
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymerization behaviour of biscarbenes derived by thermolysis of bisdiazo compounds†
Xiaosong
Liu
 a,
Mark G.
Moloney
*ab and
Koji
Okuda
c
a,
Mark G.
Moloney
*ab and
Koji
Okuda
c
aOxford Suzhou Center for Advanced Research (OSCAR), Suzhou Industrial Park, Jiangsu 215123, P. R. China. E-mail: mark.moloney@chem.ox.ac.uk
bChemistry Research Laboratory, Department of Chemistry, University of Oxford, Oxford OX1 3TA, UK
cJEOL (Beijing) Co. Ltd, Shanghai Branch, Shanghai 200335, P. R. China
First published on 23rd April 2025
Abstract
A study of the collapse of bisdiazo compounds with different terminal groups upon heating to generate reactive biscarbene intermediates has provided evidence for homopolymerization in a process that proceeds in the absence of catalysts and is tolerant of oxygen. This polymerization behaviour was monitored spectroscopically through UV-vis kinetic analysis with various combinations of temperature and solvent, and clear evidence for dimer and trimer formation was found by field desorption mass spectrometry. Oligomerization may involve the formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN–NC linkages, as studied and validated by molecular dynamics (MD) calculations, before reaching macromolecular size. In the presence of terminal NH2 groups, cross-linking resulting from carbene insertion is also observed. This unusual polymerization of diazo monomers, when conducted on a polyvinyl alcohol (PVA) surface in the open-air upon heating, creates a highly cross-linked structure that changes surface properties.
C and CN–NC linkages, as studied and validated by molecular dynamics (MD) calculations, before reaching macromolecular size. In the presence of terminal NH2 groups, cross-linking resulting from carbene insertion is also observed. This unusual polymerization of diazo monomers, when conducted on a polyvinyl alcohol (PVA) surface in the open-air upon heating, creates a highly cross-linked structure that changes surface properties.
Introduction
Diazo and bisdiazo compounds may react upon heating or activation with UV light, releasing nitrogen gas to generate highly active carbene or biscarbene intermediates. While they have been used successfully in polymer surface modification,1,2 they are more widely known for carbene insertion3,4 and coupling reactions.5,6 Diaryldiazo compounds tend to be oils or low melting solids,7,8 which are indefinitely stable while chilled but decompose on heating with an exotherm.9 Of interest is that related bis(diaryldiazo) compounds are much more stable, more convenient to use and reactive under thermal conditions, offering a convenient platform for polymer surface modification reactions.10 While the polymerization of radicals, as single-electron species, is very well known (Scheme 1a), as are the insertion and addition (Scheme 1b) reactions of carbenes, carbenic C1 polymerizations11–13 have only more recently been developed (Scheme 1c), usually occurring in the presence of metal catalysts.14 A further development of carbene reactivity involving the oligomerization and homopolymerization of biscarbenes induced by thermal decomposition of bisdiazo compounds is reported here, which can be achieved without using any catalyst (Scheme 1d), and even occurs under aerobic conditions. In this work, using a combination of experimental and theoretical methods, several aspects have been investigated, beginning with a study of the rate of collapse of bisdiazo compounds under thermal conditions, followed by a study of the possible dimerization, oligomerization or polymerization processes that could be achieved without any catalyst in both solid- and liquid phases, and lastly the identity of the possible oligomeric and polymeric products formed by such a self-reaction and in particular the nature of monomer linkages.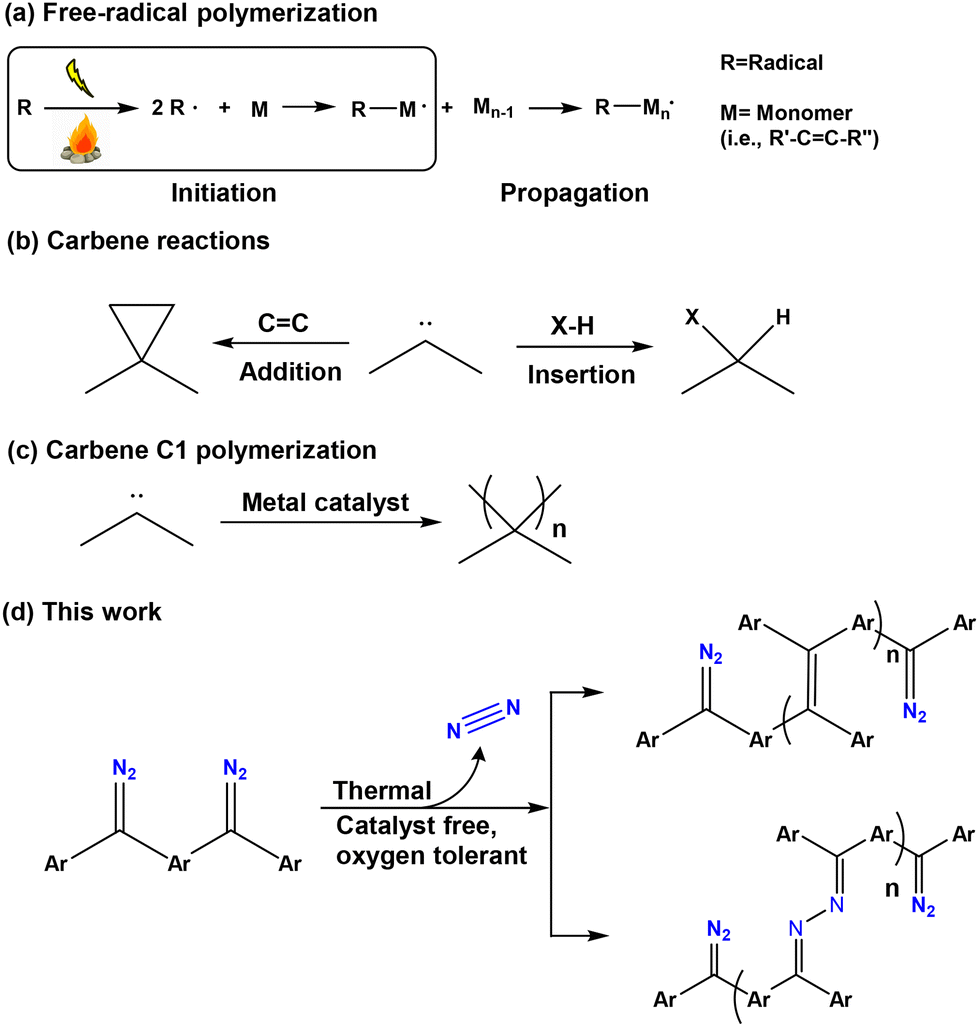 | ||
| Scheme 1 Mechanisms of (a) free-radical polymerization, (b) carbene reactions, (c) C1 polymerization, and (d) catalyst-free polymerization of bisdiazo compounds as precursors in this work. | ||
The results of this work are reported here, in which four bisdiazo compounds 1a–d with electronically neutral (R = H), donating (R = Me) or withdrawing (R = NO2) groups, along with R = NH2, which is known to both insert and cross-link, giving a polymeric material that has not been fully chemically characterized,15 were selected for the study (Fig. 1a). Then, a key question requiring an answer was whether carbenes might act as a C1 monomer, and directly polymerize, or whether polymerization might involve the diazo system alone or in combination with the carbene, with several possible coupling scenarios being illustrated in Fig. 1a. The carbene might directly couple, leading to alkene or alkane formation, or might react with the diazo starting material to give azine links, or both, and this makes for a complicated possible polymerization mechanism. Additionally, for certain potential applications, a unique outcome might be accomplished by thermally induced homopolymerization from bisdiazo-NH21d on a polyvinyl alcohol (PVA) surface when conducted with a minimized volume of the liquid phase under an open-air atmosphere.
 | ||
| Fig. 1 (a) Chemical structures of bisdiazo-R compounds 1a–d and the possible repeating units of polymers under catalyst-free polymerization. (b) Elemental analysis and (c) optical and fluorescence images of bisdiazo-R compounds 1a–d with various terminal groups (R = H, Me, NO2 and NH2). | ||
Results and discussion
Monomer synthesis
The required bisdiazo monomers, with their chemical structures shown in Fig. 1a, were readily prepared as shown in Schemes S1–S3† using reported methods;1,16 thus, ethers 2a–d were subjected to Friedel–Crafts acylation with isophthaloyl chloride to give diketones 3a–d. For 3a–c, conversion to the corresponding bishydrazones 4a–c and then oxidation to the bisdiazo species 1a–c proceeded directly. In the case of ketone 2d, reaction with hydrazine led to hydrazone formation along with hydrazinolysis of the amide, giving product 4d, which upon oxidation gave bisdiazo 1d (Scheme S3†). These were all purple-coloured solids, giving satisfactory elemental analysis (Fig. 1b) and also exhibiting fluorescence when excited by green light of λex = 460–495 nm or blue light of λex = 360–370 nm (Fig. 1c).Thermal properties of bisdiazo compounds
An initial study of bisdiazo compounds 1a–d was performed to understand thermal instability and identify the most suitable temperature range for decomposition. The thermal properties of bisdiazo compounds 1a–d were evaluated by using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), as shown in Fig. S1.† TGA revealed a weight loss of ∼10 wt% as the temperature reaches 328 °C (Fig. S1a†), which closely corresponds to the theoretical values of 11.32% and 10.68% for bisdiazo-H and bisdiazo-NH2 terminal groups after fully releasing dinitrogen (Fig. S1e and S1f†), consistent with the initial collapse to the biscarbene. Notably, the bisdiazo-NH21d reacted significantly more rapidly than the other three compounds, and N2 release upon heating continuously occurred over the temperature range up to 328 °C for all bisdiazo compounds rather than in one simple step (TGA traces in Fig. S1a†). The N2 release rate with temperature increase was studied by derivative thermogravimetric (DTG) analysis (Fig. S1b†), showing that the compounds are relatively stable at temperatures up to ∼220 °C and that at a heating rate of 5 °C min−1, the release of N2 took at least 40 min.DSC, on the other hand, was used to establish their thermal properties, i.e., a suitable temperature range for the release of N2 upon heating. The DSC trace of bisdiazo-NH2 (Fig. S1c†) shows a relatively sharp endothermic peak at around 50.5 °C, which is the expected glass transition temperature, Tg. In contrast, the Tg is even clearer for bisdiazo-H at 27.2 °C (Fig. S1c†), showing a step-like transition. These Tg points are very likely to be mislabelled as melting points solely based on observation with the naked eye.1,16 A similar pattern in Tg values is also observed in the DSC traces of bisdiazo-Me and bisdiazo-NO2, exhibiting step-like and sharp endothermic peaks (Fig. S1d†), respectively.
For all the bisdiazo compounds, decomposition leading to the release of N2 commences at 110–112 °C, but with some variation depending on the identity of the terminal groups (Fig. S1c and S1d†), in which the decomposition peak is broader for bisdiazo-NH2 than for bisdiazo-H (Fig. S1c†). Thus, it seems that the identity of the terminal group is an important determinant for stability and decomposition. The combined data from Fig. S1a–S1d† implied that subsequent mass spectral analyses would need to be conducted at temperatures lower than 60 °C to determine the necessary molecular ion, that other fragments would be observed at higher temperatures from 80 to 250 °C, and that the bisdiazo compounds would only be fully decomposed at temperatures higher than 600 °C.
To understand why the broadness of a decomposition peak in the DSC traces (Fig. S1c and S1d†) differs for each of the compounds, a stepwise isothermal DSC analysis was carried out (Fig. 2a–c and Fig. S1g†). The release of N2 started at about 60 °C for all the bisdiazo compounds, which is more significant for bisdiazo-NH2 as compared to the other three compounds (Fig. 2a–b, Fig. S1g†), and lower in area for both the bisdiazo-Me and –NO2 compounds (Fig. 2b and Fig. S1g†), consistent with the difference in activation energy. There are two major peaks for the release of N2 for bisdiazo-NH2 occurring at about 80 °C and 100 °C (Fig. 2c), respectively, while only one exists at around 100 °C and is relatively sharp for bisdiazo-H at 80 °C (Fig. 2a), but broad at 80 °C for bisdiazo-Me and –NO2 (Fig. 2b and Fig. S1g†), implying that other different terminal groups could be used to further adjust the thermal stability of this class of compounds. In addition, even at the same temperature of 100 °C, being heated for 30 min does not release all the possible N2 molecules in all the bisdiazo compounds, since there are further sharp peaks at higher temperatures of 120 °C and at 140 °C, especially noticeable for bisdiazo-NH2 (Fig. 2c).
 | ||
| Fig. 2 Stepwise isothermal DSC traces at various temperatures of (a) bisdiazo-H 1a, (b) bisdiazo-NO21c and (c) bisdiazo-NH21d. (d) Colour change of bisdiazo-H solution in THF before and after heating, (e) self-polymerization in nitrogen at 120 °C for 3 h, (f) GPC traces of molecular weight distribution and (g) UV-vis spectra of self-polymerized bisdiazo-H 1a under different conditions (in THF solution). | ||
Thermally induced catalyst-free solid-phase self-polymerization
From the visual appearance of the above thermal reactions, it was suspected that homopolymerization might occur in the molten phase of all the compounds (Fig. S2a and S2b†), and yellow-brown solids could be obtained after heating bisdiazo-H 1a and bisdiazo-NH21d at 90 °C for up to 3 h in air, conditions that were chosen to simulate the TGA and DSC measurements under nitrogen (Fig. S1a–1d†). The reaction products were only soluble in tetrahydrofuran (THF, Fig. 2d), but not methanol, ethanol or acetone (Fig. S2c†). It was also noted that the product obtained was very viscous, adhering to glassware in a manner analogous to the surface modification of such bisdiazo compounds on various materials reported previously.1,2,17Homopolymerization was conducted under air and nitrogen atmospheres at a reduced pressure of up to 0.001 mbar, leading to a colour change of bisdiazo from purple-red before polymerization to yellow after polymerization (Fig. 2d).18 Meanwhile, a foam-like structure was formed from solid-phase self-polymerization at 120 °C for 3 h under nitrogen atmospheric conditions (Fig. 2e), which does not occur in open air (Fig. S2a†). This is consistent with polymerization along with entrapment of the released N2 gas in the newly formed polymer matrix. Molecular weight (MW) distribution measurement by gel permeation chromatography (GPC, Fig. 2f and Fig. S3a†) indicated a range peaking at about 2–20 kDa and the UV-vis spectrum (Fig. 2g) further confirmed that a major peak of wavelength ∼291 nm is reduced after polymerization while a broad shoulder appeared at 320–380 nm, and complete disappearance of the diazo signal at around 2020 cm−1 by IR measurement was observed (Fig. S3b†). For a typical monomer MW of ca. 500, this would correspond to the formation of oligomers of length 6 < n < 20.
It was found that this process could be affected by both the temperature and atmospheric gas, especially oxygen (Table S1†). Overall, the MW distribution is in the range of 1–10 kDa (Fig. 2f) with a polydispersity distribution PDI (PDI = Mn/Mw) of 2.04–2.75 (Table S1†), regardless of the conditions applied, for bisdiazo-H 1a. This looks like a step-growth polymerization mechanism and these values are close to those of polymers generated from diazo compounds mediated by catalysts.19,20 Importantly, this polymerization occurred in the absence of a catalyst both in air and under an inert atmosphere like N2.
UV-vis kinetic study of thermally induced catalyst-free liquid-phase self-polymerization
The thermal decomposition reaction time is expected to be around 24 h at a lower temperature of 66 °C for bisdiazo compounds as indicated by the DSC and TGA data described above. Therefore, a THF solution of bisdiazo-NH21d was used for UV-vis spectrum kinetic analysis, as illustrated in Fig. 3a, which was also carried out in organic solvents, namely toluene at 100 °C (Fig. 3b) and chlorobenzene at 120 °C (Fig. 3c) for a systematic study. Similar UV-vis kinetic traces for the remaining bisdiazo compounds 1a–c in combinations of solvents and temperatures are shown in Fig. S4.† For the compounds in THF solutions, the UV-vis peak observed at ca. λ = 295 nm (Fig. 3a and Fig. S4a–S4c†) is ascribed to the diazo site CNN, the intensity of which decreased with polymerization time. It is also noted that there is a slight blue-shift for both starting bisdiazo-Me and bisdiazo-H compounds (Fig. S4a and S4b†), and a slight red-shift for both bisdiazo-NH2 and bisdiazo-NO2 compounds (Fig. S4c†) in THF, consistent with the changing electronics of these systems. Furthermore, the intensity of a minor peak at λ = 250 nm increases during the polymerization of bisdiazo-NH21d (Fig. 3a), which may be due to cross-linking between the carbene and the terminal amine group (vide infra).
 | ||
| Fig. 3 UV-vis kinetic traces of the polymerization of the bisdiazo-NH2 compound in (a) THF as the solvent at 66 °C, (b) toluene at 100 °C and (c) chlorobenzene at 120 °C (concentration = 0.4 mg mL−1). UV-vis kinetic rate analysis: kinetic observation of the polymerization of bisdiazo-NH2 in different solvent–temperature combinations using eqn (S11) and (S12).† | ||
In order to probe any temperature effects, UV-vis kinetic studies were also carried out in two other organic solvents, namely toluene at 100 °C and chlorobenzene at 120 °C, the patterns of which from bisdiazo-NH2 compounds are shown in Fig. 3b and c, and those from the other bisdiazo compounds are shown in Fig. S4d–S4i.† The most common observation is that the intensity of the major UV absorption peak at ∼295 nm decreased as polymerization proceeded, and a small but noticeable shoulder at ∼345 nm developed after 10 min (blue dashed-line box), although this was not so clear for bisdiazo-NO2 (Fig. S4g–S4i†).
The polymerization at 120 °C proceeded most quickly as shown via both DSC and TGA traces (Fig. S1a–S1d†) in 10 and 15 min and could also be easily perceived from the fading colour of the reacting solution (Fig. S9†). A noticeable increase in intensity of the major UV absorption band at 295 nm as the reaction continues further after 0.5 and 1 h, and a unique step-rise for bisdiazo-NH2 after the reaction for 18 h and even longer times (white arrow in Fig. 3b and c) could be seen. A pattern (Fig. S4d–S4i†) of the major band at 290 nm and the minor shoulder at around 350 nm in the UV-vis spectrum of all the bisdiazo compounds in the various combinations of solvent and temperature was also observed, as shown in Fig. S7,† except for bisdiazo-NO21c, which is similar to the polymerization of bisdiazo-Me in HPLC-grade DMSO-d6 at a higher temperature of 120 °C, as shown in Fig. S9c and S9d.†
Fig. S5† shows a simple kinetic analysis model that assumes decomposition of the diazo function directly to a carbene, and overall this model works well for diazo R = H, Me and NO2 compounds, but not for R = NH2, which shows anomalous behaviour at elevated temperatures. To study this difference more quantitatively, a consecutive reaction and/or polymerization was explored for the possible rate constants that involves the polymerization of the bisdiazo compound in various solvents at different temperatures, as shown in Fig. S6;† this is more precise than the simplified one illustrated in Fig. S5.† The results from the simplified kinetic analysis shown in Fig. S5† are acceptable for the polymerization carried out in THF at 66 °C and in toluene at 100 °C, but no fitted results could be obtained at the higher temperature of 120 °C in chlorobenzene (grey-shaded area in Fig. S6c†), or even at 100 °C in toluene for bisdiazo-NH2 alone, which does not match the case described by eqn (S10) or (S11)† (Fig. S6e†). These results also reveal a solvent–temperature combination effect on the rate constant calculation, as shown in Fig. S5e and S5f,† described using simplified eqn (S1) and (S2),† and this solvent–temperature combination effect is also clearly supported by the UV-vis spectrum observations of the resulting polymers, as illustrated in Fig. S7.†
From the rate constant data as summarized in Table S3,† there is a decrease in the rate of carbene formation with increasing temperature, along with activation for R = Me and deactivation for R = NO2, as expected at all temperatures. In a number of cases, k1 = k2, and for polymerization in chlorobenzene (PhCl, 120 °C), k2 > k1, so that the polymer is formed more effectively at higher temperature. Meanwhile, for those reactions in THF at 66 °C or toluene (PhMe) at 100 °C, k2 < k1 or k2 = k1, so that even though the rate of formation of carbene is good, polymerization is slower, since carbene does not react as quickly.
Therefore, a two-step consecutive polymerization was considered (Fig. S6a†) with more details being given in Fig. S6b.† The corresponding findings are given in Fig. 4 and Tables S2 and S3† for the possible rate constants (i.e., k1 for releasing nitrogen molecules and k2 for further reaction of the diazocarbene or biscarbene intermediates) for each step, as summarized in Tables S2 and S3.† This analysis (Tables S2 and S3†) suggests equal rate constants, k1 = k2, for both R = H and R = NH2 when polymerized in THF at 66 °C, but k1 ≫ k2 for both R = Me and R = NO2, suggesting that the second step of polymerization from the carbene species can become the rate-determining step. Second, for the polymerization carried out in toluene at 100 °C, k1 ≫ k2 for both R = H and R = Me, implying again that the second step is the rate-determining step, while k1 = k2 applies for both R = NO2 and R = NH2 (Tables S2 and S3†). More importantly, probing the rate constants for polymerization in chlorobenzene at 120 °C is even more complicated than the two cases mentioned above and the results are summarized in Fig. 4c–f, Fig. S6c and S6d.† In this scenario, both R = NO2 and R = NH2 coincidentally showed almost the same pattern (Fig. S6c†), while the polymerization process for both R = H and R = Me still fits the two-step consecutive reaction well enough, although an alternative eqn (S11)† fitting was successfully used for the polymerization of all compounds at 120 °C (Fig. 4c). Such an alternative equation, eqn (S11),† is more suitable for describing the scenario changes noticeable in the kinetics of polymer formation during the disappearance of each compound, as shown in Fig. 4d, especially for the polymerization of bisdiazo-NH21c at higher temperatures of 100 and 120 °C (Fig. 4e and f). Both Fig. 4d and f show fitted kinetic contributions from both decomposed exponential and linear parts for the polymerization. It looks like the exponential part (left sub-chart, Fig. 4d) is more likely to be the production rate of carbene species or rate of disappearance of diazo sites. The linear part (right sub-chart, Fig. 4d) seems to be the rate of polymerization with time, because it is almost the same trend for R = H, Me, and NO2 with the only difference coming from the terminal group R = NH2, most likely due to significant electronic activation. In other words, the rate of consumption of diazo sites is not necessarily equivalent to polymerization, and a two-step process, in which rate mismatch might occur, as seen in the polymerization of bisdiazo-NH2 at higher temperatures (Fig. 4e and f), suggests that a solvent–temperature combination might be important in the outcome of the polymerization. Additionally, the contribution from the linear part for bisdiazo-NH2 polymerization at lower temperature, i.e., at 66 °C, is missing (right sub-chart, Fig. 4f), implying that for heating at lower temperatures for bisdiazo-NH2, the disappearance of diazo sites is the dominant one. Furthermore, the linear part in Fig. 4f implies that cross-linking between the carbene-centred carbon atoms with the terminal –NH2 groups, and the reaction at lower temperature, i.e., at 66 °C, do not occur, but both the polymerization and cross-linking occur for reactions when carried out at both 100 °C and 120 °C (Fig. 4e). Such cross-linking between the carbene-centred carbon atoms and the terminal groups would not be able to occur for the other three compounds (i.e., R = H, Me and NO2) as shown in Fig. S6d and S6e.† However, a plot of Hammett substituent constants21 and the rate constants for the different combinations of temperature and solvent (Fig. S6f†) showed no direct correlation, underscoring the complexity of the polymerization sequence, especially so for 1d, where there are two reaction sites, at the carbene and amino centres.
 | ||
| Fig. 4 UV-vis kinetic rate analysis: (a and b) a consecutive two-step reaction for bisdiazo polymerization in THF at 66 °C and toluene at 100 °C described using eqn (S10);† (c and e) alternative descriptions of polymerization in chlorobenzene at 120 °C; (d and f) fitted kinetic contributions from both decomposed exponential and linear parts for the polymerization using eqn (S11) and (S12);† and (e and f) kinetic observation of the polymerization of bisdiazo-NH2 in different solvent–temperature combinations using eqn (S11) and (S12).† | ||
Identity of polymer products
The UV-vis spectra of the obtained polymers under various solvent–temperature combinations were studied to identify possible polymer linking groups (Fig. S7†) and several possibilities were identified (Fig. 1a). There is a broad UV-vis band at 285 nm for both polymers from R = H 1a and R = Me 1b with a shoulder at 350 nm (Fig. S8†), but those for R = NO21c and R = NH21d were closer to 295 nm. (Fig. S8†). This suggests the presence of different linking groups, and for R = H 1a and R = Me 1b, an azine linkage CN–NC is more likely to occur, as evidenced by a reported UV band λmax value of 275 nm,22 while for R = NO21c, a direct CC linkage seems more likely to occur, evidenced by a reported λmax value of 308 nm.23 That such a difference would arise is not surprising, given the different rates of collapse of diazo starting materials, the onward reaction and the stabilities of different intermediates, as indicated by the values of k1 and k2 (Table S3†). The terminal groups of the polymers could be diazo groups, stabilized within the matrix of the polymer, which would be responsible for (weak) UV absorption.
Molecular weight and distribution analysis
The molecular weight (MW) and distribution traces of the polymers derived from bisdiazo compounds 1a–d reacted in several organic solvents at various temperatures, as obtained by GPC, show at least a bimodal MW distribution in all cases (Fig. 5), with the DP higher for 1a and 1b (R = H and Me) and lower for 1c and 1d (R = NO2 and NH2). Increasing the reaction temperature leads to higher MWs for 1a and 1b, but not for 1c and 1d (R = NO2 and NH2), which are much less affected by temperature. This further supports the importance of the terminal substituent and the combination of solvent–temperature24–27 for the reaction (see Fig. S7†). The specific MW distribution pattern of each type of bisdiazo compound is significantly affected by its terminal group (especially MW subpeaks 1 and 2 shown in Fig. 5a and b for electron neutral ones). In addition, each sub-MW peak, labelled by circled numbers in Fig. 5 and summarized in Table 1 for each bisdiazo compound, could be regarded as largely monodisperse with a PDI of about 1.03–1.19. Furthermore, it is noteworthy that the MW for all of the peaks numbered 3 is the least monodisperse (i.e. PDI = Mw/Mn∼ 1.0 indicates monodisperse). Of interest is that R = NO21c gives the cleanest polymer, characterized by one major peak in the GPC trace. | ||
| Fig. 5 GPC-obtained molecular weight (MW) distribution of bisdiazo-R polymerization in different solvent–temperature combinations for 24 h: (a) R = H, (b) R = Me, (c) R = NO2 and (d) R = NH2. | ||
| M n and Mw denote the number and weight averaged molecular weight, respectively. DP is the degree of polymerization, estimated using Mn/bisdiazo-R. PDI = Mw/Mn. |
|---|

|
Moreover, the MW dispersity (PDI) obtained in the three combinations of solvent and temperature relates to the kinetics (Fig. 4). The lower MW distribution regions (MW peaks 1 and 2) of all the polymers reflect the relative rate constants that control the MW distribution for the two-step consecutive polymerization, seen for both 1a and 1d (R = H, and NH2) in THF at 66 °C (Fig. 5a and d, Tables 1, S2 and S3†), as well as both 1c and 1d (R = NO2 and NH2) in toluene at 100 °C (Fig. 5c and d) with equal rate constants. At the higher temperature of 120 °C in chlorobenzene, a mismatch in the kinetic rate, as shown in Fig. 4c–f, means that higher MW distribution dominates the resulting polymers, as clearly illustrated in Fig. 5d.
Here the MW, i.e., Mn, and degree of polymerization (DP) values are listed in Table 1, which are much better than those given in Fig. 2d and Table S1,† and this is likely to be a result of more effective liquid-phase polymerization for up to 24 h for the former, as compared to only 3 h for solid-phase polymerization for the latter. Second, for solid-phase polymerization, the honeycomb structure (Fig. 2c and Fig. S2†) hindered further heat transfer needed for activating the starting diazo compounds, giving a slower and longer reaction time, and overall, a less effective polymerization. In contrast, the liquid phase in the varied solvents allows better maintenance of the set reaction temperature, allowing effective activation of the diazo sites to carbenes.
Fig. 6 shows a comparison between the catalysed reaction process in the presence of palladium(II) (w/ Pd(DBA)2 in Fig. 6a) and without using a catalyst (i.e., w/o Pd(DBA)2 in Fig. 6b) under similar reaction conditions. It turns out that the polymer generated using the Pd catalyst is yellow (w/ Pd(DBA)2 in Fig. 6a) and this might be due to the presence of nitrogen in the product or residual Pd organic complex that can usually be removed using a strong acid such as hydrochloric acid.28–30 Furthermore, the catalyst certainly enhances the polymerization as monitored using UV-vis spectra (Fig. 6c and d), since a minor shoulder bands in the UV-vis spectra are more noticeable, taking less time to appear using the Pd catalyst than without the Pd catalyst (i.e., the blue shaded boxes in Fig. 6c and d) for bisdiazo-H (details for the other bisdiazo compounds w/ the Pd catalyst are summarized in Table S4†), indicating that the reaction of bisdiazo systems upon heating may occur in catalysed and uncatalysed scenarios.
 | ||
| Fig. 6 (a and b) Images and (c and d) UV-vis kinetic spectra of polymer solutions in toluene at 100 °C from the thermally induced polymerization of the bisdiazo-H compound (a and c) with (w/) and (b and d) without (w/o) the Pd(DBA)2 catalyst. | ||
FD MS monitored self-oligomerization
To further analyse the self-polymerization behaviour of the bisdiazo compounds with different terminal groups, upon heating alone, a high-resolution time-of-flight (HR TOF) MS system was used. The mass spectra of the material generated by the self-polymerization of bisdiazo-R compounds in THF upon heating under vacuum, and the original FD chronogram data with further analysed mass spectra are shown in Fig. S10 and Table S5.† Such field desorption (FD) chronograms (Fig. S10a†) confirmed the TGA data indicating that bisdiazo-NH21d has the highest decomposition temperature (Fig. S1b†). The exact mass of all four starting bisdiazo compounds 1a–d could be obtained with a lower capped current at two different current rates (Fig. S11†), which minimized the m/z peak of 662 at a higher current rate of 51.2 mA min−1 for bisdiazo-NH2. Along with dimer and trimer products, other decomposition fragments were observed at elevated temperatures (Fig. S10c–S10f†).The mass spectra of all four bisdiazo compounds 1a–d upon heating under vacuum exhibit a cluster pattern, as shown in Fig. 7a–d, clearly showing the monoisotopic m/z of dimer and trimer products except that for bisdiazo-NO21c (Fig. S10c–S10f†). In particular, the monoisotopic m/z values for 1a bisdiazo-H and 1d bisdiazo-NH2 (494.17 and 524.20, respectively) were easily observed (Fig. S10c, S10f, and S11a and S11d†), representing the first mass spectrometric detection of diazo compounds that are normally too unstable for successful MS analysis. For both 1a (bisdiazo-H) and 1d (bisdiazo-NH2) compounds (Fig. 7a and d), different fragments were observed at higher temperature before reaching the final decomposition, and dimers and trimers could be seen in the mass spectra, but the tetramer and higher oligomers were not observed since they are beyond the detection limit of m/z = 1600. The existence of such dimers and trimers, even tetramers, further supports the occurrence of self-polymerization of these bisdiazo compounds, giving structurally novel polymers without the need for catalysis. The possible structures of these oligomers, formed through the linkage of –CN–NC– or CC, are shown in Fig. 7e. The varied combinations of these two linkers are the most probable reason for the cluster of peaks of oligomerization and polymerization labelled as dimer and trimer in Fig. 7a–d.
 | ||
| Fig. 7 FD HR-TOF MS spectra of the self-polymerized material upon heating under vacuum for (a) bisdiazo-H 1a, (b) bisdiazo-Me 1b, (c) bisdiazo-NO21c and (d) bisdiazo-NH21d; (e) possible repeating unit of oligomerization structures with monoisotopic m/z. | ||
Additionally, the self-polymerization of bisdiazo-H and bisdiazo-NH2 is different. While for the bisdiazo with the –H terminal group, stepwise-like polymerization occurs, giving the dimer and then the trimer (Fig. S10a and S10c,† see the corresponding A# regions in the FD chronogram), the other bisdiazo with the –NH2 terminal group exhibits the dimer and the trimer within the same time frame (Fig. S10d,† D# regions noted in the FD chronogram). This difference could be due to changes in the carbene reactivity profile or the terminal group –NH2 also reacting with the biscarbene, leading to possible rapid cross-linking,15 as shown in Fig. 7e.
The predicted possible structures and m/z values of the possible fragments, dimer and trimer of bisdiazo-H upon heating are shown in Fig. S10g† (based on the corresponding data of A# regions in the FD chronogram, Fig. S10c†). This would be similar to the stepwise-like polymerization of diazo compounds arising from transition metal-catalysed carbene cross-coupling,31 so-called ‘C1 polymerization’.32 Similar C1 polymerization seems very likely for bisdiazo-H 1a upon heating (Fig. S4a and S4c†), while in the case of bisdiazo-NH21d, the presence of the terminal amine groups could lead to additional cross-linking processes as discussed above.
Furthermore, instead of releasing both dinitrogen molecules at the same time, such stepwise polymerization seems to proceed by releasing just one nitrogen at a time at lower temperature and possibly another dinitrogen at a higher temperature as exhibited in dimerization and trimerization, as shown in Fig. 7a–d and illustrated in the TGA trace, which clearly indicates a slow evolution of nitrogen as the temperature is increased. Here, a cluster of peaks for the dimer with multiple m/z values close to 14.00 is observed, equalling a nitrogen atom, which matches the continuous weight loss as perceived by TGA (Fig. S1b†) and the fact that the accumulated weight loss rate is less than 10 wt% even at 180 °C (Fig. S1a†). This is close to the trend of releasing nitrogen within 30 min at various temperatures (Fig. 2b), and heating for just tens of seconds releases one nitrogen as observed in the mass (m/z) difference of 28 (A1 region in Fig. S10c† and B1 region in Fig. S10d†). Making similar predictions for the bisdiazo-NH21d compound is more complicated, since the reaction at both carbene and amine groups is possible (Fig. S10f and S10h†); dimer and trimer fragments in the mass spectrum showed a very small time frame, as illustrated in Fig. S10f and S10h† (sub-region D2). This difference is highly likely because of possible electronic modification of the terminal group.33,34 The patterns of the dimer and trimer in the mass spectrum are similar for all reactions, indicating homo-oligomerization.
The possible monomer units of these oligomers formed through the linkage of –CN–NC– or CC is illustrated in Fig. 7e, but analysis of the data in Fig. 7a–d clearly shows that the oligomer may be clearly formulated as 5 below for all groups R and for n = 2, 3; this confirms the repeat unit to be CC, and indicates that this reaction is a C1 polymerization occurring under thermal and uncatalysed conditions.
Molecular dynamics (MD)-calculated bond length of the linkage in a dimer
To elucidate the fundamental aspects of all the bisdiazo compounds, density functional theory (DFT) calculations were performed and from the comparisons between the calculated and experimental spectra (IR and UV-vis in THF), there is a good match for IR (Fig. S14†) and also a very close one for UV-vis in THF (Fig. S15†). Similarly, the reacting carbene species leading to dimer formation involving an active singlet carbene species35 have been modelled by carbene–carbene coupling or by carbene–diazo coupling (Fig. 8a and b). The C1 polymerization mentioned above grows polymer chains in single-carbon increments,11,36 as opposed to the more usual ‘C2 polymerization’ in the preparation of saturated main-chain carbon-based polymers. In the present work, linking CC and –CN–NC– is possible. This assumption could be verified by molecular dynamics (MD) analysis (Fig. 8c and d), for which the bond length profiles are shown in Fig. 8a and b (purple dashed line). As can be seen from Fig. 8c, the calculated bond length of C⋯C from both carbene-centred carbon atoms is around 1.35 Å, which is quite closely matched with that of a typical sp2–sp2 CC bond with a length of 1.34 Å.
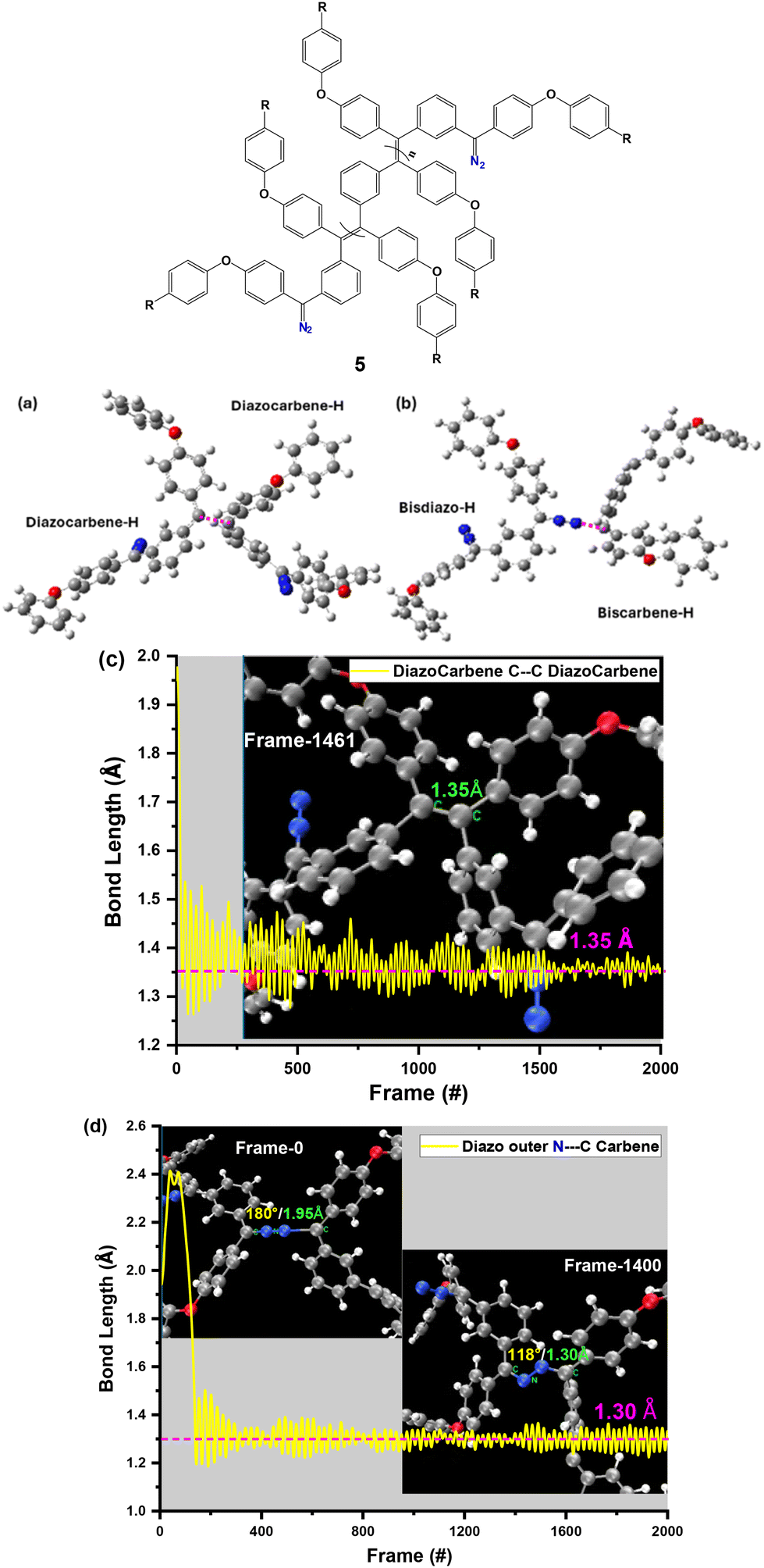 | ||
| Fig. 8 (a and b) Possible dimer structures for molecular dynamics (MD) calculations of (a) C⋯C bond formation from both carbene-centered C atoms and (b) CN–N⋯C bond formation from carbene-centred C and N atoms from a diazo site. (c and d) MD-calculated bond length profiles of (c) C⋯C and (d) CN–N⋯C bonds (here using bisdiazo-H and its carbene species, i.e., diazocarbene-H and biscarbene-H; in the inset images of the MD frame, the purple dashed line shows the possible bonds between atoms; the green labels are the bond lengths and the yellow labels are the bond angles). | ||
Similarly, the–CN–NC– linkage from a diazo site and a carbene-centred carbon atom could be confirmed with the calculated bond length of C⋯N being about 1.30 Å, close to that of a typical sp2 CN bond (Fig. 8d); at the same time, the bond angle of CN–N is reduced to around 110–130° from the previous 180° for the diazo site CNN. All these MD-calculated values confirm that both linkers are feasible.
Self-polymerization-induced nanostructure on the polymeric surface
The biscarbene polymerization reported here works without any metal-based catalyst, and tolerates oxygen conditions, in both the solid phase and liquid phase. Although diazo and similar bisdiazo compounds have been used for surface modification of various substrates,1,16 the molecular nature of these biscarbene modifications has not been determined, and the exact nature of surface modification becomes unclear. Besides, the direct carbene insertion and/or cross-linking37 at the surface, carbene polymerization and polycondensation38–40 can occur. To understand this further, the surface modification of bisdiazo-NH2 on polyvinyl alcohol (PVA) was studied through liquid-phase polymerization under open-air conditions with a minimized volume of organic solvents; noticeable colour changes were immediately observed, as shown in Fig. S12a,† and the full-range X-ray photoelectron spectroscopy (XPS) results are shown in Fig. 9a. | ||
| Fig. 9 (a) Full-range XPS scans, (b) AFM 2D FFT and phase images in tapping mode, (c) AFM-QNM results and (e) dynamic water contact angles (WCAs) of the PVA thin film before and after surface modification using bisdiazo-NH2 in different solvent systems. | ||
This outcome would be consistent with the self-polymerization of bisdiazo-NH2 upon heating. After surface modification with bisdiazo-NH2 compounds, regardless of the solvent system, significant changes and carbons from the aromatic surface are clearly visible, as evidenced by the presence of a small broad π–π* satellite peak41 (Fig. S13a†), consistent with polymerization. The conjugated-like structure form is most likely derived from carbene self-polymerization, along with surface modification, and confirms the assumed possible polymerization and cross-linking from bisdiazo-NH2-induced surface modifications.15
Atomic force microscopy (AFM) using two-dimensional fast Fourier transform (2D FFT) images shows that the surface topography and phase change significantly (Fig. 9b). Furthermore, Fig. 9c shows the surface nanomechanical properties of the surface-modified PVA thin film. As compared to the bare PVA thin film, the surface adhesion changed a little, but both the stiffness and Derjaguin–Muller–Toporov (DMT) modulus almost doubled. This could be explained by the introduction of a molecular repeat unit, which is linear for PVA but full of aromatic rings for self-polymerized biscarbene that crosslinks the PVA, giving a network which is more rigid.
The dynamic water contact angle (WCA) (Fig. 9d and e) of the unmodified PVA thin film is in the range of 75–90°, holding water for up to 15 s, although for times longer than 15 s, water penetration occurs, as illustrated in the inset image in Fig. 9d and e. After surface modification using the bisdiazo-NH2 compound, the water contact angle is quite stable with time and in the range of 75–90°, which is close to the reported WCA of different diazo-based cross-linkers applied to ultrahigh molecular weight polyethylene.42 This is likely for two reasons: bisdiazo-NH2 creates a heavily aromatic ring-based polymer-like thin film on the PVA surface upon heating at 120 °C for 1 h, leading to a hydrophobic surface, and on the other hand, the polar nature of –NH2, existing in such a polymer-like thin film, results in a hydrophilic surface. The cone-like nanostructures43,44 formed on the surface of the PVA thin film (Fig. 9b and c), as a result of self-polymerized biscarbene, might also affect the dynamic WCA,43 as shown in Fig. 9e, since the surface topology (Fig. 9b) and nanomechanical properties (Fig. 9c) are changed. This demonstrates that oxygen-tolerant surface functionalization may be achieved through a catalyst-free polymerization of biscarbene from the thermally decomposed bisdiazo compound simply by an open-air heating process with a minimized volume of liquid phase.
Experimental
Reagents, procedure and characterization of bisdiazo compounds
The bisdiazo compounds with various terminal functional groups are illustrated in Fig. 1a and the reagents and procedures for synthesizing each one are detailed in ESI Section 1.1–1.3,† along with their structural characterization, including NMR and IR vibration measurements.UV-vis kinetic study of catalyst-free polymerization upon heating
A solution of bisdiazo compounds in HPLC-grade organic solvent, such as THF, toluene and chlorobenzene, at a concentration of 0.4 mg mL−1, was prepared for the polymerization kinetics study at predetermined time intervals while sampling a 100 μL reaction mixture. For UV-vis absorption measurements, each sample was diluted with 2.5 mL of HPLC solvent and placed in a quartz cuvette. Spectra were collected (UV-1900, SHIMADZU, Japan) over the range of 200–700 nm, with scanning at an intermediate rate and data points recorded every 0.2 nm at room temperature. In this UV-vis kinetics study, the polymerization of the bisdiazo compound was conducted while refluxing in THF at temperatures of 66 °C and 100 °C in toluene and 120 °C in chlorobenzene for 24 h, respectively.A similar UV-vis kinetics study on the Pd-mediated polymerization of bisdiazo-R compounds as references for the catalyst-free polymerization was performed. Briefly, a mixture of 2.0 mol% based on bisdiazo-R compounds with the catalyst Pd(DBA)2 (0.4 mg, 0.80 μmol) in 50 mL of HPLC-grade toluene at 100 °C in a round-bottom flask was stirred at 325 rpm for 30 s at room temperature. Afterwards, 20 mg of solid powder of the bisdiazo-R compound was added and stirred for 30 s at room temperature (RT). Then, the mixture was stirred in an oil bath at 100 °C for 24 h. The liquid reaction mixture was sampled at predetermined interval times for the UV-vis kinetics analysis with the observations of the kinetics being illustrated in Fig. 6. After cooling to RT, volatile materials were removed under reduced pressure, and the residue was dissolved in 5 mL of HPCL-grade THF and precipitated in 15 mL of HPLC-grade MeOH, which was precipitated out as demonstrated in Fig. 6a.
Measurement of molecular weights via gel permeation chromatography (GPC)
Gel permeation chromatography (GPC) was conducted using a Shimadzu modular system, comprising a CBM-20A system controller, an SIL-20A automatic injector, a 10.0 μm beadsize guard column (50 × 7.5 mm) followed by KF-803L and KF-802 columns in series (both 300 × 8 mm, Shodex™), an SPD-20A ultraviolet detector, and an RID-20A differential refractive index detector, to analyse and measure the molecular weights of the self-polymerized materials from the bisdiazo precursors. The temperature of the columns was maintained at 40 °C using a CTO-20A oven, and the eluent was THF (HPLC grade) for a sample concentration of 0.5 mg mL−1. The analysis was performed using an LC-20AD pump running in isocratic flow mode at a rate of 1.0 mL min−1. A molecular weight calibration curve was produced using commercial narrow molecular weight distribution polystyrene (PS) standards with molecular weights ranging from 1300 to 1.18 × 105 Da.Homopolymerization monitored by FD HRMS
All the bisdiazo compounds were dissolved in THF (HPLC grade, 10 mg mL−1), and about 4 μL of each sample was dropped on the direct insertion probe in field desorption mode (FD) while using a high-resolution time of flight (HR-TOF) mass spectrometric system (JMS-T2000GC ‘AccuTOF™ GC Alpha’, JEOL, Japan). Ionization of the sample was then activated with a voltage of −10 kV and a current of 40 mA while the ion source was in OFF mode at a temperature of 40 °C. The temperature of the probe was programmed to correspond with an increasing rate of 25.6 mA min−1 from 0 to 40 mA. As the probe reached the target current of about 40 mA, its corresponding temperature was approximately 600 °C. An ion (m/z) range of 35–1600 was monitored using a JMS-T2000GC ‘AccuTOF™ GC Alpha’ system. The original FD chronograms and details of the mass spectrometric analysis are displayed and summarized in Fig. S9 and Table S5 (ESI†), respectively.Surface modification and characterization of PVA thin films
The preparation procedure for the as-prepared PVA film and the surface-modified PVA film is detailed in ESI Section 3,† while characterization of the films was conducted via field emission scanning electron microscopy (FE-SEM) for morphological analysis, X-ray photoelectron spectroscopy (XPS) for surface probing, atomic force microscopy (AFM, Bruker Dimension Icon) for surface topography and phase images, etc.Molecular dynamics (MD) analysis of linker formation
To acquire further theoretical understanding of the possible linkers, i.e., CC from both the carbene-centred carbon atoms and –CN–NC– from a carbene-centred carbon atom and a nitrogen atom from the diazo site, molecular dynamics (MD) calculations were performed with the free quantum computational chemistry software ORCA 5.0.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 using bisdiazo-H compounds and their carbene species as examples for the possible dimers at the theoretical level of B97-3c with default basis sets,46,47 as detailed in ESI Section 4.† The MD input file was obtained with the help of another free multifunctional wavefunction analyser, Multiwfn version 3.8 (dev),48 and the results were obtained using VMD 1.9.3 software49 for analysing and extracting the bond length profile and angle measurements, with more details provided in the ESI.†
45 using bisdiazo-H compounds and their carbene species as examples for the possible dimers at the theoretical level of B97-3c with default basis sets,46,47 as detailed in ESI Section 4.† The MD input file was obtained with the help of another free multifunctional wavefunction analyser, Multiwfn version 3.8 (dev),48 and the results were obtained using VMD 1.9.3 software49 for analysing and extracting the bond length profile and angle measurements, with more details provided in the ESI.†
Conclusions
Overall, we report a detailed thermal evaluation of the decomposition of bisdiazo compounds substituted with various terminal groups. We have demonstrated that bisdiazo compounds thermally decompose at temperatures as low as 66 °C in the absence of catalyst for initiating a possible oligomerization, and even further polymerization at higher temperatures up to 120 °C. It is also noted that such polymerizations or reactions in the solid phase without any metal catalysts give foam-like products, consistent with the loss of N2, polymerization and entrapment of the released gas in the polymer matrix, and the thermal properties and rates of the reaction are affected by substituent terminal groups. From further thorough UV-vis kinetics techniques, the reaction of nitrogen loss and catalyst-free polymerization proceeds in two steps with varied rate constants, depending on the nature of the terminal group and a combination of temperature and solvent. Both mass analysis and molecular dynamics calculations confirmed that the linking reaction is alkene formation, with diazo end groups. Importantly, the reaction is not only catalyst-free but also oxygen tolerant. Such catalyst-free oxygen-tolerant self-polymerization from biscarbene is a thermally induced reaction and this can be used for the growth of unique nanostructures on polymeric substrates, providing adjustment to surface properties. Such self-polymerization can be used to generate a new family of polymers and the mechanical characterization of these biscarbene-based polymers with different terminal groups is currently in progress.Author contributions
Dr Xiaosong Liu: conceptualization, synthesis, methodology, all measurements and data curation, formal analysis, density functional theory (DFT) and molecular dynamics (MD) calculations and result analysis, and writing, editing & reviewing – original draft. Prof./Dr Mark G. Moloney: conceptualization, investigation, supervision, project administration, and writing – review & editing. Mr Koji Okuda: methodology and data curation on the FD HRMS monitoring of bisdiazo compound oligomerization, and writing – review & editing.Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI.† Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Jiangsu Provincial Natural Science Foundation (General project, Grant Number BK20231223).References
- P. Yang and M. G. Moloney, Surface modification of polymers with bis(arylcarbene)s from bis(aryldiazomethane)s: preparation, dyeing and characterization, RSC Adv., 2016, 6(112), 111276–111290, 10.1039/C6RA24392D.
- P. Yang, Y. Wang, L. Lu, X. Yu and L. Liu, Surface hydrophobic modification of polyurethanes by diaryl carbene chemistry: Synthesis and characterization, Appl. Surf. Sci., 2018, 435, 346–351, DOI:10.1016/j.apsusc.2017.11.121.
- S. Harada, K. Tanikawa, H. Homma, C. Sakai, T. Ito and T. Nemoto, Silver,–Catalyzed Asymmetric Insertion into Phenolic O− H Bonds using Aryl Diazoacetates and Theoretical Mechanistic Studies, Chem. – Eur. J., 2019, 25(52), 12058–12062, DOI:10.1002/chem.201902126.
- H. M. L. Davies and K. Liao, Dirhodium tetracarboxylates as catalysts for selective intermolecular C–H functionalization, Nat. Rev. Chem., 2019, 3(6), 347–360, DOI:10.1038/s41570-019-0099-x.
- D. Wang and K. J. Szabó, Copper-Catalyzed, Stereoselective Cross-Coupling of Cyclic Allyl Boronic Acids with α-Diazoketones, Org. Lett., 2017, 19(7), 1622–1625, DOI:10.1021/acs.orglett.7b00433.
- H. R. Allcock, W. C. Hymer and P. E. Austin, Diazo coupling of catecholamines with poly(organophosphazenes), Macromolecules, 1983, 16(9), 1401–1406, DOI:10.1021/ma00243a001.
- S. Chng, E. M. Parker, J.-P. Griffiths, M. G. Moloney and L. Y. L. Wu, A study of diazonium couplings with aromatic nucleophiles both in solution and on a polymer surface, Appl. Surf. Sci., 2017, 401, 181–189, DOI:10.1016/j.apsusc.2017.01.017.
- P. J. Davis, L. Harris, A. Karim, A. L. Thompson, M. Gilpin, M. G. Moloney, M. J. Pound and C. Thompson, Substituted diaryldiazomethanes and diazofluorenes: structure, reactivity and stability, Tetrahedron Lett., 2011, 52(14), 1553–1556, DOI:10.1016/j.tetlet.2011.01.116.
- X. Yu, P. Yang, M. G. Moloney, L. Wang, J. Xu, Y. Wang, L. Liu and Y. Pan, Electrospun Gelatin Membrane Cross-Linked by a Bis(diarylcarbene) for Oil/Water Separation: A New Strategy To Prepare Porous Organic Polymers, ACS Omega, 2018, 3(4), 3928–3935, DOI:10.1021/acsomega.8b00162.
- S. Iqbal, Y. Lui, J. G. Moloney, E. M. Parker, M. Suh, J. S. Foord and M. G. Moloney, A comparative study of diaryl carbene insertion reactions at polymer surfaces, Appl. Surf. Sci., 2019, 465, 754–762, DOI:10.1016/j.apsusc.2018.09.182.
- E. Jellema, A. L. Jongerius, J. N. H. Reek and B. de Bruin, C1 polymerisation and related C–C bond forming ‘carbene insertion’ reactions, Chem. Soc. Rev., 2010, 39 (5), 1706–1723, 10.1039/B911333A.
- C. R. Cahoon, K. Goossens and C. W. Bielawski, Poly(carbyne)s via reductive C1 polymerization, Polym. Int., 2021, 70(1), 34–40, DOI:10.1002/pi.6115 , (acccessed 2025/03/12).
- S. Kang, S. J. Lu, M. Cho, Y. Cho, P. R. Sultane and C. W. Bielawski, N-heterocyclic carbene copper complexes catalyze the C1 polymerization of substituted diazomethanes, J. Polym. Sci., 2024, 62(24), 5553–5561, DOI:10.1002/pol.20240591 , (acccessed 2025/03/12).
- E. Ihara, Development of polymer syntheses using diazocarbonyl compounds as monomers, Polym. J., 2025, 57(1), 1–23, DOI:10.1038/s41428-024-00954-1.
- D. Wang, W. F. Hartz, K. E. Christensen and M. G. Moloney, Surface modification of glass fiber membrane via insertion of a bis(diarylcarbene) assisted with polymerization and cross-linking reactions, Surf. Interfaces, 2022, 32, 102155, DOI:10.1016/j.surfin.2022.102155.
- P. Yang and M. G. Moloney, Surface modification using crosslinking of diamine and a bis(diarylcarbene): synthesis, characterization, and antibacterial activity via binding hydrogen peroxide, RSC Adv., 2017, 7(47), 29645–29655, 10.1039/C7RA05258H.
- K. A. Mix, M. R. Aronoff and R. T. Raines, Diazo Compounds: Versatile Tools for Chemical Biology, ACS Chem. Biol., 2016, 11(12), 3233–3244, DOI:10.1021/acschembio.6b00810.
- R. Tian, X. Ren, P. Niu, L. Yang, A. Sun, Y. Li, X. Liu and L. Wei, Development of chromenoquinoline-fused coumarin dyes and their application in bioimaging, Dyes Pigm., 2022, 205, 110530, DOI:10.1016/j.dyepig.2022.110530.
- J. Wang, Diazo compounds: Recent applications in synthetic organic chemistry and beyond, Tetrahedron Lett., 2022, 108, 154135, DOI:10.1016/j.tetlet.2022.154135.
- Q. Zhou, Y. Gao, Y. Xiao, L. Yu, Z. Fu, Z. Li and J. Wang, Palladium-catalyzed carbene coupling of N-tosylhydrazones and arylbromides to synthesize cross-conjugated polymers, Polym. Chem., 2019, 10(5), 569–573, 10.1039/C8PY01529E.
- D. H. McDaniel and H. C. Brown, An Extended Table of Hammett Substitutent Constants Based on the Ionization of Substituted Benzoic Acids, J. Org. Chem., 1958, 23(3), 420–427, DOI:10.1021/jo01097a026.
- L. N. Ferguson and T. C. Goodwin, Absorption Spectra of Azines and Dianils, J. Am. Chem. Soc., 1949, 71(2), 633–637, DOI:10.1021/ja01170a069.
- Z. Fang, F. Wu, Q. Tao, Q. Qin, C. Au, Y. Li, H. Zhang, N. Wang and B. Yi, Substituent effects on the ultraviolet absorption properties of stilbene compounds-Models for molecular cores of absorbents, Spectrochim. Acta, Part A, 2019, 215, 9–14, DOI:10.1016/j.saa.2019.02.072.
- L. Sipos, P. De and R. Faust, Effect of Temperature, Solvent Polarity, and Nature of Lewis Acid on the Rate Constants in the Carbocationic Polymerization of Isobutylene, Macromolecules, 2003, 36(22), 8282–8290, DOI:10.1021/ma034581z.
- R. E. Cais and W. L. Brown, Stereoconfigurations of Poly(vinyl bromide) and Poly(vinyl chloride). An Examination by Carbon-13 NMR of the Effects of Temperature and n-Butyraldehyde, Macromolecules, 1980, 13(4), 801–806, DOI:10.1021/ma60076a006.
- J. Krstina, G. Moad and D. H. Solomon, Effects of solvent on model copolymerization reactions. A 13C-NMR study, Eur. Polym. J., 1992, 28(3), 275–282, DOI:10.1016/0014-3057(92)90189-9.
- B. Liu, L. Wei, N.-N. Li, W.-P. Wu, H. Miao, Y.-Y. Wang and Q.-Z. Shi, Solvent/Temperature and Dipyridyl Ligands Induced Diverse Coordination Polymers Based on 3-(2′,5′-Dicarboxylphenyl)pyridine, Cryst. Growth Des., 2014, 14(3), 1110–1127, DOI:10.1021/cg401599x.
- E. Ihara, T. Hiraren, T. Itoh and K. Inoue, Palladium-mediated Polymerization of Diazoacetamides, Polym. J., 2008, 40(11), 1094–1098, DOI:10.1295/polymj.PJ2008145.
- E. Ihara, N. Haida, M. Iio and K. Inoue, Palladium-Mediated Polymerization of Alkyl Diazoacetates To Afford Poly(alkoxycarbonylmethylene)s. First Synthesis of Polymethylenes Bearing Polar Substituents, Macromolecules, 2003, 36(1), 36–41, DOI:10.1021/ma021169v.
- E. Ihara, Y. Goto, T. Itoh and K. Inoue, Palladium-Mediated Polymerization of Bifunctional Diazocarbonyl Compounds: Preparation of Crosslinked Polymers by Copolymerization of Bi- and Monofunctional Diazocarbonyl Compounds, Polym. J., 2009, 41 (12), 1117–1123, DOI:10.1295/polymj.PJ2009065R.
- Q. Xiao, Y. Zhang and J. Wang, Diazo Compounds and N-Tosylhydrazones: Novel Cross-Coupling Partners in Transition-Metal-Catalyzed Reactions, Acc. Chem. Res., 2013, 46(2), 236–247, DOI:10.1021/ar300101k.
- E. Jellema, A. L. Jongerius, J. N. H. Reek and B. de Bruin, C1 polymerisation and related C-C bond forming ‘carbeneinsertion’ reactions reactions, Chem. Soc. Rev., 2010, 39 (5), 1706–1723, 10.1039/B911333A.
- H. M. L. Davies and J. R. Denton, Application of donor/acceptor-carbenoids to the synthesis of natural products, Chem. Soc. Rev., 2009, 38(11), 3061–3071, 10.1039/B901170F.
- H. M. L. Davies and D. Morton, Guiding principles for site selective and stereoselective intermolecular C-H functionalization by donor/acceptor rhodium carbenes, Chem. Soc. Rev., 2011, 40(4), 1857–1869, 10.1039/C0CS00217H.
- R. C. Dobson, D. M. Hayes and R. Hoffmann, Potential surface for the insertion of singlet methylene into a carbon-hydrogen bond, J. Am. Chem. Soc., 1971, 93(23), 6188–6192, DOI:10.1021/ja00752a033.
- C. R. Cahoon and C. W. Bielawski, Metal-promoted C1 polymerizations, Coord. Chem. Rev., 2018, 374, 261–278, DOI:10.1016/j.ccr.2018.06.017.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Stable Carbenes, Chem. Rev., 2000, 100(1), 39–92, DOI:10.1021/cr940472u.
- H. Shimomoto, Synthesis of functional polymers by the Pd-mediated polymerization of diazoacetates and polycondensation of bis(diazocarbonyl) compounds, Polym. J., 2020, 52(3), 269–277, DOI:10.1038/s41428-019-0271-7.
- H. Shimomoto, H. Mukai, H. Bekku, T. Itoh and E. Ihara, Ru-Catalyzed Polycondensation of Dialkyl 1,4-Phenylenebis(diazoacetate) with Dianiline: Synthesis of Well-Defined Aromatic Polyamines Bearing an Alkoxycarbonyl Group at the Adjacent Carbon of Each Nitrogen in the Main Chain Framework, Macromolecules, 2017, 50(23), 9233–9238, DOI:10.1021/acs.macromol.7b01994.
- Y.-C. Wang, Y.-J. Shao, G.-S. Liou, S. Nagao, Y. Makino, E. Akiyama, M. Kato, H. Shimomoto and E. Ihara, Synthesis and electrochromic properties of polyamines containing a 4,4′-diaminotriphenylamine-N,N’-diyl unit in the polymer backbone: Ru-catalyzed N-H insertion polycondensation of 1,4-phenylenebis(diazoacetate) with 4,4′-diaminotriphenylamine derivatives, Polym. Chem., 2022, 13(46), 6369–6376, 10.1039/D2PY01118B.
- L. Zhang, Z. Li, Y. Tan, G. Lolli, N. Sakulchaicharoen, F. G. Requejo, B. S. Mun and D. E. Resasco, Influence of a Top Crust of Entangled Nanotubes on the Structure of Vertically Aligned Forests of Single-Walled Carbon Nanotubes, Chem. Mater., 2006, 18(23), 5624–5629, DOI:10.1021/cm061783b.
- S. Yang, S. Yi, J. Yun, N. Li, Y. Jiang, Z. Huang, C. Xu, C. He and X. Pan, Carbene-Mediated Polymer Cross-Linking with Diazo Compounds by C-H Activation and Insertion, Macromolecules, 2022, 55(9), 3423–3429, DOI:10.1021/acs.macromol.2c00527.
- M. S. Ambrosia, M. Y. Ha and S. Balachandar, The effect of pillar surface fraction and pillar height on contact angles using molecular dynamics, Appl. Surf. Sci., 2013, 282, 211–216, DOI:10.1016/j.apsusc.2013.05.104.
- M. J. Yoo, M. S. Ambrosia, T. W. Kwon, J. Jang and M. Y. Ha, Wetting characteristics of a water droplet on solid surfaces with various pillar surface fractions under different conditions, J. Mech. Sci. Technol., 2018, 32(4), 1593–1600, DOI:10.1007/s12206-018-0314-6.
- U. Ekstrom, L. V. R. Bast, A. J. Thorvaldsen and K. Ruud, Arbitrary-Order Density Functional Response Theory from Automatic Differentiation, J. Chem. Theory Comput., 2010, 6(7), 1971–1980, DOI:10.1021/ct100117s.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32(7), 1456–1465, DOI:10.1002/jcc.21759 , (acccessed 2025/03/03).
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104, DOI:10.1063/1.3382344 , (acccessed 3/3/2025).
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592, DOI:10.1002/jcc.22885 , (acccessed 2025/03/03).
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graph., 1996, 14(1), 33–38, DOI:10.1016/0263-7855(96)00018-5.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01474j |
| This journal is © The Royal Society of Chemistry 2025 |