DOI:
10.1039/D5RA03541D
(Paper)
RSC Adv., 2025,
15, 20712-20722
Probing the sensing potential of Nb- and Mo-based MXene nanosheets and their heterostructures for air pollutants: a DFT insight
Received
20th May 2025
, Accepted 31st May 2025
First published on 18th June 2025
Abstract
In this study, we utilized density functional theory (DFT) with plane-wave calculations to investigate the gas-sensing capabilities of Mo- and Nb-based MXene nanosheets, including Nb2C, Nb2N, Mo2C, and Mo2N, as well as their heterostructures (NbMoC and NbMoN). We focused on assessing their effectiveness in detecting hazardous airborne gases, such as carbon monoxide (CO) and nitrogen monoxide (NO). The adsorption energy, charge transfer, work function, and electronic properties of the nanosheets and their heterostructures were examined to understand their adsorption behavior. Molecular dynamics and phonon calculations confirmed the thermal and mechanical stability of the nanosheets, with NbMoC being more stable than NbMoN. The band structures and density of states (DOS) indicate the metallic behavior of the nanosheets. CO and NO were adsorbed on Nb2C, with adsorption energies of −3.014 and −4.479 eV, respectively. A similar adsorption phenomenon was found for Nb2N. The adsorption of CO and NO on Mo2C occurred with adsorption energies of −2.456 and −2.984 eV, respectively. For heterostructures, gas molecules were adsorbed on the Mo and Nb sites of the nanosheets, with the Nb site being more favorable. Therefore, all MXenes exhibit strong sensitivity towards gas molecules, high interaction properties in the chemisorption range, short adsorption distance, and a significant amount of charge transfer to the gases. Although Mo2N interacts with gas molecules at exceptionally high adsorption energies, it is unsuitable for gas adsorption because of its high recovery time and high structural deformation upon adsorption. Therefore, all nanosheets, except for Mo2N, were considered promising candidates for detecting CO and NO gas molecules.
1. Introduction
Air quality is a serious global issue impacting the delicate ecosystem and human health. Global, social, and economic stability are essential for the maintenance of renewable energy, social health, sustainable growth, and environmental protection. Fossil fuel burning, motor vehicle emissions, industrial manufacturing processes, particulate matter from traffic, road dust, and soil are some sources of air pollution. During these activities, toxic gases such as nitrogen dioxide (NO2), carbon dioxide (CO2), carbon monoxide (CO), and mono-nitrogen oxides (NO) are released into the atmosphere.1,2 The nature of greenhouse gases has changed because of the emission of CO2 and CO pollutants. In particular, the emission of carbon monoxide is a significant contributor to climate change and global warming. At high concentrations, CO, a colorless and tasteless gas, can cause rhythm disorders and breathing issues.3 These pollutants are extremely harmful, severely deteriorating air quality and causing major damage to ecosystems. Nitrogen-containing gases in the ecosystem, such as NOx (a combination of NO and NO2), contribute to acid rain, which causes breathing issues, damages visual organs in humans, and ultimately harms the planet.4,5 NO indirectly causes climate change by promoting the formation of ozone (O3), the third-largest driver of positive solar radiation. N2O acts as a powerful greenhouse gas. They are also a major part of the nitrogen cycle, affecting climate and ecosystem productivity.4 Therefore, toxic gases must be filtered out, or the pollutants must be eradicated after identification. Searching for practical solutions is crucial because the identification and filtration of these gases have become important in the past few decades. In this process, gas sensors play an important role in developing sensors with high sensitivity, rapid response, selectivity, and low operating temperatures for real-time monitoring and damage prevention.
In 2011, the Gogotsi group at Drexel University in the United States proposed MXenes.6 Over the past century, MXenes and their composites have attracted much interest in sensitive electronics,7 filed emitter,8 catalysis,9 electromagnetic shielding,10 and energy conversion and storage.11 The scientific community is very interested in the newly discovered MXene family of two-dimensional (2D) materials due to its numerous uses and exceptional properties. The high surface-to-volume ratio, outstanding electronic conductivity, superior mechanical strength, and adjustable surface chemistry make it an ideal candidate for advanced technologies.12,13 The general formula for MXenes is Mn+1XnTx, where M denotes a transition metal (such as Ti, V, Nb, or Mo), X represents carbon or nitrogen, and Tx denotes surface groups (such as –O, –OH, and –F).14 The capacity of MXenes to identify harmful gases has recently led to a surge in interest in gas sensing. They exhibit promise for real-time environmental monitoring and safety applications because their sensitivity depends on the adsorption behavior, charge transfer, recovery time, and changes in electronic properties. In 2017, Lee et al. reported the remarkable gas-sensing capabilities of titanium carbide at room temperature, unlike standard semiconductors that require 200–400 °C.15 Thomas et al. investigated the sensing performance of Mo2CTx MXene for CO2 and found high degree of stability with fast response/recovery times of 32/45 seconds.16 The superior performance of MoTe2-based sensors for detecting NO2 and NH3 was demonstrated by Feng et al. They showed excellent reversibility at room temperature without sensitivity loss.17 Furthermore, Cho et al. and Zhao et al. discovered that MoS2 nanosheets made by CVD efficiently sense NO2, NH3, NO, and SO2 because of charge-transfer interactions, highlighting the wide range of applications for 2D materials in gas sensing.18,19 Another DFT study by Altarawneh et al. explored NO adsorption and separation on Mo2N surfaces, highlighting the possible process of converting NO into N2. They found that Mo2N surfaces were more conducive to the dissociative adsorption of NO.20 By depositing potassium on Mo2C/Mo(100), Solymosi et al. adapted the work function and studied the adsorption of CO and CO2.21 Wang et al. investigated the stability, CO adsorption, activation of CO and H2, and their co-adsorption on the CdI2-antitype metallic Mo2C(001) surface.22 Using DFT, Dou et al. investigated the gas sensing capabilities of Janus WSTe monolayers for CO, NO, and O2. They discovered that Te-vacancies improve the sensing performance and that the adsorption strength follows O2 > NO > C.23 Yang et al. found that Nb2C can capture SOF2 and SO2F2 gases and release H2S (in 4.69 s) and SO2 (in 2.26 s) upon increasing the temperature. However, Nb2C is best suited as a low-power sensor for detecting H2S gas.24 Lv et al. demonstrated that Nb2CO2 is a promising material for CH4 capture and release, where charge regulation enhances adsorption from weak (physisorption) to strong (chemisorption).25
Using first-principles calculations based on the DFT method, we examined the adsorption behavior of Nb- and Mo-based MXene nanosheets, specifically, Nb2C, Nb2N, Mo2C, Mo2N, and their heterostructures (NbMoC and NbMoN) towards toxic gases (CO and NO). The adsorption distances, adsorption energies, charge transfer, and charge difference density (CDD) were the primary subjects of the analysis to develop a better understanding of the adsorption behavior of these gas molecules. Ab initio molecular dynamics (AIMD) simulations and phonon calculations were used to investigate the thermal and dynamical stability of the heterostructures. We also calculated the material recovery time to better understand the adsorption mechanism.
2. Computational details
To explore the relationship between molecules of gases (CO and NO) and several nanosheets (Nb2C, Nb2N, Mo2C, Mo2N, NbMoC, and NbMoN), a calculation based on density functional theory (DFT) was conducted employing the DMol3 module within the Material Studio software. The Perdew–Burke–Ernzerhof (PBE) functional was used in the generalized gradient approximation (GGA) for electron-exchange–correlation due to its combined accuracy and computational efficiency, particularly for investigating the geometry and surface reactivity of periodic systems.26,27 The DFT semi-core pseudopotential (DSPP) was used with a double numerical basis set with polarization (DNP) to consider relativistic effects in the core region. In this research, we used the spin-unrestricted method so that spin-up and spin-down electrons were given different orbitals to better represent systems with unpaired electrons better,28 and the Tkatchenko–Scheffler (TS) dispersion correction was used to correct for interactions generated by van der Waals.29 We determined the convergence limits of the geometry optimization as 10−5 Ha for energy, 0.002 Ha Å−1 for force, and 0.005 Å for displacement. The calculations were performed using a Ha-Fermi smearing parameter of 0.005. We examined a 3 × 3 × 1 supercell with 34 atoms and a 20 Å vacuum region for this investigation. Studies have confirmed that a 3 × 3 × 1 supercell is sufficient for gas adsorption, while 20 Å spacing inhibits interactions between nearest units. All calculations were performed on a k-point grid set at 4 × 4 × 1 and a global cut-off radius set at 5.6 Å. To gain insight into the interactions between the gas molecules and the nanosheets, calculated the adsorption energy.30| | |
Eads = Ecomplex − Enanosheets − Egas
| (1) |
where Ecomplex is the total energy of the nanosheets containing the absorbed gas molecules, Enanosheets and Egas are the energies of the nanosheets and gas molecules before adsorption, respectively.
To observe the method of charge transfer behavior properly, CDD maps were generated via CASTEP using nanosheets according to the following equation:31
| | |
Δρ = ρtotal − ρnanosheet − ρgas
| (2) |
The charge density of the nanosheets containing the absorbed gas molecules is denoted by
ρtotal,
ρnanosheet, and
ρgas, respectively. The gas molecules were CO and NO, and the nanosheets were Nb
2C, Nb
2N, Mo
2C, Mo
2N, NbMoC, and NbMoN.
3. Results and discussion
3.1 Geometry and structural stability
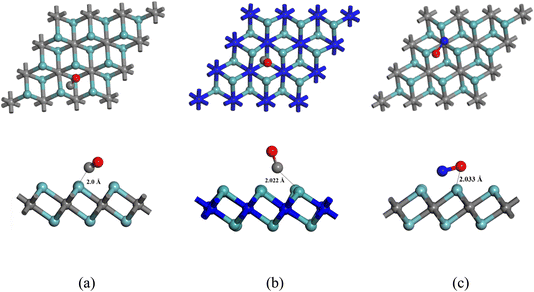
In this investigation, we initially selected four different MXenes: Nb2N, Nb2C, Mo2C, and Mo2N. To enhance their surface reactivity and sensing capabilities towards toxic gas molecules, we then created heterostructures by substituting one Mo/Nb layer with Nb/Mo, forming NbMoC and NbMoN. A 3 × 3 × 1 supercell of each of these six nanosheets consisted of 34 atoms with 16 N or C atoms, and their optimized structures are shown in Fig. 1. After optimization, The Nb–C and Nb–N bond lengths were identical at 2.141 Å, which is consistent with the previous findings of Hu et al. and Wang et al. for Nb2C and Nb2N nanosheets, respectively.32,33 The bond angles of Nb–C–Nb and Nb–N–Nb are both 89.6° for these nanosheets. In Mo2C, the Mo–C bond length is 2.098 Å, and the Mo–C–Mo bond angle is 87.252°, closely matching the values reported by Hassan et al., who found the Mo–C bond length to be 2.10 Å.34 The Mo–N bond length of 2.124 Å is in good agreement with previous findings, and the calculated Mo–N–Mo bond angle is 88.7° in Mo2N.35 Similarly, for the NbMoC heterostructure, the calculated Mo–Nb and Mo–C bond lengths in NbMoC are 2.939 Å and 2.078 Å, respectively, with a Mo–C–Nb bond angle of 88.046°. The Mo–N and Mo–Nb bond lengths are 2.105 Å and 2.983 Å, respectively, with a Mo–N–Nb bond angle of 88.933° for NbMoN. We also relaxed the gas molecules (CO and NO) to the ground state to obtain stable geometries. The C–O and N–O bond lengths in CO and NO are 1.142 Å and 1.164 Å; these findings are consistent with earlier research by Khoshnobish et al.36 Cohesive energies were determined to examine the structural stability of the materials. The cohesive energy is a key parameter for quantifying the bond strength of crystal structures. The cohesive energies of Nb2C, Nb2N, Mo2C, Mo2N, NbMoC, and NbMoN MXenes were measured using the following equation:37| |
 | (3) |
where N represents the overall number of atoms within the nanosheets and N = x + y + z; EMxene denotes the total energy of MXene, Ea, Eb, and Ec are the energies of the individual constituent atoms (e.g., Mo, Nb, N, C). x, y, and z indicate the number of atoms. The cohesive energies were determined to be −6.99 eV, −6.33 eV, −6.14 eV, −5.24 eV, −6.58 eV, and −5.79 eV for Nb2C, Nb2N, Mo2C, Mo2N, NbMoC, and NbMoN, respectively. Shein et al. observed that the cohesive energies of nitride MXenes are generally lower than those of carbide MXenes.38 The negative cohesive energy values confirm that all structures are stable, with more negative values indicating higher structural stability.39 The cohesive energies increase progressively in the order of Mo2N < NbMoN < Mo2C < Nb2N < NbMoC < Nb2C, indicating that Nb2C exhibits the highest thermal stability among the studied materials.
 |
| | Fig. 1 Top and side views of the optimized structure of the nanosheets. (a) Nb2C, (b) Nb2N, (c) Mo2C, (d) Mo2N, (e) NbMoC and (f) NbMoN. | |
Previous studies indicate that Nb2N and Mo2N monolayers are thermodynamically and dynamically stable.30,37 Furthermore, to evaluate the thermodynamic stability of the two heterostructures (NbMoC and NbMoN), we performed ab initio molecular dynamics (AIMD) simulations in the Nosé–Hoover thermostat (NVT) ensemble at 300 K, with an SCF energy convergence tolerance of 10−6 eV. The variation in potential energy with respect to time at 300 K temperatures of NbMoC and NbMoN is shown in Fig. 2. The time step of the AIMD simulation was set to 1 fs (1000 steps). For NbMoC, the potential energy exhibited minor fluctuations ranging from −0.04 eV to +0.01 eV. The potential energy of NbMoN varied between −0.001 eV and +0.0005 eV, showing minor variations. With such minimal energy fluctuations at room temperature (300 K), both NbMoC and NbMoN exhibit strong thermodynamic stability. Subsequently, to confirm the dynamical stability of the predicted heterostructures, we calculated their vibrational spectra through accurate phonon calculations. As shown in Fig. 2, the NbMoC monolayers are dynamically stable because they are free of imaginary phonon modes across the entire Brillouin zone. Conversely, For NbMoN, most phonon modes are positive, but imaginary modes appear along the M–K path, Frequency range: near −10 THz (imaginary) to +20 THz (optical modes). The presence of fewer transverse bands travels into the negative frequency range, indicating minor dynamic instability in NbMoN. The instability may arise from weaker bonding in the nitride structure than in its carbide counterpart, or unresolved strain in the lattice.
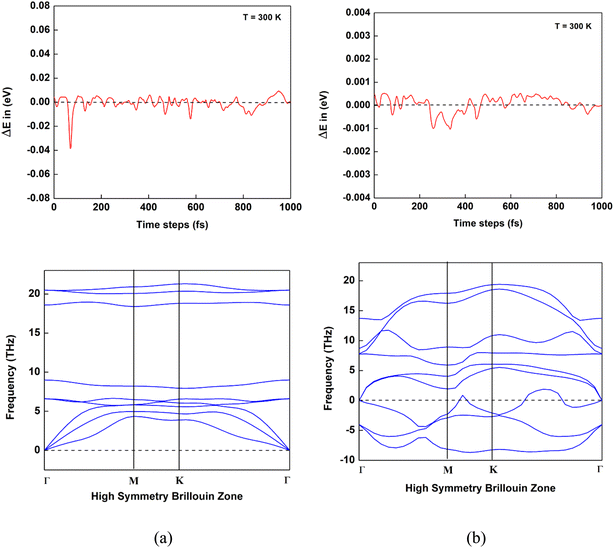 |
| | Fig. 2 Variation in potential energy as a function of time and phonon spectra of heterostructures. (a) NbMoC and (b) NbMoN. | |
3.2 Adsorption behavior of CO and NO on Nb2C and Nb2N
To examine the adsorption characteristics of these gas molecules on Nb2C and Nb2N, we calculated the adsorption energy (Eads) with respect to minimum adsorption distances (dmin) and charge transfer (ΔQ) between the gas molecules and nanosheets, as listed in Table 1. The orientations of the gas molecules and the different adsorption sites on the nanosheets were studied to determine the most favorable adsorption sites. At this site, the nanosheets and gases were aligned in parallel and optimized in their ground state (Fig. 3). After optimization, a strong interaction between the toxic gases and the nanosheets was observed with negative adsorption energies. A negative adsorption value indicates both the attractive interaction between the gas molecules, MXenes, and the exothermic reaction of the adsorption process.41 The gas molecules preferentially adsorb at distances of 2.052 Å and 2.006 Å from Nb2C, with adsorption energies of approximately −3.014 eV and −4.479 eV for CO and NO, respectively. The adsorption energies of CO and NO molecules on Nb2N are −3.136 eV and −4.397 eV, respectively, while the minimum distances for both are 2.017 Å and 1.988 Å. Li et al. observed that the adsorption energies for CO and NO on the neutral surface of 2D-WO3 were 0.28 eV and 0.75 eV, respectively. This shows that Nb2N reflects significantly stronger interactions with these gas molecules than the neutral surface of WO3.42 The value of Eads is >0.8 eV, suggesting chemisorption, whereas a value under 0.6 eV indicates physisorption.43,44 Such high adsorption energies and small adsorption distances indicate a strong chemisorption interaction between the gas molecules and both nanosheets. Numerically, the absolute values of adsorption energies are Nb2C (CO) < Nb2N (CO) < Nb2N (NO) < Nb2C (NO). According to these values, NO adsorbs onto these nanosheets more strongly than CO does. According to Pham et al., CO molecules adsorb at the A site of the O-vacancy-containing Sc2CO2 monolayer with an adsorption energy of −1.07 eV, whereas NO exhibits an adsorption energy of −3.19 eV.45 Kalwar et al. discovered that the adsorption energies for CO and NO on a Ti-doped hBN monolayer are −1.66 eV and −1.82 eV, respectively.46 Junkaew et al. reported adsorption energies of −0.12 eV for CO and −0.21 eV for NO on the Nb2CO2.47 Thus, the proposed nanosheets (Nb2C and Nb2N) exhibit significantly stronger adsorption energies than those reported in their study.
Table 1 Calculated adsorption energy (Eads), minimum adsorption distance (dmin), and charge transfer (Q) of the gas molecules adsorbed on Nb2C and Nb2N nanosheets
| Gas molecules |
Nanosheets |
Eads (eV) |
dmin (Å) |
Q (e) (Mulliken) |
Q (e) (Hirshfeld) |
| CO |
Nb2C |
−3.014 |
2.052 |
−0.776 |
−0.368 |
| Nb2N |
−3.136 |
2.017 |
−0.767 |
−0.359 |
| NO |
Nb2C |
−4.479 |
2.006 |
−0.898 |
−0.367 |
| Nb2N |
−4.397 |
1.988 |
−0.881 |
−0.351 |
 |
| | Fig. 3 Top and side views of the CO and NO gas molecules adsorbed on the Nb2C and Nb2N nanosheets. The green, grey, blue and red spheres indicate Nb, C, N and O atoms, respectively. (a) Nb2C_CO, (b) Nb2N_CO, (c) Nb2C_NO and (d) Nb2N_NO. | |
Mulliken and Hirshfeld charge analyses were conducted to investigate the net charge transfer between the gas molecules and the nanosheets. The calculation for the charge difference (ΔQ) is as follows:48
| | |
ΔQ = Qafter − Qbefore
| (4) |
where
Qafter and
Qbefore denote the net charges on the gas molecules after and before adsorption on the nanosheets, respectively. Typically, the gas molecules are assumed to have no net charge before adsorption. The gas molecules either gain or lose charge from the nanosheets following adsorption. When Δ
Q is positive, the electrons move from the gas molecules to the surface of the nanosheet, indicating that the nanosheets accept electrons.
49 The nanosheets act as electron donors when the Δ
Q is negative, which denotes electron transfer from the nanosheet surface to the gas molecules.
49,50 According to
Table 1, Mulliken and Hirshfeld charge analyses indicated a significant amount of charge transfer between CO and NO gas molecules and the Nb
2C and Nb
2N nanosheets. Specifically, the Mulliken charge analysis shows charge transfers of 0.776
e and 0.898
e, and the Hirshfeld analysis indicates transfers of 0.368
e and 0.367
e from Nb
2C to CO and NO, respectively. For Nb
2N, the charge transfer to CO and NO gas molecules was 0.767
e and 0.881
e according to Mulliken analysis, whereas Hirshfeld analysis showed transfers of 0.359
e and 0.351
e, respectively. Both nanosheets act as electron donors, and the toxic gas molecules serve as electron acceptors, suggesting a strong interaction between them. According to our analysis, the charge transfer for NO was larger than that for CO in the case of both nanosheets. Similarly, Pham
et al. reported a charge transfer of 0.989
e from the O-vacancy-containing Sc
2CO
2 monolayer to the CO molecule, while the NO molecule acts as an acceptor with a charge transfer of 2.051
e from the monolayer, which is consistent with our findings.
45 In contrast, Kalwar
et al. observed a smaller charge transfer of 0.21
e from the Ti atom to the O atom of the CO molecule and 0.28
e donated by the Ti atom to the NO molecule in a Ti-doped hBN monolayer, both of which are significantly lower than the values obtained in our study.
46
3.3 Adsorption behavior of CO and NO on Mo2C and Mo2N
We also examined the adsorption capacity of Mo2C and Mo2N nanosheets upon exposure to the gas molecules. Following a similar optimization process, we analyzed the amount of adsorption energy using the minimum adsorption distance and charge transfer between the gas molecules and the nanosheets Table 2. According to our calculations, after geometrical optimization, the Mo2N nanosheets were destroyed by NO adsorption. This deformation is attributed to strong chemical interactions between the NO molecule and the surface Mo atoms. The NO molecule acts as an electron acceptor, withdrawing electron density from the Mo atoms and weakening the Mo–N bonds. This resulted in significant charge redistribution and structural instability, causing the Mo2N monolayer to deform. However, a strong interaction occurs between Mo2N and CO, with an adsorption energy of −8.137 eV and an adsorption distance of 2.033 Å. On the other hand, we found that Mo2C interacts with gas molecules without any structural deformation (Fig. 4). The Mo–C bonds in Mo2C are less polar and more stable due to carbon's lower electronegativity compared to nitrogen. As a result, Mo2C remains structurally intact during gas adsorption. The adsorption distances for CO and NO gas molecules on Mo2C nanosheets are 2.00 Å and 2.033 Å, respectively, and the corresponding adsorption energies are −2.456 eV and −2.984 eV. The adsorption energies indicate that the gases interact with Mo2C through chemisorption, with NO exhibiting a slightly stronger interaction than CO, as reflected in the trend Eads (NO) > Eads (CO).
Table 2 Calculated adsorption energy (Eads), minimum adsorption distances (dmin), and charge transfer (Q) of the gas molecules adsorbed on Mo2C and Mo2N nanosheets
| Gas molecules |
Nanosheets |
Eads (eV) |
dmin (Å) |
Q (e) (Mulliken) |
Q (e) (Hirshfeld) |
| CO |
Mo2C |
−2.456 |
2.00 |
−0.375 |
−0.239 |
| Mo2N |
−8.137 |
2.033 |
−0.32 |
−0.238 |
| NO |
Mo2C |
−2.984 |
2.033 |
−0.656 |
−0.333 |
| Mo2N |
−9.938 |
1.909 |
−1.488 |
−0.679 |
 |
| | Fig. 4 Top and side views of the CO and NO gas molecules adsorbed on the Mo2C and Mo2N nanosheets. The cyan, grey, blue and red spheres indicate Mo, C, N and O atoms, respectively. (a) Mo2C_CO, (b) Mo2N_CO and (c) Mo2C_NO. | |
The highest adsorption energies were found at the Mo site, with values of −2.172 eV for CO and −3.406 eV for NO on Mo2C, as reported by Hassan et al.34 Jing-yun et al. observed an adsorption energy of −2.5 eV for CO on α-Mo2C. Both studies are consistent with our findings.51 In terms of Mulliken and Hirshfeld charge analysis, a Mulliken charge transfer of 0.32e and a Hirshfeld charge transfer of 0.238e occurred from Mo2N to CO. A significant amount of Hirshfeld and Mulliken charges are transferred from Mo2C to CO and NO, respectively, and are approximately 0.375e and 0.656e for Mulliken and 0.239e and 0.333e for Hirshfeld during the interaction between the nanosheets and gases. This highlights that Mo2C shared more electrons with NO than CO. Hassan et al. reported a net charge transfer of 0.360e from the Mo2C monolayer to the CO molecule and 0.423e to the NO molecule at the Mo site, which is consistent with our results.34 Similarly, Zaman et al. observed a charge transfer of +0.457e for a CO molecule adsorbed on the Mo(4) atom of γ-Mo2N(111), which also aligns with our findings.52
3.4 Adsorption behavior of CO and NO on NbMoC and NbMoN
To gain deeper insights into the nanosheets of NbMoC and NbMoN, which have equal contributions of Mo and Nb atoms, we constructed these materials. After optimization, gas molecules were adsorbed in parallel onto the NbMoC and NbMoN nanosheets at the Mo and Nb sites. Fig. 5 illustrates the interaction configurations between the gas molecules and the nanosheets. To evaluate the adsorption capacity of CO and NO gas molecules on these nanosheets, the adsorption energies were calculated using eqn (1), the net charge transfer (ΔQ) was determined using eqn (4), and the minimum adsorption distances were analysed, and the results are presented in Table 3. By exploring these nanosheets, it is evident that NbMoC and NbMoN exhibit adsorption behaviour nearly similar to that of our proposed Mo- and Nb-based nitrides and carbides. These findings indicate that the Nb site exhibits stronger adsorption and is more attractive than the Mo site, with adsorption at both sites occurring via chemisorption. The Mo sites of the NbMoC nanosheet exhibited adsorption energies of −2.376 eV for CO and −3.341 eV for NO with adsorption distances of 2.019 Å and 2.082 Å, respectively. Meanwhile, the Nb sites exhibited stronger adsorption, with energies of −3.034 eV for CO and −4.457 eV for NO at corresponding distances of 2.034 Å and 2.015 Å. These findings indicate that the gases interact with NbMoC through strong chemisorption, with NO exhibiting slightly stronger adsorption than CO. Significant adsorption energies were observed, with −2.653 eV and −3.464 eV for CO and NO molecules adsorbed on the Mo site of the NbMoN nanosheet, at adsorption distances of 2.005 Å and 1.853 Å, respectively. On the other hand, at the Nb site, the calculated adsorption energies are −3.314 eV and −8.581 eV for CO and NO molecules, with corresponding adsorption distances of 2.017 Å and 2.008 Å. Similar to NbMoC, the interaction between the gases and NbMoN occurs via chemisorption, with NO interacting more strongly than CO at both sites. The adsorption energies on Mo and Nb sites follow the following trend: for CO, NbMoC (Mo) < NbMoN (Mo) < NbMoC (Nb) < NbMoN (Nb), and a similar trend is observed for NO. Recent studies have identified several promising materials for toxic gas detection. Huzaifa et al. demonstrated that gold-functionalized Ti3C2 acts as an efficient CO sensor with an adsorption energy of −0.14 eV.53 Similarly, Weng et al. reported that Ti3C2O2 adsorbs both CO (−0.162 eV) and NO (−0.348 eV).54 For transition metal-doped systems, Sambare et al. found that Mo-doped bismuth ferrite oxide (BiFeO3) shows strong CO adsorption (1.0532 eV).55 Naqvi et al. reported that Ti2NS2 shows particularly strong adsorption for both CO (−0.172 eV) and NO (−0.406 eV).56 Similarly, Wang et al. suggested that Fe- and Co-doped VBF can be used as effective materials for capturing the toxic NO gas.57 In comparison, the significantly more negative adsorption energies observed for NbMoC and NbMoN indicate that these materials exhibit much stronger gas adsorption, suggesting that they are more promising candidates for gas sensor applications than those reported in previous studies.
 |
| | Fig. 5 Top and side views of the CO and NO gas molecules adsorbed at two possible sites (Mo and Nb) on the heterostructures NbMoC and NbMoN. Green, cyan, grey, blue and red spheres indicate Nb, Mo, C, N and O atoms, respectively. (a) NbMoC(Nb)_CO, (b) NbMoC(Mo)_CO, (c) NbMoC(Nb)_NO, (d) NbMoC(Mo)_NO, (e) NbMoN(Nb)_CO, (f) NbMoN(Mo)_CO, (g) NbMoN(Nb)_NO and (h) NbMoN(Mo)_NO. | |
Table 3 Calculated adsorption energy (Eads), minimum adsorption distance (dmin), and charge transfer (Q) of the gas molecules adsorbed on NbMoC and NbMoN nanosheets
| Gas molecules |
Nanosheets |
Eads (eV) |
dmin (Å) |
Q (e) (Mulliken) |
Q (e) (Hirshfeld) |
| CO |
NbMoC(Nb) |
−3.034 |
2.034 |
−0.755 |
−0.369 |
| NbMoC(Mo) |
−2.376 |
2.019 |
−0.348 |
−0.234 |
| NbMoN(Nb) |
−3.314 |
2.017 |
−0.764 |
−0.368 |
| NbMoN(Mo) |
−2.653 |
2.005 |
−0.038 |
−0.139 |
| NO |
NbMoC(Nb) |
−4.457 |
2.015 |
−0.900 |
−0.365 |
| NbMoC(Mo) |
−3.341 |
2.082 |
−0.701 |
−0.364 |
| NbMoN(Nb) |
−8.581 |
2.008 |
−1.684 |
−0.742 |
| NbMoN(Mo) |
−3.464 |
1.853 |
−0.232 |
−0.170 |
We also noticed that, especially at the Nb sites, the charge transfer for NO is larger than that for CO when considering Mulliken and Hirshfeld charge analysis. However, a considerable amount of charge is transferred from the heterostructure to the gas molecules. For NbMoC, Mulliken charge transfer to gas molecules was significant, with approximately 0.755e for CO and 0.900e for NO at the Nb site and 0.348e for CO and 0.701e for NO at the Mo site during the interaction between the nanosheets and gas molecules. For the Hirshfeld charge analysis, charge transfers of 0.369e and 0.365e occurred at the Nb site, whereas 0.234e and 0.364e were transferred at the Mo site from NbMoC to CO and NO, respectively. For NbMoN, a notable amount of charge is transferred from the nanosheets to the toxic gases. At the Nb site, Mulliken charge transfers of 0.764e and 1.684e, and Hirshfeld charge transfers of 0.368e and 0.742e are transferred to CO and NO, respectively. At the Mo site, the charge transfer values were 0.038e and 0.232e for Mulliken and 0.139e and 0.170e for Hirshfeld, respectively, transferred to CO and NO. The charge transfer (Mulliken) trends for CO and NO adsorbed on both Mo and Nb sites show the following characteristics: NbMoN (Mo) < NbMoC (Mo) < NbMoC (Nb) < NbMoN (Nb). Notably, the NO adsorption exhibited a qualitatively similar charge-transfer trend across these sites. Huzaifa et al. demonstrated that Au-functionalized Ti3C2 transfers 0.01e to CO, whereas Weng et al. reported a larger charge transfer from Ti3C2O2 to NO (0.226e) compared to CO (0.002e).53,54 Naqvi et al. observed the reverse trend in Ti2NS2, with CO and NO donating 0.01e and 0.134e, respectively.56 For TM-doped VBF systems, Wang et al. found that Fe-doped sites exhibited stronger charge transfer than Co-doped ones: for CO, Fe-VBF showed +0.146e (TM) and −0.047e (gas) versus Co-VBF's −0.022e (TM) and −0.052e (gas); for NO, Fe-VBF had +0.269e (TM) and −0.282e (gas) compared to Co-VBF's +0.032e (TM) and −0.212e (gas).57 The NbMoC and NbMoN heterostructures exhibit much stronger charge transfer and are a few times larger than those of the aforementioned structures. This suggests that the proposed nanosheets interact more strongly with gas molecules.
3.5 Electronic properties
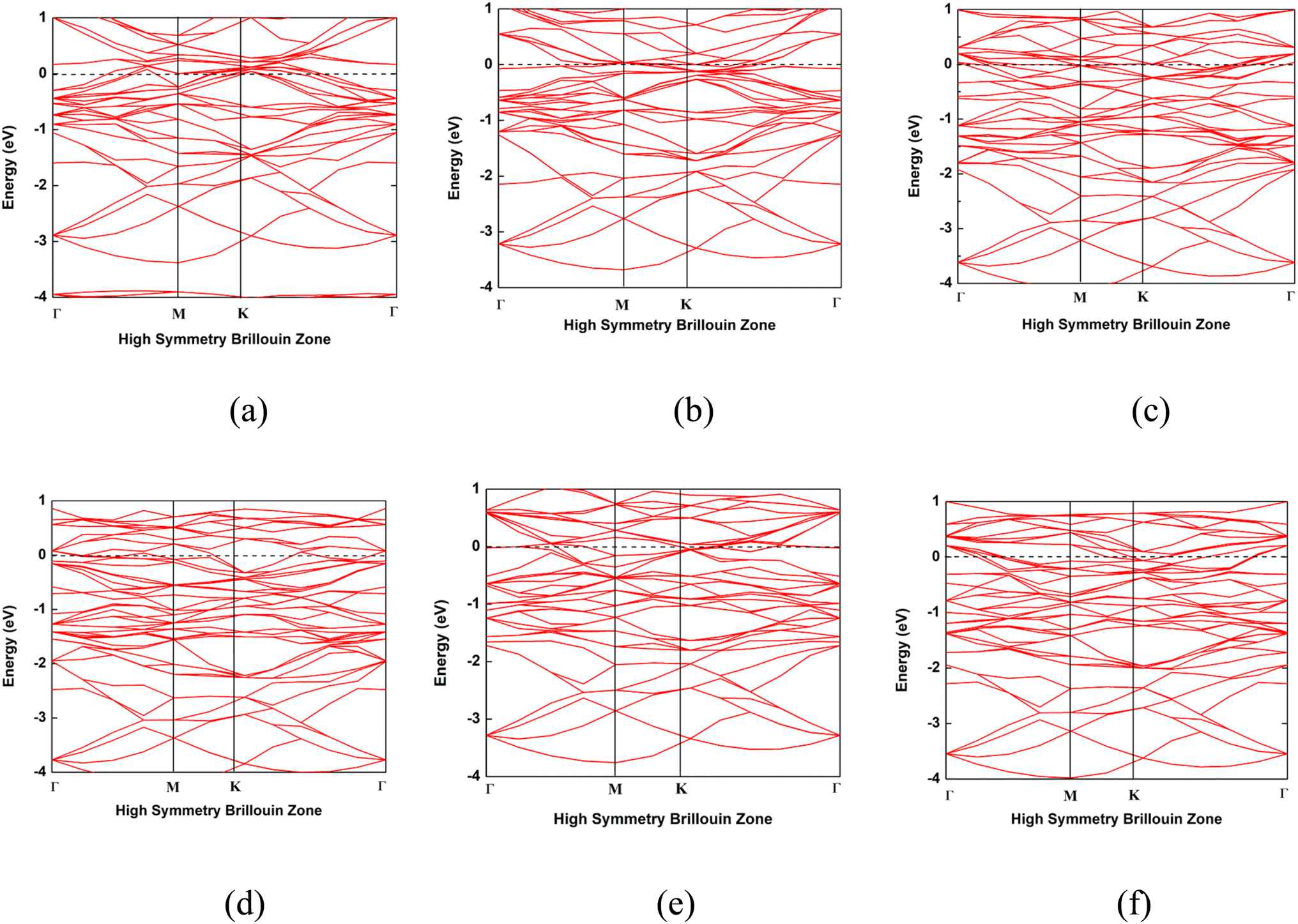
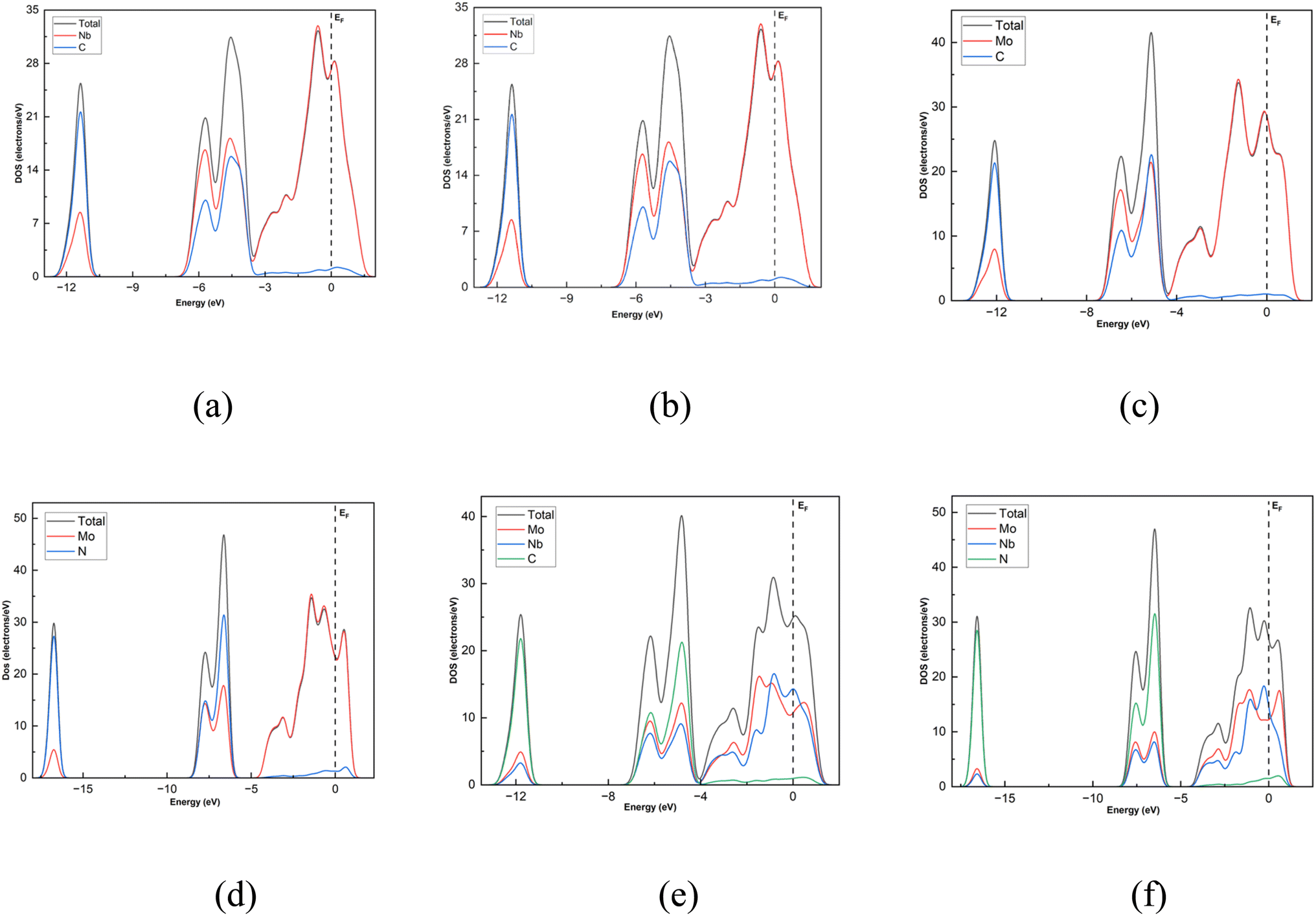
The band structures of the Nb2C, Nb2N, Mo2C, Mo2N, NbMoC, and NbMoN nanosheets are shown in Fig. 6 and were calculated to better understand the electrical features. The bandgap is indicated by the difference between the valence and conduction bands, which characterizes the conductivity of the materials. In our investigation, the conduction band crossed the Fermi level, indicating that the nanosheets were metallic, consistent with previous findings.33,40 The electronic band structures were computed along the high-symmetry Γ–M–K–Γ path in reciprocal space, with coordinates Γ (0,0,0) → M (0,0.5,0) → K (−0.333,0.667,0) → Γ (0,0,0). We also analyzed the DOS of the studied systems (Nb2N, Nb2C, Mo2N, Mo2C, MoNbN, and MoNbC), as shown in Fig. 7. The metallic nature of these materials was confirmed by the finite density of states (DOS) at the Fermi level (EF). Near the Fermi level, the electronic states of Nb2C and Nb2N are dominated by Nb d-states, whereas Mo d-states dominate in Mo2C and Mo2N. In contrast, MoNbC and MoNbN exhibit nearly equal contributions from both the Mo and Nb d-states. We calculated the CDD of the proposed materials to understand how electrons move between the gas molecules (CO and NO) and the material surfaces. CDD analysis helps visualize the spatial distribution of charge transfer after molecular adsorption. In the CDD map (Fig. 8), the yellow region indicates an increase in electron density and the green region represents a decrease in electron density. After adsorption, the charge density of the gas molecules increased, and that of the nanosheets decreased. This indicates that the substrate loses electrons to molecules, resulting in significant charge transfer between them. Table 3 shows that for NbMoC, the Mulliken charge transfer with CO is −0.348e at the Mo site, while −0.755e at the Nb site. In contrast, NbMoN exhibited even greater charge transfer with NO at the Nb site (−0.900e). For CO adsorption, NbMoN exhibits a charge transfer of 0.764e to CO; for Nb2N, 0.767e is transferred to CO. The negative values of ΔQ verify that the nanosheets are positively charged; they lose electrons, and gas molecules gain electrons. Therefore, the population analysis results agree with the CDD findings.
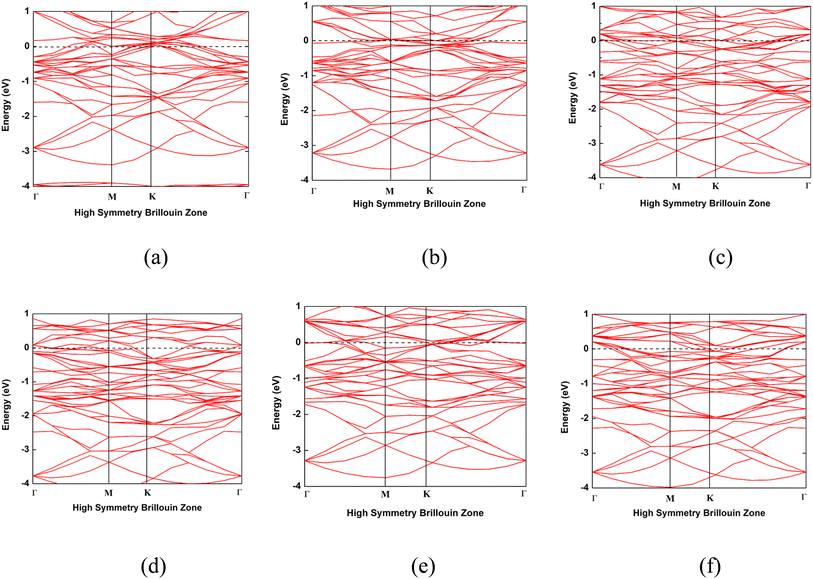 |
| | Fig. 6 Band structures of the nanosheets. (a) Nb2C, (b) Nb2N, (c) Mo2C, (d) Mo2N, (e) NbMoC and (f) NbMoN. | |
 |
| | Fig. 7 Total and partial DOS of the nanosheets. (a) Nb2C, (b) Nb2N, (c) Mo2C, (d) Mo2N, (e) NbMoC and (f) NbMoN. | |
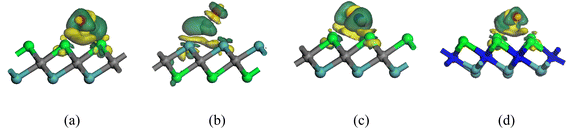 |
| | Fig. 8 CDD maps of the CO and NO gas molecules adsorbed on heterostructures. Green, cyan, grey, and blue spheres indicate Nb, Mo, C and N atoms, respectively. (a) NbMoC(Nb)_CO, (b) NbMoC(Mo)_CO, (c) NbMoC(Nb)_NO and (d) NbMoN(Nb)_CO. | |
3.6 Recovery time
The recovery time (τ) represents an important parameter for evaluating the performance of gas sensors. The recovery time is the duration required for gas molecules to desorb from the material surface. Based on van't Hoff–Arrhenius's expression in transition state theory, recovery time is defined as58| |
 | (5) |
where ϑ is the attempt frequency, kB denotes the Boltzmann constant with a value of 8.62 × 10−5 eV K−1, and T is the temperature in K. To reuse the sensing device, recovery time should be rapid. From this equation, it is evident that τ depends on both the adsorption energy (Eads) and the temperature (T). This result also indicates that stronger adsorption leads to longer recovery times, hinders the desorption process, and increases the sensor recovery time. The experimental results demonstrated that sensor recovery can be significantly accelerated by exposure to either an electric field or vacuum UV light, with tested frequencies of 1012, 3 × 1014, and 1018 s−1 and temperatures of 300, 400, and 500 K.59–62 This acceleration likely occurs because varying UV wavelengths modify molecular vibrational frequencies, influencing the attempt frequency and ultimately shortening the recovery time.58 The highest value of the recovery time was observed when NO was adsorbed on the NbMoN surface because of the highest adsorption energy (−8.581 eV) at the Nb site without structural deformation. This finding suggests that NO exhibits strong adsorption stability toward NbMoN, which inhibits the desorption process from occurring efficiently. In our observations, all the nanosheets exhibited stronger adsorption towards the gases, leading to a significantly larger recovery time.
4. Conclusions
DFT investigations were conducted to determine the gas sensing capabilities of Mo- and Nb-based MXene nanosheets, such as Nb2C, Nb2N, Mo2C, and Mo2N, and their heterostructures (NbMoC and NbMoN) functionalized with carbon and nitrogen. We focus on assessing their effectiveness in detecting hazardous airborne gases, such as CO and NO. In this regard, we examined the adsorption energy, adsorption distance, charge transfer, recovery time, and electronic properties to understand the adsorption behavior. Initially, we observed the structural properties of the nanosheets using phonon and AIMD simulations and found that the NbMoC heterostructure showed higher stability than the NbMoN. The band structures and DOS indicate the metallic behavior of the nanosheets. Subsequently, we adsorbed the gas molecules on the nanosheets to calculate the adsorption behavior. Surprisingly, all of the MXenes exhibited strong sensitivity towards the gas molecules and high interaction properties, especially in the chemisorption range with short adsorption distance, and a significant amount of charge was transferred to the gases. Although Mo2N interacts with gas molecules at exceptionally high adsorption energies, it is unsuitable for gas adsorption because of its high recovery time and high structural deformation upon adsorption. The sensitivity and recovery time were also evaluated. Therefore, all nanosheets, except for Mo2N, were considered promising candidates for detecting CO and NO gas molecules.
Data availability
The data supporting this article have been included in the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thankfully acknowledge the Bangladesh Research and Education Network (BdREN) for providing computational access to their data.
References
- P. Khakbaz, M. Moshayedi, S. Hajian, M. Soleimani, B. B. Narakathu, B. J. Bazuin, M. Pourfath and M. Z. Atashbar, J. Phys. Chem. C, 2019, 123, 29794–29803 CrossRef CAS.
- T. Van Mourik and R. J. Gdanitz, J. Chem. Phys., 2002, 116, 9620–9623 CrossRef CAS.
- I. Manisalidis, E. Stavropoulou, A. Stavropoulos and E. Bezirtzoglou, Front. Public Health, 2020, 8, 14 CrossRef PubMed.
- K. Pilegaard, Philos. Trans. R. Soc., B, 2013, 368, 20130126 CrossRef PubMed.
- J. D. Gouveia, A. Morales-Garcia, F. Vines, J. R. B. Gomes and F. Illas, ACS Nano, 2022, 16, 12541–12552 CrossRef CAS PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- J. T. Mazumder, M. M. Hasan, F. Parvez, T. Shivam, D. Pamu, A. Kabir, M. Hossain and R. K. Jha, Phys. Chem. Chem. Phys., 2025, 27, 10506 RSC.
- Y. Xin and Y.-X. Yu, Mater. Des., 2017, 130, 512–520 CrossRef CAS.
- Y.-X. Yu, J. Colloid Interface Sci., 2025, 695, 137799 CrossRef CAS PubMed.
- Q. T. H. Ta, M. Jianbin, N. T. Chau, N. H. Nguyen, D. L. Tran, T. M. H. Nguyen, M. H. Tran, V.-Q. Hoang, S. Seo and D. H. Nguyen, Nanoscale Adv., 2025 10.1039/D5NA00021A.
- Y.-X. Yu, J. Phys. Chem. C, 2016, 120, 5288–5296 CrossRef CAS.
- W. Wang, Z. Li, T. Jiang, Z. Zhao, Y. Li, Z. Wang and C. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 6080–6084 CrossRef CAS PubMed.
- Y. Yan, G. Yang, J.-L. Xu, M. Zhang, C.-C. Kuo and S.-D. Wang, Sci. Technol. Adv. Mater., 2020, 21, 768–786 CrossRef CAS PubMed.
- I. Z. Papp, A. Szerlauth, T. Szűcs, P. Bélteky, J. F. G. Perez, Z. Kónya and Á. Kukovecz, Thin Solid Films, 2023, 778, 139908 CrossRef CAS.
- E. Lee, A. VahidMohammadi, B. C. Prorok, Y. S. Yoon, M. Beidaghi and D. J. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 37184 CrossRef CAS PubMed.
- T. Thomas, J. A. Ramon, V. Agarwal, A. Álvarez-Méndez, Y. Kumar and K. C. Sanal, Microporous Mesoporous Mater., 2022, 336, 111872 CrossRef CAS.
- Z. Feng, Y. Xie, J. Chen, Y. Yu, S. Zheng, R. Zhang, Q. Li, X. Chen, C. Sun and H. Zhang, 2D Mater., 2017, 4, 25018 CrossRef.
- B. Cho, M. G. Hahm, M. Choi, J. Yoon, A. R. Kim, Y.-J. Lee, S.-G. Park, J.-D. Kwon, C. S. Kim and M. Song, Sci. Rep., 2015, 5, 8052 CrossRef CAS PubMed.
- S. Zhao, J. Xue and W. Kang, Chem. Phys. Lett., 2014, 595, 35–42 CrossRef.
- M. Altarawneh, Z. Jaf, H. Oskierski, Z. T. Jiang, J. Gore and B. Z. Dlugogorski, J. Phys. Chem. C, 2016, 120, 22270–22280 CrossRef CAS.
- F. Solymosi and L. Bugyi, Catal. Lett., 2000, 66, 227–230 CrossRef CAS.
- T. Wang, Q. Luo, Y. W. Li, J. Wang, M. Beller and H. Jiao, Appl. Catal., A, 2014, 478, 146–156 CrossRef CAS.
- H. Dou, B. Yang, X. Hu, C. Huo, X. Wang and C. Shi, Comput. Theor. Chem., 2021, 1195, 113089 CrossRef CAS.
- D. Yang, D. He, D. Lu, M. Yuan, W. Zeng and Q. Zhou, Phys. Scr., 2024, 99, 125404 CrossRef CAS.
- X. Lv, S. Zhang, J. Wang, M. Wang, J. Shan and S. Zhou, J. Mol. Graphics Modell., 2022, 110, 108056 CrossRef CAS PubMed.
- A. Tkatchenko, R. A. DiStasio, M. Head-Gordon and M. Scheffler, J. Chem. Phys., 2009, 131, 094106 CrossRef PubMed.
- M. Ropo, K. Kokko and L. Vitos, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 195445 CrossRef.
- R. Pollet and H. Amara, J. Chem. Theory Comput., 2009, 5, 1719–1722 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- A. A. Piya, T. Ahmed, M. A. Khaleque, K. Ahmed and S. U. D. Shamim, Comput. Theor. Chem., 2022, 1217, 113902 CrossRef PubMed.
- S. U. Daula Shamim, M. K. Hossain, S. M. Hasan, A. Hossain and F. Ahmed, Mol. Simul., 2020, 46, 1135–1145 CrossRef CAS.
- J. Hu, B. Xu, C. Ouyang, Y. Zhang and S. A. Yang, RSC Adv., 2016, 6, 27467–27474 RSC.
- Y. Wang, W. Tian, H. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2021, 23, 12288–12295 RSC.
- A. Hassan, S. Z. Ilyas, S. Ahmed, F. Niaz, A. Jalil and Q. Khan, Phys. Lett. A, 2021, 392, 127119 CrossRef CAS.
- V. Mehta, H. S. Saini, S. Srivastava, M. K. Kashyap and K. Tankeshwar, Mater. Today Commun., 2021, 26, 102100 CrossRef CAS.
- S. R. Khoshnobish, T. Ahmed, T. Arefin, A. A. Piya and S. U. D. Shamim, Appl. Surf. Sci., 2025, 679, 161301 CrossRef.
- S. U. D. Shamim, A. A. Piya, M. S. Rahman, S. M. Hasan, M. K. Hossain and F. Ahmed, Phys. Chem. Chem. Phys., 2023, 25, 4047–4061 RSC.
- I. R. Shein and A. L. Ivanovskii, Comput. Mater. Sci., 2012, 65, 104–114 CrossRef CAS.
- Y. Wang, W. Tian, H. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2021, 23, 12288–12295 RSC.
- Y. Yu, Z. Guo, Q. Peng, J. Zhou and Z. Sun, J. Mater. Chem. A, 2019, 7, 12145–12153 RSC.
- Y. Yong, H. Cui, Q. Zhou, X. Su, Y. Kuang and X. Li, Appl. Surf. Sci., 2019, 487, 488–495 CrossRef CAS.
- J.-H. Li, J. Wu and Y.-X. Yu, Appl. Surf. Sci., 2021, 546, 149104 CrossRef CAS.
- M. G. Ahangari, A. H. Mashhadzadeh, M. Fathalian, A. Dadrasi, Y. Rostamiyan and A. Mallahi, Vacuum, 2019, 165, 26–34 CrossRef CAS.
- T. Pakornchote, A. Ektarawong, B. Alling, U. Pinsook, S. Tancharakorn, W. Busayaporn and T. Bovornratanaraks, Carbon, 2019, 146, 468–475 CrossRef CAS.
- D. K. Pham, L. H. Luong, T. T. H. Nguyen and V. T. Vu, VNU J. Sci.: Math., 2022, 38(1), 32–43 Search PubMed.
- B. A. Kalwar, W. Fangzong, A. M. Soomro, M. R. Naich, M. H. Saeed and I. Ahmed, RSC Adv., 2022, 12, 34185–34199 RSC.
- A. Junkaew and R. Arroyave, Phys. Chem. Chem. Phys., 2018, 20, 6073–6082 RSC.
- K. N. Munny, T. Ahmed, A. A. Piya and S. U. D. Shamim, Struct. Chem., 2023, 34, 2089–2105 CrossRef CAS.
- S. U. D. Shamim, D. Roy, S. Alam, A. A. Piya, M. S. Rahman, M. K. Hossain and F. Ahmed, Appl. Surf. Sci., 2022, 596, 153603 CrossRef CAS.
- H. Mi, Q. Zhou and W. Zeng, Appl. Surf. Sci., 2021, 563, 150329 CrossRef CAS.
- Y. Jing-yun, Z. Tian-yu, X. Ling-yun, Y. Shu-xia, K. Weerasinghe, P. Ubaldo, H. Ping and G. Qing-feng, J. Electrochem., 2017, 23, 371–380 Search PubMed.
- S. F. Zaman, Bull. Chem. Commun., 2015, 47, 125–132 Search PubMed.
- M. Huzaifa, M. Shafiq, N. Ali, C. Cocchi, M. Nur-e-Alam and Z. Ul-Haq, ACS omega, 2024, 10, 1562–1570 CrossRef PubMed.
- K. Weng, J. Peng, Z. Shi, A. Arramel and N. Li, ACS Omega, 2023, 8, 4261–4269 CrossRef CAS PubMed.
- A. A. Sambare, R. Pawar and M. Shirsat, Theor. Chem. Acc., 2023, 142, 61 Search PubMed.
- S. R. Naqvi, V. Shukla, N. K. Jena, W. Luo and R. Ahuja, Appl. Mater. Today, 2020, 19, 100574 Search PubMed.
- L. Wang, Q. Zhou, W. Feng, W. Ju, D. Kang and Y. Wang, Vacuum, 2024, 224, 113152 CrossRef CAS.
- Y.-H. Zhang, Y.-B. Chen, K.-G. Zhou, C.-H. Liu, J. Zeng, H.-L. Zhang and Y. Peng, Nanotechnology, 2009, 20, 185504 CrossRef PubMed.
- F. Behmagham, E. Vessally, B. Massoumi, A. Hosseinian and L. Edjlali, Superlattices Microstruct., 2016, 100, 350–357 CrossRef CAS.
- M. P. Hyman and J. W. Medlin, J. Phys. Chem. B, 2005, 109, 6304–6310 CrossRef CAS PubMed.
- S. M. Aghaei, M. M. Monshi, I. Torres, S. M. J. Zeidi and I. Calizo, Appl. Surf. Sci., 2018, 427, 326–333 CrossRef.
- Z. Zhao, Y. Yong, Q. Zhou, Y. Kuang and X. Li, ACS Omega, 2020, 5, 12364–12373 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Taslima Akter,
Aditi Ahmed Ananna
*,
Taslima Akter,
Aditi Ahmed Ananna