DOI:
10.1039/D5RA00457H
(Paper)
RSC Adv., 2025,
15, 20760-20769
Influence of a spin crossover iron(III) complex on the detection of phenylenediamines of graphene-modified screen printed electrodes†
Received
19th January 2025
, Accepted 9th June 2025
First published on 19th June 2025
Abstract
We developed a sensitive voltammetric sensor for detecting aromatic phenylenediamine (PD) isomers: para-phenylenediamine (p-PD), ortho-phenylenediamine (o-PD), and meta-phenylenediamine (m-PD). This sensor uses a screen-printed graphene electrode (SPGE) modified with a new spin crossover (SCO) FeIII-complex [Fe(salEen-5-I)2]NCS 1. The structure of 1·0.3CH2Cl2·0.5H2O has been determined by single crystal X-ray crystallography and confirmed by spectroscopic techniques. Magnetic susceptibility measurements reveal a gradual SCO around room temperature. SEM-EDX and XPS analysis confirm the smooth surface morphology and uniform distribution of the SCO molecule on the modified 1-SPGE. Cyclic voltammetry (CV) shows that the modified 1-SPGE exhibited higher sensitivity to PDs than the bare SPGE. Differential pulse voltammetry (DPV) provided linear responses for p-PD (0.3–150 μM), o-PD (0.5–100 μM), and m-PD (1–50 μM), with corresponding limits of detection (LODs, S/N = 3) of 0.062 μM, 0.20 μM, and 0.41 μM, respectively. Furthermore, we successfully employed 1-SPGE to simultaneously detect a mixture of all three PD isomers. Remarkably, these isomers displayed distinct and well-defined electrochemical signals, with comparable electrochemical parameters to those observed in individual measurements, allowing us to successfully use the modified 1-SPGE sensor to monitor PDs in real samples.
Introduction
Phenylenediamines (PDs) are important ingredients often present in permanent hair dyes and used by the polymer and textile industries.1–3 Oxidative coupling of the PD dye precursors and dihydroxybenzene couplers are involved in the dying process.4 While the toxicity of these ingredients is well established,5 PDs are of particular concern.6–10 To mitigate their adverse effects on human health, PDs concentration in hair dye products is regulated by the European Council Directive (76-768-EEC).3 Moreover, o-phenylenediamine (o-PD) and m-phenylenediamine (m-PD) have been classified as banned ingredients. These dyes, particularly para-phenylenediamine (p-PD), possess carcinogenic and mutagenic properties and can generate mutagenic and allergenic intermediates during the dyeing process.9 Therefore, determining the levels of toxic PDs in hair dyes is crucial for quality control, ensuring product reliability, and meeting legal requirements.
The complex composition of hair dyes presents a challenge for analytical chemists, as determining their components often requires separation techniques. Current methods for the determination of PDs include high-performance chromatography (HPLC),11 mass spectrometry (MS),12 capillary zone electrophoresis coupled with amperometric detection (CZE-AD),13 and gas chromatography-mass chromatography (GC-MS).14 However, these analytical methods are complex and suffer from issues like poor resolution and peak tailing and often cannot provide real-time analysis. Electrochemical sensors offer a compelling alternative due to their operational simplicity, low cost, ability to perform real-time qualitative and quantitative analysis, and, are suitable for determining multiple analytes, both individually and simultaneously.5,11,15–17
Among electrochemical sensors, screen-printed carbon electrodes (SPCEs) have attracted increasing attention because of their multiple advantages.18 In recent years, many nanomaterials such as carbon nanotubes, nanoparticles and graphene have been extensively used for electrode modification.19,20 Graphene, a two-dimensional material, is particularly prominent due to its high surface-to-volume ratio and excellent electrical conductivity, making it a favored electrode modification material.21 While simple drop-casting is a common method for modifying screen-printed electrodes, it often leads to poor reproducibility and is unsuitable for large-scale production. Inkjet printing offers a promising alternative as it is a rapid and straightforward manufacturing process that enables low-cost, high-volume production of sensors with improved precision and reproducibility.22–24 However, bare screen-printed electrodes, including SPCEs, have limitations in certain applications due to their fixed surface area and limited catalytic activity. Screen-printed graphene electrodes (SPGEs) have demonstrated superior electrochemical performance compared to SPCEs.25 Moreover, the electrocatalytic performance of graphene-based electrodes can be further improved by incorporating materials like metal nanoparticles, conducting polymers, and ionic liquids.26 These modifications increase the active surface area and the number of electrocatalytic sites on the electrode, facilitating interactions with the analyte molecules, which ultimately leads to stronger electrical signals.
Spin crossover (SCO) materials are well-known bistable molecular systems. Their ability to abruptly switch spin states in response to external stimuli like temperature, pressure, electric fields, or light makes them promising for a variety of applications.27 The accompanying changes in structural, magnetic, dielectric, and optical absorption properties make them attractive candidates for data storage, optoelectronics, and sensors.28 However, like many molecular compounds, SCO materials are typically non-conductive, which hinders their integration into electrical devices.29 In contrast, graphene is a highly conductive material that is very sensitive to its surrounding environment.30
Combining the unique properties of SCO materials and graphene, we prepared a modified a screen-printed graphene electrode (SPGE) by blending it with a novel SCO complex, [Fe(salEen-5-I)2]NCS (salEen-5-I = 2-{[(2-(ethylamino)ethyl]-imino)methyl}-4-iodophenolate; 1). This modified electrode is referred to as 1-SPGE. Complex 1 shows a gradual spin crossover around room temperature. The resulting 1-SPGE demonstrated improved sensitivity for all three phenylenediamine isomers compared to unmodified SPGE. Notably, this work is the first to use a molecular SCO complex to modify an electrochemical electrode and achieve quantitative electrochemical detection of PD isomers, both individually and simultaneously.
Experimental section
Materials
All reactions were carried out in aerobic conditions using commercial grade solvents for the synthesis of all compounds. All chemicals were purchased from TCI Chemicals or Sigma-Aldrich and used as received. A 0.1 M phosphate buffer saline (PBS) of pH 7 was prepared by mixing the solutions of NaH2PO4·2H2O (0.042 M) and Na2HPO4 (0.058 M) with 0.1 M KCl in 1 L distilled water. The pH was adjusted by using diluted HCl or NaOH if required followed by autoclaving at 120 °C. Deionized distilled water was used to prepare standards and regents by diluting appropriate quantities for voltammetry experiments. Three hair dyes (HC-1, HC-2 and HC-3; low to high price) were chosen for real sample analysis which were purchased from local market, “Saveone Night Market”, Nakhon Ratchasima, Thailand.
Synthesis of [Fe(salEen-5-I)2]NCS 1
A solution of 5-iodosalicylaldehyde (0.496 g, 2 mmol) in ethanol (6 mL) was charged with N-ethylethylenediamine (0.210 mL, 2 mmol) and stirred at ambient condition for 2 h to produce HsalEen-5-I. Triethylamine (0.278 mL, 2 mmol) was then added and left for 15 minutes for deprotonation. To this mixture, a mixture of iron(III) chloride (0.170 g, 1.0 mmol) and potassium thiocyanate (0.296 g, 3.0 mmol) in ethanol (6 mL) was added through a Celite® pad. The reaction was stirred at room temperature for 48 h. A blue tint characteristic of 1 gradually appeared. The solution was filtered and reduced to dryness in vacuo to give a black residue. The residue was dissolved in 20 mL CH2Cl2 and washed twice with water (20 mL). The resulting CH2Cl2 solution was dried with MgSO4 and filtered. Hexane (10 mL) was added to the blue solution and volume slowly reduced under vacuum to give black microcrystals. The crystals were filtered, rinsed with cold Et2O (5 mL) and dried in air. 1 was collected as a black microcrystalline solid (576 mg, 73%). IR spectroscopy: 3130 (νN–H), 2945, 2939, 2872 (νC–H), 2044 (νC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1608 (νC
N), 1608 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). UV-visible absorption (DMF): 367 nm (7293 M−1 cm−1), 544 nm (2676 M−1 cm−1), 650 nm (2060 M−1 cm−1). Elemental analysis: calcd (found) for C23H28N5O2I2SFe: C, 36.92 (37.23); H, 3.77 (3.93); N, 9.36 (9.28).
N). UV-visible absorption (DMF): 367 nm (7293 M−1 cm−1), 544 nm (2676 M−1 cm−1), 650 nm (2060 M−1 cm−1). Elemental analysis: calcd (found) for C23H28N5O2I2SFe: C, 36.92 (37.23); H, 3.77 (3.93); N, 9.36 (9.28).
Physical measurements
Infrared spectra (4000–400 cm−1) were recorded as KBr pellets on a Bruker Tensor 27 FT-IR spectrometer with OPUS data collection program (Table S1; Fig. S1†). Absorption spectra was collected on PG Instruments T80+ UV/Vis spectrometer from a solution of 1 in CH2Cl2 (Fig. S2†). Elemental analyses were carried out by using a Eurovector EA3000 analyzer. ESI-MS were carried out on a Bruker AmaZon X LCMS mass spectrometer. Thermogravimetric analyses were performed on Mettler Toledo TGA/DSC1 in the range of 35–300 °C (Fig. S4†). The magnetic susceptibility data of 1 was collected with Microsense EZ7 Vibrating Sample Magnetometer under applied field of 10 kOe, sweeping from 220 to 430 K in the rate of 10 K min−1. The powdered sample was placed in a sealed aluminum capsule and attached to a 3 mm diameter quartz rod.
X-ray crystallography
The diffraction data of 1·0.3CH2Cl2·0.5H2O was collected at 150 K on a Bruker Nonius Kappa CCD diffractometer using MoKα radiation (λ = 0.71073 Å; Table S2†). The structure was solved, and the space group P21/c was determined by intrinsic phasing using ShelXT and refined by full matrix least-squares minimization on F2 using SHELXL.31,32 All non-hydrogen atoms were refined anisotropically. Disorder was found in the NCS anion, and this was modelled in two parts with appropriate SADI and RIGU restraints. CH2Cl2 and H2O with partial occupancies of 0.3 and 0.5 were also found in the asymmetric unit and modelled in their appropriate parts. Hydrogen atoms were included in calculated positions and refined with isotropic thermal parameters, which were 1.2× or 1.5× the equivalent isotropic thermal parameters of their parent carbon or oxygen atoms. All pictures were generated with OLEX2.33 Crystallographic data for the structure have been deposited with the Cambridge Crystallographic Data Centre (CCDC) 2417988. Powder X-ray diffraction data were measured on a Rigaku SuperNova diffractometer with a HyPix 3000 detector using Cu α radiation (λ = 1.54184 Å). The samples were ground and then suspended in Fomblin Y oil, and the data were collected between 2θ = 5–80°.
Fabrication of 1-SPGE
In this work, we used a screen-printed electrode (SPGE), developed based on previous reports by W. Kamsong and coworkers. The electrode fabrication involved a carbon ink to graphene to [Fe(salEen-I)2]NCS (1) ratio of 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:1% (w/w). This composited ink was mixed by using a ball-milling machine at 800 rpm for 1 h. A homogeneous conductive ink was then screened by using an automatic screen-printing machine. Briefly, the homogeneous conductive ink was screened on polyethylene terephthalate (PET) substrates to obtain working and counter electrodes. Next, Ag/AgCl paste ink was screened on the same substrates to create reference electrodes. Afterward, insulating ink was screened to define the working area of the electrodes. Each fabrication step of the electrodes was baked in an oven at 60–100 °C for 15–20 min. Finally, this modified electrode is referred to as 1-SPGE. The studied parameters included a viscosity range of 4.0–7.0 Pa s, a thickness range of 20–25 μm, a resistance of 347.53 ± 30.50 Ω, and a conductivity of 261.67 ± 64.33 Ω−1.
:5:1% (w/w). This composited ink was mixed by using a ball-milling machine at 800 rpm for 1 h. A homogeneous conductive ink was then screened by using an automatic screen-printing machine. Briefly, the homogeneous conductive ink was screened on polyethylene terephthalate (PET) substrates to obtain working and counter electrodes. Next, Ag/AgCl paste ink was screened on the same substrates to create reference electrodes. Afterward, insulating ink was screened to define the working area of the electrodes. Each fabrication step of the electrodes was baked in an oven at 60–100 °C for 15–20 min. Finally, this modified electrode is referred to as 1-SPGE. The studied parameters included a viscosity range of 4.0–7.0 Pa s, a thickness range of 20–25 μm, a resistance of 347.53 ± 30.50 Ω, and a conductivity of 261.67 ± 64.33 Ω−1.
Electrochemical measurements
Electrochemical measurements were conducted with a SPELEC-RAMAN bipotentiostat (Metrohm Dropsens, Oviedo, Asturias, Spain). An unmodified and modified (1-SPGE) working electrode, a Ag/AgCl reference electrode, and a SPGE counter electrode were used for all measurements. Inert gas was purged to remove dissolved oxygen for a couple of minutes before each experiment. The deaerated electrolyte solution (14 mL) and the desired concentration of analyte was added to the electrochemical cell before performing cyclic voltammetry (CV) or differential pulse voltammetry (DPV) analysis and then electrochemically cleaned by running 3 CV/DPV cycles to remove contaminants on the electrode surface. CV experiments were performed at a scan rate of 50 mV s−1, by selecting a potential window suitable for the corresponding analyte. DPV experiments were conducted at a scan rate of 10 mV s−1, Epulse step of 2 mV s−1 and tpulse of 50 ms within the appropriate potential range of each analyte. The electrochemical stability of modified 1-SPGE was tested in a 5.0 mM K3[Fe(CN)6] solution with 0.1 M KCl. All other electrochemical experiments for selected PDs and real sample analysis were examined in 0.1 M PBS buffer (pH 7) in aq. 0.1 M KCl electrolyte. All peaks from raw data were extracted using DropView SPELEC software and figures were drawn in OriginPro-8.5.
Results and discussion
Synthesis and characterization
The synthesis of [Fe(salEen-5-I)2]NCS 1 involved the reaction of Fe(NCS)3, prepared by anion metathesis, with the ligand HsalEen-5-I prepared in situ34,35 in the presence of NEt3 (Scheme 1). Long reaction times of not less than 48 h were required to isolate the targeted bis-tridentate complex which appeared as an intense blue solution. Washing the crude mixture with water and recrystallizing from CH2Cl2:hexane gave 1 as black crystals in good yield. This reaction procedure is highly scalable up to 1.5 mmol. The complex was fully characterized by IR and UV-visible spectroscopy, mass spectrometry, elemental analysis and thermogravimetric analysis.
 |
| | Scheme 1 Synthesis of 1. | |
The IR spectrum of 1 reveals peaks corresponding to the amino group, aromatic rings, aliphatic backbone, coordinating imine and the thiocyanate anion. The imine stretch is comparable to the reported value for [Fe(salEen-5-I)2]ClO4 (ref. 36) consistent with the proposed structure. The absorption spectrum in dimethylformamide (DMF) exhibits strong peaks at approximately 544 nm and 650 nm, matching the LMCT band reported for [Fe(salEen-5-I)2]ClO4,36 [Fe(salEen-5-Br)2]ClO4 (ref. 37) and [Fe(salMeen-5-Br)2]ClO4 (salMeen = N-methyl-N-(2-aminoethyl)salicylaldiminate).38 The λmax of 544 nm and 650 nm is also consistent with HS and LS state Fe(III) as found in a range of [Fe(naphRen)2]+ (naphRen = 1-{[2-(alkylamino)ethylimino]-methyl}-2-naphtholate) complexes respectively, demonstrated by [Fe(naphEen)2]halide (naphEen = 1-{[2-(ethylamino)-ethylimino]methyl}-2-naphtholate; halide = Cl, Br).39
Solid-state structure and packing
Suitable black crystals of 1 were grown by slow diffusion of a CH2Cl2 solution of the complex into hexane. At 150 K the structure is found in monoclinic P21/c (Fig. 1). The asymmetric unit reveals one cationic complex with an octahedral Fe(III) center exhibiting a mer geometry, a disordered thiocyanate anion and CH2Cl2 and water molecules. Despite the disorder, the thiocyanate forms strong N–H⋯N hydrogen bonds as is well documented for this class of complexes.34,40,41 The Fe–N/O distances are typical of the LS state, supported by the octahedral distortion parameters Σ and θ.40,42
 |
| | Fig. 1 Asymmetric unit of 1 at 150 K. | |
Solid-state packing of 1 reveals a 1-D ladder coincident with the c-axis composed of pairs of cationic complexes, with the counteranion and solvent layers connected to this layer via amine N–H⋯NCS, S⋯Cl and C–H⋯I interactions. These chains are linked into 2-D planes within the ab plane through multiple C–H⋯I, C–H⋯S and C–H⋯N interactions (Fig. 2). While there are extensive interactions these are not unusually short and suggest that the overall cooperativity in the complex is weak.
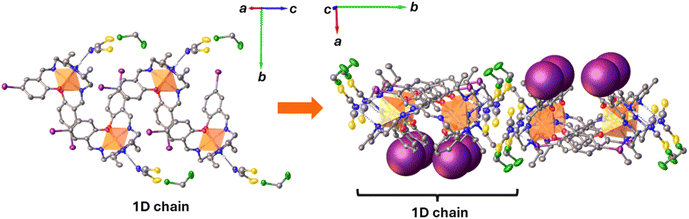 |
| | Fig. 2 Schematic illustration of the 1D chain in [Fe(salEen-5-I)2]NCS 1 at 150 K (right) and the arrangement of the 1D chains into 2D planes (left). | |
Magnetic studies
Temperature dependent solid-state magnetism of [Fe(salEen-I)2]NCS 1 was studied by vibrating sample magnetometry (VSM) and is displayed as a plot of magnetic susceptibility (χMT) and temperature (K) in Fig. 3. It's important to note that the sample on which magnetic studies were conducted did not contain solvent as shown by elemental analysis. The magnetic susceptibility increased from 0.60 cm3 K mol−1 to 3.50 cm3 K mol−1 upon heating from 220–430 K, indicating an incomplete SCO from the LS to a mostly HS state (ca. 80% HS), indicating gradual SCO and hence weak cooperativity. The magnetic profile upon cooling is similar to that above, with a slight increase in the magnetic susceptibility to approximately 0.75 cm3 K mol−1 at 220 K, indicating no noticeable thermal hysteresis.
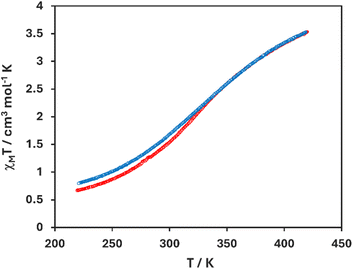 |
| | Fig. 3 Plot of χMT against temperature in [Fe(salEen-I)2]NCS 1. | |
Surface morphology and elemental composition
The surface morphology of SPGE and 1-SPGE electrodes was analysed by SEM-EDX (Fig. 4) at 15 kV. Fig. 4A and B shows the SEM images of both electrodes and EDX elemental mapping analysis of 1-SPGE. From the surface morphology images, regular dispersed surface of the working electrode area was conformed for SPGE (Fig. 4A) and 1-SPGE (Fig. 4B). Elemental color mapping confirms that 1 is evenly distributed in 1-SPGE as shown in Fig. 4C. X-ray photoelectron spectroscopy (XPS) using Al-K radiation was employed to determine the chemical states and elemental composition of elements in the SPGE and 1-SPGE electrodes, as shown in Fig. 4D–I. The XPS scans confirm that the two main elements, are carbon (C 1s, Fig. 4D) and oxygen (O 1s, Fig. 4E) with C:O atomic compositions of 87.5:5.3 and 71.4:23.5 for SPGE and 1-SPGE, respectively. It suggests that 1-SPGE has a slightly higher concentration of oxygen as it contains 1 and the decrease in carbon composition is due to the presence of other atoms in the SCO compound. From the C 1s core level in Fig. 4D the spectra of SPGE and 1-SPGE could be deconvoluted into six Gaussian components and their main peaks at 285.1 and 285 eV that are assigned to sp2 C–C bonding of graphitic lattice. While, minor peaks are assigned to CO, C–O and C–OH surface carbon function groups as indicated in the inset of Fig. 4D, respectively.43 Fig. 4E displays the core level O 1s spectra of SPGE and 1-SPGE, with the signals deconvoluted into four Gaussian peaks centered 531.2–535.1 and 532.1–535.4, respectively (see inset of Fig. 4E for detailed surface functional groups). It could be observed that the XPS peaks of 1-SPGE were positively shifted compared with those of SPGE, presumably due to the presence of 1.43 Furthermore, the XPS spectra of the other elements (Fig. 4F–I, N, S, I and Fe) were also confirmed with their corresponding Gaussian peaks. It was very difficult to locate Fe peaks (Fig. 4I) due to their relatively very low atomic percentile compositions in the blended complex. Collectively, SEM-EDX and XPS analysis are broadly consistent with the calculated and observed compositions of the corresponding elements in the modified 1-SPGE.
 |
| | Fig. 4 SEM images of SPGE (A) and 1-SPGE (B); EDX, elemental mapping of 1-SPGE (C); SPGE vs. 1-SPGE XPS spectra comparisons of C 1s (D) and O 1s (E); XPS spectra of other elements in complex 1 of 1-SPGE (F–I) as indicated in the inset of each figure. | |
Electrochemical stability of modified 1-SPGE
The electrochemical stability of the modified 1-SPGE electrode was investigated in the presence of 5 mM redox probe K3[Fe(CN)6] in aqueous 0.1 M KCl as a supporting electrolyte solution by cyclic voltammetry between −0.4 to 0.7 V at a scan rate of 50 mV s−1 in an inert environment. Fig. 5A shows that the redox peak positions and current response are virtually identical for all 20 voltammograms. Further studies on 10 different 1-SPGEs reveal that the calculated average peak to peak potential separation was ΔEp = 0.164 V with 3.38% relative standard deviation between the electrodes (Fig. 5B), signifying that 1-SPGE is extremely stable.44–46 As expected varying the scan rate (Fig. 5C) results in a corresponding change in the peak current, which is proportional to the scan rate, indicating a diffusion controlled redox process (Fig. 4D).
 |
| | Fig. 5 (A) 20 CV-cycles of modified 1-SPGE at the scan rate of 50 mV s−1 in 5.0 mM in K3[Fe(CN)6] solution added with 0.1 M KCl from −0.4 V to 0.7 V potential range. (B) calculated peak to peak potential separation (ΔEp) of 10 different 1-SPGEs with 3.38% relative standard deviation and y-bar error represents the standard deviation in ΔEp of 20 CV-cycles for each 1-SPGE electrode. (C) and (D) Different scan rates and linear fitting, respectively. | |
Electrochemical performance of 1-SPGE versus SPGE
The performance of 1-SPGE and SPGE against the PD isomers, p-PD, o-PD and m-PD was examined by cyclic voltammetry. Three CVs were performed by adding 100 μM of each analyte in deaerated 0.1 M PBS buffer (pH 7) in aq. 0.1 M KCl electrolyte at 50 mV s−1 for both electrodes (Fig. 6). The modified 1-SPGE shows enhanced redox peak currents as compared to bare SPGE for p-PD. In addition, the modified electrode also reveals a slightly smaller peak potential separation (ΔEp = 40 mV) compared with the bare electrode (ΔEp = 49 mV), although both are typical of a reversible redox couple (Fig. 6A).17,44–47 This reversibility is mirrors previous reports.48 Similarly, 1-SPGE also revealed better electrochemical activity for irreversible oxidation of o-PD and m-PD under identical CV conditions as the peak current increased for both analytes compared to the unmodified SPGEs (Fig. 6A and B). The o-PD isomer in Fig. 6B shows two distinct oxidation–oxidation process peaks at around 0.25 and 0.36 V, respectively. Hereafter, only the first oxidation peak at 0.25 V will be discussed. The voltammograms in Fig. 6C, show irreversible oxidation of m-PD, and again show a well-defined oxidation peak at ≈0.51 V in 1-SPGE with enhanced oxidation current compared to bare SPGE. Fig. S6† reveals that the peak current is proportional to the scan rate, indicating diffusion controlled redox processes.16 Overall, 1-SPGE demonstrated significantly better electrochemical performance toward all three PD isomers compared to bare SPGE suggesting that the inclusion of the SCO complex in modified 1-SPGE enhances the sensitivity of the electrode to the analyte molecules.49
 |
| | Fig. 6 Cyclic voltammograms of 1-SPGE vs. SPGE added 100 μM concentration of indicated analyte in 0.1 M PBS buffer (pH 7) in aq. 0.1 M KCl at 50 mV s−1, (A) p-PD (B) o-PD and (C) m-PD. | |
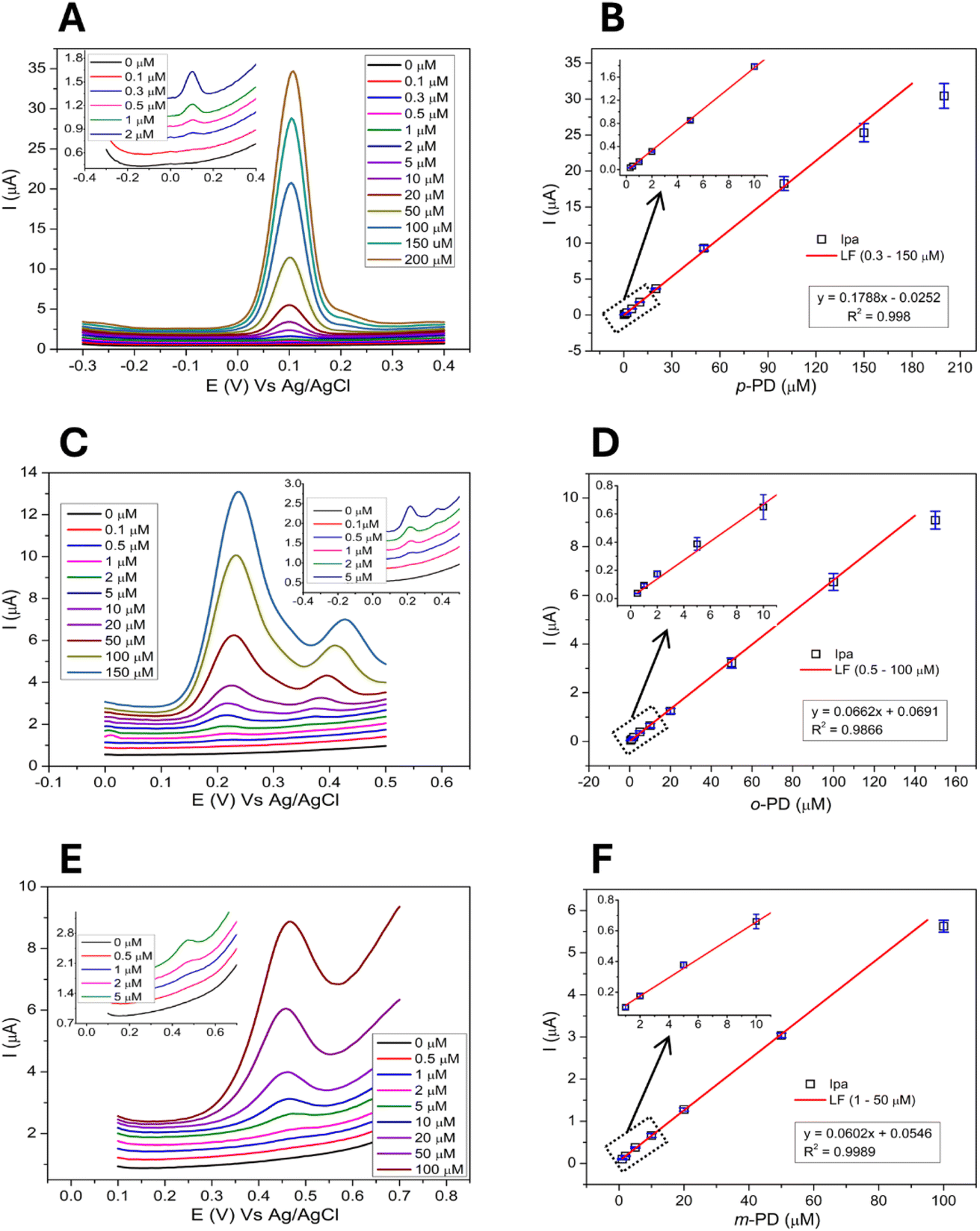
The electrochemical response of the modified 1-SPGE was also investigated using DPV. Successive addition of calculated concentrations of p-PD (Fig. 7A), o-PD (Fig. 7C) and m-PD (Fig. 7E) allowed detection of all three analytes. The peak current of the modified 1-SPGE was found to exhibit a linear relationship with the concentration of the analyte, with the correlation coefficient and detection limit summarized in the insets of Fig. 7 and in Table 1. The limit of detection (LOD) was calculated using eqn (1) based on three replicates,50
| |
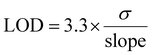 | (1) |
where
σ is the standard deviation of the linear regression plot.
 |
| | Fig. 7 Electrochemical response of the 1-SPGE for (A) p-PD, (C) o-PD, and (E) m-PD at different concentrations and the calibration curve (horizontally parallel), error bars represent the standard deviation among three replicates (SD, n = 3) (B) p-PD, (D) o-PD, and (F) m-PD respectively. | |
Table 1 Electrochemical comparison of the separate and simultaneous detection of PDsa
| Parameters |
Separate detection of PDs |
Simultaneous detection of PDs |
| p-PD |
o-PD |
m-PD |
p-PD |
o-PD |
m-PD |
| Detection limit = (3.3 × σ)/slope (where, σ = standard deviation of the slope; and n = 3). |
| Linear range (μM) |
0.3–150 |
0.5–100 |
0.5–50 |
0.3–30 |
2–50 |
5–50 |
| Detection limit (μM) |
0.062 |
0.20 |
0.41 |
0.08 |
0.25 |
0.55 |
| Peak potential (V) |
0.09 ± 0.01 |
0.24 ± 0.01 |
0.48 ± 0.02 |
0.07 ± 0.01 |
0.24 ± 0.01 |
0.52 ± 0.02 |
For the determination of PDs isomers, the widest linear range of 0.3–150 μM was achieved for p-PD with the lowest detection limit of 0.062 μM. By comparison, the concentration range and detection limit of o-PD, was 0.5–100 and 0.20 μM and m-PD, 1–50 and 0.41 μM, respectively. These results indicate that 1-SPGE possesses larger linear ranges and lower limit of detections than most reported PDs biosensors (Table S3.1†).2,3,16,17 Moreover, the peak potential of the three PD analytes varies sufficiently from each other (p-PD, o-PD, and m-PD; 0.09, 0.24 (first oxidation) and 0.48 V, accordingly) providing an opportunity for selective and simultaneous detection of a mixture of PDs isomers.
Simultaneous detection of phenylenediamine isomers
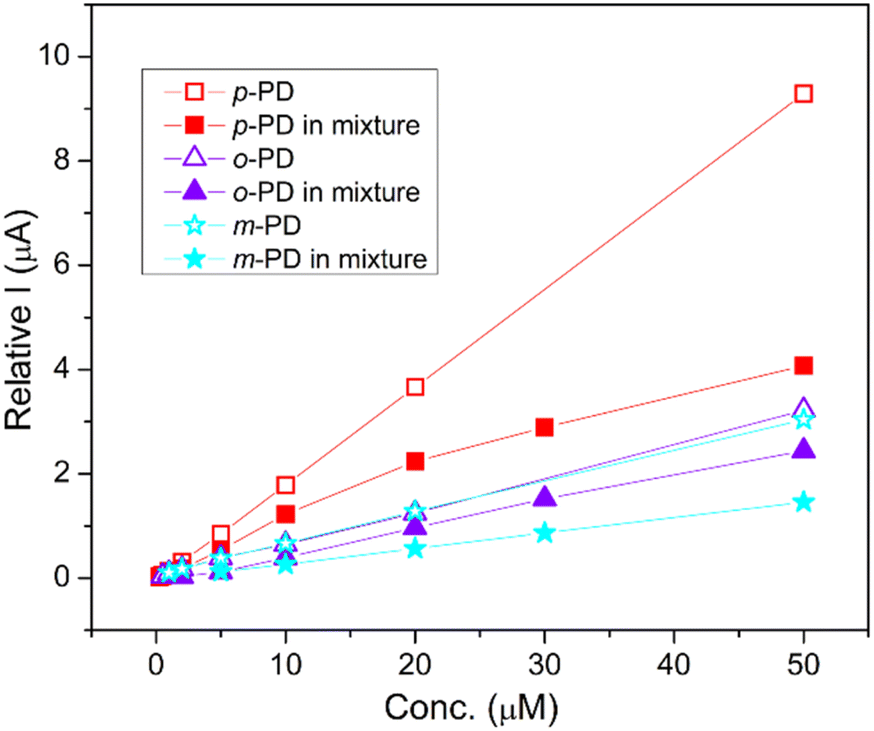
The simultaneous detection of the three PD isomers is shown in Fig. 8. As before, we used a deaerated saline PBS (0.1 M, pH 7) and chose a potential range from −0.2 to +0.65 V which contains the peak potential of all three analytes. Pleasingly, the oxidation peaks of p-PD, o-PD and m-PD were clearly separated at 0.07 V, 0.24 V and 0.52 V, respectively, with the peak current proportional to the concentration to the analyte. The linear concentration range of p-PD, o-PD and m-PD was 0.3–30 μM, 2–50 μM and 5–50 μM with an LOD of 0.08 μM, 0.25 μM and 0.55 μM, respectively. Simultaneous detection of the analytes results in a slightly lower peak current when compared with individual detection as shown in Fig. 9. This decrease may be due to the intermolecular interactions between the PD isomers which might hinder their mobility towards electrode surface. This is supported by the fact that this effect is very negligible at lower concentrations. Despite the lower current, the limits of detections are quite similar whether the analytes are detected individually or together (Table 1). We also note that the 1-SPGE is more sensitive for p-PD than o-PD and m-PD isomers, irrespective of the experimental conditions, presumably as the redox process is fully reversible in the case of p-PD.
 |
| | Fig. 8 Electrochemical response of 1-SPGE in the presence of p-PD, o-PD, and m-PD (A) and their calibration curves (B), error bars represent the standard deviation among three replicates (SD, n = 3). | |
 |
| | Fig. 9 Comparison of the electrochemical response of 1-SPGE for the separate and simultaneous detection of PD isomers. Error bars are hidden for clarity. | |
Real sample determination of phenylenediamine isomers
To explore the potential of 1-SPGE for the detection of PDs we tested the electrode on three real hair dye (HD) samples. Firstly, 1 g of each HD was dissolved in 20 mL of water and then 10 μL aliquots from that solution were added to the electrochemical cell containing PBS to give a total volume of 14 mL for DPV measurements. Then the solution was spiked with a known concentration of each PD isomer with three determinations per sample. Comparison with standard analytical techniques reveals that 1-SPGE was able to detect all three PDs very accurately (Fig. S7†). The calculated concentrations of PDs in the HDs and the percent recovery are summarized in Table 2 which clearly demonstrate the utility of 1-SPGE as a sensor.
Table 2 PDs determination in three different hair dyes
| Hair dye |
PDs |
Founda (mM) |
Added (μM) |
Recovery (μM) |
RSD (%) |
Recovery (%) |
| Concentration found in 20 mL solution containing 1 g HD. |
| HD-1 |
p-PD |
32 ± 0.6 |
10 |
10.15 |
2.0 |
101.5 |
| o-PD |
3.2 ± 0.1 |
10 |
9.97 |
2.9 |
99.7 |
| m-PD |
7.5 ± 0.2 |
10 |
10.02 |
3.3 |
100.2 |
| HD-2 |
p-PD |
35 ± 0.2 |
10 |
9.17 |
0.2 |
91.7 |
| o-PD |
2.2 ± 0.1 |
10 |
9.62 |
4.4 |
96.2 |
| m-PD |
5.3 ± 0.3 |
10 |
9.53 |
4.5 |
95.3 |
| HD-3 |
p-PD |
14 ± 0.2 |
10 |
10.4 |
2.1 |
104 |
| o-PD |
3.0 ± 0.3 |
10 |
10.41 |
5.0 |
104.1 |
| m-PD |
0.0 |
— |
— |
— |
— |
Conclusions
In summary, blending a FeIII-based SCO complex with graphene allows fabrication of a modified 1-SPGE electrode for the reliable detection of three phenylenediamine isomers. Electrochemical studies reveal that the modified electrode exhibits better performance than the unmodified electrode. We are also able, for the first time, to demonstrate simultaneous detection of all three phenylenediamine isomers indicating that 1-SPGE is an excellent electrode for such aromatic amines. Finally, analysis of real samples highlights the applicability of 1-SPGE for electrochemical monitoring of these analytes in real-world applications. Future work will focus on determining whether this approach is more broadly applicable and develop a deeper understanding of how the inclusion of such metal complexes is able to enhance electrochemical performance.
Data availability
All underlying data for this work is available from the authors upon request. Data relating to the crystal structure is available from the CCDC, 2417988.
Author contributions
Basharat Ali: investigation, formal analysis, data curation, visualization, writing. Theerapoom Boonprab: investigation, formal analysis, data curation, visualization, writing. Warisa Thammasangwan: investigation, formal analysis, data curation. Sittipong Kaewmorakot: investigation, formal analysis, data curation. Chanpen Karuwan: investigation, formal analysis, data curation. Adisorn Tuantranont: investigation, formal analysis, data curation. Guillaume Chastanet: investigation, formal analysis, data curation, writing – review & editing. David J. Harding: conceptualization, resources, writing – review & editing, supervision. Phimphaka Harding: conceptualization, resources, writing – review & editing, supervision, funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the National Research Council of Thailand (NRCT) grant number NRCT5-RSA63019-02 for funding. This research has received funding support from the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation [grant number B13F660056]. The Development and Promotion of Science and Technology Talents Project (DPST) is thanked for scholarships to W. T. and S. K. The National Science Technology and Innovation Policy Office for Integrated Research and Innovation Plan (Grant No. 256113A3050001) is thanked for funds to purchase an X-ray diffractometer.
Notes and references
- Y.-H. Chen, Y.-Y. Liu, R.-H. Lin and F.-S. Yen, J. Hazard. Mater., 2009, 163, 973–981 CrossRef CAS PubMed.
- M. Singh, S. R. Bhardiya, A. Rai and V. K. Rai, Sens. Diagn., 2022, 1, 376–386 RSC.
- Y. H. Bai, J. Y. Li, Y. h. Zhu, J. J. Xu and H. Y. Chen, Electroanalysis, 2010, 22, 1239–1247 CrossRef CAS.
- G. P. Hooff, N. A. van Huizen, R. J. Meesters, E. E. Zijlstra, M. Abdelraheem, W. Abdelraheem, M. Hamdouk, J. Lindemans and T. M. Luider, PLoS One, 2011, 6, e22191 CrossRef CAS PubMed.
- N. S. Lawrence, E. L. Beckett, J. Davis and R. G. Compton, Analyst, 2001, 126, 1897–1900 RSC.
- K. Öllinger and A. Brunmark, Biochem. Pharmacol., 1994, 48, 1707–1715 CrossRef PubMed.
- T. Harauchi and M. Hirata, Toxicol. Lett., 1993, 66, 35–46 CrossRef CAS PubMed.
- K.-T. Chung, J. Environ. Sci. Health, Part C, 2016, 34, 233–261 CrossRef CAS PubMed.
- S. Xu, H. Wang, Y. Liu, C. Zhang, Y. Xu, F. Tian and L. Mei, PLoS One, 2021, 16, e0243792 CrossRef CAS PubMed.
- G. J. Nohynek, R. Fautz, F. Benech-Kieffer and H. Toutain, Food Chem. Toxicol., 2004, 42, 517–543 CrossRef CAS PubMed.
- N. A. Penner and P. N. Nesterenko, Analyst, 2000, 125, 1249–1254 RSC.
- W. Chen, T. A. Nkosi, S. Combrinck, A. M. Viljoen and C. Cartwright-Jones, J. Pharm. Biomed. Anal., 2016, 128, 119–125 CrossRef CAS PubMed.
- S. Dong, L. Chi, S. Zhang, P. He, Q. Wang and Y. Fang, Anal. Bioanal. Chem., 2008, 391, 653–659 CrossRef CAS PubMed.
- Y. Bousliman, R. Eljaoudi, M. A. Elcadi, T. Basset, J.-P. Gay-Montchamp, A. A. Zeggwagh, A. Bouklouze, Y. Cherrah and M. Ollagnier, 2012.
- J. P. Winiarski, B. F. Tavares, K. de Fátima Ulbrich, C. E. M. de Campos, A. A. Souza, S. M. G. U. Souza and C. L. Jost, Microchem. J., 2022, 175, 107141 CrossRef CAS.
- R. Sasikumar, P. Ranganathan, S.-M. Chen and S.-P. Rwei, J. Colloid Interface Sci., 2017, 504, 149–157 CrossRef CAS PubMed.
- C. Wu, Z. Liu, H. Sun, X. Wang and P. Xu, Biosens. Bioelectron., 2016, 79, 843–849 CrossRef CAS PubMed.
- Z. Taleat, A. Khoshroo and M. Mazloum-Ardakani, Microchim. Acta, 2014, 181, 865–891 CrossRef CAS.
- J. N. Tiwari, V. Vij, K. C. Kemp and K. S. Kim, ACS Nano, 2016, 10, 46–80 CrossRef CAS PubMed.
- C. Zhu, G. Yang, H. Li, D. Du and Y. Lin, Anal. Chem., 2015, 87, 230–249 CrossRef CAS PubMed.
- P. Bollella, G. Fusco, C. Tortolini, G. Sanzò, G. Favero, L. Gorton and R. Antiochia, Biosens. Bioelectron., 2017, 89, 152–166 CrossRef CAS PubMed.
- H. Xing, X. Zhang, S. Zhai, W. Mu, C. Li and X. Han, J. Electroanal. Chem., 2024, 118222 CrossRef CAS.
- C. D. Lima, L. M. de Almeida Melo, L. C. Arantes, N. dos Santos Conceição, I. de França Schaffel, L. L. Machado, R. de Queiroz Ferreira and W. T. P. Dos Santos, Talanta, 2024, 269, 125375 CrossRef CAS PubMed.
- M. R. Islam, S. Afroj, J. Yin, K. S. Novoselov, J. Chen and N. Karim, Adv. Sci., 2024, 11, 2304140 CrossRef CAS PubMed.
- C. Karuwan, A. Wisitsoraat, D. Phokharatkul, C. Sriprachuabwong, T. Lomas, D. Nacapricha and A. Tuantranont, RSC Adv., 2013, 3, 25792–25799 RSC.
- W. Kamsong, V. Primpray, P. Pasakon, C. Sriprachuabwong, S. Pakapongpan, J. P. Mensing, A. Wisitsoraat, A. Tuantranont and C. Karuwan, Electrochem. Commun., 2022, 135, 107209 CrossRef CAS.
- G. Molnár, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2018, 30, 1703862 CrossRef PubMed.
- R. Torres-Cavanillas, M. Gavara-Edo and E. Coronado, Adv. Mater., 2024, 36, 2307718 CrossRef CAS PubMed.
- F. Prins, M. Monrabal-Capilla, E. A. Osorio, E. Coronado and H. S. van der Zant, Adv. Mater., 2011, 23, 1545–1549 CrossRef CAS PubMed.
- V. Dhinakaran, M. Lavanya, K. Vigneswari, M. Ravichandran and M. Vijayakumar, Mater. Today: Proc., 2020, 27, 824–828 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- T. Boonprab, P. Harding, K. S. Murray, W. Phonsri, S. G. Telfer, A. Alkaş, R. Ketkaew, Y. Tantirungrotechai, G. N. Jameson and D. J. Harding, Dalton Trans., 2018, 47, 12449–12458 RSC.
- T. Boonprab, S. J. Lee, S. G. Telfer, K. S. Murray, W. Phonsri, G. Chastanet, E. Collet, E. Trzop, G. N. Jameson and P. Harding, Angew. Chem., 2019, 131, 11937–11941 CrossRef.
- F. F. Martins, A. Joseph, H. P. Diogo, M. E. Minas da Piedade, L. P. Ferreira, M. D. Carvalho, S. Barroso, M. J. Romão, M. J. Calhorda and P. N. Martinho, Eur. J. Inorg. Chem., 2018, 2018, 2976–2983 CrossRef CAS.
- P. N. Martinho, A. I. Vicente, S. Realista, M. S. Saraiva, A. I. Melato, P. Brandão, L. P. Ferreira and M. de Deus Carvalho, J. Organomet. Chem., 2014, 760, 48–54 CrossRef CAS.
- M. A. Al-Azzani, F. Al-Mjeni, R. Mitsuhashi, M. Mikuriya, I. A. Al-Omari, C. C. Robertson, E. Bill and M. S. Shongwe, Chem.–Eur. J., 2020, 26, 4766–4779 CrossRef CAS PubMed.
- R. H. Petty, E. V. Dose, M. F. Tweedle and L. J. Wilson, Inorg. Chem., 1978, 17, 1064–1071 CrossRef CAS.
- A. Tissot, P. Fertey, R. Guillot, V. Briois and M. L. Boillot, Eur. J. Inorg. Chem., 2014, 2014, 101–109 CrossRef CAS.
- R. T. Marques, L. P. Ferreira, C. S. Gomes, C. S. Lopes, C. E. Bernardes, N. K. Sarangi, T. E. Keyes and P. N. Martinho, Cryst. Growth Des., 2023, 23, 3222–3229 CrossRef CAS.
- D. J. Harding, P. Harding and W. Phonsri, Coord. Chem. Rev., 2016, 313, 38–61 CrossRef CAS.
- K. Ugsornrat, A. Saenkhamai, A. Chantaraklud, P. Pasakon, C. Karuwan, C. Sriprachuabwong, N. Atthi, A. Wisitsoraat and A. Tuantranont, IEEE Sens. J., 2023, 23, 28613–28625 CAS.
- C. I. Ogbu, X. Feng, S. N. Dada and G. W. Bishop, Sensors, 2019, 19, 3741 CrossRef CAS PubMed.
- E. P. Randviir, Electrochim. Acta, 2018, 286, 179–186 CrossRef CAS.
- P. Chen and R. L. McCreery, Anal. Chem., 1996, 68, 3958–3965 CrossRef CAS.
- D. Dechtrirat, B. Sookcharoenpinyo, P. Prajongtat, C. Sriprachuabwong, A. Sanguankiat, A. Tuantranont and S. Hannongbua, RSC Adv., 2018, 8, 206–212 RSC.
- S. A. Da França, M. F. Dario, V. B. Esteves, A. R. Baby and M. V. R. Velasco, Cosmetics, 2015, 2, 110–126 CrossRef.
- J. Thangphatthanarungruang, P. Pasakon, A. Wisitsoraat, A. Tuantranont, V. Intasanta and C. Karuwan, Surf. Interfaces, 2024, 104145 CrossRef CAS.
- J. He, J. Sunarso, J. Miao, H. Sun, J. Dai, C. Zhang, W. Zhou and Z. Shao, J. Hazard. Mater., 2019, 369, 699–706 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Theerapoom Boonprab
a,
Theerapoom Boonprab