodmeal over CdS QDs. (d) Effect of the length of surface ligands on photocatalytic valorization of birch woodmeal over CdS QDs. (e) Transient absorption spectroscopy. Reprinted with permission from ref. 30. | ||
Metal sulfide QDs, which can effectively catalyze the breaking of C–C or C–O bonds in lignocellulosic biomass, have made some progress in photocatalytic conversion of natural lignin.145,152 For instance, Reisner et al. proposed the application of semiconductor CdS QDs for light-driven transformation of hemicellulose, lignin, and cellulose to H2 in alkaline aqueous solution.148 Recently, Wang et al. developed a method of solubilizing a catalyst to convert natural lignin in birch wood meal with the help of the colloidal nature of CdS QDs.150 Under mild reaction conditions, the selective cleavage of the β-O-4 bond can be induced by CdS QDs with the generation of a Cα radical intermediate via the electron–hole coupled (EHCO) mechanism (Fig. 13b),145,150 which consuming both photogenerated electrons and holes. The main products found in the liquid phase after reaction are functionalized aromatic monomers such as syringyl-derived ketones (S-ketones) and guaiacyl-derived ketones (G-ketones). Subsequently, Wang and co-workers also explored with different surface ligands the effect of different surface ligands for photocatalytic conversion of native lignin over QDs.30 Specifically, the hydrophilic organic ligands make the CdS QDs to be pseudo homogeneous in polar CH3OH/H2O solution, which allows the intimate contact of QDs with the reactant, thus increasing the conversion efficiency of native birch lignin. Instead, the hydrophobic ligands cause CdS QDs to readily aggregate in solvents so that the native lignin conversion rate is not satisfactory (Fig. 13c). In addition, with the decrease in the length of mercaptoalkanoic acid ligands on CdS QDs, the production of aromatic monomers increases (Fig. 13d). The kinetic curves of electron decay indicate that the length of mercaptoalkanoic acid ligands is firmly relevant to the photocatalytic performance of CdS QDs (Fig. 13e). Further kinetic studies reveal that the charge carriers of CdS QDs can be transferred to the β-O-4 bond of native lignin via organic ligands.30
In another example, Cossairt et al. demonstrated that the QDs are versatile for Cα–O bond cleavage of lignin model substrates.73 The lignin model substrates are oxidized or photochemically reduced from benzylic alcohol to 4′-methoxyacetophenone or guaiacol by phenyl ketone intermediates. Moreover, with the help of QD photocatalysts, the lignin model substrates can be successfully converted to obtain the cleavage products without unnecessary purification steps.
Recently, Wu's group demonstrated that semiconductor QDs can interact with tetrahydrofuran (THF) to form CdSe QDs/THF conjugates for photocatalytic selective direct α-C–H bond activation of THF under mild conditions.161 Control experiments and spectroscopic studies prove that the QDs/THF conjugates are formed by combining the oxygen atom of THF with the “vacant sites” of the QD surface. The mechanism for photocatalytic conversion of THF over QDs/THF conjugates is depicted in Fig. 14a. The adsorbed THF molecules are oxidized by photogenerated holes into alkoxyalkyl radicals, while the protons produced by oxidative cleavage of the α-C–H bonds of THF are reduced by photogenerated electrons into H atoms. Then, the produced alkoxyalkyl radical directly undergoes radical cross-coupling with an α-amino radical from the amino C(sp3)–H bond or radical addition with alkene or phenylacetylene (Fig. 14a). Besides the coupling and addition reactions of THF, Wu et al. also presented the ZnSe QDs in the C–C coupling of furfural and revealed that furfural at exposed QD metal sites blocks the competitive reaction (H2 evolution) in photocatalytic CO2 reduction (Fig. 14b).162 According to the mechanistic analysis, the furfural coupling occurs at the exposed surface Zn-sites of QDs, consuming the electrons and protons initially devoted to produce H2. Fig. 14c shows a positive shift in the Zn 2p XPS binding energy after adding furfural into the solution of ZnSe QDs, indicating that the charge density decreases. That is attributed to the migration of electrons from surface Zn to furfural, that is, furfural competes with protons for photogenerated electrons simultaneously. According to the above results, furfural can effectively inhibit the production of H2 over ZnSe QDs.
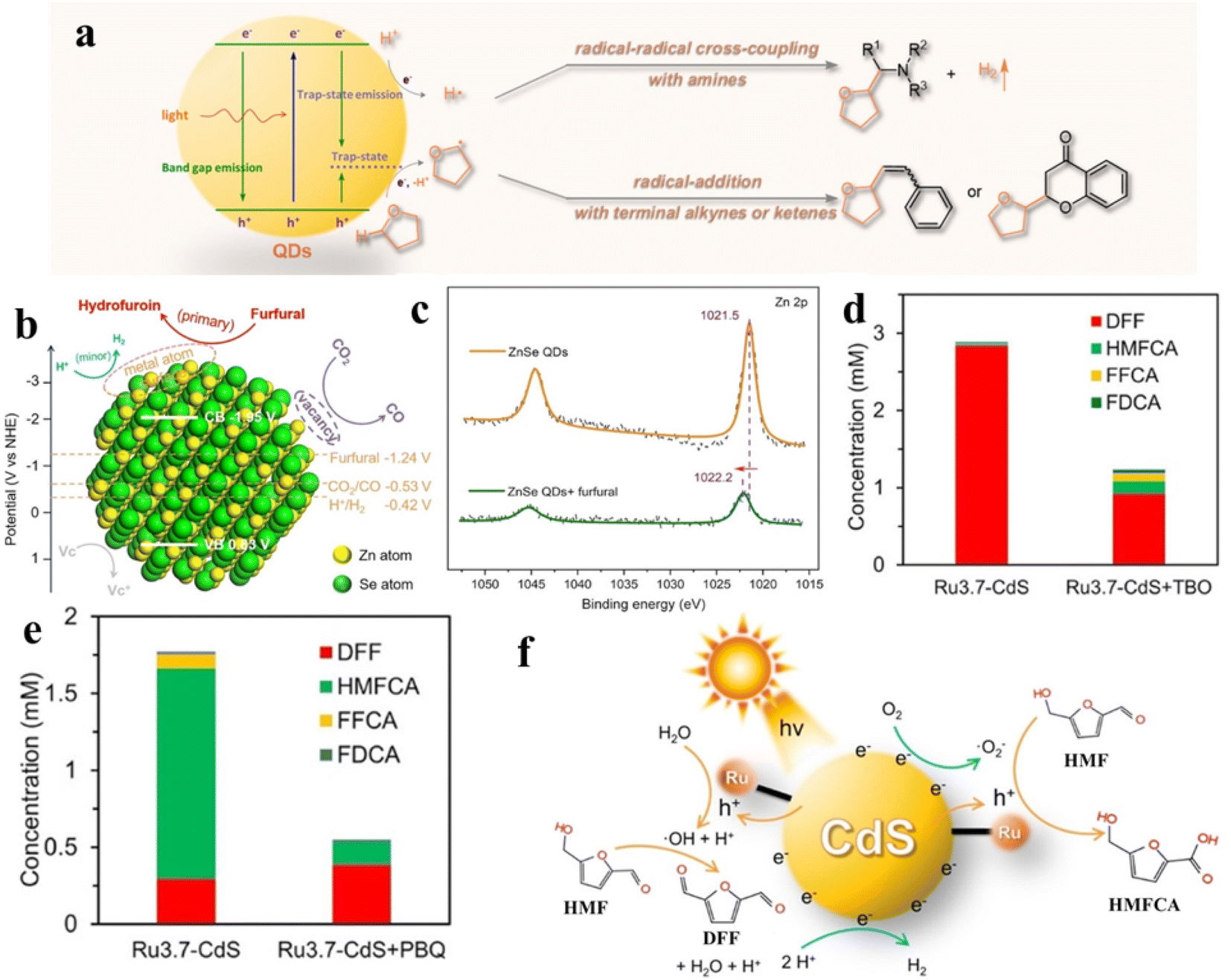 | ||
| Fig. 14 (a) Schematic diagram of the activation of THF on QD surfaces. Reprinted with permission from ref. 161. (b) Mechanism of furfural inhibiting H2 production over ZnSe QDs. (c) X-ray photoelectron spectroscopy (XPS) spectra. Reprinted with permission from ref. 162. Concentration of the products from photocatalytic HMF oxidation over Ru-CdS catalysts (d) under an Ar atmosphere and (e) under an air atmosphere. (f) The mechanism of photocatalytic oxidation of HMF over Ru-CdS. Reprinted with permission from ref. 41. | ||
In addition, Xiong and co-workers designed a novel photocatalyst by anchoring a Ru complex on CdS QDs for selective oxidation of HMF with excellent photocatalytic activity and selectivity.41 The Ru complex can effectively extract photogenerated holes of CdS QDs, thus remarkably improving the photostability of CdS and providing moderate oxidation capacity. By adding different radical scavengers in different atmospheres, it is further proved that the product selectivity of HMF oxidation is intimately connected to ˙OH and ˙O2− radicals. As displayed in Fig. 14d, under an Ar atmosphere, when tert-butanol (TBO) consuming ˙OH is added to the reaction solution, the conversion of HMF to DFF is significantly suppressed. The results suggest that DFF is produced by ˙OH. Similarly, p-benzoquinone (PBQ) is added to consume ˙O2− under an air atmosphere. It can be found that ˙O2− has an impact on the generation of HMFCA and FFCA (Fig. 14e). Through further experimental studies, the catalytic mechanism schematic as shown in Fig. 14f is proposed. The photogenerated holes on CdS QDs are transferred to the Ru complex for the oxidation of water to generate ˙OH under an Ar atmosphere. Instead, ˙O2− is produced by combining photogenerated electrons with O2 on catalysts under an air atmosphere.
4. Conclusions and perspectives
In this review, we discuss the current status of photocatalysis in selective organic synthesis over semiconductor QD-based materials. After briefly introducing the fundamental principles of photoredox organic transformation of QD-based catalysts and the effects of surface chemistry on photocatalytic performance of QDs, we focus on the recent advances in various QD-mediated photoredox organic conversion reactions including nitroaromatic reduction, selective alcohols oxidation, C–H functionalization, sulfide oxidation, amine oxidation, and biomass valorization. Photocatalytic organic conversion based on QDs has made plenty of achievements and has a broad prospect of research. However, research in this area is still in its infancy with both opportunities and several long-standing challenges needing continuous efforts to promote future innovation in organic synthesis on semiconductor QDs.First, the performance of QD catalysts currently used in photoredox catalysis is unsatisfactory. By integrating QDs with cocatalysts, semiconductors, and/or carbon materials, decorating with organic/inorganic ligands, or constructing core/shell hybrid composites, it is promising to develop methods of QD functionalization for more efficient photocatalysts with structural modifiability, exciton controllability, and surface tunability. Generally, improving the activity and selectivity of organic conversion over QD-based photocatalysts can be roughly started from the following aspects: (i) enhancing light absorption and improving light utilization efficiency, (ii) boosting carrier separation/transfer and facilitating surface reaction kinetics, and (iii) manipulating radicals and/or intermediates to form target products. For example, QDs can recycle the scattered light in the near-field of SiO2 through interface interaction to enhance light-harvesting112 or can realize effective charge immigration by engineering intimate interfacial contact via constructing a type II heterostructure or forming a QD conjugate, thus further promoting the photocatalytic performance over QD-based catalysts.
Second, the construction of a cooperative photoredox reaction system for combining oxidative organic synthesis with H2 evolution or CO2 reduction deserves more attention due to the limited activity and selectivity of traditional single-functional photocatalytic reactions. On one hand, in order to select suitable catalysts and construct an efficient catalytic system, the band structure suitable for both oxidation and reduction reactions, the active sites for this dual-functional photoredox reactions, and the efficiency of electron and hole separation and transfer need to be considered. On the other hand, it is necessary to screen the organic substrates matching the energy band of catalysts and take the interaction among organic substrates, radical intermediates, and the photocatalyst surface into consideration to allow the photoexcited holes and electrons on the QD-based catalysts to be consumed simultaneously to produce value-added chemicals and solar fuels such as CO and H2.
Third, continues efforts are needed to enhance the selectivity of target products in the dual-functional photocatalytic system. The selectivity of an organic synthesis reaction can be modulated by optimizing the external reaction conditions including temperature, solvent, light source, reaction time and so on. For example, the ˙O2− derived from O2 in air can manipulate the organic intermediates to produce specific products, which cannot occur in an inert atmosphere. The catalyst modification strategy is also an effective approach to improve the selectivity of organic synthesis. Since the interaction between semiconductor QDs and photogenerated radicals is unselective, the radical conversion can be manipulated by rational design of QD-based catalysts to accurately control the selectivity of organic synthesis reactions, such as cationic/anionic ratio variation, ligand exchange, doping with heteroatoms, and hybridization with cocatalysts or other matchable semiconductors to construct stable heterostructures. The photoredox reaction kinetics can be promoted by regulating the band structure, adjusting the molecular adsorption sites and improving the surface chemical structure.
Last but not least, QDs-based catalysts are multicomponent materials in that a layer of organic ligands inevitably exists on the surface of inorganic core, thus making the system much more complex. To this end, the potential mechanism of various photocatalytic organic transformations over QDs remains to be explored. In situ comprehensive studies can monitor the changes of the structure and surface valence states of the photocatalyst, explore the intermediates formed during the photocatalytic organic conversion reaction, and identify the rate-limiting step, which assist the exploration of the reaction mechanism of diverse redox organic transformations over QD-based photocatalysts. Further study of the organic synthesis mechanism is practically necessary to reveal the influence of material morphology, structure, composition, reaction conditions and other related factors on the efficiency and selectivity of organic conversion of QD-based catalysts, and offers a new sparkling idea for the more rational design and development of QD-based artificial photocatalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support from the National Natural Science Foundation of China (NSFC) (22172030, 22072023, 21872029, U1463204, and 21173045), the Program for National Science and Technology Innovation Leading Talents (00387072), the Program for Leading Talents of Fujian Universities, the first Program of Fujian Province for Top Creative Young Talents, and the Natural Science Foundation of Fujian Province (2017J07002 and 2019J01631) is acknowledged.References
- P. Riente and T. Noël, Catal. Sci. Technol., 2019, 9, 5186–5232 RSC.
- J.-Y. Li, Y.-H. Li, M.-Y. Qi, Q. Lin, Z.-R. Tang and Y.-J. Xu, ACS Catal., 2020, 10, 6262–6280 CrossRef CAS.
- M.-Y. Qi, M. Conte, M. Anpo, Z.-R. Tang and Y.-J. Xu, Chem. Rev., 2021, 121, 13051–13085 CrossRef CAS PubMed.
- R. Akiyama and S. Kobayashi, Chem. Rev., 2009, 109, 594–642 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812–847 CrossRef CAS PubMed.
- M. Cherevatskaya, M. Neumann, S. Füldner, C. Harlander, S. Kümmel, S. Dankesreiter, A. Pfitzner, K. Zeitler and B. König, Angew. Chem., Int. Ed., 2012, 51, 4062–4066 CrossRef CAS PubMed.
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Nat. Rev. Chem., 2018, 2, 160–173 CrossRef CAS.
- Y. Yuan, N. Jin, P. Saghy, L. Dube, H. Zhu and O. Chen, J. Phys. Chem. Lett., 2021, 12, 7180–7193 CrossRef CAS PubMed.
- H. H. Wei, C. M. Evans, B. D. Swartz, A. J. Neukirch, J. Young, O. V. Prezhdo and T. D. Krauss, Nano Lett., 2012, 12, 4465–4471 CrossRef CAS PubMed.
- R. D. Harris, S. Bettis Homan, M. Kodaimati, C. He, A. B. Nepomnyashchii, N. K. Swenson, S. Lian, R. Calzada and E. A. Weiss, Chem. Rev., 2016, 116, 12865–12919 CrossRef CAS PubMed.
- J. Luo, S. Zhang, M. Sun, L. Yang, S. Luo and J. C. Crittenden, ACS Nano, 2019, 13, 9811–9840 CrossRef CAS PubMed.
- M. S. Kodaimati, K. P. McClelland, C. He, S. Lian, Y. Jiang, Z. Zhang and E. A. Weiss, Inorg. Chem., 2018, 57, 3659–3670 CrossRef CAS PubMed.
- K. V. Vokhmintcev, P. S. Samokhvalov and I. Nabiev, Nano Today, 2016, 11, 189–211 CrossRef CAS.
- H. Lu, Z. Huang, M. S. Martinez, J. C. Johnson, J. M. Luther and M. C. Beard, Energy Environ. Sci., 2020, 13, 1347–1376 RSC.
- Q. Shen, K. Katayama and T. Toyoda, J. Energy Chem., 2015, 24, 712–716 CrossRef.
- Y. Barak, I. Meir, A. Shapiro, Y. Jang and E. Lifshitz, Adv. Mater., 2018, 30, 1801442 CrossRef PubMed.
- W. M. Witzel, A. Shabaev, C. S. Hellberg, V. L. Jacobs and A. L. Efros, Phys. Rev. Lett., 2010, 105, 137401 CrossRef PubMed.
- R. J. Ellingson, M. C. Beard, J. C. Johnson, P. Yu, O. I. Micic, A. J. Nozik, A. Shabaev and A. L. Efros, Nano Lett., 2005, 5, 865–871 CrossRef CAS PubMed.
- C. R. Kagan, L. C. Bassett, C. B. Murray and S. M. Thompson, Chem. Rev., 2021, 121, 3186–3233 CrossRef CAS PubMed.
- H. Zhu, Y. Yang and T. Lian, Acc. Chem. Res., 2013, 46, 1270–1279 CrossRef CAS PubMed.
- A. J. Nozik, M. C. Beard, J. M. Luther, M. Law, R. J. Ellingson and J. C. Johnson, Chem. Rev., 2010, 110, 6873–6890 CrossRef CAS PubMed.
- Y. Yang, W. Rodríguez-Córdoba and T. Lian, Nano Lett., 2012, 12, 4235–4241 CrossRef CAS PubMed.
- S. J. O. Hardman, D. M. Graham, S. K. Stubbs, B. F. Spencer, E. A. Seddon, H.-T. Fung, S. Gardonio, F. Sirotti, M. G. Silly, J. Akhtar, P. O'Brien, D. J. Binks and W. R. Flavell, Phys. Chem. Chem. Phys., 2011, 13, 20275–20283 RSC.
- S. C. Jensen, S. Bettis Homan and E. A. Weiss, J. Am. Chem. Soc., 2016, 138, 1591–1600 CrossRef CAS PubMed.
- S. Hou, M.-H. Huang, Y.-B. Li, S. Xu, X. Lin, X.-Y. Fu and F.-X. Xiao, Inorg. Chem., 2020, 59, 16654–16664 CrossRef CAS PubMed.
- Y. Li, T.-T. Zhuang, F. Fan, O. Voznyy, M. Askerka, H. Zhu, L. Wu, G.-Q. Liu, Y.-X. Pan, E. H. Sargent and S.-H. Yu, Nat. Commun., 2018, 9, 4947 CrossRef PubMed.
- L.-M. Zhao, Q.-Y. Meng, X.-B. Fan, C. Ye, X.-B. Li, B. Chen, V. Ramamurthy, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 3020–3024 CrossRef CAS PubMed.
- X. Wu, S. Xie, C. Liu, C. Zhou, J. Lin, J. Kang, Q. Zhang, Z. Wang and Y. Wang, ACS Catal., 2019, 9, 8443–8451 CrossRef CAS.
- A. Pal, I. Ghosh, S. Sapra and B. König, Chem. Mater., 2017, 29, 5225–5231 CrossRef CAS.
- M.-H. Huang, X.-C. Dai, T. Li, Y.-B. Li, Y. He, G. Xiao and F.-X. Xiao, J. Phys. Chem. C, 2019, 123, 9721–9734 CrossRef CAS.
- F.-X. Xiao, J. Miao, H.-Y. Wang and B. Liu, J. Mater. Chem. A, 2013, 1, 12229–12238 RSC.
- Y. Jiang, C. Wang, C. R. Rogers, M. S. Kodaimati and E. A. Weiss, Nat. Chem., 2019, 11, 1034–1040 CrossRef CAS PubMed.
- J. Hu, T.-J. Pu, Z.-W. Xu, W.-Y. Xu and Y.-S. Feng, Adv. Synth. Catal., 2019, 361, 708–713 CrossRef CAS.
- C. Huang, X.-B. Li, C.-H. Tung and L.-Z. Wu, Chem.–Eur. J., 2018, 24, 11530–11534 CrossRef CAS PubMed.
- Y. Yuan, H. Zhu, K. Hills-Kimball, T. Cai, W. Shi, Z. Wei, H. Yang, Y. Candler, P. Wang, J. He and O. Chen, Angew. Chem., Int. Ed., 2020, 59, 22563–22569 CrossRef CAS PubMed.
- M.-Y. Qi, Q. Lin, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2022, 307, 121158 CrossRef CAS.
- Z.-W. Xi, L. Yang, D.-Y. Wang, C.-D. Pu, Y.-M. Shen, C.-D. Wu and X.-G. Peng, J. Org. Chem., 2018, 83, 11886–11895 CrossRef CAS PubMed.
- X. Li, J. Wang, Y. Men and Z. Bian, Appl. Catal., B, 2016, 187, 115–121 CrossRef CAS.
- T. Xia, W. Gong, Y. Chen, M. Duan, J. Ma, X. Cui, Y. Dai, C. Gao and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202204225 CAS.
- M.-Y. Qi, M. Conte, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2022, 16, 17444–17453 CrossRef CAS PubMed.
- S. Wang, S. Tang and A. Lei, Sci. Bull., 2018, 63, 1006–1009 CrossRef CAS PubMed.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- S. Ji, Y. Chen, S. Zhao, W. Chen, L. Shi, Y. Wang, J. Dong, Z. Li, F. Li, C. Chen, Q. Peng, J. Li, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2019, 58, 4271–4275 CrossRef CAS PubMed.
- L. Wang, W. Zhang, S. Wang, Z. Gao, Z. Luo, X. Wang, R. Zeng, A. Li, H. Li, M. Wang, X. Zheng, J. Zhu, W. Zhang, C. Ma, R. Si and J. Zeng, Nat. Commun., 2016, 7, 14036 CrossRef CAS PubMed.
- Z. Liu, F. Huang, M. Peng, Y. Chen, X. Cai, L. Wang, Z. Hu, X. Wen, N. Wang, D. Xiao, H. Jiang, H. Sun, H. Liu and D. Ma, Nat. Commun., 2021, 12, 6194 CrossRef PubMed.
- A. M. Smith and S. Nie, Acc. Chem. Res., 2010, 43, 190–200 CrossRef CAS PubMed.
- D. A. Wheeler and J. Z. Zhang, Adv. Mater., 2013, 25, 2878–2896 CrossRef CAS PubMed.
- S. K. Haram, A. Kshirsagar, Y. D. Gujarathi, P. P. Ingole, O. A. Nene, G. B. Markad and S. P. Nanavati, J. Phys. Chem. C, 2011, 115, 6243–6249 CrossRef CAS.
- F.-K. Shang, M.-Y. Qi, C.-L. Tan, Z.-R. Tang and Y.-J. Xu, ACS Phys. Chem. Au, 2022, 2, 216–224 CrossRef CAS.
- X. Lin, S.-H. Li, K.-Q. Lu, Z.-R. Tang and Y.-J. Xu, New J. Chem., 2018, 42, 14096–14103 RSC.
- M.-H. Sun, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2023, 321, 122019 CrossRef CAS.
- S. V. Kilina, P. K. Tamukong and D. S. Kilin, Acc. Chem. Res., 2016, 49, 2127–2135 CrossRef CAS PubMed.
- J. Jasieniak and P. Mulvaney, J. Am. Chem. Soc., 2007, 129, 2841–2848 CrossRef CAS PubMed.
- S. Chen, Y. Qi, C. Li, K. Domen and F. Zhang, Joule, 2018, 2, 2260–2288 CrossRef CAS.
- E. Hofman, R. J. Robinson, Z.-J. Li, B. Dzikovski and W. Zheng, J. Am. Chem. Soc., 2017, 139, 8878–8885 CrossRef CAS PubMed.
- S. V. Kershaw, L. Jing, X. Huang, M. Gao and A. L. Rogach, Mater. Horiz., 2017, 4, 155–205 RSC.
- D. Liu, J. Nyakuchena, R. Maity, X. Geng, J. P. Mahajan, C. C. Hewa-Rahinduwage, Y. Peng, J. Huang and L. Luo, Chem. Commun., 2022, 58, 11260–11263 RSC.
- E. A. Weiss, ACS Energy Lett., 2017, 2, 1005–1013 CrossRef CAS.
- Z. Zhang, K. Edme, S. Lian and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 4246–4249 CrossRef CAS PubMed.
- Y. Zhu, T. Jin, T. Lian and E. Egap, J. Chem. Phys., 2021, 154, 204903 CrossRef CAS PubMed.
- K. A. Perez, C. R. Rogers and E. A. Weiss, Angew. Chem., Int. Ed., 2020, 59, 14091–14095 CrossRef CAS PubMed.
- A. Nag, D. S. Chung, D. S. Dolzhnikov, N. M. Dimitrijevic, S. Chattopadhyay, T. Shibata and D. V. Talapin, J. Am. Chem. Soc., 2012, 134, 13604–13615 CrossRef CAS PubMed.
- X.-B. Li, B. Liu, M. Wen, Y.-J. Gao, H.-L. Wu, M.-Y. Huang, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Adv. Sci., 2016, 3, 1500282 CrossRef PubMed.
- Y. Bao, J. Wang, Q. Wang, X. Cui, R. Long and Z. Li, Nanoscale, 2020, 12, 2507–2514 RSC.
- B. M. Graff, B. P. Bloom, E. Wierzbinski and D. H. Waldeck, J. Am. Chem. Soc., 2016, 138, 13260–13270 CrossRef CAS PubMed.
- P. R. Brown, D. Kim, R. R. Lunt, N. Zhao, M. G. Bawendi, J. C. Grossman and V. Bulović, ACS Nano, 2014, 8, 5863–5872 CrossRef CAS PubMed.
- L. K. Putri, B.-J. Ng, W.-J. Ong, H. W. Lee, W. S. Chang, A. R. Mohamed and S.-P. Chai, Appl. Catal., B, 2020, 265, 118592 CrossRef CAS.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef CAS PubMed.
- T. X. Ding, J. H. Olshansky, S. R. Leone and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 2021–2029 CrossRef CAS PubMed.
- J. H. Olshansky, T. X. Ding, Y. V. Lee, S. R. Leone and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 15567–15575 CrossRef CAS PubMed.
- M. J. Enright, K. Gilbert-Bass, H. Sarsito and B. M. Cossairt, Chem. Mater., 2019, 31, 2677–2682 CrossRef CAS.
- X.-B. Fan, S. Yu, X. Wang, Z.-J. Li, F. Zhan, J.-X. Li, Y.-j. Gao, A.-D. Xia, Y. Tao, X.-B. Li, L.-P. Zhang, C.-H. Tung and L.-Z. Wu, Adv. Mater., 2019, 31, 1804872 CrossRef PubMed.
- X. Lin, Z.-Q. Wei, Q.-L. Mo, S. Hou, S. Xu, X.-Y. Fu and F.-X. Xiao, J. Catal., 2021, 400, 28–39 CrossRef CAS.
- P. Wang, D. Li, J. Chen, X. Zhang, J. Xian, X. Yang, X. Zheng, X. Li and Y. Shao, Appl. Catal., B, 2014, 160–161, 217–226 CrossRef CAS.
- P. Wang, X. Li, J. Fang, D. Li, J. Chen, X. Zhang, Y. Shao and Y. He, Appl. Catal., B, 2016, 181, 838–847 CrossRef CAS.
- D. I. Gittins and F. Caruso, Angew. Chem., Int. Ed., 2001, 40, 3001–3004 CrossRef CAS PubMed.
- M.-Q. Yang, N. Zhang, M. Pagliaro and Y.-J. Xu, Chem. Soc. Rev., 2014, 43, 8240–8254 RSC.
- F.-K. Shang, Y.-H. Li, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Catal. Today, 2023, 410, 85–101 CrossRef CAS.
- A. Li, W. Zhu, C. Li, T. Wang and J. Gong, Chem. Soc. Rev., 2019, 48, 1874–1907 RSC.
- H.-L. Wu, X.-B. Li, C.-H. Tung and L.-Z. Wu, Adv. Mater., 2019, 31, 1900709 CrossRef PubMed.
- G. Gao, Q. Xi, H. Zhou, Y. Zhao, C. Wu, L. Wang, P. Guo and J. Xu, Nanoscale, 2017, 9, 12032–12038 RSC.
- R. S. Selinsky, Q. Ding, M. S. Faber, J. C. Wright and S. Jin, Chem. Soc. Rev., 2013, 42, 2963–2985 RSC.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843 CrossRef PubMed.
- B. Pal, T. Torimoto, K.-i. Okazaki and B. Ohtani, Chem. Commun., 2007, 5, 483–485 RSC.
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2019, 58, 10804–10811 CrossRef CAS PubMed.
- C. Chu, S. Rao, Z. Ma and H. Han, Appl. Catal., B, 2019, 256, 117792 CrossRef CAS.
- C. Zhang, T. Li, J. Zhang, S. Yan and C. Qin, Appl. Catal., B, 2019, 259, 118030 CrossRef CAS.
- C. M. Malengreaux, S. L. Pirard, J. R. Bartlett and B. Heinrichs, Chem. Eng. J., 2014, 245, 180–190 CrossRef CAS.
- W.-Z. Gao, Y. Xu, Y. Chen and W.-F. Fu, Chem. Commun., 2015, 51, 13217–13220 RSC.
- H.-U. Blaser, Science, 2006, 313, 312–313 CrossRef CAS PubMed.
- C. Yu, B. Liu and L. Hu, J. Org. Chem., 2001, 66, 919–924 CrossRef CAS PubMed.
- P. Eskandari, F. Kazemi and Z. Zand, J. Photochem. Photobiol., A, 2014, 274, 7–12 CrossRef CAS.
- N.-N. Chai, H.-X. Wang, C.-X. Hu, Q. Wang and H.-L. Zhang, J. Mater. Chem. A, 2015, 3, 16613–16620 RSC.
- Y.-C. Nie, F. Yu, L.-C. Wang, Q.-J. Xing, X. Liu, Y. Pei, J.-P. Zou, W.-L. Dai, Y. Li and S. L. Suib, Appl. Catal., B, 2018, 227, 312–321 CrossRef CAS.
- H. Liu, H. Wang, Y. Qian, J. Zhuang, L. Hu, Q. Chen and S. Zhou, ACS Appl. Nano Mater., 2019, 2, 7043–7050 CrossRef CAS.
- A. W. Mureithi, Y. Sun, T. Mani, A. R. Howell and J. Zhao, Cell Rep. Phys. Sci., 2022, 3, 100889 CrossRef CAS.
- P. Wang, X. Zhou, Y. Shao, D. Li, Z. Zuo and X. Liu, J. Colloid Interface Sci., 2021, 601, 186–195 CrossRef CAS PubMed.
- C. Han, Z. Chen, N. Zhang, J. C. Colmenares and Y.-J. Xu, Adv. Funct. Mater., 2015, 25, 221–229 CrossRef CAS.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2011, 5, 7426–7435 CrossRef CAS PubMed.
- G. Palmisano, E. García-López, G. Marcì, V. Loddo, S. Yurdakal, V. Augugliaro and L. Palmisano, Chem. Commun., 2010, 46, 7074–7089 RSC.
- M. Addamo, V. Augugliaro, M. Bellardita, A. Di Paola, V. Loddo, G. Palmisano, L. Palmisano and S. Yurdakal, Catal. Lett., 2008, 126, 58–62 CrossRef CAS.
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed.
- G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425–3437 RSC.
- M. Zhang, Q. Wang, C. Chen, L. Zang, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2009, 48, 6081–6084 CrossRef CAS PubMed.
- Q. Wang, M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7976–7979 CrossRef CAS PubMed.
- C. Aellig, C. Girard and I. Hermans, Angew. Chem., Int. Ed., 2011, 50, 12355–12360 CrossRef CAS PubMed.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, Chem. Sci., 2012, 3, 2812–2822 RSC.
- M.-Y. Qi, Y.-H. Li, M. Anpo, Z.-R. Tang and Y.-J. Xu, ACS Catal., 2020, 10, 14327–14335 CrossRef CAS.
- K. P. McClelland and E. A. Weiss, ACS Appl. Energy Mater., 2019, 2, 92–96 CrossRef CAS.
- I. Tamiolakis, I. N. Lykakis and G. S. Armatas, Catal. Today, 2015, 250, 180–186 CrossRef CAS.
- H. Li, R. Liu, S. Lian, Y. Liu, H. Huang and Z. Kang, Nanoscale, 2013, 5, 3289–3297 RSC.
- Q. Guo, F. Liang, X.-B. Li, Y.-J. Gao, M.-Y. Huang, Y. Wang, S.-G. Xia, X.-Y. Gao, Q.-C. Gan, Z.-S. Lin, C.-H. Tung and L.-Z. Wu, Chem, 2019, 5, 2605–2616 CAS.
- S. Yu, Z.-J. Li, X.-B. Fan, J.-X. Li, F. Zhan, X.-B. Li, Y. Tao, C.-H. Tung and L.-Z. Wu, ChemSusChem, 2015, 8, 642–649 CrossRef CAS PubMed.
- G. Borah, A. Borborah and N. Gogoi, Bull. Mater. Sci., 2022, 45, 136 CrossRef CAS.
- T. G. Frihed, M. Bols and C. M. Pedersen, Eur. J. Org. Chem., 2016, 2016, 2740–2756 CrossRef CAS.
- N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS.
- J. A. Caputo, L. C. Frenette, N. Zhao, K. L. Sowers, T. D. Krauss and D. J. Weix, J. Am. Chem. Soc., 2017, 139, 4250–4253 CrossRef CAS PubMed.
- C. Huang, J. Qiao, R.-N. Ci, X.-Z. Wang, Y. Wang, J.-H. Wang, B. Chen, C.-H. Tung and L.-Z. Wu, Chem, 2021, 7, 1244–1257 CAS.
- C. Huang, R.-N. Ci, J. Qiao, X.-Z. Wang, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 11779–11783 CrossRef CAS PubMed.
- Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- Q.-J. Yao and B.-F. Shi, Chem, 2021, 7, 1405–1406 CAS.
- N. Sundaravelu, S. Sangeetha and G. Sekar, Org. Biomol. Chem., 2021, 19, 1459–1482 RSC.
- C.-F. Lee, R. S. Basha and S. S. Badsara, Top. Curr. Chem., 2018, 376, 25 CrossRef PubMed.
- G. Liang, J.-H. Wang, T. Lei, Y.-Y. Cheng, C. Zhou, Y.-J. Chen, C. Ye, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2021, 23, 8082–8087 CrossRef CAS PubMed.
- G. Zhang, C. Liu, H. Yi, Q. Meng, C. Bian, H. Chen, J.-X. Jian, L.-Z. Wu and A. Lei, J. Am. Chem. Soc., 2015, 137, 9273–9280 CrossRef CAS PubMed.
- J. Kim, B. Kang and S. H. Hong, ACS Catal., 2020, 10, 6013–6022 CrossRef CAS.
- T. Kondo and T.-a. Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS PubMed.
- J. V. Burykina, N. S. Shlapakov, E. G. Gordeev, B. König and V. P. Ananikov, Chem. Sci., 2020, 11, 10061–10070 RSC.
- N. Velasco, C. Virumbrales, R. Sanz, S. Suárez-Pantiga and M. A. Fernández-Rodríguez, Org. Lett., 2018, 20, 2848–2852 CrossRef CAS PubMed.
- R. B. Wagh, S. H. Gund and J. M. Nagarkar, J. Chem. Sci., 2016, 128, 1321–1325 CrossRef CAS.
- H. Shinkai, K. Maeda, T. Yamasaki, H. Okamoto and I. Uchida, J. Med. Chem., 2000, 43, 3566–3572 CrossRef CAS PubMed.
- F. Mangiavacchi, L. Crociani, L. Sancineto, F. Marini and C. Santi, Molecules, 2020, 25, 2711 CrossRef CAS PubMed.
- J. K. Vandavasi, W.-P. Hu, C.-Y. Chen and J.-J. Wang, Tetrahedron, 2011, 67, 8895–8901 CrossRef CAS.
- C. M. Bernt, P. T. Burks, A. W. DeMartino, A. E. Pierri, E. S. Levy, D. F. Zigler and P. C. Ford, J. Am. Chem. Soc., 2014, 136, 2192–2195 CrossRef CAS PubMed.
- S.-J. Jeon, T.-W. Kang, J.-M. Ju, M.-J. Kim, J. H. Park, F. Raza, J. Han, H.-R. Lee and J.-H. Kim, Adv. Funct. Mater., 2016, 26, 8211–8219 CrossRef CAS.
- C. Han, Y.-H. Li, J.-Y. Li, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Angew. Chem., Int. Ed., 2021, 60, 7962–7970 CrossRef CAS PubMed.
- X.-B. Li, Z.-J. Li, Y.-J. Gao, Q.-Y. Meng, S. Yu, R. G. Weiss, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2014, 53, 2085–2089 CrossRef CAS PubMed.
- R. Ray, A. S. Hazari, G. K. Lahiri and D. Maiti, Chem.–Asian J., 2018, 13, 2138–2148 CrossRef CAS PubMed.
- Y. Xu, Y. Chen and W.-F. Fu, Appl. Catal., B, 2018, 236, 176–183 CrossRef CAS.
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198–6223 RSC.
- J. Ye, K. Ni, J. Liu, G. Chen, M. Ikram and Y. Zhu, ChemCatChem, 2018, 10, 259–265 CrossRef CAS.
- X. Tu, Q. Wang, F. Zhang, F. Lan, H. Liu and R. Li, Nanoscale, 2020, 12, 4410–4417 RSC.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- X. Wu, S. Xie, H. Zhang, Q. Zhang, B. F. Sels and Y. Wang, Adv. Mater., 2021, 33, 2007129 CrossRef CAS PubMed.
- S.-H. Li, S. Liu, J. C. Colmenares and Y.-J. Xu, Green Chem., 2016, 18, 594–607 RSC.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- N. Luo, M. Wang, H. Li, J. Zhang, H. Liu and F. Wang, ACS Catal., 2016, 6, 7716–7721 CrossRef CAS.
- N. Luo, M. Wang, H. Li, J. Zhang, T. Hou, H. Chen, X. Zhang, J. Lu and F. Wang, ACS Catal., 2017, 7, 4571–4580 CrossRef CAS.
- I. Bosque, G. Magallanes, M. Rigoulet, M. D. Kärkäs and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef CAS PubMed.
- I. F. Teixeira, B. T. W. Lo, P. Kostetskyy, M. Stamatakis, L. Ye, C. C. Tang, G. Mpourmpakis and S. C. E. Tsang, Angew. Chem., Int. Ed., 2016, 55, 13061–13066 CrossRef CAS PubMed.
- N. Luo, T. Montini, J. Zhang, P. Fornasiero, E. Fonda, T. Hou, W. Nie, J. Lu, J. Liu, M. Heggen, L. Lin, C. Ma, M. Wang, F. Fan, S. Jin and F. Wang, Nat. Energy, 2019, 4, 575–584 CrossRef CAS.
- Y.-H. Li, F. Zhang, Y. Chen, J.-Y. Li and Y.-J. Xu, Green Chem., 2020, 22, 163–169 RSC.
- J. Qiao, Z.-Q. Song, C. Huang, R.-N. Ci, Z. Liu, B. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 27201–27205 CrossRef CAS PubMed.
- Z.-K. Xin, M.-Y. Huang, Y. Wang, Y.-J. Gao, Q. Guo, X.-B. Li, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2022, 61, e202207222 CAS.
| This journal is © The Royal Society of Chemistry 2023 |