
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign strategy and research progress of NaTi2(PO4)3 anode/electrolyte interface in aqueous sodium ion batteries
Chenyang Zhaoa,
Ying Liua,
Yang Shaoa,
Feng Lia,
Dong Zhaob,
Xiaoping Yang *b,
Fang Cheng*b and
Zhengfu Zhang*a
*b,
Fang Cheng*b and
Zhengfu Zhang*a
aFaculty of Materials Science and Engineering, Kunming University of Science and Technology, Kunming, 650093, P. R. China. E-mail: zhang-zhengfu@163.com
bFaculty of Metallurgical and Energy Engineering, Kunming University of Science and Technology, Kunming, 650093, P. R. China. E-mail: yangxiaoping@kust.edu.cn; chengfang@kust.edu.cn
First published on 18th June 2025
Abstract
Aqueous sodium-ion batteries (ASIBs) have emerged as promising candidates for large-scale energy storage systems due to their superior safety, cost-effectiveness and environmental friendliness. Among various anode materials, sodium titanium phosphate (NaTi2(PO4)3, NTP) as a NASICON-type compound with its high theoretical capacity, excellent sodium ion conductivity and good structural stability. However, the electrochemical performance of NTP anodes used for ASIBs is significantly hindered by electrode–electrolyte interface instability resulting from the hydrogen evolution reaction (HER), electrode dissolution and unstable solid electrolyte interphase (SEI) in aqueous electrolytes. This review systematically outlines recent advances and technological innovations in the design strategies of NTP anode/electrolyte interfaces to address the previously underexplored interfacial challenges between NTP anode materials and aqueous electrolytes in ASIBs. Subsequently, the proposed solutions, including electrolyte compositional optimization, interfacial coating modification and SEI interface modulation, to the abovementioned issues are correspondingly summarized and discussed. Finally, the development direction and future prospective of NTP anode/electrolyte interface research is further discussed, providing a guidance for the design of high-performance ASIBs.
1 Introduction
Throughout the industrialization process, traditional fossil fuels, such as coal, oil, and natural gas have long dominated the global energy supply. These fuels were formed over geological timescales and release greenhouse gases including carbon dioxide, methane, and nitrous oxide during combustion, which are considered to be major contributors to global warming.1,2 It is worth noting that the depletion of these non-renewable resources exhibits a significant difference from the increasing trend of global energy demand. In this context, the development and utilization of green renewable energy sources, such as wind, solar, and tidal power, are increasing due to their environmental benefits and widespread availability.3–5However, these energy sources face critical challenges, including uneven geographical distribution and inherent intermittency, which render their generated electricity incompatible with direct grid integration.6 Dynamic characteristics such as diurnal cycles and seasonal fluctuations further impose stringent demands on existing energy storage technologies. Due to the global energy structure transition and the rapid development of renewable energy, efficient, safe, and low-cost energy storage technologies have become a research hotspot.6,7 Energy storage can be achieved through various methods, including chemical, electrochemical, electrical, mechanical, and thermodynamic energy storage.8 Unlike conventional methods such as pumped hydro, electrochemical energy storage technology has become a critical component of modern energy systems due to its advantages, including adaptability to different locations, short construction times, quick response, and high efficiency.6,9,10 These attributes make it essential for integrating renewable energy sources and enhancing grid stability in modern power infrastructures. Electrochemical energy storage uses reversible chemical reactions to convert chemical energy into electrical energy and vice versa, thus enabling energy storage.6,11 Based on electrode materials, these systems are mainly categorized into sodium-ion batteries (SIBs), lithium-ion batteries (LIBs), lead–acid batteries (LAs), sodium–sulfur batteries (NaS), nickel–cadmium batteries (Ni–Cd), and nickel–zinc batteries (Ni–Zn). Table 1 compares the performance characteristics of these secondary batteries. Among the secondary battery systems being investigated, LIBs are widely used in energy storage systems due to their high energy density, large capacity, long cycle life, and fast charge/discharge capabilities.12 However, the sustainable development of this technology faces geological constraints due to lithium's relatively low crustal abundance of only 20 ppm. In contrast, sodium is the fourth most abundant element in Earth's crust.13 Therefore, SIBs have emerged as promising candidates for large-scale energy storage systems, owing to their abundant sodium resources and economic viability.14–16 Their electrochemical mechanisms, analogous to LIBs, further enhance their practicality.17,18
| Secondary battery types | Specific energy [Wh kg−1] | Efficiency [%] | Lifetime (cycles) | Nominal cell voltage |
|---|---|---|---|---|
| SIBs | 80–150 | 80–85 | 3000 | 2.8–3.5 |
| LIBs | 200 | 85–100 | 1000–4500 | 3.6–3.8 |
| LAs | 35 | 70–90 | 500–1000 | 2.1 |
| NaS | 100–250 | 75–90 | 2500 | 2.78–3.2 |
| Ni–Cd | 50 | 70–80 | 1500 | 1.2 |
| Ni–Zn | 75 | 70 | 500 | 1.73 |
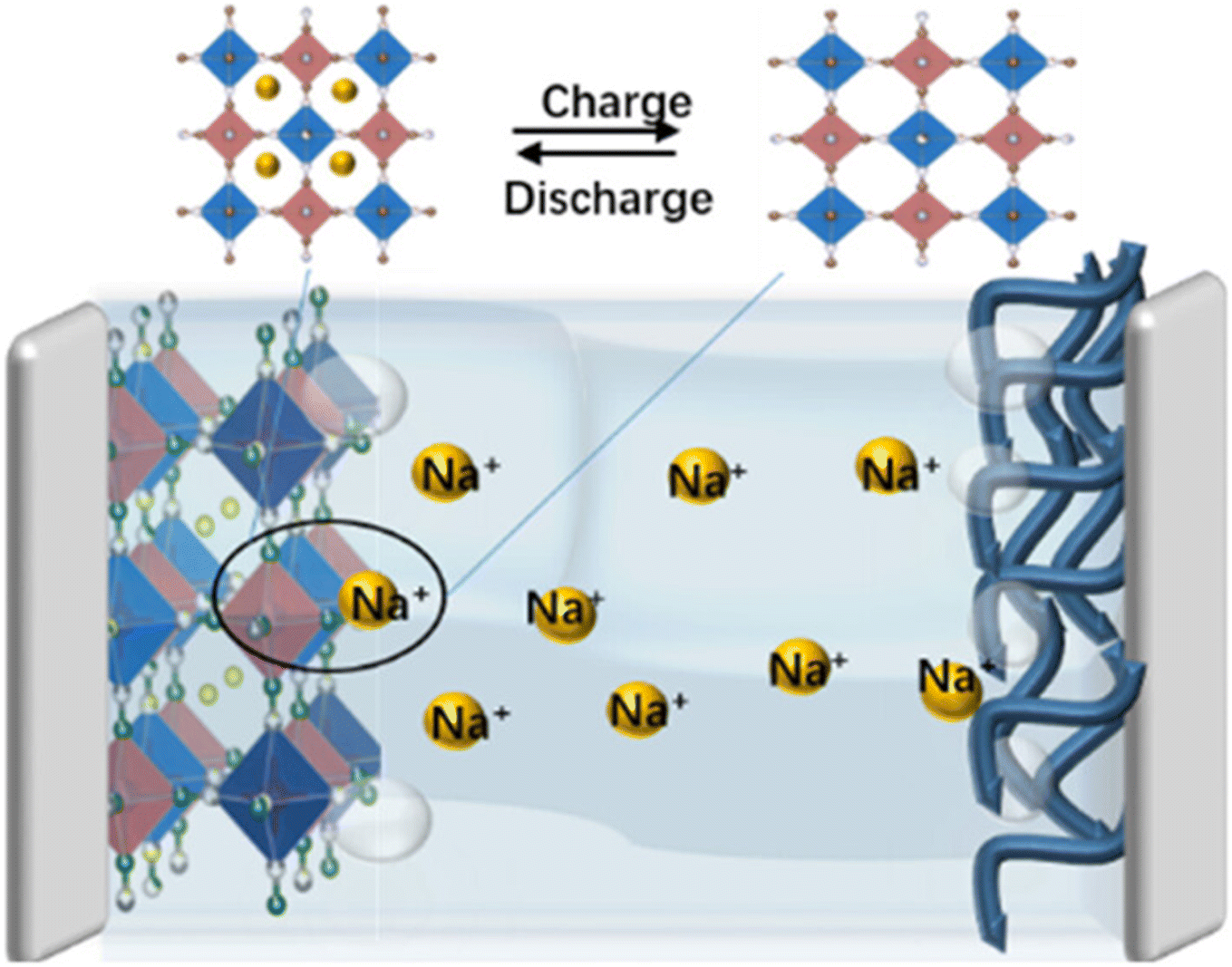
Among various SIBs configurations, aqueous SIBs (ASIBs) utilizing aqueous electrolytes demonstrate distinct advantages, including enhanced safety, superior ionic conductivity, environmental compatibility, and simplified assembly conditions compared to their organic electrolyte counterparts.20–22 Specifically, the advantages and disadvantages of ASIBs and other aqueous batteries are presented in Table 2. ASIBs exhibit remarkable merits compared to other aqueous battery technologies, particularly in the context of large-scale energy storage applications. Firstly, the elemental abundance of sodium fundamentally mitigates reliance on scarce resources such as lithium, nickel, and rare earth elements, ensuring sustainable supply chains for grid-scale deployment. Secondly, the inherent safety advantages stem from dual mechanisms: (i) utilization of non-flammable aqueous electrolytes eliminate combustion risks, and (ii) the absence of dendritic sodium deposition mechanisms prevents internal short circuits-a critical advancement over conventional alkali metal batteries. Thirdly, the system demonstrates superior environmental compatibility through elimination of heavy metal contaminants (e.g., plumbum (Pb), Cd), aligning with global sustainability initiatives and carbon neutrality objectives. Finally, the high ionic conductivity of aqueous media enables exceptional rate performance and low-temperature operation, overcoming limitations of organic electrolyte systems. These synergistic advantages position ASIBs as a transformative technology for next-generation low-cost, safe and sustainable energy storage solutions. In addition, ASIBs operate via a reversible sodium-ion intercalation/deintercalation mechanism within electrode materials, analogous to the “rocking-chair” paradigm observed in lithium-ion systems (as shown in Fig. 1).
| Aqueous battery types | Advantages | Disadvantages |
|---|---|---|
| ASIBs | Rich resources, low-cost, superior ionic conductivity, superior safety performance, environment-friendly, low production cost | Narrow electrochemical window, hydrogen evolution reaction and oxygen evolution reaction, high solubility of sodium-based compounds |
| AZIBs | Environmentally friendly, sufficient energy density, superior safety performance | Zinc dendrite growth, cathode material dissolution, low coulombic efficiency, hydrogen evolution reaction |
| ALIBs | Environmentally friendly, excellent kinetic properties, superior safety performance | Narrow electrochemical window, severe hydrogen evolution reaction, current collector corrosion |
| LAs | Highly recycled, straightforward charging mechanism, advantageous cost/performance ratio | High weight, low energy density, short cycle life, poor low-temperature performance |
| NiMH | Environmentally friendly, long cyclic life, good tolerance to overcharge and overdischarge | Low energy density, large volume, memory effect |
 | ||
| Fig. 1 Schematic diagram illustrating the working principle of an ASIB during charge/discharge cycles.28 | ||
Despite these merits, ASIBs face critical challenges that hinder practical implementation. The narrow thermodynamic stability window of aqueous electrolytes (1.23 V)29 imposes stringent voltage limitations, beyond which detrimental water electrolysis occurs through hydrogen evolution reaction (HER).30,31 Secondly, the high solubility and chemical instability of numerous sodium-containing compounds in aqueous electrolytes significantly compromise electrode stability, energy density, and cycle life. In addition, the original SEI may be predominantly composed of inorganic species such as Na2CO3, NaF, or sodium oxides in aqueous electrolyte environments.32,33 These materials exhibit high brittleness and poor ionic conductivity, coupled with inadequate mechanical compliance to accommodate electrode volume variations during cycling, ultimately leading to detrimental structural fractures within the SEI layer. These issues further restrict viable anode material selection.29,34,35 Collectively, these limitations underscore the imperative to develop innovative strategies for electrode material optimization and robust electrode–electrolyte interface engineering in ASIB systems.
Among various anode candidates, sodium superionic conductor (NASICON)-type materials have attracted significant attention due to their unique crystallographic configuration.36,37 The NASICON-type phase consists of an open three-dimensional framework of TiO6 octahedra and PO4 tetrahedra, where 2 TiO6 octahedra are separated by 3 PO4 tetrahedra, with which they have shared all their corner oxygens but no edges or faces (Fig. 2a).38,39 This material demonstrates exceptional chemical stability, extended cycling durability, and inherent safety advantages, positioning it as a superior anode candidate for ASIBs.40 Specifically, sodium titanium phosphate (NaTi2(PO4)3, NTP) has emerged as a particularly promising anode material for ASIBs,41,42 owing to its unique structural characteristics.43,44 The three-dimensional open framework of NTP contains large internal pore channels. Each NTP unit cell can accommodate two Na+ ions through intercalation, forming the Na3Ti2(PO4)3 phase (Fig. 2b), corresponding to a theoretical specific capacity of 132.8 mAh g−1,39 which is conducive to the rapid diffusion of Na+ and high ionic conductivity,45,46 thereby enabling long-term cycling stability.39 Additionally, NTP exhibits an appropriate negative voltage window. Electrochemical tests reveal that the redox potential of the Ti4+/Ti3+ couple in NTP is −0.6 V versus the standard hydrogen electrode (SHE) (typically ∼0 V vs. SHE under neutral conditions), slightly higher than the hydrogen evolution potential. This characteristic maximizes the working voltage of ASIBs from the anode perspective.47 Despite these advantages, the NTP anode/electrolyte interface in ASIBs suffers from severe side reactions and poor interfacial stability,30,48 resulting in low practical specific capacity and short cycle life, which critically limits further improvement of its electrochemical performance.
 | ||
| Fig. 2 Schematic illustration of the crystal structure. (a) Schematic illustration of the NASICON-type structure.39 (b) Schematic illustration of the NTP structure.47 | ||
The rational design and optimization of the anode/electrolyte interface represent a critical pathway to address the aforementioned challenges. This interface not only governs the Na+ transport kinetics, but also dictates the cycling stability and rate capability of the battery. Despite the growing interests in ASIBs, regarding the interfacial issues between NTP anode materials and aqueous electrolytes, there has been a notable lack of comprehensive review articles systematically addressing the abovementioned critical challenges (i.e., HER, electrode dissolution, and unstable SEI). In this sence, this review systematically summarizes recent advances and technological innovations in the design strategies of NTP anode/electrolyte interfaces to address the previously underexplored interfacial challenges between NTP anode materials and electrolytes in ASIBs. Subsequently, the proposed solutions, including electrolyte compositional optimization, interfacial coating modification and SEI interface modulation (Fig. 3), to the abovementioned issues are correspondingly summarized and discussed. Based on a critical study of existing literature, potential solutions are proposed to guide future interface design and performance optimization of NTP anode materials for high-performance ASIBs.
 | ||
| Fig. 3 Schematic illustration of the challenges and design strategy of NaTi2(PO4)3 anode/electrolyte interface in ASIBs. | ||
2 Design strategy
To address the abovementioned issues, researchers have proposed various design strategies for the NTP anode/electrolyte interface, including electrolyte compositional optimization, interfacial coating strategies and SEI interface modulation.2.1 Electrolyte compositional optimization
Electrolytes are the key ion pipeline connecting the cathode and the anode in the battery system.49 Its composition (e.g., solvents, salts, and additives) and physicochemical properties (e.g., viscosity, ionic conductivity, and thermal stability) significantly affect the performance and life of the battery. In particular, the stability of the NTP anode/electrolyte interface depends on the ability of the electrolyte to promote rapid transport of Na+, while inhibiting parasitic side reactions and degradation. Electrolytes with custom compositions can facilitate the formation of a robust SEI layer, mitigate the interfacial impedance, and inhibit the decomposition of the electrolyte on the electrode surface. The optimized electrolyte can enhance the formation of a stable SEI layer, reduce the impedance, and prevent the electrolyte decomposition on the electrode surface, thus significantly improving the stability and reliability of the NTP anode/electrolyte interface.Suo et al.50 pioneered the concept of water-in-salt (WIS) electrolyte, which is defined as an aqueous solution with a weight-to-water ratio of dissolved salt to water higher than 1. The application of WIS electrolyte has successfully widened the ESPW of aqueous electrolyte to 3.0 V and enabled the stable operation of high-voltage aqueous batteries. This concept began to be used in aqueous lithium-ion batteries. This concept, initially applied in aqueous lithium-ion batteries, has gained traction due to its promising results. With the extensive research on ASIBs in recent years, this method has also been successfully applied to ASIBs, further expanding its applicability and practicability in the field of advanced battery technology.51,52 However, the formation of WIS electrolytes is heavily dependent on the solubility of the salt. The relatively lower solubility of sodium salts compared to other alkali metal salts may limit the range of applications of WIS electrolytes in ASIBs. For example, the solubility (9–10 mol kg−1) of typical sodium salts with fluorinated anions, sodium triflate (NaOTF) and sodium (bis(trifluoromethane sulfonyl)imide) (NaTFSI), is less than half that of their Li or K counterparts at room temperature.52,53 Therefore, to address this issue, Jiang et al.54 designed a new ultra-high salt concentration WIS electrolyte with inert cations by dissolving tetraethylammonium triflate (TEAOTF) and NaOTF salts in water (9 mol NaOTF + 22 mol TEAOTF). The electrolyte not only realizes a wide voltage window of 3.3 V (Fig. 4a), but also effectively inhibits the dissolution of electrode materials during the cycling process. Due to the large radius of the TEA+ cations (calculated value of 3.6 Å), they are not embedded in the positive and negative electrode materials, avoiding the cation co-embedding problem commonly found in mixed cation electrolytes. In addition, the TEA+ and OTF− anions interact weakly in this new type of ultra-high salt concentration electrolyte, resulting in a relatively low viscosity and high conductivity even at 31 m ultra-high salt concentration. The Na1.88Mn[Fe(CN)6]0.97⋯1.35H2O (NaMnHCF)//NaTiOPO4 full battery assembled with this electrolyte was cycled between 0.7 and 2.6 V (Fig. 4b). And it showed excellent electrochemical performance at both low multiplicity (0.25C) and high multiplicity (1C), with capacity retention of 90% after 200 cycles at 0.25C and capacity retention of 76% at 1C after 800 cycles (Fig. 4c and d).
 | ||
| Fig. 4 Battery performance after optimization of an inert WIS high-concentration electrolyte. (a) The electrochemical voltage window of 9 mol per kg NaOTF electrolyte and Na inert-cation-assisted WIS electrolyte (9 mol per kg NaOTF + 22 mol per kg TEAOTF) at the scanning rate of 10 mV s−1.54 (b) The 1st, 4th, and 10th charge–discharge curves of full battery from 0.7 to 2.6 V at 0.25C.54 (c) The long-term cycling performance of the full battery at 0.25C.54 (d) Long-term cycling performance of the NaMnHCF//NaTiOPO4 full battery.54 | ||
Therefore, there is an urgent need for a balanced approach to address the inherent stability and efficiency of the water battery system without compromising the inherent economic and operational advantages of the water battery system. To address the above limitations of WIS electrolytes while simultaneously expanding the ESPW of aqueous electrolytes, researchers have developed bisolvent-in-salt electrolytes incorporating organic cosolvents. Nian et al.58 demonstrated that introducing dimethyl sulfoxide (DMSO) with a molar fraction of 0.5 into aqueous electrolytes (e.g., 2 mol NaClO4 in H2O) could significantly delay the HER onset potential by up to 1.0 V. DMSO as a hydrogen bond acceptor replaces the weak hydrogen-bonding interactions between H2O–H2O with stronger DMSO-H2O hydrogen bonds (Fig. 5a and b), thereby eliminating free water molecules in the electrolyte. This reduction in water activity effectively suppresses HER. This strategy demonstrates that manipulating hydrogen bonding between water molecules represents an effective and versatile strategy to suppressing the HER in aqueous electrolytes. Shang et al.59 proposed a general strategy for selecting hydrogen-bond-capturing solvents, wherein the solvents must possess lone electron pairs to serve as hydrogen bond acceptors and exhibit stability in aqueous environments.
 | ||
| Fig. 5 Schematic illustration showing the design strategy and battery performance after optimizing the electrolyte. (a) Schematic diagram of the HER process.58 (b) H-bond patterns between DMSO and water molecules.58 (c and d) Schematic showing design strategies for aqueous electrolytes with a wide ESPW.60 (e) Linear sweep voltammetry curves of different electrolytes at a scanning rate of 10 mV s−1.60 | ||
Approaching from the perspective of weakening hydrogen bonding interactions experienced by water molecules, Peng et al.60 proposed the use of weak-polar molecules as co-solvents of aqueous electrolytes to achieve a wide ESPW by comparing the effects of strong and weak-polar solvents on the ESPW of aqueous electrolytes. They prepared aqueous electrolytes with weak-polar co-solvent (AEWPS-5.3 m) as an example of a weakly polar molecule, 1,3,6-hexanetricarbonitrile (HTCN), to achieve an ESPW of 3.5 V at a relatively low concentration (Fig. 5e). In Fig. 5e, the blue curve corresponding to the 17 mol WIS electrolyte exhibits two small kinks at −1.5 V and 1.8 V, potentially indicative of decomposition processes involving residual impurities or side reactions within the high-concentration electrolyte system. As one of the weak-polar co-solvents, the weak interaction between HTCN and water helps to weaken the hydrogen bonding of water molecules and indirectly enhances the O–H bonding and stability of water molecules (Fig. 5c and d), thus effectively broadening the ESPW of the aqueous electrolyte. In addition, HTCN can also act at the electrode–electrolyte interface inhibiting the dissolution of transition metal ions and stabilizing the structure of electrode materials. Based on the designed electrolyte, Na2FeFe(CN)6‖NaTi2(PO4)3 achieves an ultra-long cycle life of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, and at the same time, the energy density reaches 71 Wh kg−1, showing the great potential of aqueous sodium electrodes for large-scale energy storage.
000 cycles, and at the same time, the energy density reaches 71 Wh kg−1, showing the great potential of aqueous sodium electrodes for large-scale energy storage.
2.2 Interfacial coating modification
Interfacial coating modification is a sophisticated technique aimed at enhancing the performance and longevity of NTP negative electrodes in battery systems. This process involves the precise construction of a protective layer on the surface of the NTP electrode, which serves as a barrier to isolate the direct contact between the electrolyte and the electrode material. The primary objectives of this protective layer are to enhance other interfacial properties, such as improving the electronic conductivity and structural stability of the NTP anode interface. Furthermore, the protective layer can inhibit the interfacial side reactions, leading to the degradation of the electrode and electrolyte against dissolving the electrode material. | ||
| Fig. 6 Electrode performance after carbon coating. (a) Discharge capacity versus cycle number of NTP/G + CNTs.61 (b) The first cycle of the cyclic voltammetry curves of NTP, NTP/G and NTP/C composites (scan rate of 0.5 mV s−1).62 (c) Discharge capacity versus cycle number at 2C for NTP/C and NTP/G composites.62 (d and e) Scanning electron microscopy (SEM) images at different magnifications (inset in e: magnified SEM).63 (f) High-resolution transmission electron microscopy (TEM) image.63 (g) CV curves at a scan rate of 0.5 mV s−1.63 (h) Typical charge/discharge profiles for the initial few cycles at a rate of 0.2C.63 (i) Rate-capabilities of hollow and bulk NTP@C nanocubes.63 (j) Long cycling performance of hollow NTP@C nanocubes at different rates.63 (k) Cycling performance of full cell at the current rate of 5C.63 | ||
Alexander I. Mohamed et al.68 introduced polypyrrole (PPy) as a coating for NTP through a high-energy ball milling process and found that a 5 wt% PPy coating (∼8 nm thick) significantly inhibited the unwanted side reactions with the electrolyte, increasing the capacity retention rate from 10% (uncoated) to 57% after 50 cycles and stabilizing the electrochemical impedance; whereas, an excessively thick coating (20 wt%, ∼23 nm) can hinder Na+ diffusion and lead to material deactivation. Despite the problems of limited sodium ion diffusion and the need to optimize the coating thickness, this study provides new ideas for the development of more efficient energy storage materials.
 | ||
| Fig. 7 Synergistic effects of nitridation and carbon–nitrogen co-coating on electrode performance. (a) HR-TEM images of the TiN modified NaTi2(PO4)3 (nitriding for 1.5 h).47 (b) The profiles of cyclic voltammetry (CV) of the TiN modified NaTi2(PO4)3 electrodes in 1 M Na2SO4 at 0.5 mV s−1 sweep rate.47 (c) The cycling performance of samples at 2C.47 (d) Schematic fabrication process of HNTP@PNC film.69 (e) Cyclic voltammetry (CV) curves comparison at 5 mV s−1 of PNC film and HNTP@PNC film.69 (f) Galvanostatic charge–discharge (GCD) curves at 1.10 A g−1.69 (g) Long-term cycling performance with the corresponding coulombic efficiency at 5.50 A g−1 of HNTP@PNC film.69 | ||
He et al.69 developed a facial template-free solvothermal method to synthesize rugby-like NTP nanoparticles with well-defined hollow structures. These hollow-structured NTP (HNTP) particles were uniformly encapsulated within cross-linked porous N-doped carbon nanofibers (denoted as HNTP@PNC) via electrospinning and carbonization processes (Fig. 7d), directly serving as a binder-free anode for flexible ASIBs. Theoretical calculations revealed that the N-doped carbon (NC) coating on NTP significantly enhances electronic conductivity and accelerates Na+ diffusion kinetics (Fig. 7e and f). Benefiting from its unique hollow architecture, continuous conductive network, and favorable synergistic effects, the HNTP@PNC electrode delivers a high capacity of 108.3 mAh g−1 at 5.5 A g−1 and exhibits impressive cycling stability with 97.2% capacity retention after 3000 cycles (Fig. 7g). Furthermore, a flexible quasi-solid-state ASIB was successfully assembled by employing the optimized HNTP@PNC film as the anode and a CNTF@KZHCF (potassium zinc hexacyanoferrate-coated carbon nanotube films) electrode as the cathode. This device achieves a high output voltage plateau of 1.6 V, along with a remarkable volumetric capacity of 24.5 mAh cm−3 and an energy density of 39.2 mWh cm−3, outperforming most reported flexible aqueous rechargeable energy storage systems. These exciting results provide valuable new insights into the design of advanced binder-free NTP-based anodes for next-generation wearable ASIBs.
2.3 SEI interface modulation
The modulation of the SEI interface is crucial for the performance of drainage sodium-ion batteries. A well-formed SEI layer effectively prevents unwanted reactions between the electrode material and the electrolyte, reducing dissolution–precipitation behavior during charging and discharging, further enhancing the stability and reliability of the battery.70 Given the narrow ESPW of ASIBs, the stability and composition of the SEI layer are particularly important. By optimizing the SEI layer, the electrochemical performance of the NTP negative electrode can be significantly improved, leading to better charge and discharge efficiency as well as higher energy density.:1:x (x = 1, 2, etc.) (abbreviated as AWE). With this hybrid electrolyte, almost all water molecules are confined in the primary solvation shell of Na+ (Fig. 8a). This unique solvated structure lacking free water molecules not only enables the formation of NaF-rich SEI, but also effectively inhibits HER. In addition, the oxidative decomposition potential of the electrolyte is also elevated due to the inherent high anodic stability of ADN, which ultimately widens the ESPW to 2.75 V (Fig. 8b). Hybrid electrolytes can also alleviate electrode dissolution problems due to the lack of free water molecules. Paired with this electrolyte, the Na3V2(PO4)3/NaTi2(PO4)3 full cell has good cycling stability, achieving an average coulombic efficiency of 99.6% and a capacity retention of 71% over 1000 cycles (Fig. 8c).
 | ||
| Fig. 8 Mechanism and performance of realizing in situ SEI protection in ASIBs. (a) Unique solventized structure lacking free water molecules.72 (b) ESPW of AWE and 9 m NaTFSI obtained by linear sweep voltammetry on Pt foil electrodes.72 (c) Long term cycling performance of Na3V2(PO4)3/NaTi2(PO4)3 full cell at 5C.72 | ||
Xu et al.33 used the iron-based Prussian blue system as a model and chose Na2C4O4 (NCO) as a functional additive, which successfully solved two key problems. For one thing, NCO effectively compensates the sodium defect of the anode material and realizes the two-electron reaction of iron-based Prussian blue. Secondly, the SEI on the NTP particles showed a denser layer with an increase in thickness of 10 nm after the addition of NCO (Fig. 9a and b). The decomposition product of NCO, CO2, reacted with Na+ and was deposited on the surface of the NTP anode, and a Na2CO3-rich and stable SEI layer was constructed in situ on the surface of the NTP anode. The robust SEI protective layer can effectively alleviate the continuous Na consumption and H2 precipitation from the NTP anode in the high-concentration electrolyte system, which is often regarded as a key factor for the excellent cycling stability of ASIBs. In addition, compared to the iron-based hexacyanoferrates (FeHCF)‖NTP counterpart, the full cell constructed using FeHCF and NTP anodes along with an appropriate amount of NCO has a reversible capacity of 144 mAh g−1 at 0.2C and an excellent cycling stability of 15000 cycles at 10C with a retention rate of 85% (Fig. 9c–e).
 | ||
| Fig. 9 Mechanism and performance evaluation of in situ SEI protection in ASIBs. (a) Schematic illustration of NCO additives for sodium supplementation with simultaneous construction of SEI interface in PBA-based full ASIBs.33 (b) The cryo-TEM images of and elemental EDS mapping.33 (c and d) The initial three charge–discharge profiles of FeHCF‖NTP and FeHCF–NCO‖NTP ASIBs at 0.2C. (e) The long-term cycling performance of FeHCF–NCO‖NTP ANIBs at 10C.33 | ||
For instance, Liu et al.75 developed a composite material comprising a sodiated perfluorosulfonic acid polymer (Nafion-Na) and NaX zeolite molecular sieve (Fig. 10a), which was coated onto the surface of an NTP anode. This approach established a Na+-conductive or ion-sieving interphase protective layer. The engineered interphase selectively enables the permeation of dehydrated sodium ions while effectively suppressing water decomposition and dissolution of electrode materials, thereby leading to a marked improvement in the cycling lifespan of the battery (Fig. 10b). The electrode coated with this ion-sieving interphase layer extends the ESPW of a 2 mol kg−1 (2 m) NaOTF low-concentration electrolyte to 2.70 V (Fig. 10c). Furthermore, the Na2MnFe(CN)6//NaTi2(PO4)3 battery retains 94.9% of its initial capacity after 200 charge–discharge cycles at 1C, demonstrating exceptional cycling stability. When integrated with emerging electrolyte modifications, this molecular sieve-derived interphase delivers amplified benefits for the long-term operation of ASIBs.
 | ||
| Fig. 10 Schematic illustration and electrochemical performance of artificial SEI protected ASIBs full cells. (a) Molecular-sieving process for desolvation of hydrated Na+ ions.75 (b) TEM spectra and of Nafion-Na/NaX.75 (c) ESPWs of 9 m NaOTF on the unprotected working electrode and 2 m NaOTF, on unprotected, Nafion-Na protected, and Nafion-Na/NaX protected working electrodes at a scan rate of 1.0 mV s−1.75 (d) Schematic for the charge/discharge processes involving possible re-action pathways at the modified NTP/C anode with the PPY protective interphase layer.76 (e) The NTP/C anodes modified under 0.9 V in Na2SO4 solutions with varying amounts of PY at 5C rate.76 | ||
Polypyrrole (PPY) conductive polymers have been used to coat NTP in ASIBs due to their excellent electrical conductivity and stability. This coating aims to enhance the electrochemical performance of NTP by improving charge transfer and providing structural stability during cycling.68 However, due to the uncontrolled preparation process, PPY conductive polymers may have some difficulties in forming a uniformly distributed coating on the NTP surface. Therefore, the in situ synthesis of PPY on the NTP surface may not be easy, further promotion of PPY attachment on the NTP surface is needed.
For that reason, Wang et al.76 attempted electrochemical oxidative polymerization to construct a protective interphase layer of artificial PPY on the carbon-coated NTP (NTP/C) anode surface in an electrolyte solution containing pyrrole (PY) additives. This interphase layer can inhibit the side reactions at the electrode/electrolyte interface and improve the chemical stability of the anode. By optimizing the polarization time and the amount of PY additive, a uniform and dense interfacial layer was ex situ obtained, which effectively prevented the direct contact between the NTP active substance and the aqueous electrolyte, thus improving the chemical stability of the anode. The modified NTP/C anode exhibited higher discharge capacity, better cycling stability, and higher coulombic efficiency during the charge–discharge cycle (the charging and discharging process is shown in Fig. 10d). The modified anode with a PPY coating thickness of about 24 μm exhibited higher charge–discharge capacity (228.6 mAh g−1), higher coulombic efficiency (>95%) (Fig. 10e), and better cycling stability. When the oxidation modification time was 30 or 60 min, the PPY layer was thicker and denser, which could better cover the NTP/C substrate and improve the chemical stability of the anode.
The above strategies aim to address the aforementioned challenges faced by NTP materials in aqueous electrolytes from various angles, and have all demonstrated their good effects. In order to comparative study these NTP anode/electrolyte interface electrocatalytic systems for ASIBs, their categories, advantages and challenges were comprehensive summarized in Table 3.
| Categories | Representative materials/strategies | Advantages | Challenges |
|---|---|---|---|
| Surface coating modification | Nafion-Na/NaX molecular sieve composite75 | Suppressing HER | High cost (e.g., Nafion materials) |
| Structural design optimization | Hollow carbon-coated NTP nanocubes63 | Shorter ion diffusion pathway, buffering volume expansion, better rate performance | Complex fabrication process, challenges in controlling nanostructures |
| Composite material synergy | PPy coating,76 N-doped carbon nanofibers encapsulated hollow NTP structure69 | Good electronic conductivity and cycling stability | Poor interfacial compatibility, material ratios require precise adjustment |
| Electrolyte engineering | High-concentration NaOTF electrolyte,54 weakly polar molecule-HTCN60 | Lower water reactivity, minimal side reactions, wider ESPW | High costs, inferior compatibility with electrodes |
| Interface SEI layer regulation | Constructed NaF-rich SEI and Na2CO3-rich SEI33,72 | Stable SEI, inhibiting electrodes dissolution | Long-term stability of SEI to be verified |
3 Conclusion and outlook
In summary, the interface problems between the NTP anode and aqueous electrolyte in ASIBs are mainly characterized by HER triggered by the narrow ESPW, and electrode dissolution and unstable SEI formation, resulting in low energy density and short cycle life. Significant progresses have been made in the design of advanced NTP anode–electrolyte interface in ASIBs through electrolyte compositional optimization, interfacial coating modification and SEI interface modulation. In terms of electrolyte system innovation, the electrolyte developed can extend the voltage window to more than 3.3 V, which not only inhibits the electrode dissolution and hydrogen precipitation side reaction, but also realizes a high capacity retention of the full battery after many cycles. For NTP anode materials, the surface coating strategy effectively improves the electronic conductivity and structural stability to achieve an excellent capacity and longer duration. Regarding SEI regulation, the strategies of in situ formation of stabilized SEI and construction of artificial SEI are favor of inhibiting side reactions and improving battery performances. These proposed solutions are of great significance for addressing the aforementioned challenges faced by NTP in aqueous electrolytes.However, these efforts still face challenges: (1) although the surface coating of NTP material can inhibit the side reaction in the short term, the uniformity of coating, long-term stability and bonding strength with NTP body are insufficient. It is easy to peel or rupture during the cycling process, which leads to the reappearance of interfacial failure. (2) While the optimized electrolyte can widen the electrochemical window, it may decompose with cycling or participate in irreversible reactions, leading to the failure of interface protection after electrolyte depletion. (3) In situ SEI is difficult to form a stable and uniform protective layer due to the conflict between the high activity of water molecules in the aqueous electrolyte and the low potential of NTP operation, while artificial SEI is limited by the weak bonding between the coating and the substrate, the contradiction between the ion transport efficiency and the mechanical strength, as well as the imbalance between the complex preparation process and the cost of scaling up.
In conclusion, nevertheless aqueous NTP anode presents certain challenges, their electrochemical performance can be substantially enhanced through targeted strategies such as interface modification, electrolyte optimization, and SEI membrane modulation. Future research should continue to focus on the development of new materials and electrolytes, as well as the application of advanced interface engineering methods to promote the further development and application of ASIBs. In sight of the future development of ASIBs, this paper proposes the following prospects:
(i) Development of new materials: the new material is of great significance for improving the overpotential of hydrogen evolution and stabilizing the contact with the aqueous electrolytes. With its unique chemical structure and physical properties, the composite NTP material can effectively reduce the activation energy barrier of hydrogen evolution reaction, thus significantly increasing the overpotential of hydrogen evolution, so then to improve their stability and electrochemical properties in aqueous electrolytes.
(ii) Electrolyte optimization: explore and optimize new alkaline or neutral electrolytes to inhibit hydrogen and electrode dissolution and improve the overall performance and life of the battery. Specifically, in the future, we need to focus on the synergistic optimization of electrode and electrolyte to develop a new composite system with a wide voltage window and low cost. The electrolyte formulation of ASIB should be designed to disrupt the hydrogen bonding network, thus widening the ESPW of the electrolyte, inhibiting hydrogen precipitation by the electrolyte on the NTP surface, and stabilizing the electrode/electrolyte interface.
(iii) Interface engineering: the employment of sophisticated interface engineering techniques, such as atomic layer deposition (ALD) and spray coating technology, facilitates the formation of a stable artificial SEI with high ionic conductivity on the anode material surface. This artificial SEI established between the electrolyte and the electrode interface, effectively alleviates the problems such as the hydrogen precipitation reaction and the electrode solubility. Therefore, these improvements can help to significantly improve the cycle life and coulomb efficiency of the battery, thus improving the overall performance and reliability of the energy storage system.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Chenyang Zhao: literature review, performed study, data analysis and writing original draft, Ying Liu: literature review, data curation, discussion, Yang Shao: literature review, data curation, discussion, Feng Li: literature review, data curation, discussion, Dong Zhao: literature review, data curation, Xiaoping Yang: conceptualization, supervision, project administration, writing & review and editing, Fang Cheng: conceptualization, supervision, writing & review and editing, Zhengfu Zhang: supervision, writing & review and editing.Conflicts of interest
The authors declare that they have no known competing financial interests.Acknowledgements
This work was supported by Yunnan Fundamental Research Projects (Grant Number: 202301BE070001-004) and the National Natural Science Foundation (Grant Number: 52464035).References
- S. Asad, A. Zahoor and F. A. Butt, et al., Recent advances in titanium carbide MXene (Ti3C2Tx) cathode material for lithium-air battery, ACS Appl. Energy Mater., 2022, 5, 11933–11946 CrossRef CAS
.
- N. Imanishi and O. Yamamoto, Perspectives and challenges of rechargeable lithium-air batteries, Mater. Today Adv., 2019, 4, 100031 CrossRef
- A. Jilani, Z. Awan and S. A. A. Taqvi, et al., Recent advances in the development of Li-Air batteries, experimental and predictive approaches-prospective, challenges, and opportunities, ChemBioEng Rev., 2024, 11, 95–114 CrossRef CAS
- R. Siregar, Environmentally friendly alternative energy development policies in overcoming the energy crisis, British J. Environ. Stud., 2021, 1, 44–50 Search PubMed
- Z. Yuan, H. J. Peng and J. Q. Huang, et al., Hierarchical Free-Standing Carbon-Nanotube Paper Electrodes with Ultrahigh Sulfur-Loading for Lithium-Sulfur Batteries, Adv. Funct. Mater., 2014, 24, 6105–6112 CrossRef CAS
- B. Dunn, H. Kamath and J.-M. Tarascon, Electrical energy storage for the grid: a battery of choices, Science, 2011, 334, 928–935 CrossRef CAS PubMed
- Z. Yang, J. Zhang and M. C. W. Kintner-Meyer, et al., Electrochemical Energy Storage for Green Grid, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed
- J. Mitali, S. Dhinakaran and A. Mohamad, Energy storage systems: A review, Energy Storage Sav., 2022, 1, 166–216 CrossRef CAS
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Sodium-ion batteries: present and future, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC
- H. Pan, Y.-S. Hu and L. Chen, Room-temperature stationary sodium-ion batteries for large-scale electric energy storage, Energy Environ. Sci., 2013, 6, 2338–2360 RSC
- D. R. Rolison and L. F. Nazar, Electrochemical energy storage to power the 21st century, MRS Bull., 2011, 36, 486–493 CrossRef CAS
- M. Li, J. Lu and Z. Chen, et al., 30 years of lithium-ion batteries, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed
- N. Yabuuchi, K. Kubota and M. Dahbi, et al., Research development on sodium-ion batteries, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed
- J. Deng, W. B. Luo and S. L. Chou, et al., Sodium-ion batteries: from academic research to practical commercialization, Adv. Energy Mater., 2018, 8, 1701428 CrossRef
- R.-M. Gao, Z.-J. Zheng and P.-F. Wang, et al., Recent advances and prospects of layered transition metal oxide cathodes for sodium-ion batteries, Energy Storage Mater., 2020, 30, 9–26 CrossRef
- H. Xu, K. Jiang and X. Zhang, et al., Sodium alginate enabled advanced layered manganese-based cathode for sodium-ion batteries, ACS Appl. Mater. Interfaces, 2019, 11, 26817–26823 CrossRef CAS PubMed
- T. Kulova and A. Skundin, From lithium-ion to sodium-ion battery, Russ. Chem. Bull., 2017, 66, 1329–1335 CrossRef CAS
- X. Yang and Z. Zhang, Carbon-coated vanadium selenide as anode for lithium-ion batteries and sodium-ion batteries with enhanced electrochemical performance, Mater. Lett., 2017, 189, 152–155 CrossRef CAS
- M. D. Slater, D. Kim and E. Lee, et al., Sodium-ion batteries, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS
- Y. Liang and Y. Yao, Designing modern aqueous batteries, Nat. Rev. Mater., 2023, 8, 109–122 CrossRef
- J. Whitacre, A. Tevar and S. Sharma, Na4Mn9O18 as a positive electrode material for an aqueous electrolyte sodium-ion energy storage device, Electrochem. Commun., 2010, 12, 463–466 CrossRef CAS
- Z. Zhu, T. Jiang and M. Ali, et al., Rechargeable batteries for grid scale energy storage, Chem. Rev., 2022, 122, 16610–16751 CrossRef CAS PubMed
- U. Fegade, G. Jethave and F. Khan, et al., Recent development of aqueous zinc-ion battery cathodes and future challenges, Int. J. Energy Res., 2022, 46, 13152–13177 CrossRef CAS
- M. Fetcenko, S. Ovshinsky and B. Reichman, et al., Recent advances in NiMH battery technology, J. Power Sources, 2007, 165, 544–551 CrossRef CAS
- P. P. Lopes and V. R. Stamenkovic, Past, present, and future of lead-acid batteries, Science, 2020, 369, 923–924 CrossRef CAS PubMed
- A. von Wald Cresce and K. Xu, Aqueous lithium-ion batteries, Carbon Energy, 2021, 3, 721–751 CrossRef CAS
- D. Bin, F. Wang and A. G. Tamirat, et al., Progress in aqueous rechargeable sodium-ion batteries, Adv. Energy Mater., 2018, 8, 1703008 CrossRef
- X. Zhang, Y. Hong and T. Zhang, et al., Toward High-Energy and Long-Cycle-Life Prussian Blue-Based Aqueous Sodium-Ion Batteries, ACS Appl. Nano Mater., 2024, 8, 733–740 CrossRef
- J. Liu, C. Xu and Z. Chen, et al., Progress in aqueous rechargeable batteries, Green Energy Environ., 2018, 3, 20–41 CrossRef
- Z. Liu, Y. Huang and Y. Huang, et al., Voltage issue of aqueous rechargeable metal-ion batteries, Chem. Soc. Rev., 2020, 49, 180–232 RSC
- H. Zhang, B. Qin and J. Han, et al., Aqueous/nonaqueous hybrid electrolyte for sodium-ion batteries, ACS Energy Lett., 2018, 3, 1769–1770 CrossRef CAS
- W. Sun, C. Hao and X. Zhang, et al., Robust Solid Electrolyte Interphase Engineered by Catalysis Chemistry toward Durable Anode-Free Sodium Metal Batteries, Angew. Chem., Int. Ed., 2025, 64, e202503691 CrossRef PubMed
- C. Xu, Y. Liu and S. Han, et al., Rational Design of Aqueous Na Ion Batteries Toward High Energy Density and Long Cycle Life, J. Am. Chem. Soc., 2025, 147, 7039–7049 CrossRef CAS PubMed
- D. Chao and S.-Z. Qiao, Toward high-voltage aqueous batteries: super-or low-concentrated electrolyte?, Joule, 2020, 4, 1846–1851 CrossRef
- H. Kim, J. Hong and K.-Y. Park, et al., Aqueous rechargeable Li and Na ion batteries, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed
- Y. Wu, X. Meng and L. Yan, et al., Vanadium-free NASICON-type electrode materials for sodium-ion batteries, J. Mater. Chem. A, 2022, 10, 21816–21837 RSC
- L. Zhang, Y. Liu and Y. You, et al., NASICONs-type solid-state electrolytes: The history, physicochemical properties, and challenges, Interdiscip. Mater., 2023, 2, 91–110 Search PubMed
- C. Delmas, F. Cherkaoui and A. Nadiri, et al., A nasicon-type phase as intercalation electrode: NaTi2 (PO4)3, Mater. Res. Bull., 1987, 22, 631–639 CrossRef CAS
- G. Pang, C. Yuan and P. Nie, et al., Synthesis of NASICON-type structured NaTi2(PO4)3–graphene nanocomposite as an anode for aqueous rechargeable Na-ion batteries, Nanoscale, 2014, 6, 6328–6334 RSC
- Z. Li, D. Young and K. Xiang, et al., Towards high power high energy aqueous sodium-ion batteries: the NaTi2 (PO4)3/Na0. 44MnO2 system, Adv. Energy Mater., 2013, 3, 290–294 CrossRef CAS
- C. Delmas, F. Cherkaoui and A. Nadiri, et al., A nasicon-type phase as intercalation electrode: NaTi2(PO4)3, Mater. Res. Bull., 1987, 22, 631–639 CrossRef CAS
- S. I. Park, I. Gocheva and S. Okada, et al., Electrochemical properties of NaTi2(PO4)3 anode for rechargeable aqueous sodium-ion batteries, J. Electrochem. Soc., 2011, 158, A1067 CrossRef CAS
- J. B. Goodenough, H.-P. Hong and J. Kafalas, Fast Na+-ion transport in skeleton structures, Mater. Res. Bull., 1976, 11, 203–220 CrossRef CAS
- X. Li, X. Zhu and J. Liang, et al., Graphene-supported NaTi2(PO4)3 as a high rate anode material for aqueous sodium ion batteries, J. Electrochem. Soc., 2014, 161, A1181 CrossRef CAS
- N. Anantharamulu, K. Koteswara Rao and G. Rambabu, et al., A wide-ranging review on Nasicon type materials, J. Mater. Sci., 2011, 46, 2821–2837 CrossRef CAS
- H. Kabbour, D. Coillot and M. Colmont, et al., α-Na3M2 (PO4)3 (M = Ti, Fe): absolute cationic ordering in NASICON-type phases, J. Am. Chem. Soc., 2011, 133, 11900–11903 CrossRef CAS PubMed
- Z. Liu, Y. An and G. Pang, et al., TiN modified NaTi2(PO4)3 as an anode material for aqueous sodium ion batteries, Chem. Eng. J., 2018, 353, 814–823 CrossRef CAS
- X. Li, P. Liu and C. Han, et al., Corrosion of metallic anodes in aqueous batteries, Energy Environ. Sci., 2025, 18, 2050–2094 RSC
- Y.-S. Hu and H. Pan, Solvation Structures in Electrolyte and the Interfacial Chemistry for Na-ion Batteries, ACS Publications, 2022, pp. 450–4503 Search PubMed
- L. Suo, O. Borodin and T. Gao, et al., “Water-in-salt” electrolyte enables high-voltage aqueous lithium-ion chemistries, Science, 2015, 350, 938–943 CrossRef CAS PubMed
- K. Nakamoto, R. Sakamoto and Y. Sawada, et al., Over 2 V aqueous sodium-ion battery with Prussian blue-type electrodes, Small Methods, 2019, 3, 1800220 CrossRef CAS
- L. Suo, O. Borodin and Y. Wang, et al., “Water-in-salt” electrolyte makes aqueous sodium-ion battery safe, green, and long-lasting, Adv. Energy Mater., 2017, 7, 1701189 CrossRef
- L. Jiang, Y. Lu and C. Zhao, et al., Building aqueous K-ion batteries for energy storage, Nat. Energy, 2019, 4, 495–503 CrossRef CAS
- L. Jiang, L. Liu and J. Yue, et al., High-voltage aqueous Na-ion battery enabled by inert-cation-assisted water-in-salt electrolyte, Adv. Mater., 2020, 32, 1904427 CrossRef CAS PubMed
- Y. Yamada, J. Wang and S. Ko, et al., Advances and issues in developing salt-concentrated battery electrolytes, Nat. Energy, 2019, 4, 269–280 CrossRef CAS
- D. Bin, F. Wang and A. G. Tamirat, et al., Progress in aqueous rechargeable sodium-ion batteries, Adv. Energy Mater., 2018, 8, 1703008 CrossRef
- R. Rao, L. Chen and J. Su, et al., Issues and challenges facing aqueous sodium-ion batteries toward practical applications, Battery Energy, 2024, 3, 20230036 CrossRef CAS
- Q. Nian, X. Zhang and Y. Feng, et al., Designing electrolyte structure to suppress hydrogen evolution reaction in aqueous batteries, ACS Energy Lett., 2021, 6, 2174–2180 CrossRef CAS
- Y. Shang, S. Chen and N. Chen, et al., A universal strategy for high-voltage aqueous batteries via lone pair electrons as the hydrogen bond-breaker, Energy Environ. Sci., 2022, 15, 2653–2663 RSC
- D. Peng, R. Sun and J. Han, et al., A Low-Concentrated Electrolyte with a 3.5 V Electrochemical Stability Window, Made by Restructuring the H-Bond Network, for High-Energy and Long-Life Aqueous Sodium-Ion Batteries, ACS Energy Lett., 2024, 9, 6215–6224 CrossRef CAS
- W. Wu, J. Yan and A. Wise, et al., Using intimate carbon to enhance the performance of NaTi2(PO4)3 anode materials: carbon nanotubes vs graphite, J. Electrochem. Soc., 2014, 161, A561 CrossRef CAS
- Y. He, H. Yuan and Y. Wu, et al., NaTi2(PO4)3/carbon and NaTi2(PO4)3/graphite composites as anode materials for aqueous rechargeable Na-ion batteries, Electrochemistry, 2016, 84, 705–708 CrossRef
- Z. Hou, X. Zhang and J. Chen, et al., Towards high-performance aqueous sodium ion batteries: constructing hollow NaTi2(PO4) 3@C nanocube anode with Zn metal-induced pre-sodiation and deep eutectic electrolyte, Adv. Energy Mater., 2022, 12, 2104053 CrossRef CAS
- F. Han, D. Li and W. C. Li, et al., Nanoengineered polypyrrole-coated Fe2O3@C multifunctional composites with an improved cycle stability as lithium-ion anodes, Adv. Funct. Mater., 2013, 23, 1692–1700 CrossRef CAS
- Y. Liu, B. Zhang and Y. Yang, et al., Polypyrrole-coated α-MoO3 nanobelts with good electrochemical performance as anode materials for aqueous supercapacitors, J. Mater. Chem. A, 2013, 1, 13582–13587 RSC
- Q. Qu, Y. Zhu and X. Gao, et al., Core-shell structure of polypyrrole grown on V2O5 nanoribbon as high performance anode material for supercapacitors, Adv. Energy Mater., 2012, 2, 950–955 CrossRef CAS
- M. Wu, X. Xiao and N. Vukmirovic, et al., Toward an ideal polymer binder design for high-capacity battery anodes, J. Am. Chem. Soc., 2013, 135, 12048–12056 CrossRef CAS PubMed
- A. I. Mohamed, N. J. Sansone and B. Kuei, et al., Using polypyrrole coating to improve cycling stability of NaTi2(PO4)3 as an aqueous Na-ion anode, J. Electrochem. Soc., 2015, 162, A2201 CrossRef CAS
- B. He, K. Yin and W. Gong, et al., NaTi2(PO4)3 hollow nanoparticles encapsulated in carbon nanofibers as novel anodes for flexible aqueous rechargeable sodium-ion batteries, Nano Energy, 2021, 82, 105764 CrossRef CAS
- E. Umeshbabu, B. Zheng and J. Zhu, et al., Stable cycling lithium–sulfur solid batteries with enhanced Li/Li10GeP2S12 solid electrolyte interface stability, ACS Appl. Mater. Interfaces, 2019, 11, 18436–18447 CrossRef CAS PubMed
- S. Chen, J. Ishii and S. Horiuchi, et al., Difference in chemical bonding between lithium and sodium salts: influence of covalency on their solubility, Phys. Chem. Chem. Phys., 2017, 19, 17366–17372 RSC
- H. Wang, T. Liu and X. Du, et al., Hybrid Electrolytes Enabling in-situ Interphase Protection and Suppressed Electrode Dissolution for Aqueous Sodium-Ion Batteries, Batteries Supercaps, 2022, 5, e202200246 CrossRef CAS
- Z. Hou, M. Dong and Y. Xiong, et al., Formation of solid-electrolyte interfaces in aqueous electrolytes by altering cation-solvation shell structure, Adv. Energy Mater., 2020, 10, 1903665 CrossRef CAS
- J. Yue, L. Lin and L. Jiang, et al., Interface concentrated-confinement suppressing cathode dissolution in water-in-salt electrolyte, Adv. Energy Mater., 2020, 10, 2000665 CrossRef CAS
- T. Liu, H. Wu and H. Wang, et al., A Molecular-Sieving Interphase Towards Low-Concentrated Aqueous Sodium-Ion Batteries, Nano-Micro Lett., 2024, 16, 144 CrossRef CAS PubMed
- W. Wang, J. Wu and C. Zeng, New construction of polypyrrole interphase layers to improve performance stability of NaTi2(PO4)3 anode for aqueous Na-ion batteries, Solid State Ionics, 2023, 397, 116259 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |