
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monodisperse amphiphilic double-crystalline block oligomers composed of linear alkyl chains and poly(vinyl alcohol) segments prepared by aldol-group transfer polymerization and recycling size-exclusion chromatography separation†
Toa
Nakane
a,
Hironobu
Watanabe
 a,
Mineto
Uchiyama
a,
Fabrice
Muller
b,
Nathalie
Mathieu
b,
Christophe
Sinturel
*b and
Masami
Kamigaito
*a
a,
Mineto
Uchiyama
a,
Fabrice
Muller
b,
Nathalie
Mathieu
b,
Christophe
Sinturel
*b and
Masami
Kamigaito
*a
aDepartment of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: kamigait@chembio.nagoya-u.ac.jp
bUniversité d'Orléans, CNRS, ICMN, UMR 7374, Orléans, France. E-mail: christophe.sinturel@univ-orleans.fr
First published on 5th April 2025
Abstract
As a model of block copolymers of ethylene and vinyl alcohol, monodisperse amphiphilic double crystalline diblock oligomers composed of linear alkyl chains and poly(vinyl alcohol) segments were prepared using aldol-group transfer polymerization (aldol-GTP) and recycling size-exclusion chromatography (SEC) separation. The aldol-GTP of tert-butyldimethylsilyl vinyl ether (TBSVE) was conducted using octadecyl aldehyde as an initiator and ZnBr2 as a catalyst for the synthesis of oligo(TBSVE) with controlled molecular weights and a C16 alkyl chain and an aldehyde group at the α- and ω-chain ends, respectively. The obtained oligomers were separated into monodisperse unimers, dimers, trimers, tetramers, and pentamers via preparative recycling SEC. Reduction of the terminal aldehyde group followed by deprotection of the silyl groups resulted in monodisperse amphiphilic oligomers composed of C16 alkyl chains and poly(vinyl alcohol) (PVA) units. The effects of the number of PVA units on the thermal properties were characterized using differential scanning calorimetry. Furthermore, the morphology of the discrete block oligomer was examined by optical microscopy, atomic force microscopy, and small- and wide-angle X-ray scattering. Owing to the high incompatibility between the hydrophobic and hydrophilic blocks, self-assembly was observed despite the very low molar weights of the polymers. Based on our results, we propose that a double-crystalline lamellar structure is formed and consists of alternating hydrophobic (alkyl chains) and hydrophilic (PVA segments) domains (with a period of 6.8 nm), showing a distinct crystallization/melting process.
Introduction
Block polymers composed of immiscible segments can self-assemble to form well-defined nanostructures that can be used for wide applications, such as thermoplastic elastomers, microelectronics, and drug delivery.1–6 In particular, hydrophobic and hydrophilic segments in amphiphilic block copolymers strongly segregate in both solid and solution states to construct unique morphologies. Recent developments in controlled/living polymerization have enabled the synthesis of a variety of block polymers in a controlled manner, in which the polymer chains grow in a controlled fashion due to reversible deactivation of the propagating species into dormant species.7 However, distributions in the chain lengths of the resulting polymers are inevitable because they are kinetically or statistically controlled by the deactivation and propagation rates. Since the morphology is governed by the Flory–Huggins interaction parameter (χ) and the chain length (N), a narrower dispersity has been targeted to create more distinct nanostructures.8–19On the other hand, recent developments in high-performance chromatography techniques and apparatus have enabled easy purification of various products. In particular, the use of chromatographic separation of the products obtained by controlled/living or reversible deactivation polymerization allows the preparation of discrete or monodisperse oligomers from monomers used for common synthetic polymers.20–27 Separation is accomplished by the differentiation based on either the size or polarity of the products, such as the use of size-exclusion chromatography (SEC) or adsorption column chromatography, and is applicable for both homopolymers and block polymers. Although chromatographic separation of synthetic polymers can become difficult with increasing molecular weights of the products due to the decrease in relative differences in size and polarity, this method has enabled the unprecedented preparation of a series of monodisperse oligomers, and thus, the effects of the degree of polymerization (n) on the properties of the synthetic oligomers have been revealed and could further contribute to the design of functional materials.
In this study, we focused on the preparation and characterization of monodisperse block oligomers composed of long linear alkyl chains and poly(vinyl alcohol) (PVA) segments as discrete model compounds for block polymers of ethylene and vinyl alcohol. Polyethylene (PE) and PVA are representative hydrophobic and hydrophilic vinyl polymers, respectively, with the simplest structures, and both are crystalline polymers. The different crystallinities caused by hydrophobic long alkyl chains and hydrophilic hydrogen-bonding interactions can lead to strong segregation of each segment and are expected to induce distinct phase separation. Although the organic synthesis and characterization of discrete amphiphilic molecules composed of long alkyl chains and sugar-derived polyol units have been performed,8,9,17,18 PVA-based monodisperse diblock oligomers have not been synthesized and characterized. In addition, non-monodisperse block copolymers of ethylene and vinyl acetate, which are precursor monomers of vinyl alcohol, are also challenging to synthesize.28–33
To construct PVA units with controlled molecular weights, we selected a method based on aldol-group transfer polymerization (GTP) of silyl vinyl ether followed by deprotection of the silyl groups.34–41 Although vinyl acetate is a more common precursor of the PVA units, irreversible termination, chain-transfer reactions, and head-to-head linkages prone to radical polymerization are inevitable even in the reversible deactivation radical polymerization (RDRP), namely reversible addition–fragmentation chain transfer (RAFT) polymerization. In this study, tert-butyldimethylsilyl vinyl ether (TBSVE) was polymerized using octadecyl aldehyde (1-octadecanal or stearyl aldehyde; CH3(CH2)16CHO) as a hydrophobic initiator in the presence of a zinc bromide (ZnBr2) catalyst to obtain controlled oligo(TBSVE) (CH3(CH2)15(CH2CH(OSiMe2t-Bu))nCH2CHO) possessing a C16 alkyl chain and an aldehyde group at the α- and ω-chain ends, respectively (Scheme 1). The obtained products were separated into monodisperse oligomers (n = 1–5) using preparative recycling SEC. After the reduction of the terminal aldehyde group into the primary alcohol, the silyl groups in the discrete oligo(TBSVE) were deprotected, which resulted in monodisperse amphiphilic oligomers composed of C16 alkyl chains and PVA units (CH3(CH2)15(CH2CH(OH))nCH2CH2OH). Interestingly, the reduction of the terminal aldehyde resulted in one additional PVA unit (–CH2CH2OH) as the hydrophilic terminal, which is also a benefit of aldol-GTP, and completed the synthesis of discrete hydrophilic and hydrophobic block oligomers. Thus, the obtained block oligomers should have eight ethylene and (n + 1) vinyl alcohol units. The effects of the degree of polymerization (n) on the thermal properties were characterized using differential scanning calorimetry (DSC). Furthermore, the morphology of the discrete block oligomer with n = 5 was analyzed by optical microscopy (OM), atomic force microscopy (AFM), and small- and wide-angle X-ray scattering (SAXS and WAXS).
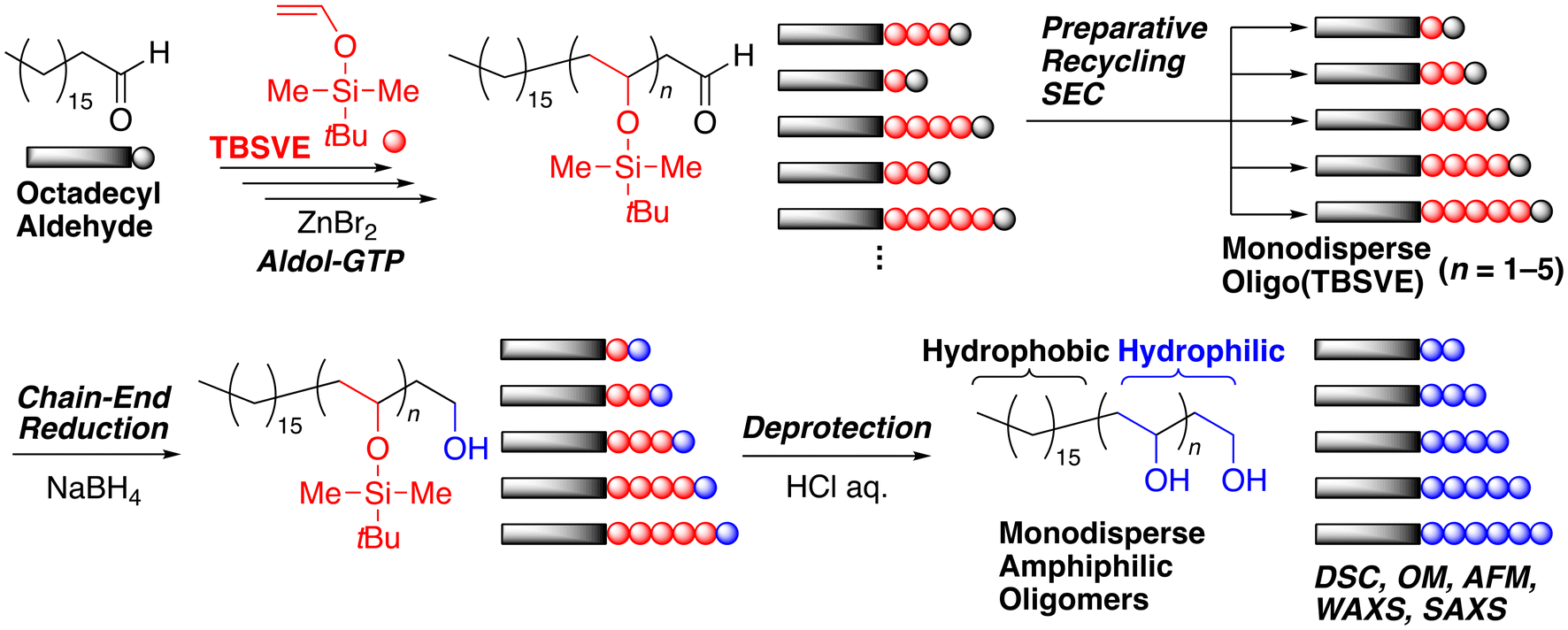 | ||
| Scheme 1 Preparation of monodisperse amphiphilic block oligomers composed of linear C16 alkyl chains and poly(vinyl alcohol) segments by aldol-group transfer polymerization and recycling size-exclusion chromatography. | ||
Results
Aldol-GTP of TBSVE with octadecyl aldehyde
Aldol-GTP of TBSVE was investigated using octadecyl aldehyde and ZnBr2 as an initiator and a catalyst, respectively, in dichloromethane at 0 °C (Fig. 1). Here, the targeted number-average degree of polymerization (DPn) was set to 5 ([TBSVE]0/[octadecyl aldehyde]0 = 500/100 mM). The monomer conversion reached 81% in 10 min, resulting in oligomers with a controlled molecular weight (Mn = 1400) and relatively narrow dispersity (Đ) (Đ = 1.16). The Mn values of the resulting oligomers were higher than the calculated values, but the discrepancies decreased with increasing monomer conversion. Similarly, as the polymerization proceeded, the height of the sharp peak of the remaining octadecyl aldehyde in the low-molecular-weight region of the SEC curves decreased. These results indicated that the initiation from octadecyl aldehyde was slower than the propagation. This could be attributed to the lower coordination ability of the carbonyl group of the alkyl aldehyde to ZnBr2 than that of the oligomer terminal, which had an adjacent silyloxyl group of the penultimate TBSVE unit. These results indicated that an alkyl aldehyde was less efficient as an initiator than an aromatic aldehyde namely benzaldehyde, which is generally used in aldol-GTP (Scheme S1 in the ESI†).34–41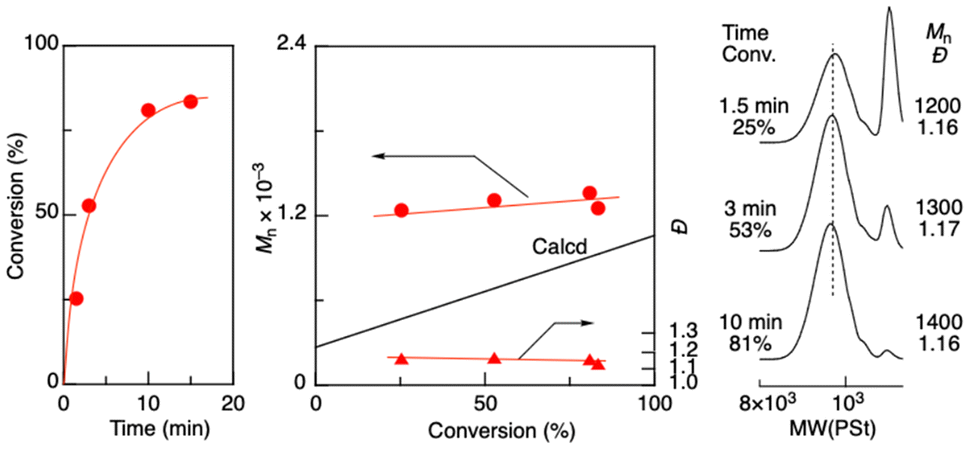 | ||
| Fig. 1 Aldol-GTP of TBSVE with octadecyl aldehyde as an initiator and ZnBr2 as a catalyst: [TBSVE]0/[octadecyl aldehyde]0/[ZnBr2]0 = 500/100/20 mM in CH2Cl2 at 0 °C. | ||
The 1H NMR spectrum of the resultant oligomer showed each characteristic signal of the alkyl chain (α1–α3), the aldehyde group (ω1), and the TBSVE units (a–d) (Fig. 2a). The DPn(NMR) determined from the peak intensity ratio of the TBSVE units to the alkyl chain (c to α1) was 4.26. The Mn(NMR) was determined to be 940; this value was close to the theoretical value (Mn(calcd) = 930), assuming that one molecule of octadecyl aldehyde generated one oligomer chain. Furthermore, the chain-end functionality of the aldehyde terminal (Fn(ω1)) calculated from the peak intensity ratio of ω1 to α1 was 0.90. These results indicated that the aldol-GTP of TBSVE was initiated from octadecyl aldehyde to result in a targeted oligo(TBSVE) with a controlled molecular weight and an alkyl chain and aldehyde group at the α- and ω-chain ends, respectively. In addition to these main peaks, olefin peaks were slightly observed at around 5.5 ppm, which were most likely formed by the elimination of TBSOH from the polymer during polymerization with ZnBr2 as a Lewis acid catalyst (Fig. S1†).42 The amount of the olefin units was calculated to be 2.5% per TBSVE unit.
 | ||
| Fig. 2 1H NMR (in CDCl3 at 55 °C) (a) and MALDI-TOF-MS (b) spectra of oligo(TBSVE) obtained by aldol-GTP of TBSVE. | ||
Furthermore, the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) spectrum of the oligomers revealed a series of peaks separated by the molar mass of the TBSVE units (158.32 Da) (Fig. 2b). The main series of peaks were assigned to oligo(TBSVE) with a C16 alkyl chain and an aldehyde group. Furthermore, the highest peak was assigned to the tetramer (n = 4) of TBSVE. On the other hand, a minor series of peaks located in the lower-molecular-weight regions away from the main peaks by ∼132 Da could be assigned to oligomers with one TBSOH group eliminated, consistent with the olefin peaks observed in the 1H NMR spectrum (Fig. S1†). Thus, the MALDI-TOF-MS analysis also revealed that the main products obtained by aldol-GTP were oligo(TBSVE)s with targeted average degrees of polymerization, a C16 alkyl chain, and an aldehyde terminal.
Preparation of monodisperse oligo(TBSVE) by recycling SEC separation
The obtained oligo(TBSVE) (DPn(NMR) = 4.26, Đ = 1.16) was fractionated using preparative recycling SEC with CHCl3 as the eluent to obtain a series of monodisperse oligomers with discrete degrees of polymerization (n). The fractionation was examined for oligomers with n from 1 to 5. The SEC curves of a series of fractionated oligomers exhibited extremely narrow dispersity (Đ = 1.01–1.02) in all cases, where the Mn values measured by SEC were 630, 830, 1000, 1100, and 1300 (Fig. 3a). All 1H NMR spectra showed similar characteristic peaks attributed to the alkyl chain, aldehyde group, and TBSVE units (Fig. 3b). Interestingly, these peaks of each discrete oligomer were sharper than those of the original oligomers, which were mixtures of these oligomers with different n values. Furthermore, the peak shapes of the main-chain methylene (a) and methine (b) of the TBSVE units and methylene (ω2) adjacent to the terminal aldehyde were slightly different from each other because they were oligomers with different n values. In all cases, each DPn(NMR) calculated from the peak intensity ratio of the TBSVE units to the alkyl chain was close to n. In addition, the aldehyde group remained almost quantitatively at the oligomer terminal, as indicated by the Fn(ω1) values close to unity. Furthermore, the MALDI-TOF-MS spectra showed nearly one peak, particularly for the oligomers with n = 4 and 5 (Fig. 3c). The results of the SEC, 1H NMR, and MALDI-TOF-MS analyses support the preparation of monodisperse oligo(TBSVE) with a C16 alkyl chain and an aldehyde terminal using preparative recycling SEC separation. | ||
| Fig. 3 SEC (a), 1H NMR (b), and MALDI-TOF-MS (c) spectra of monodisperse oligo(TBSVE) fractionated by preparative recycling SEC. *CHCl3 and †water in (b). The peaks marked with “x” in (c) are derived from trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) as the ionizing matrix. | ||
Reduction of the aldehyde terminal group and deprotection of the silyl group
A series of fractionated oligo(TBSVE)s (n = 1–5) with C16 alkyl chains and aldehyde terminals were then converted to a series of monodisperse amphiphilic oligomers with a C16 alkyl chain and PVA units by the reduction of the aldehyde group at the ω-chain end and subsequent deprotection of the TBS groups. The solubility of the discrete oligomers depended on the number of original TBSVE units (Table S1†). Fig. 4 shows the 1H NMR spectra for the discrete oligomer with n = 5 (Fig. 4a) and those obtained after the reduction (Fig. 4b) and subsequent deprotection (Fig. 4c). The reduction of the aldehyde was conducted by treating the oligomer with NaBH4 in THF/MeOH (4/1 v/v) at 0 °C. The aldehyde proton (ω1) at 9.8 ppm completely disappeared (Fig. 4b), indicating that the reduction was successful. Then, the deprotection of the TBS groups in the side chains was examined using HCl in THF/water (20/1 v/v) at room temperature. The methyl protons (c) of the TBS groups at 0 ppm nearly completely disappeared (conv. > 99%) (Fig. 4c); these results indicated that they were removed and resulted in the OH groups located in the side chains. These results demonstrated that the monodisperse amphiphilic block oligomers composed of C16 alkyl chains and oligo(vinyl alcohol) segments were successfully synthesized by a combination of aldol-GTP, the preparative recycling SEC technique, and modification reactions. In particular, the oligomer with n = 5 could be considered a discrete linear block oligomer composed of eight ethylene and six vinyl alcohol units (PE8-PVA6).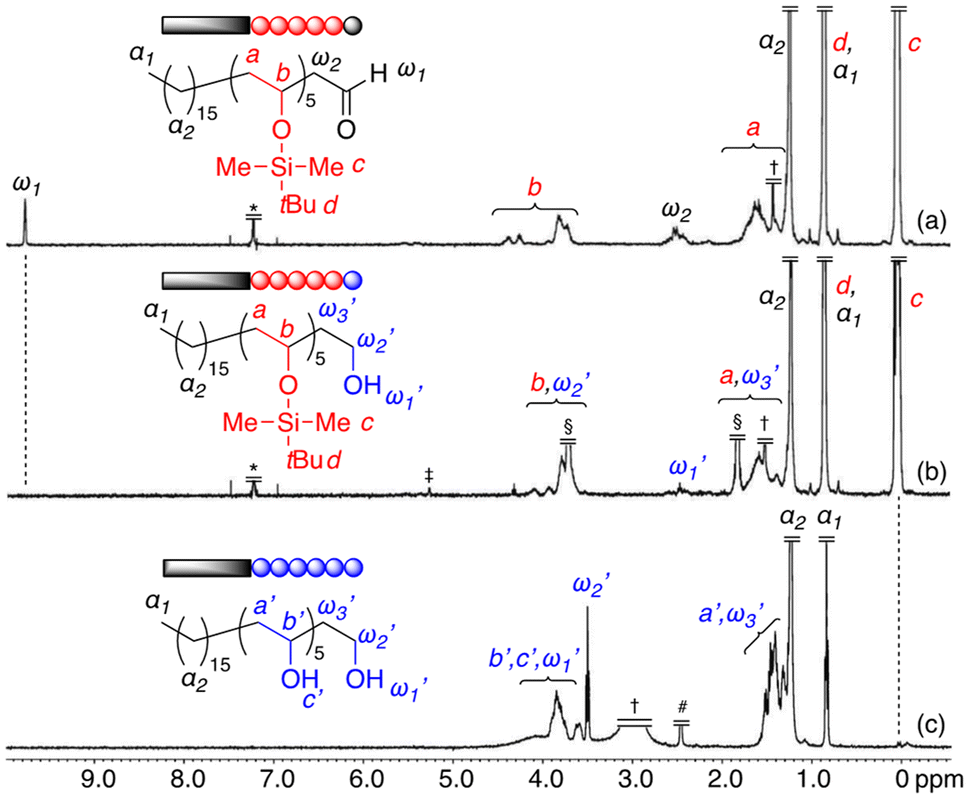 | ||
| Fig. 4 1H NMR spectra of the original oligomer (n = 5) (a) and the oligomer obtained after reduction (b) and deprotection (c) (CDCl3 at 55 °C for a and b, DMSO-d6 at 100 °C for c). *CHCl3, †water, ‡CH2Cl2, §THF, and #DMSO. | ||
The solubility of the discrete oligomers depended on the number of original TBSVE units (Table S1†). The oligomer with n = 1, which consisted of a hydrophobic C16 alkyl chain and only two hydrophilic PVA units, was soluble in nonpolar solvents such as toluene and chloroform at room temperature, but insoluble in polar solvents such as acetone and methanol. The oligomers with n = 2 and n = 3, having longer hydrophilic PVA units, were soluble in both nonpolar and polar solvents. In contrast, the oligomer with n = 4 dissolved in THF, acetone, methanol, and DMSO only at elevated temperatures. Furthermore, the oligomer with n = 5 was insoluble in most of these solvents at any temperature. Notably, 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), a good solvent for highly crystalline polymers such as polyamides, was able to dissolve the oligomers with n = 4 and n = 5 at room temperature. The low solubility of the oligomers with n = 4 and n = 5 suggested high crystallinity of these oligomers.
DSC
Fig. 5 shows the thermograms of the monodisperse oligomers bearing 1 to 5 vinyl alcohol (VA) units (Fig. 5c–l), the mixture of the oligomers (Fig. 5a and b) and the pure 1-octadecanol (Fig. 5m and n), which can be obtained by the reduction of 1-octadecanal used as the initiator. Under our conditions, 1-octadecanol has one melting point at 58.9 °C upon heating and two exotherms at 55.0 and 47.4 °C upon cooling; this represents a common behavior observed in fatty alcohols and is attributed to the formation of a transient rotator phase before a transition toward a stable crystalline state.43,44 | ||
| Fig. 5 DSC thermograms of the unseparated oligomers (a and b), separated monodisperse oligomers bearing 1 to 5 original TBSVE units (c–l) and 1-ocatadecanol (m and n). | ||
As the number of VA units increases in the oligomers, the single endotherm (upon heating) of 1-octadecanol evolves toward two separate signals, indicating two melting temperatures (Tm1 and Tm2, see Table S2 and Fig. S6 in the ESI†); these presumably originate from the successive melting of the alkyl chains (Tm1, close to the melting temperature of 1-octadecanol) and the VA segments (Tm2). This assumption is confronted with the behavior of related compounds in the Discussion section of this manuscript. The oligomer with only one VA unit shows intermediate behavior; this result indicates that the number of VA units in the sample is not sufficiently high to form a crystalline phase. With an increasing number of VA units, the value of Tm1 (alkyl chains) only slightly decreases (from 56.4 °C for n = 2 to 48.6 °C for n = 5), whereas Tm2 tends to increase (from 45.9 °C for n = 2 to 112.1 °C for n = 5). These results are consistent with the structure of the oligomers, which originate from the same alkyl initiator but have increasing numbers of VA units. Evidently, the highest Tm2 (n = 5) is much lower than the value typically reported for PVA,45 reflecting the very small DP in our case (only 5 VA units). Notably, the VA segment crystallizes even if the tacticity is not controlled. This is caused by the strong hydrogen bonding between the hydroxyl groups.46–49 Interestingly, the mixture of the different oligomers (DPn = 2.9) shows broad crystallization; these results indicate more complex behavior resulting from the presence of various VA segments with different unit numbers (the same for melting).
Upon cooling, the double exotherm of 1-octadecanol first evolves into a single peak (n = 1) before splitting into two distinct signals; these results indicate the existence of two distinct crystallization processes, starting with the crystallization of the VA segments (Tc2) followed by the crystallization of the alkyl chains (Tc1). The value of Tc1 (alkyl chains) is slightly affected by the number of VA units (a limited decrease from 56.4 °C for n = 2 to 48.6 °C for n = 5). In contrast, Tc2 (VA segments) significantly increases (from 40.4 °C for n = 2 to 106.3 °C for n = 5). These results are consistent with the increase in the number of VA units while keeping the size of the alkyl chain constant.
For both the heating and cooling processes, an analysis of the thermograms enables the differentiation of a third type of signal for n = 3, 4 and 5 above Tm2 and Tc2. Here, both parts of the oligomers (alkyl chains and VA segments) are assumed to be in the molten state at this temperature; thus, this transition may tentatively be attributed to an order–disorder temperature between an ordered melt and a disordered (isotropic) state, indicating liquid crystalline (LC) thermotropic behavior. This hypothesis is supported by the optical microscopy measurements showing macroscopic flow, with the formation of liquid droplets, only at temperatures above the order–disorder transition temperature (TODT) (Fig. S7†). For temperatures above Tm2 but below TODT, oligomer mobility is hindered, preventing any macroscopic flow. As discussed later, this kind of order–disorder transition has already been observed in similar amphiphilic oligomers composed of alkyl chains coupled to a polyol segment.9,10,19
SAXS–WAXS characterization
The sample with n = 5 units of VA was subjected to combined SAXS–WAXS analysis at room temperature. The SAXS pattern (Fig. 6a) shows the typical characteristics of a lamellar structure with a scattering peak series at q*, 2, 3, 4, and 5, with q* = 0.92 nm−1 (d = 6.8 nm). The simulation of the scattering profile assuming a lamellar structure is consistent with the experimental data (Fig. S8†); these results indicated that a lamellar structure is likely to be formed at room temperature. With the assumption of a double crystalline structure deduced from the DSC result, this SAXS result is consistent with alternating crystalline layers composed of alkyl chains and VA segments, with a period of 6.8 nm. The dimension of a single oligomer in a fully extended conformation is 3.4 nm; thus, the oligomers are packed into a head-to-head arrangement, with no interdigitation (Fig. 6c).49 | ||
| Fig. 6 SAXS (a) and WAXS (b) patterns at room temperature of oligomers with n = 5 VA units and the corresponding proposed structure (c) formed in the solid state (the dimensions shown are calculated using the C–C bond length, assuming a fully extended conformation). | ||
The WAXS pattern (Fig. 6b) confirms the existence of a fine crystalline structure, with strong peaks at 14.2 and 15.5 nm−1. Alkyl chains exhibit scattering peaks in this region, typically at 15.0 and 17.0 nm−1; these peaks are associated with the (110) and (200) reflections of the orthorhombic structure.50,51 The positions of these scattering peaks can be shifted due to the functionalization of the chains,43 and this can be used to explain the shift observed here. PVA also shows diffraction peaks in this region, typically at 13.6 and 14.6 nm−1; these peaks are associated with the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and (101) reflections of the monoclinic cell.45,48 The experimental broad scattering peak observed in our case can be attributed to the superposition of the scattering peaks from the diffraction of the alkyl chains and VA segments.
) and (101) reflections of the monoclinic cell.45,48 The experimental broad scattering peak observed in our case can be attributed to the superposition of the scattering peaks from the diffraction of the alkyl chains and VA segments.
AFM
To verify the characteristic dimensions of the self-assembled structures formed at room temperature (double crystalline lamellar structure), a solution of oligomers in HFIP was spin-coated onto a silicon wafer, annealed above the TODT and slowly cooled. The resulting morphology was then characterized by AFM. As shown in Fig. 7, stacked flat terraces are observed, which is in agreement with the lamellar structures of the oligomers. From the profile line, a mean step height of 3.2 ± 0.2 nm was measured; this value is in good agreement with the dimension of the length of the molecule in the all-trans extended conformation (3.4 nm). These results indicate that the oligomers adopt a perpendicular orientation to the substrate (Fig. S9†), forming incomplete lamellae that are stacked in flat terraces. This type of structure has already been observed in thin films of alkanes deposited using vapor deposition.52 In our case, the vertical orientation is in good agreement with the distinct chemistries of the two sides of the molecule, favoring the selective positioning of one side of the oligomers at the surface of the substrate (most likely the hydrophilic VA segments, considering the presence of native SiO2 on the silicon substrate). Therefore, the horizontal positioning of the oligomers (i.e., with the two sides of the oligomers being in contact with the surface of the substrate) is unlikely to occur in this case. It is important to mention that this specific self-assembled structure was obtained for a given set of experimental conditions. We expect that the conditions of self-assembly (spin-coating speed, type of solvent, and the surface energy of the substrate) and the effect of post-treatment (solvent annealing and thermal annealing) will deeply affect the properties of the self-assembled structures. | ||
| Fig. 7 AFM image (7 × 7 μm) (left) and the corresponding height line profile (right) showing flat terraces for oligomers with n = 5 VA units, spin coated on a glass substrate and annealed above TODT. | ||
Discussion
Based on our results, we propose that a double-crystalline lamellar structure is formed and is composed of alternating hydrophobic (alkyl chains) and hydrophilic (VA segments) domains, resulting in a distinct crystallization/melting process. This type of behavior is often encountered in block polymers.51,53 For blocks that crystallize at lower temperatures (therefore in a confined environment), a change in the crystallization mechanism is often encountered. This process includes a change from heterogeneous nucleation in the bulk to surface or homogeneous nucleation in small domains; this leads to greater supercooling for crystallization (i.e., at lower temperatures). Specifically, the crystallization in small domains requires more energy because of the lack of nucleation species in the domains (the number of domains surpasses the number of nucleation species in the materials). Therefore, a common feature of crystallization in confined environments is a decrease in the crystallization temperature (whereas in some cases, the crystallization temperature increases due to the nucleation effect of the surface).54 In the present study, the crystallization peak of the alkyl group continuously decreases when the number of VA units increases; these results indicate that the crystallization mechanism progressively shifts from heterogeneous to homogeneous (with no surface nucleation effect). In contrast, the crystallization temperature of the VA segments continuously increases with increasing DP due to the increase in molecular weight and hydroxy group density.For temperatures above the second melting temperature (above the melting temperature of the VA segments), we proposed that the oligomers form a liquid crystalline (LC) thermotropic state. This hypothesis is in good agreement with the behavior reported by others on very similar structures.9,10,19 In these studies, oligomers composed of alkyl chains (C12 and C14) coupled to an open-chain monosaccharide-based polyol unit (one hydroxy group per carbon atom) were considered. The detailed structural characterization based on temperature (SAXS and WAXS) conducted in these studies demonstrated the formation of a liquid crystalline phase above the melting point of the compounds, followed by the formation of an isotropic state (disordered) at higher temperatures. The LC behavior of these amphiphilic oligomers was attributed to a “microphase separation” in the molten state (very similar to the block copolymer situation) of the two incompatible parts of the molecule. This defines an amphiphilic LC, in contrast to a monophilic LC, where the ordering is driven by the shape of the mesogenic building blocks.55 In our case, even if the density of hydroxy groups is lower (only one hydroxy group per two carbon atoms in the hydrophilic part), this microphase separation is likely to occur, leading to the formation of self-assembled structures in the molten state (confirmed by optical microscopy). However, the temperature observed for the final transition (order/disorder) is lower in our case (a maximum of 112 °C for n = 5 instead of T > 200 °C for the monosaccharide derivatives); these results are in agreement with the lower hydroxy group density of the oligomers prepared in this work (leading to a lower aggregation strength due to less hydrogen bonding).
Notably, these two papers reported only one melting for the entire oligomer, instead of the successive melting of the alkyl chain and polyol segments, as we proposed in our study. For oligomers showing three transitions (as in our system), the simultaneous melting of the alkyl and polyol segments was reported; here, this melting led to the first cubic LC phase (first transition), before transitioning to the second LC lamellar phase (second transition) and eventually transitioning toward the LC isotropic phase (third transition). Although we do not have complete evidence for the behavior proposed in our study (this would have required a detailed structural analysis upon heating/cooling, which was beyond the scope of this chemistry-oriented study), evidence for this interpretation does exist.
First, the position of the lowest melting peak is herein observed in the range of 55–50 °C (very similar to 1-octadecanol), whereas this peak shifts to much higher temperatures in Barreda's work9 (114 °C) and in Mattsson's work19 (70–180 °C), farther from the melting temperature of the corresponding alkyl initiator. In our study, since the alkyl chains are longer (C18 here vs. C12 and C14 in the cited work), they are more likely to show distinct crystallization. In the cited work, the crystallization is in fact driven by the hydrophilic group, which has a high hydroxy group density (one hydroxy group per carbon atom with controlled stereochemistry, favoring hydrogen bonding), prohibiting the crystallization of the shorter alkyl chains.
Second, Mattsson's work19 demonstrated that oligomers exhibiting three transitions did not form lamellar crystalline structures in the solid state; their results were consistent with the formation of a cubic LC thermotropic phase after melting. The formation of the first LC phase after melting was clearly driven by the morphology of the original solid crystalline phase (the lamellar crystalline phase yields the lamellar LC phase). Only oligomers showing two transitions displayed lamellar crystalline structures in the solid state (lam crystal/lam ordered LC/isotropic). These results indicated that in our case, the behavior was different since a lamellar crystalline structure was found in the solid state, and three transitions occurred. In our case, if an LC thermotropic phase was directly formed after the first melting, a lamellar phase would more likely be formed, as previously mentioned. The second transition (order/order transition) would then be difficult to explain since the lamellar phase was the most stable phase at high temperatures.
Conclusions
The combination of the aldol-GTP of silyl vinyl ether initiated with a long-chain alkyl aldehyde and chromatographic separation enabled the preparation of a series of monodisperse diblock oligomers composed of long alkyl chains and PVA units. At room temperature, the discrete block oligomers had a double-crystalline lamellar structure composed of alternating hydrophobic and hydrophilic domains. Upon heating, we hypothesize that the compounds underwent distinct melting processes (Tm1 and Tm2) of the alkyl chains and polyol segments, respectively, to form a crystalline (LC) thermotropic state above Tm2 and eventually a disordered phase for T > TODT.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the CNRS International Research Project (IRP) entitled “Block Copolymer Ordering and Crystallization in Extreme Conditions of Confinement”-“Extreme”, the JSPS KAKENHI Grant-in-Aid for Scientific Research (A) (No. JP22H00333 and JP25H00894), a project (JPNP18016) commissioned by the New Energy and Industrial Technology Development Organization (NEDO), and the Deep Tech Serial Innovation Center VBL fellowship of Nagoya University. Nicolas Gouillon is thanked for the SAXS measurements.References
- N. Hadjichristidis, S. Pispas and G. A. Floudas, Block Copolymers: Synthetic Strategies, Physical Properties, and Applications, Wiley-Interscience, New Jersey, 2003 Search PubMed.
- Block Copolymers in Nanoscience, ed. M. Lazzari, G. Liu and S. Lecommandoux, Wiley-VCH, Weinheim, 2006 Search PubMed.
- Block Copolymers: Phase Morphology, Material Applications and Future Challenges, ed. R. H. Diaz and A. P. Ferguson, Nova Science Publishers, New York, 2014 Search PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- E. A. Murphy, S. J. Skala, D. Kottage, P. A. Kohl, Y. Li, C. Zhang, C. J. Hawker and C. M. Bates, Phys. Rev. Mater., 2024, 8, 015602 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- C. Sinturel, F. S. Bates and M. A. Hillmyer, ACS Macro Lett., 2015, 4, 1044–1050 CrossRef CAS PubMed.
- L. Barreda, Z. Shen, Q. P. Chen, T. P. Lodge, J. I. Siepmann and M. A. Hillmyer, Nano Lett., 2019, 19, 4458–4462 CrossRef CAS PubMed.
- Z. Shen, J. L. Chen, V. Vernadskaia, S. P. Ertem, M. K. Mahanthappa, M. A. Hillmyer, T. M. Reineke, T. P. Lodge and J. I. Siepmann, J. Am. Chem. Soc., 2020, 142, 4352–9362 Search PubMed.
- B. vas Genabeek, B. F. M. de Waal, J. M. J. Gosens, L. M. Pitet, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 4210–4218 CrossRef PubMed.
- B. vas Genabeek, B. F. M. de Waal, B. Ligt, A. R. A. Palmans and E. W. Meijer, ACS Macro Lett., 2017, 6, 674–678 CrossRef PubMed.
- B. vas Genabeek, B. A. G. Lamers, B. F. M. de Waal, M. H. C. van Son, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2017, 139, 14869–17872 CrossRef PubMed.
- A. Das, K. Petkau-Milroy, G. Klerks, B. vas Genabeek, R. P. M. Lafleur, A. R. A. Palmans and E. W. Meijer, ACS Macro Lett., 2018, 7, 546–550 CrossRef CAS PubMed.
- B. A. G. Lamers, A. Herdlitschka, T. Schnitzer, M. F. J. Mabesoone, S. M. C. Schoenmakers, B. F. M. de Waal, A. R. A. Palmans, H. Wennemers and E. W. Meijer, J. Am. Chem. Soc., 2021, 143, 4032–4042 CrossRef CAS PubMed.
- B. Oschmann, J. Lawrence, M. W. Schluze, J. M. Ren, A. Anastasaki, Y. Luo, M. D. Nothling, C. W. Pseter, K. T. Delaney, L. A. Connal, A. J. McGrath, P. G. Clark, C. M. Bates and C. J. Hawker, ACS Macro Lett., 2017, 6, 668–673 CrossRef CAS PubMed.
- T. Isono, R. Komaki, C. Lee, N. Kawakami, B. J. Ree, K. Watanabe, K. Yoshida, H. Mamiya, T. Yamamoto, R. Borsali, K. Tajima and T. Satoh, Commun. Chem., 2020, 3, 135 CrossRef CAS PubMed.
- K. Chen, C. Lee, C.-Y. Chen, T. Satoh, T. Isono and H.-L. Chen, Giant, 2024, 19, 100308 CrossRef CAS.
- I. Mattsson, J. Majoinen, M. Lahtinen, T. Sandberg, A. Fogde, T. Saloranta-Simell, O. J. Rojas, O. Ikkala and R. Leino, Soft Matter, 2023, 19, 8360–8377 RSC.
- E. A. Murphy, C. Zhang, C. M. Bates and C. J. Hawker, Acc. Chem. Res., 2024, 57, 1202–1213 CrossRef CAS PubMed.
- B. vas Genabeek, B. A. G. Lamers, C. J. Hawker, E. W. Meijer, W. R. Gutekunst and B. V. K. J. Schmidt, J. Polym. Sci., 2021, 59, 373–403 CrossRef.
- M. G. Carstens, C. F. van Nostrum, A. Ramzi, J. D. Meeldijk, R. Verrijk, L. L. de Leede, D. J. A. Crommelin and W. E. Hennink, Langmuir, 2005, 21, 11446–11454 CrossRef CAS PubMed.
- J. Lawrence, S.-H. Lee, A. Abdilla, M. D. Nothling, J. M. Ren, A. S. Knight, C. Fleischmann, Y. Li, A. S. Abrams, B. V. K. Schmidt, M. C. Hawker, L. A. Connal, A. J. McGrath, P. G. Clark, W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2016, 138, 6306–6310 CrossRef CAS PubMed.
- J. Lawrence, E. Goto, J. M. Ren, B. McDearmon, D. S. Kim, Y. Ochiai, P. G. Clarik, D. Laitar, T. Higashihara and C. J. Hawker, J. Am. Chem. Soc., 2017, 139, 13735–13739 CrossRef CAS PubMed.
- Y. Hoshino, S. Taniguchi, H. Takimoto, S. Akashi, S. Katakami, Y. Yonamine and Y. Miura, Angew. Chem., Int. Ed., 2020, 59, 679–683 CrossRef CAS PubMed.
- J. D. Neve, J. J. Haven, S. Harrison and T. Junkers, Angew. Chem., Int. Ed., 2019, 58, 13869–13873 CrossRef PubMed.
- A. Kleine, J. Kimming, C. Mankel, U. S. Schubert and M. Jäger, Macromolecules, 2024, 57, 6090–6099 CrossRef CAS.
- Z. Chen and M. Brookhart, Acc. Chem. Res., 2018, 51, 1831–1839 CrossRef CAS PubMed.
- J. Demarteau, P. B. V. Sholten, A. Kermagoret, J. D. Winter, M. A. R. Meier, V. Monteil, A. Debuigne and C. Detrembleur, Macromolecules, 2019, 52, 9053–9063 CrossRef CAS.
- A. Wolpers, F. Baffie, L. Verrieux, L. Perrin, V. Monteil and F. D'Agosto, Angew. Chem., Int. Ed., 2020, 59, 19304–19310 CrossRef CAS PubMed.
- A. Wolpers, F. Baffie, V. Monteil and F. D'Agosto, Macromol. Rapid Commun., 2021, 42, 2100270 CrossRef CAS PubMed.
- F. Baffie, M. Lansalot, V. Monteil and F. D'Agosto, Polym. Chem., 2022, 13, 2469–2476 RSC.
- C. Zhao and E. Harth, Eur. Polym. J., 2024, 220, 113460 CrossRef CAS.
- D. Y. Sogah and O. W. Webster, Macromolecules, 1986, 19, 1775–1777 CrossRef CAS.
- F. P. Boettcher, Makromol. Chem., Macromol. Symp., 1988, 13/14, 193–202 CrossRef.
- Y. Kawakami, T. Aoki and Y. Yamashita, Polym. Bull., 1987, 18, 473–477 Search PubMed.
- B. Charleux and C. Pichot, Polymer, 1993, 34, 195–203 CrossRef CAS.
- A. Takasu, S. Ohmori, Y. Yamauchi and T. Hirabayashi, J. Polym. Sci., Part A:Polym. Chem., 2002, 40, 4477–4484 CrossRef CAS.
- J. Zhou, A. Kaga, A. Takasu, Y. Inai and T. Hirabayashi, Polym. J., 2003, 35, 757–765 CrossRef CAS.
- J. Zhou, A. Takasu, Y. Inai and T. Hirabayashi, Polym. J., 2004, 36, 182–189 CrossRef CAS.
- T. Moyori, T. Hayashi and A. Takasu, J. Polym. Sci., Part A:Polym. Chem., 2013, 51, 3516–3522 CrossRef CAS.
- A. Kanazawa, S. Kanaoka and S. Aoshima, Macromolecules, 2009, 42, 3965–3972 CrossRef CAS.
- F. M. A. Leyva-Gutierrez and T. Wang, Ind. Eng. Chem. Res., 2021, 60, 14651–14663 CrossRef CAS.
- F. M. A. Leyva-Gutierrez and T. Wang, Cryst. Growth Des., 2023, 23, 2351–2360 CrossRef CAS.
- H. E. Assendert and A. H. Windle, Polymer, 1998, 39, 4295–4302 CrossRef.
- C. W. Bunn, Nature, 1948, 161, 929–930 CrossRef CAS.
- I. Sakurada, K. Fuchino and N. Okada, Bull. Inst. Chem. Res., Kyoto Univ., 1950, 23, 78 CAS.
- B. G. Colvin, Nature, 1974, 248, 756–759 CrossRef CAS.
- A. D. Bond, New J. Chem., 2004, 28, 104–114 RSC.
- M. C. Luyten, G. O. R. Alberda van Ekenstein, G. ten Brinke, J. Ruokolainen, O. Ikkala, M. Torkkeli and R. Serimaa, Macromolecules, 1999, 32, 4404–4410 CrossRef CAS.
- L. Sun, Y. Liu, L. Zhu, B. S. Hsiao and C. A. Avila-Orta, Polymer, 2004, 45, 8181–8193 CrossRef CAS.
- K. Nozaki, R. Saihara, K. Ishikawa and T. Yamamoto, Jpn. J. Appl. Phys., 2007, 46, 761–769 CrossRef CAS.
- R. V. Castillo and A. J. Müller, Prog. Polym. Sci., 2009, 34, 516–560 CrossRef CAS.
- V. S. D. Voet, G. O. R. Alberda van Ekenstein, N. L. Meereboer, A. H. Hofman, G. ten Brinke and K. Loos, Polym. Chem., 2014, 5, 2219–2230 RSC.
- V. Vill and R. Hashimb, Curr. Opin. Colloid Interface Sci., 2002, 7, 395–409 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00145e |
| This journal is © The Royal Society of Chemistry 2025 |