
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBasic concepts and tools of artificial intelligence in polymer science
Khalid
Ferji

Lorraine university-CNRS Laboratoire de Chimie Physique Macromoleculaire (LCPM), France. E-mail: khalid.ferji@univ-lorraine.fr
First published on 28th April 2025
Abstract
In recent years, artificial intelligence (AI) has emerged as a transformative force across scientific disciplines, offering new ways to analyze data, predict material properties, and optimize processes. Yet, its integration into polymer science remains a challenge, as the field has traditionally relied on empirical methods and intuition-driven discovery. The complexity of polymer systems, combined with technical barriers and a lack of interdisciplinary training, has slowed AI adoption, leaving many researchers uncertain about where to begin. This perspective serves as an entry point for polymer scientists, introducing AI's real-world applications, accessible tools, and key challenges. Rather than an exhaustive review for specialists, it aims to lower entry barriers and spark interdisciplinary dialogue, bridging the gap between conventional polymer research and data-driven innovation. As AI reshapes material discovery, those who embrace this transformation today will define the future of polymer science.
 Khalid Ferji | Khalid Ferji is an associate professor in polymer chemistry at the Laboratoire de Chimie Physique Macromoléculaire (LCPM) – CNRS, Université de Lorraine, Nancy. His research focuses on the design and self-assembly of functional polymers, with an emerging specialization in integrating machine learning approaches into polymer science. With a multidisciplinary background, he promotes the development of accessible AI tools and fosters collaboration between experimentalists and data scientists to accelerate the digital transformation of materials discovery. |
1. Introduction
Imagine a scientific assistant that never sleeps—an intelligent system operating 24/7, continuously analyzing vast amounts of data, identifying research gaps, and pinpointing pressing industrial and societal needs. This assistant could suggest innovative polymers tailored for specific applications, recommend optimal synthesis pathways, predict degradation behavior, and propose strategies for enhanced recyclability and sustainability. But its role would go far beyond theoretical predictions. It could directly interact with an automated laboratory, executing real-time experiments and dynamically adjusting reaction parameters to optimize synthesis conditions. Such a system could self-adapt by learning from experimental feedback, iteratively refining reaction conditions to minimize material waste, enhance polymer properties, and accelerate discovery cycles. Practical tasks such as sourcing reagents, ensuring quality control, storing and organizing data, as well as maintaining experiment logs would be seamlessly managed.This vision, once considered science fiction, is now on the verge of becoming reality, made possible by Artificial Intelligence (AI).1,2 The concept of AI-driven “self-driving laboratories” is no longer speculative.3,4 The technologies required for seamless integration of AI, automation, and laboratory workflows are already emerging or actively under development.
AI refers to a broad set of computational techniques that enable machines to analyze data, recognize patterns, and make predictions beyond human capabilities. At the core of this revolution is Machine Learning (ML), a subset of AI that empowers computers to learn from data and refine predictions without explicit programming.5,6 ML has already revolutionized materials science and biology,7–11 as evidenced by DeepMind's AlphaFold, which solved the long-standing protein folding problem.12
The adoption of AI in polymer science has surged exponentially in recent years, as reflected in the increasing number of publications on the topic (Fig. 1). In this research field—where traditional trial-and-error methods struggle to navigate the immense combinatorial complexity, ML is unlocking new possibilities by predicting material properties, designing novel polymers, and optimizing synthesis conditions with unprecedented efficiency.13–18 Despite the rapid progress of AI in polymer science,13–19 significant challenges remain. Many researchers, while intrigued by AI's potential, find themselves overwhelmed by its complexity and the lack of clear entry points. How does AI truly work? What ML techniques are most relevant to polymer research? And how can these tools be effectively implemented?
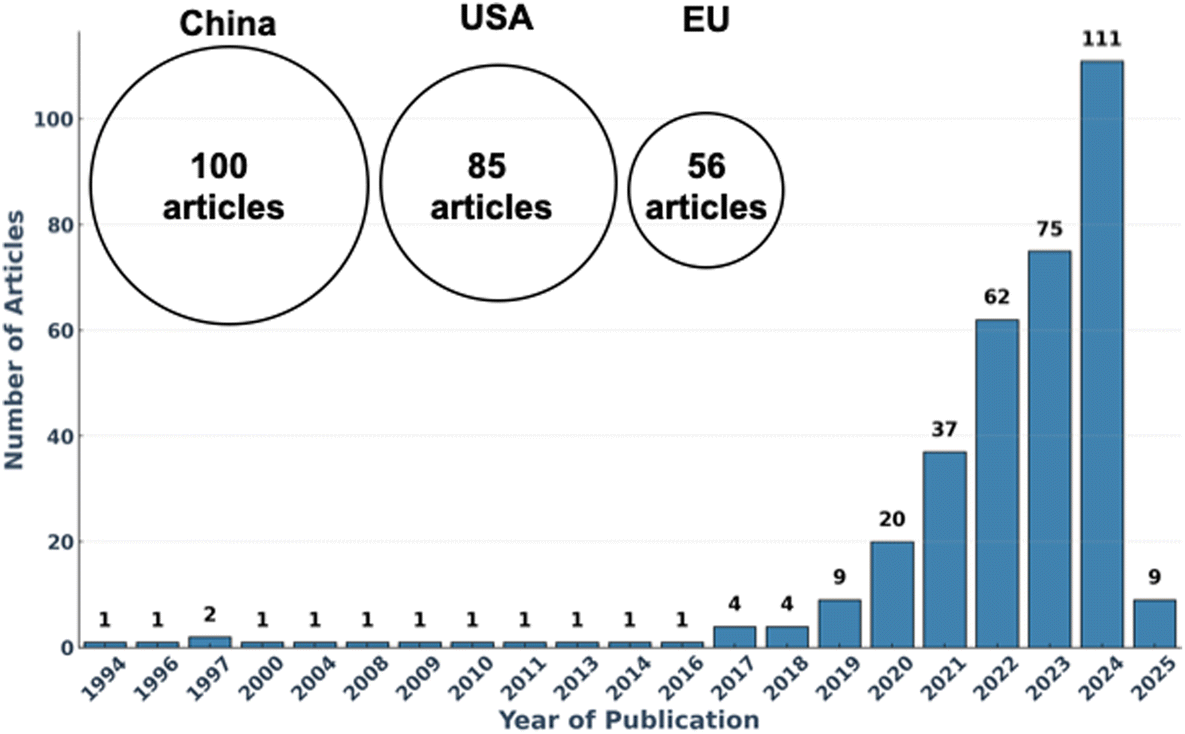 | ||
| Fig. 1 Number of publications related to AI in polymer science, extracted from Web of Science using the keywords (‘machine learning’ AND ‘polymer’) OR (‘artificial intelligence’ AND ‘polymer’) within the research areas: Materials Science, Polymer Science, and Chemistry. The geographical distribution highlights the leading contributors to this emerging field. EU: European union. | ||
This perspective seeks to bridge the gap between polymer science and AI by offering researchers a practical starting point. By focusing on key applications, foundational ML methodologies, and accessible tools, we aim to demystify AI and lower the barriers to its adoption. Rather than presenting a complete mastery of the subject, this work serves as a stepping stone—a first step in a learning journey that will require further exploration. For readers seeking deeper technical detail, several recent reviews provide complementary insights. For instance, the work by Aspuru-Guzik and coll.4 explores the integration of machine learning in self-driving laboratories, with a particular focus on Bayesian optimization, autonomous experiment loops, and decision-making algorithms for molecular and materials discovery. Meanwhile, Stenzel and coll.20 offer a polymer-focused perspective, addressing challenges in data curation, the translation of chemical structure into machine-readable descriptors, and the practical use of ML for property prediction, synthesis planning, and emerging biomedical applications. These resources are valuable for polymer chemists aiming to move beyond introductory concepts and explore more advanced or specialized AI-driven strategies.
2. AI as a new scientific paradigm in polymer science
As AI gains traction, its role may be misunderstood, especially for new users in fields like polymer science, where it could be easily confused with conventional modeling techniques or industrial automation. Researchers accustomed to traditional computational methods may struggle to differentiate AI from explicitly programmed simulations or automated control systems. This could lead to misconceptions about what truly defines AI and how it differs from other digital tools. As a result, many may assume that any computational system, from molecular simulations to factory sensors, qualifies as AI—a misunderstanding that could blur the true distinction between data-driven intelligence and traditional computing.21,22For example, a scientific calculator could be mistaken for AI because it performs complex calculations. However, it simply follows predefined mathematical rules and provides deterministic outputs—meaning the same input will always yield the same result. It does not learn from data, adapt to user behavior, or refine its responses over time. In contrast, an AI-powered system—such as an adaptive math assistant—could recognize handwritten equations, suggest alternative solutions, and improve its predictions based on past interactions. This distinction highlights a fundamental aspect of AI: it is not just about performing computations but about learning, adapting, and making independent decisions based on data.
Similarly, in industrial settings, the presence of sensors on a machine does not necessarily mean AI is at play. While automation and control systems follow pre-programmed instructions, AI must learn from data, identify patterns, and adapt to new conditions dynamically. True AI in polymer research extends beyond basic automation—it involves self-optimizing synthesis, predictive modeling, and data-driven material discovery.
Unlike molecular dynamics (MD)23 simulations or density functional theory (DFT),24 which rely on explicit physical equations to predict behaviors like phase transitions, chain conformations, or mechanical properties, AI offers an entirely new paradigm by extracting patterns directly from data. This enables accurate predictions even when the underlying physics is not fully understood.25 However, unlike traditional models, many AI techniques function as “black boxes”, making predictions based on complex statistical correlations rather than explicit physical laws.26 This lack of interpretability can lead to skepticism within the scientific community, as AI-generated results may be difficult to explain using physical principles.
For example, Bhattacharya and Patra27 demonstrated that AI could accurately predict polymer phase transitions, such as the coil-to-globule transition, while significantly reducing the computational cost compared to MD simulations. Rather than replacing these traditional methods, AI serves as a powerful complement, enhancing our ability to explore complex polymer systems efficiently.28 However, AI is not a magic solution—its accuracy depends directly on the quality and diversity of the data it is trained on. Poorly curated datasets can result in misleading predictions, ultimately limiting AI's reliability in real-world applications.
3. Key machine learning techniques
Machine Learning (ML) is a core subset of AI, designed to develop models that learn from data to make predictions or decisions without explicit programming.29–31 In polymer science, ML is increasingly leveraged to predict properties,32–45 optimize synthesis,46–51 and guide material discovery.52–57 This section introduces the key ML techniques used in polymer science, outlines their core principles, and provides selected examples of their applications. As summarized in Fig. 2, ML can be broadly categorized into three main classes: supervised learning (SL),58 unsupervised learning (UL),59 and reinforcement learning (RL).60 These approaches differ in how they process data, the level of human supervision required, and the types of problems they solve.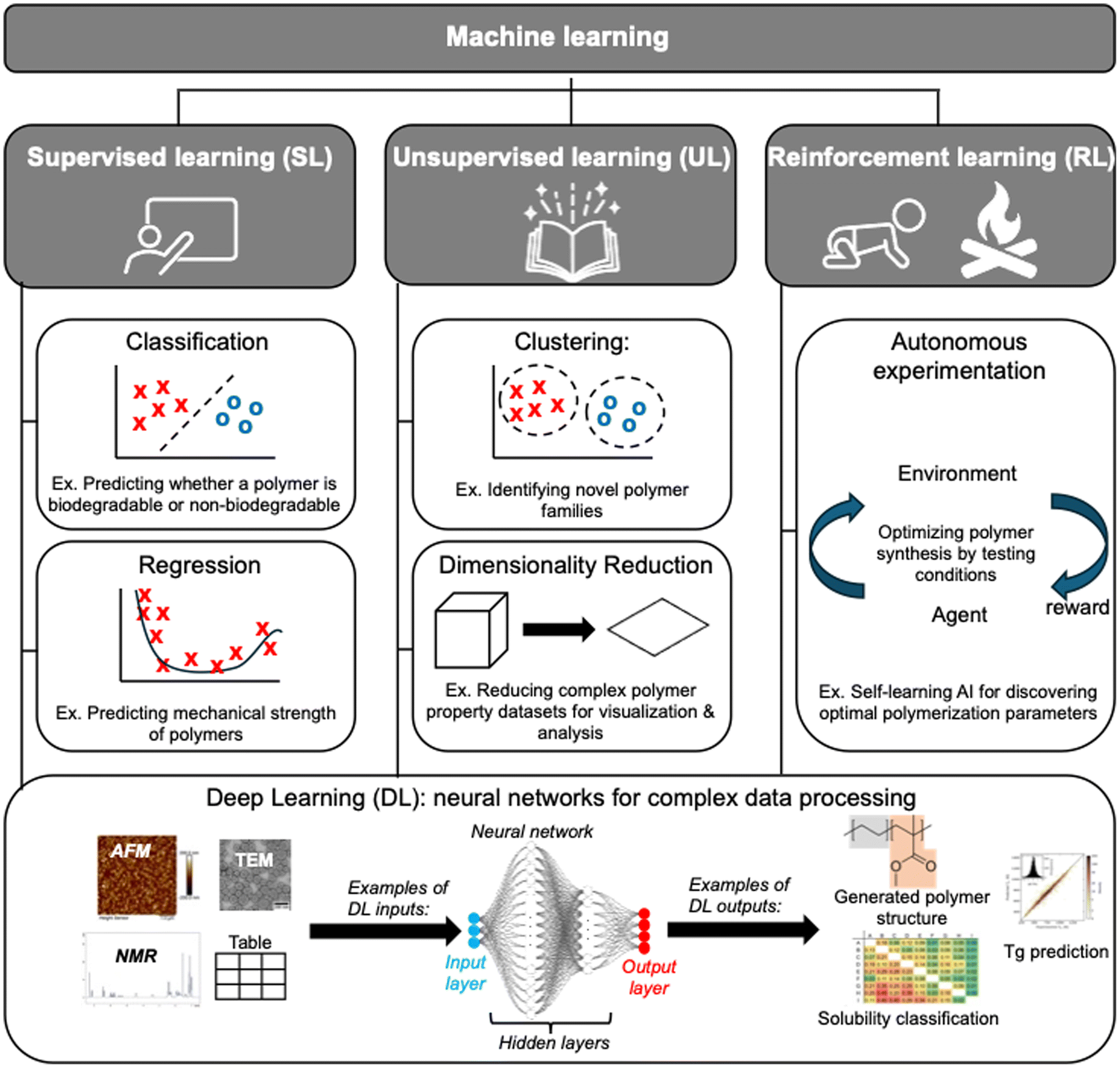 | ||
| Fig. 2 Overview of main machine learning methods and their applications in polymer science. Deep learning (DL) can be applied across all three categories (supervised, unsupervised, and reinforcement learning) to analyze complex polymer data, predict properties, and optimize synthesis. Example of experimental data sources used in ML driven polymer research include Atomic Force Microscopy (AFM), Transmission Electron Microscopy (TEM), and Nuclear Magnetic Resonance (NMR) spectroscopy. | ||
For those looking to quickly apply these methods, numerous detailed and practical resources are available.61–67 These guides introduce chemists to ML through hands-on examples, often requiring minimal setup, with pre-written code that can be downloaded and executed easily.
Before introducing each category and providing comprehensive examples, it is important to note that ML encompasses a broad range of algorithms. While deep learning (DL) has gained prominence, particularly for complex polymer datasets, traditional ML algorithms such as Random Forests,68 Support Vector Machines,69 Principal Component Analysis,70 and k-Means Clustering71 remain widely used in materials science. These methods are particularly effective for small datasets, structured tabular data, and explainable models, where interpretability is crucial. While a thorough exploration of their mathematical foundations and methodological workflows is beyond the scope of this work, readers can refer to authoritative resources for deeper insights.72,73
At its core, DL relies on neural networks (NN), a mathematical model inspired by the human brain.2,74 Like biological neurons, artificial neural networks consist of layers of interconnected neurons that process and learn from data (Fig. 2). The input layer receives raw data, which is then processed through hidden layers where patterns are identified, before reaching the output layer, which generates predictions or classifications. Each artificial neuron refines its parameters over time through training, improving the model's accuracy. One way to visualize this process is to think of a team of specialists solving a complex puzzle: the first layer gathers basic clues, the middle layers analyze deeper relationships between the clues, and the final layer makes an informed conclusion.
Several types of neural networks exist, each suited to specific tasks. Fully Connected Neural Networks (FCNNs)75 are commonly used for classification and regression in structured datasets. Convolutional Neural Networks (CNNs)76 excel at image processing by detecting spatial hierarchies of patterns. Recurrent Neural Networks (RNNs)77 and Long Short-Term Memory (LSTM)78 networks handle sequential data, making them ideal for time-series analysis and language modeling. Lastly, Deep Neural Networks (DNNs)79 with multiple hidden layers are used for highly complex, nonlinear problems. Given the complexity of polymer characterization and property prediction, DL is increasingly being integrated into polymer informatics workflows to enhance efficiency and accuracy.
3.1. Supervised learning
In supervised learning (SL), models learn from labeled datasets, where each input is associated with a known output. This approach is similar to traditional classroom teaching, where a teacher provides examples and corrections to guide the student's learning. However, unlike human learning, an SL model continuously evaluates its performance, adjusting its parameters iteratively until it reaches a high-performance threshold, ensuring reliable predictions.SL is used for two major types of tasks: classification and regression. In classification problems, the model predicts categorical outcomes, such as distinguishing between biodegradable and non-biodegradable polymers. By analyzing a dataset containing chemical structures and degradation properties, an SL model can learn patterns that enable it to predict the biodegradability of new polymers with high accuracy. In contrast, regression tasks involve predicting continuous values, such as the glass transition temperature (Tg) of a polymer. By identifying relationships between molecular structure and thermal properties, an SL model can estimate Tg for novel polymers, helping to accelerate materials discovery.
SL has been adopted in polymer science35–41,45–53,80–83 to address complex material challenges by leveraging large experimental datasets. One such application is in predicting polymer–solvent compatibility. Chandrasekaran et al.41 demonstrated a powerful application of SL to enhance polymer–solvent compatibility predictions. Their model was trained on a dataset of over 4500 polymers and 24 solvents, using experimental data that classified each polymer–solvent pair as either compatible (good solvent) or incompatible (non-solvent). As summarized in Fig. 3, the neural network model first converts the chemical structures of polymers and solvents into numerical descriptors that encode key molecular properties such as size, polarity, and functional groups. These descriptors are then compressed into a simplified mathematical representation (known as a latent space), where the neural network detects patterns that govern polymer–solvent interactions. Finally, the trained model predicts whether a new polymer–solvent pair will be compatible. This approach achieved an impressive 93% accuracy—significantly outperforming traditional heuristic methods such as the Hildebrand and Hansen solubility parameters. Such advancements are particularly valuable in plastics recycling, membrane science, and drug delivery, where selecting the appropriate solvent is essential for material processing and performance.
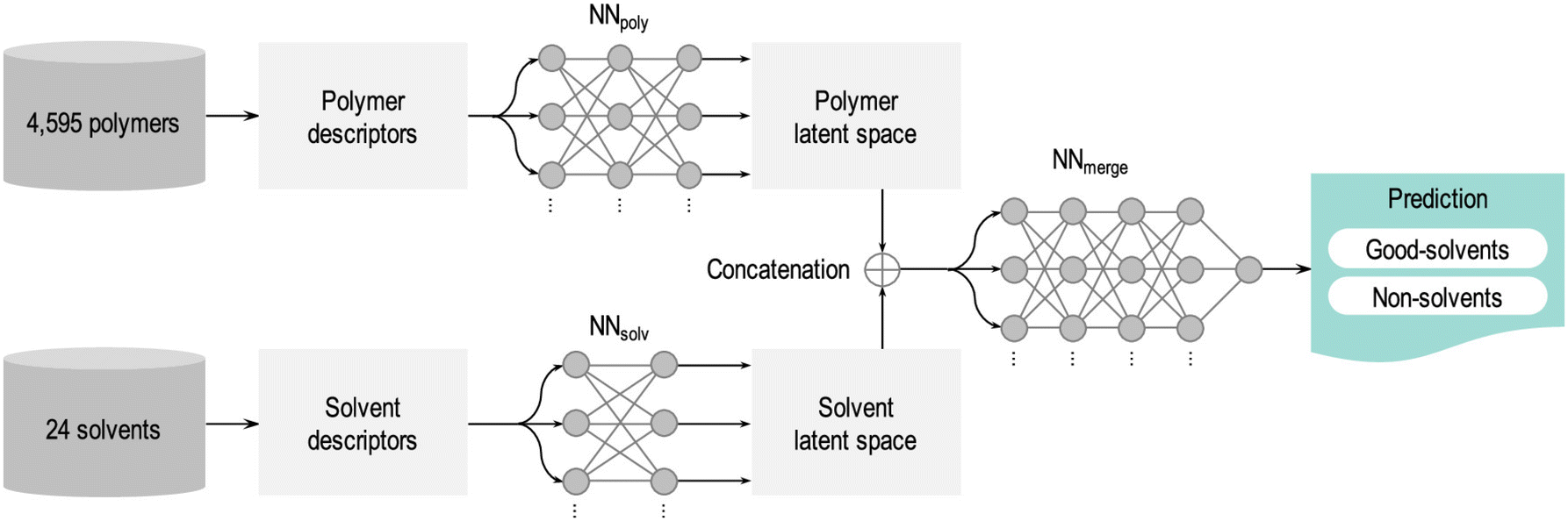 | ||
| Fig. 3 Machine learning workflow for predicting polymer–solvent compatibility. The trained neural network model processes polymer and solvent descriptors separately, transforming them into latent space representations before merging them for final classification. The model evaluates a given polymer structure against 24 solvents and predicts whether they act as good solvents or non-solvents based on learned compatibility patterns. Reproduced with permission from ref. 41 Copyright 2020, American Chemical Society. | ||
In another application, Lu et al.36 employed SL to predict phase behavior in polymerization induced self-assembly (PISA) using random forest models, a widely used decision tree-based algorithm for classification tasks. Their model was trained on a dataset of 592 experimental data points, where each entry was labeled with the experimentally observed morphology (e.g., spheres, worms, or vesicles). By analyzing key features such as monomer composition, polymerization conditions, and block ratio, the algorithm learned to classify new PISA systems with high accuracy. A key advantage of this approach is its interpretability, allowing researchers to identify which molecular parameters most influence phase transitions.
Building on this foundation, Fonseca Parra et al.37 employed DL framework to construct 3D pseudo-phase diagrams for block copolymers (Fig. 4). Their approach utilized a deep neural network trained on literature data to capture complex morphology transitions. Unlike traditional 2D phase diagrams that only consider a few experimental variables, their model incorporates multiple processing parameters simultaneously, offering a predictive understanding of phase behavior. The neural network learns nonlinear relationships between polymer composition, concentration, and self-assembly behavior, making it a more powerful tool for predicting morphologies that may not follow simple heuristic rules.
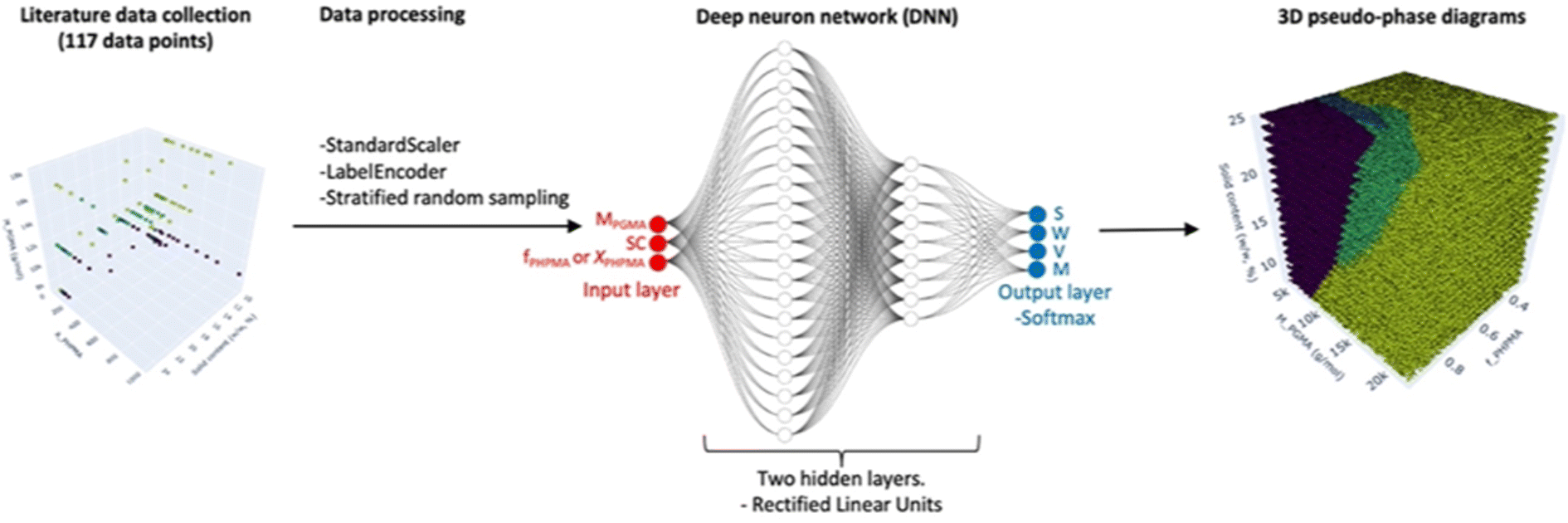 | ||
| Fig. 4 Deep learning workflow for predicting 3D pseudo-phase diagrams of copolymer self-assembly. Experimental data were collected from the literature and processed to ensure consistency before being used to train a deep neural network. The model classifies polymer compositions into different self-assembled morphologies—spheres (S), worms (W), or vesicles (V)—and generates high-resolution 3D pseudo-phase diagrams. Reproduced with permission from ref. 37 Copyright 2025, American Chemical Society. | ||
SL has been used to automate complex data analysis tasks, particularly in microscopy image processing. A significant challenge in polymer nanocomposite research is the precise localization and characterization of nanoparticles within polymer matrices, which is traditionally done manually or with labor-intensive image analysis techniques. To address this, Qu et al.82 developed a deep learning-based method to detect and quantify nanoparticles in transmission electron microscopy (TEM) images. Their approach, summarized in Fig. 5, involves a SL pipeline where a Convolutional Neural Networks (CNNs), a specific type of neural network, model is trained on labeled datasets of nanoparticle positions and sizes. The dataset consists of 72 TEM images, from which 279![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 057 labeled sub-images were extracted using an automated cropping and labeling method (DOPAD). Once trained, the model accurately predicts the positions and sizes of nanoparticles in new TEM images, significantly improving the speed and precision of nanoparticle characterization compared to manual methods. This technique enhances polymer nanocomposite analysis, facilitating research in advanced materials, coatings, and functional polymer-based nanotechnologies.
057 labeled sub-images were extracted using an automated cropping and labeling method (DOPAD). Once trained, the model accurately predicts the positions and sizes of nanoparticles in new TEM images, significantly improving the speed and precision of nanoparticle characterization compared to manual methods. This technique enhances polymer nanocomposite analysis, facilitating research in advanced materials, coatings, and functional polymer-based nanotechnologies.
 | ||
| Fig. 5 Supervised learning workflow for nanoparticle detection in polymer nanocomposites using a Convolutional Neural Network (CNN). A dataset of 72 TEM images was processed into 279057 labeled sub-images using automated labeling and cropping (DOPAD). The trained CNN model detects and localizes nanoparticles in new images, predicting their positions and sizes with high accuracy, thereby streamlining the characterization process. Reproduced with permission from ref. 82 Copyright 2021, American Chemical Society. | ||
It is important to differentiate between types of input data when designing supervised learning pipelines. While property prediction tasks (e.g., Tg, solubility) typically rely on structured chemical descriptors derived from SMILES or molecular fingerprints, image-based analyses (e.g., TEM, AFM) require entirely different approaches. These involve models such as CNNs or object detection architectures like YOLOv, which operate directly on pixel-level information. Each data modality presents unique challenges: image data often demands extensive annotation and data augmentation strategies, while descriptor-based models are sensitive to the choice and quality of input features. Recognizing and adapting to these differences is crucial for model performance and interpretability.
3.2. Unsupervised learning
Unsupervised learning (UL) is a powerful approach that identifies patterns in unlabeled data, meaning that no predefined outputs are available.59 Unlike supervised learning, which relies on explicit input–output pairs, UL models explore data autonomously to detect hidden structures, clusters, or relationships. In other words, it is like a student analyzing books independently to identify common themes without a teacher guiding them.This makes UL particularly valuable for understanding complex polymer datasets where experimental labels may be scarce or difficult to define. UL is particularly useful for clustering, where polymers with similar chemical properties or structural characteristics are grouped together, and for dimensionality reduction, which simplifies high-dimensional polymer datasets while preserving essential information.84,85
UL techniques have been successfully applied in polymer research to extract meaningful insights from complex datasets. Ziolek et al.55 used UL methods to investigate the nanoscale structure of micelles formed by four-arm and linear block copolymers. By clustering molecular conformations, they identified groups of micelle structures with similar corona arrangements, while dimensionality reduction helped simplify the complex structural variations. Their approach provided deeper insights into self-assembly mechanisms, which are crucial for drug delivery and biomaterials development.
Another interesting example is the work of Sutliff et al.,33 who applied UL to analyze near-infrared (NIR) spectra of polyolefins. NIR spectroscopy generates rich spectral data containing valuable chemical information, but interpreting this data manually is challenging due to its complexity. To simplify the analysis, the researchers used functional principal component analysis (fPCA), a mathematical technique, that transforms the original complex data into a smaller number of new variables called principal components. These components are calculated in such a way that they retain most of the variability present in the original data. In simpler terms, fPCA acts like a “compression” method that keeps the most important chemical signals while filtering out noise and redundancy. In this case, each spectrum was treated as a function across wavelengths, and fPCA identified common patterns (or “shapes”) across the spectra. This allowed the researchers to cluster the polyolefins based on similarities in their spectral fingerprints, without requiring prior labeling of the samples (Fig. 6). This dimensionality reduction not only made the dataset easier to visualize and interpret, but also highlighted meaningful groupings linked to polymer composition and structure. As a result, UL revealed chemical trends that would have been difficult to extract using traditional analysis methods.
 | ||
| Fig. 6 Workflow of the unsupervised learning (UL) approach applied to polyolefins using near-infrared (NIR) spectroscopy. (1) Raw NIR spectra of different polymer types: polypropylene (PP), low-density polyethylene (LDPE), linear low-density polyethylene (LLDPE), medium-density polyethylene (MDPE), high-density polyethylene (HDPE), and polypropylene-co-polyethylene (PP-co-PE). (2) Functional principal component analysis (fPCA) reduces the spectral data into a low-dimensional space, clustering samples based on spectral similarities. (3) The extracted principal components correlate with crystallinity, demonstrating how UL can reveal hidden relationships in polymer data without predefined labels. Reproduced with permission from ref. 33 Copyright 2024, American Chemical Society. | ||
3.3. Reinforcement learning and closed-loop optimization
Reinforcement Learning (RL) is a distinct category of machine learning in which models learn by interacting with an environment and receiving rewards for taking optimal actions.60 Unlike SL, where models are trained on labeled datasets, RL algorithms discover optimal strategies through trial and error, making them particularly suited for tasks requiring sequential decision-making. A useful analogy is that of a child learning that fire is dangerous only after touching it—the knowledge is gained through direct experience rather than prior instruction.Compared to supervised and unsupervised learning, RL is significantly more complex as it involves sequential decision-making, long-term reward optimization, and an exploration–exploitation trade-off. Unlike models that learn from static datasets, RL dynamically adjusts strategies based on continuous feedback, requiring extensive computational resources and advanced algorithms. These properties make RL a powerful tool for optimizing polymerization processes and autonomous experimental control, but they also contribute to its greater mathematical and implementation complexity.3,48,86,87
Li et al.87 developed a strategy to regulate the molecular weight distribution (MWD) in atom transfer radical polymerization (ATRP). Instead of relying on predefined reaction protocols, their model learns dynamically by interacting with the polymerization process. As illustrated in Fig. 7, the system follows a classic RL framework, where the reactor acts as the environment, and the AI agent (policy network and value network) selects reagent addition strategies based on observed reaction states (e.g., monomer and initiator concentrations). The model continuously compares the current MWD to the target distribution (e.g., Gaussian or bimodal profiles) and updates its decision-making policy based on rewards received for achieving optimal polymer properties. By iteratively refining reagent addition, the RL-based system optimizes ATRP conditions in real time, improving precision in molecular weight control and enabling the design of custom polymer architectures with minimal experimental trials.
 | ||
| Fig. 7 Reinforcement learning framework for optimizing molecular weight distribution (MWD) in atom transfer radical polymerization (ATRP). The AI agent observes the reaction state, selects actions (adjusting reagent addition), and updates its strategy based on real-time feedback and reward evaluation, iteratively improving polymerization outcomes. Reproduced with permission from ref. 87 Copyright 2018, Royal Society of Chemistry. | ||
While RL holds great promise, its application in polymer science remains limited by several factors. RL typically requires either extensive real-time experimentation or high-fidelity simulation environments, both of which are resource-intensive. Moreover, defining suitable reward functions and action spaces for polymer systems can be non-trivial. As such, RL may be best suited for narrowly defined problems (e.g., optimizing a specific polymerization protocol) rather than broad exploratory tasks. Hybrid strategies that combine RL with Bayesian optimization (BO)88 or SL may offer more practical solutions in the near term. A recent example by Pittaway et al.89 illustrates how such hybrid strategies can be implemented in practice, combining multi-objective BO with real-time analytical feedback (DLS) to enable closed-loop self-optimization of emulsion polymerization in a continuous-flow reactor platform.
Warren et al.48 developed an AI-driven closed-loop polymerization system to optimize reversible-addition fragmentation chain transfer (RAFT) polymerization conditions, achieving targeted molecular weight and dispersity with minimal experimental trials. Their approach (Fig. 8) integrates real-time experimental feedback with BO, specifically the Thompson Sampling Efficient Multi-Objective Optimization (TSEMO) algorithm. The system iteratively tests reaction conditions, evaluates the results, and refines its strategy based on real-time feedback from nuclear magnetic resonance (NMR) and gel permeation chromatography (GPC). Instead of relying on predefined datasets, the platform learns from its own experiments, systematically adjusting temperature and reaction time to maximize monomer conversion while minimizing dispersity. To make informed decisions, it builds a predictive model that estimates the outcome of untested reaction conditions, and uses this model to select the most informative next experiments. The algorithm balances exploration (testing uncertain regions of the parameter space) and exploitation (focusing on promising conditions), enabling efficient optimization across multiple objectives.
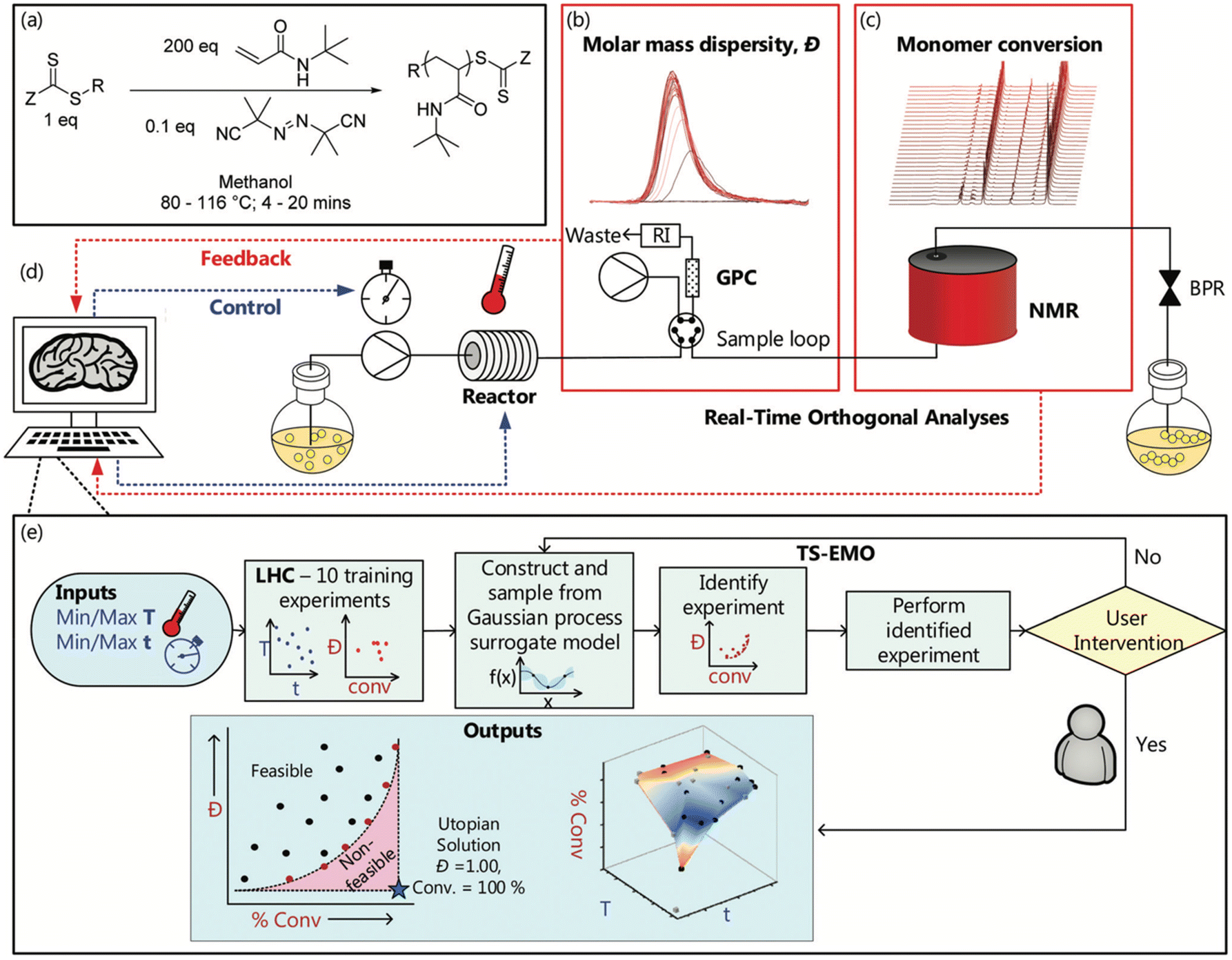 | ||
| Fig. 8 AI-guided closed-loop optimization of reversible addition–fragmentation chain transfer (RAFT) polymerization using Bayesian optimization. The system integrates real-time feedback from nuclear magnetic resonance (NMR) and gel permeation chromatography (GPC) to dynamically adjust reaction parameters such as temperature and time, optimizing monomer conversion and controlling molar mass dispersity (Đ). The panels show (a) a generalized scheme for the RAFT synthesis platform, (b) representative GPC chromatograms, (c) 1H NMR spectra, (d) a schematic of the automated platform, and (e) an overview of the structure of the Thompson-sampling efficient multi-objective optimisation (TSEMO) algorithm-based experiments. Reproduced with permission from ref. 48 Copyright 2022, Royal Society of Chemistry. | ||
Despite not being a pure RL system, the work by Warren et al. compellingly demonstrates how autonomous experimentation and adaptive optimization can be applied to complex polymer synthesis challenges. This approach lays the groundwork for semi-autonomous, self-learning platforms that reduce human workload and enable more precise control over polymerization processes. It represents a significant step forward toward fully integrated AI-driven material discovery.
Through these simplified examples, we have demonstrated the diverse potential of ML in polymer science, from predicting polymer properties to autonomously optimizing synthesis conditions. Each ML technique—supervised, unsupervised and reinforcement learning—offers distinct capabilities, whether for making accurate property predictions, uncovering hidden patterns, or enabling self-learning experimental workflows. These methods differ in learning process, computational complexity, and scope of application. To provide a structured comparison, Table 1 summarizes the key characteristics of each ML approach, highlighting their data requirements, optimization strategies, and relevance to polymer research.
| Feature | Supervised learning (SL) | Unsupervised learning (UL) | Reinforcement learning (RL) |
|---|---|---|---|
| Data type | Labeled data (input–output pairs) | Unlabeled data (finding patterns) | No predefined labels, learns from interaction |
| Goal | Predict outputs (classification/regression) | Cluster/group similar data or reduce dimensions | Learn a sequence of actions to maximize rewards |
| Learning process | Learns from explicit examples | Identifies hidden structures autonomously | Learns by trial & error via environment feedback |
| Optimization focus | Minimize loss (error) | Find clusters, patterns, representations | Maximize long-term rewards |
| Computational complexity | Moderate | Moderate to high | Very high (complex decision-making) |
While machine learning offers powerful tools to accelerate discovery and optimize polymer systems, it is important to emphasize that it is not always the most effective or appropriate solution. In certain contexts, especially when the system is well-characterized or the design space is limited, simpler programmatic screening approaches may outperform more sophisticated ML-based optimization methods. As such, comparative benchmarking and critical method selection should remain integral to any data-driven strategy in polymer science.
4. Real-world ML tools
To facilitate the integration of ML in polymer science, numerous AI tools and platforms are available to support data management, analysis, and modeling. Table 2 organizes these resources by functionality, from no-code ML platforms and execution environments to data handling libraries, visualization tools, cheminformatics toolkits, and polymer-specific repositories. Open-source tools play a central role in this ecosystem, fostering transparency, reproducibility, and accessibility, empowering a broader scientific community to engage in AI-driven materials discovery.| Category | Tools/platforms (access) | Functionality |
|---|---|---|
| No-code/low-code ML platforms | Teachable machine (FT), Weka (OS), KNIME (OS), Google AutoML (P), Azure ML (P) | ML without coding via graphical interfaces; ideal for classification, clustering, basic workflows. |
| Programming and execution environments | Google Colab (FT), Jupyter Notebooks (OS), Anaconda (FT), Python (OS) | Interactive coding, script execution, environment management for data science workflows |
| Data manipulation & preprocessing | Numpy (OS), Pandas (OS) | Efficient handling of arrays, tables, and structured experimental data |
| Data visualization | Matplotlib (OS), Seaborn (OS) | Graphical representation of data and model outputs for analysis and communication |
| Machine learning libraries | Scikit-learn (OS), TensorFlow (OS), PyTorch (OS) | Libraries for classical machine learning and deep learning: regression, classification, neural networks |
| Chemical representation & descriptors | SMILES, BigSMILES (OS), RDKit (OS) | Encoding of molecular/polymeric structures and generation of chemical descriptors |
| Polymer data repositories | Polymer Genome (FT), PoLyInfo, PI1M, CROW, NIST DB (R/FT), Materials Project (FT) | Databases of experimental and computational polymer properties |
| Collaboration & sharing platforms | GitHub (OS), Zenodo (OS), Hugging Face (OS), Figshare (OS) | Hosting of code, datasets, and trained models; support for version control and DOI-based citation |
For researchers new to ML, Python has become the primary programming language due to its simplicity, flexibility, and extensive ecosystem of scientific libraries. User-friendly platforms like Google Colab and Jupyter Notebooks provide interactive coding environments, allowing researchers to write and execute Python code without requiring advanced computational resources or complex installations. These tools facilitate key tasks such as loading datasets, cleaning and preprocessing data, as well as applying ML models. Open-source libraries such as Pandas and Numpy streamline data handling and numerical processing, while visualization libraries such as Matplotlib and Seaborn enable researchers to generate high-quality scientific graphs and complex data visualizations.
For researchers who prefer minimal coding, no-code or low-code platforms provide an alternative entry point. KNIME, for instance, offers a drag-and-drop interface for building ML workflows, making it possible to preprocess data, train models, and evaluate predictions without writing code. Similarly, Teachable Machine by Google simplifies classification tasks, while platforms like Google AutoML and Azure ML enable researchers to train custom models through intuitive web interfaces.
A significant application of ML in polymer science is the processing of molecular representations using cheminformatics tools. RDKit converts chemical structures into machine-readable formats, such as SMILES strings or molecular fingerprints, which serve as inputs for ML models. BigSMILES90 extends this functionality to stochastic polymers, allowing for the representation of structural variations in polymer chains. Meanwhile, Polymer Genome offers pre-trained models for polymer property prediction, facilitating rapid screening of polymer candidates based on molecular descriptors.
Navigating and analyzing large polymer datasets is another common challenge that ML tools effectively address. For example, using Python's Pandas library, a researcher can filter polymers based on molecular weight, calculate property correlations, or generate statistical insights within seconds—tasks that would be time-consuming with traditional tools like Excel. These workflows accelerate analysis, improve reproducibility, and enhance data-driven decision-making.
With the growing accessibility of open-source libraries, user-friendly platforms, and pre-trained ML models, integrating ML into polymer research has never been more feasible. Researchers can start with beginner-friendly tools such as Scikit-learn for predictive modeling or KNIME for workflow automation, progressively expanding their expertise into deep learning frameworks like TensorFlow and PyTorch as needed.
5. Challenges and considerations
While AI holds great promise for transforming polymer science, its integration into the field requires overcoming several key challenges. The growing number of AI-driven studies (Fig. 1) reflects increasing interest, particularly in machine learning techniques. However, despite this surge in research, significant barriers still hinder widespread ML adoption in experimental and industrial settings.These challenges stem from data availability and quality issues, the learning curve for polymer scientists, computational constraints, and the lack of standardized frameworks for integrating ML into polymer research. Addressing these obstacles is essential to ensuring that ML evolves from a promising concept into an accessible, widely used tool. The following sections outline key hurdles and potential solutions to bridge the gap between AI potential and real-world implementation in polymer science.
5.1. Data resources: availability, accessibility, and challenges
The integration of ML into polymer science relies heavily on the availability of structured, high-quality datasets and collaborative coding platforms. Several initiatives (Table 2) have been developed to support researchers by providing curated databases, machine-readable polymer representations, and repositories for sharing ML models. These resources enable scientists to train and fine-tune ML models effectively, accelerating both material discovery and AI-driven innovation. Among these, Polymer Genome offers ML-driven polymer property predictions, Materials Project includes computationally derived polymer-related data, and the NIST Polymer Database compiles experimentally validated polymer properties, serving as a benchmark for AI applications. Other domain-specific resources such as PoLyInfo and PI1M, also offer structured datasets of polymer structures and properties, though often with limited interoperability.Although these platforms are growing in number, most available datasets in polymer science still fall into the “small data” category—typically comprising dozens to hundreds of entries, often collected manually or extracted from literature. This contrasts sharply with big data contexts and limits the scope and robustness of ML models, particularly for deep learning applications. Addressing this issue requires both community-driven data generation and improved access to standardized, high-volume datasets.
Beyond polymer-specific databases, various general platforms facilitate collaborative coding, AI model sharing, and data accessibility, which can be leveraged by the polymer science community (Table 2). These platforms not only facilitate interdisciplinary collaboration but also serve as prototypes for developing specialized equivalents tailored to polymer research. Hugging Face is widely recognized for its repository of pre-trained ML models, including polymer-specific tools, while Zenodo serves as an open-access repository for structured datasets and ML models, ensuring proper attribution through Digital Object Identifiers (DOIs). Meanwhile, GitHub remains an essential platform for collaborative coding, dataset hosting, and version-controlled AI workflows, enhancing transparency and reproducibility.
Despite the increasing availability of these resources, significant challenges persist in data standardization and accessibility. Many studies still suffer from fragmented, inconsistent, or inaccessible datasets, often lacking sufficient metadata or omitting critical details about synthesis conditions, characterization techniques, and experimental outcomes. Without standardized data-sharing protocols, polymer science risks falling behind disciplines such as biology and materials science, where open data practices have already enabled rapid AI and ML adoption. Scientific journals and funding agencies should take an active role in addressing this issue by mandating structured dataset publication alongside research articles to enhance reproducibility and accessibility. Establishing community-wide norms for data collection, annotation, and dissemination is essential for creating interoperable datasets that serve as a foundation for ML-driven polymer research.
To move from raw data to ML-ready datasets, researchers are encouraged to consider the following workflow: (i) standardize chemical representation (e.g., using SMILES or BigSMILES), (ii) enrich datasets with metadata (synthesis conditions, characterization techniques), (iii) perform basic data cleaning (handling missing values, duplicates), and (4) publish structured datasets via open platforms such as Zenodo, GitHub, or the Polymer Genome repository. Ensuring datasets are machine-readable (CSV, JSON, HDF5) and version-controlled is essential for reproducibility. Additionally, researchers are encouraged not only to share datasets but also to publish their ML workflows, and if possible pre-trained models to foster transparency and collaboration. Open-source initiatives and collaborative coding environments have the potential to reduce redundancy, improve model accuracy, and create a shared knowledge base that benefits the entire field. Whenever applicable, both data and code should comply with the FAIR principles (Findable, Accessible, Interoperable, and Reusable). By moving toward a more open and collaborative research culture, the polymer community can fully harness ML's potential, ensuring that data is widely available, standardized, and effectively utilized for accelerating material discovery and polymer informatics.
5.2. Educational gaps in polymer science: the need for interdisciplinarity
The adoption of AI in polymer science represents a fundamental shift for many researchers accustomed to empirical methods or traditional computational approaches. While short-term collaborations between polymer scientists and AI experts help bridge this gap, the long-term solution lies in integrating AI and ML education into polymer science curricula. Given the specialized nature of polymer science and its experimental nuances, teaching AI and ML to polymer researchers is often more practical than training computer scientists in polymer chemistry and engineering.Despite the growing impact of AI on materials research, structured AI education within polymer science curricula remains scarce. Few master's programs offer specialized training that integrates polymer science and data-driven approaches, limiting the number of researchers capable of advancing ML-driven polymer research. This educational gap not only slows academic progress but also affects the polymer industry, where demand for interdisciplinary expertise is increasing.
Several industrial leaders have already integrated AI-driven strategies into their research and development efforts. BASF has invested in AI for materials discovery, Dow Chemical is exploring ML for process optimization, Covestro is leveraging AI for sustainable polymer design, and Arkema has initiated AI-based material innovation programs. However, the full potential of AI in the polymer industry remains underutilized, largely due to the limited availability of professionals who can bridge the gap between data science and polymer engineering.
To close this gap, universities should introduce ML, data science, and AI courses specifically tailored to polymer science applications. Early exposure to AI tools and computational methods will enable future polymer researchers to integrate these techniques into their workflows with confidence. Additionally, workshops, summer schools, and online training programs should be expanded to provide current researchers and industry professionals with foundational ML and AI skills. These initiatives will ensure that AI adoption in polymer science is not limited to a small group of interdisciplinary experts but becomes a standard component of both academic and industrial education.
5.3. Computational costs
Integrating AI into polymer research requires substantial computational power, particularly for deep learning and other data-intensive techniques. Training large neural networks or analyzing high-dimensional datasets from molecular simulations or spectroscopy can be highly resource-intensive, making access to high-performance computing (HPC) infrastructure a limiting factor for many academic and industrial laboratories.To address these challenges, government-led initiatives worldwide provide researchers with access to advanced computing facilities:
6. Future outlook
The convergence of AI and polymer science is unlocking new possibilities, from predictive modeling to autonomous research workflows. As AI-driven methodologies mature, the next frontier will not only be about discovering novel macromolecules but about designing the algorithms that lead to their discovery. Moving forward, reinforcement learning and unsupervised learning are expected to play a larger role in self-optimizing polymerization processes and unraveling complex structure–property relationships. Instead of merely accelerating workflows, AI will fundamentally transform how polymer research is conducted, shifting from empirical iteration to data-driven hypothesis generation.AI-driven autonomous laboratories will continue evolving, but the true revolution lies in their decision-making capabilities rather than just automation. With advances in robotic synthesis, in situ characterization, and self-learning models, these labs will refine materials in real time, dynamically adjusting synthesis conditions to optimize properties with minimal human intervention. This shift will redefine the role of polymer scientists, moving from experiment control to precision-driven material design.
Beyond discovery, AI's role in sustainable polymer development will be crucial. By enabling rational design of biodegradable polymers, AI-assisted recycling strategies, and energy-efficient synthesis, AI can help drive the transition toward a circular polymer economy.
7. Conclusion
AI is no longer just an emerging tool in polymer science—it is a strategic necessity for accelerating discovery, optimizing materials, and enabling sustainable innovation. However, AI's full potential will only be realized if polymer scientists actively engage in its development rather than relying on AI specialists to drive innovation.This perspective serves as an introduction to AI's capabilities in polymer research, aiming to motivate scientists to explore machine learning techniques as a complement to traditional approaches. To ensure AI benefits the broader polymer community, universities, research institutions, and industries must accelerate efforts to incorporate AI training, develop standardized polymer datasets, and promote interdisciplinary collaboration. The barriers to AI adoption are real—but they are not insurmountable. Each step taken today—whether running a first ML model, sharing a dataset, or integrating AI-driven synthesis—contributes to shaping polymer science as an AI-augmented discipline. As AI-driven laboratories take over routine experimentation, polymer scientists will transition from the era of control to one of fine precision, where AI refines synthesis conditions beyond human intuition.
And if tomorrow's revolution in polymer science is no longer about the macromolecule itself, but about the algorithm that led to its discovery?
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflits to declare.References
- S. J. Russell and P. Norvig, Artificial intelligence: a modern approach, Pearson, 2016 Search PubMed.
- Y. LeCun, Y. Bengio and G. Hinton, Nature, 2015, 521, 436–444 CrossRef PubMed.
- P. A. Beaucage, D. R. Sutherland and T. B. Martin, Macromolecules, 2024, 57, 8661–8670 CrossRef.
- G. Tom, S. P. Schmid, S. G. Baird, Y. Cao, K. Darvish, H. Hao, S. Lo, S. Pablo-García, E. M. Rajaonson, M. Skreta, N. Yoshikawa, S. Corapi, G. D. Akkoc, F. Strieth-Kalthoff, M. Seifrid and A. Aspuru-Guzik, Chem. Rev., 2024, 124, 9633–9732 CrossRef.
- A. L. Samuel, IBM J. Res. Dev., 1959, 3, 210–229 Search PubMed.
- V. Vapnik, The nature of statistical learning theory, Springer science & business media, 2013 Search PubMed.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef.
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah, M. Spitzer and S. R. Zhao, Nat. Rev. Drug Discovery, 2019, 18, 463–477 CrossRef.
- R. Ramprasad, R. Batra, G. Pilania, A. Mannodi-Kanakkithodi and C. Kim, npj Comput. Mater., 2017, 3, 54 CrossRef.
- A. K. Cheetham and R. Seshadri, Chem. Mater., 2024, 36, 3490–3495 CrossRef CAS.
- A. Rajkomar, J. Dean and I. Kohane, N. Engl. J. Med., 2019, 380, 1347–1358 CrossRef PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- L. Chen, G. Pilania, R. Batra, T. D. Huan, C. Kim, C. Kuenneth and R. Ramprasad, Mater. Sci. Eng., R, 2021, 144, 100595 CrossRef.
- G. Ignacz, L. Bader, A. K. Beke, Y. Ghunaim, T. Shastry, H. Vovusha, M. R. Carbone, B. Ghanem and G. Szekely, J. Membr. Sci., 2025, 713, 123256 CrossRef.
- A. J. Gormley and M. A. Webb, Nat. Rev. Mater., 2021, 6, 642–644 CrossRef CAS.
- W. Sha, Y. Li, S. Tang, J. Tian, Y. Zhao, Y. Guo, W. Zhang, X. Zhang, S. Lu, Y.-C. Cao and S. Cheng, InfoMat, 2021, 3, 353–361 CrossRef.
- T. B. Martin and D. J. Audus, ACS Polym. Au, 2023, 3, 239–258 CrossRef.
- J. Wang, K. Tian, D. Li, M. Chen, X. Feng, Y. Zhang, Y. Wang and B. Van der Bruggen, Sep. Purif. Technol., 2023, 313, 123493 CrossRef.
- Y. K. Zhao, R. J. Mulder, S. Houshyar and T. C. Le, Polym. Chem., 2023, 14, 3325–3346 RSC.
- W. Ge, R. De Silva, Y. Fan, S. A. Sisson and M. H. Stenzel, Adv. Mater., 2025, 37, 2413695 CrossRef.
- P. V. Coveney and R. Highfield, J. Chem. Inf. Model., 2024, 64, 5739–5741 CrossRef.
- A. Bewersdorff, X. Zhai, J. Roberts and C. Nerdel, Comput. Educ.: Artif. Intell., 2023, 4, 100143 Search PubMed.
- S. A. Hollingsworth and R. O. Dror, Neuron, 2018, 99, 1129–1143 CrossRef PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- M. Krenn, R. Pollice, S. Y. Guo, M. Aldeghi, A. Cervera-Lierta, P. Friederich, G. dos Passos Gomes, F. Häse, A. Jinich, A. Nigam, Z. Yao and A. Aspuru-Guzik, Nat. Rev. Phys., 2022, 4, 761–769 CrossRef PubMed.
- V. Hassija, V. Chamola, A. Mahapatra, A. Singal, D. Goel, K. Huang, S. Scardapane, I. Spinelli, M. Mahmud and A. Hussain, Cognit. Comput., 2024, 16, 45–74 CrossRef.
- D. Bhattacharya and T. K. Patra, Macromolecules, 2021, 54, 3065–3074 CrossRef.
- Y. Wang, J. M. Lamim Ribeiro and P. Tiwary, Curr. Opin. Struct. Biol., 2020, 61, 139–145 CrossRef.
- M. I. Jordan and T. M. Mitchell, Science, 2015, 349, 255–260 CrossRef.
- P. Domingos, Commun. ACM, 2012, 55, 78–87 CrossRef.
- L. Zdeborová, Nat. Phys., 2017, 13, 420–421 Search PubMed.
- Y. Aoki, S. Wu, T. Tsurimoto, Y. Hayashi, S. Minami, O. Tadamichi, K. Shiratori and R. Yoshida, Macromolecules, 2023, 56, 5446–5456 CrossRef.
- B. P. Sutliff, S. Goyal, T. B. Martin, P. A. Beaucage, D. J. Audus and S. V. Orski, Macromolecules, 2024, 57, 2329–2338 CrossRef CAS.
- C. Kuenneth, W. Schertzer and R. Ramprasad, Macromolecules, 2021, 54, 5957–5961 CrossRef CAS.
- S. Wu, Y. Kondo, M.-a. Kakimoto, B. Yang, H. Yamada, I. Kuwajima, G. Lambard, K. Hongo, Y. Xu, J. Shiomi, C. Schick, J. Morikawa and R. Yoshida, npj Comput. Mater., 2019, 5, 66 CrossRef.
- Y. Lu, D. Yalcin, P. J. Pigram, L. D. Blackman and M. Boley, J. Chem. Inf. Model., 2023, 63, 3288–3306 CrossRef CAS PubMed.
- E. P. Fonseca Parra, J. Oumerri, A. A. Arteni, J.-L. Six, S. P. Armes and K. Ferji, Macromolecules, 2025, 58, 61–73 CrossRef CAS.
- B. Ma, N. J. Finan, D. Jany, M. E. Deagen, L. S. Schadler and L. C. Brinson, Macromolecules, 2023, 56, 3945–3953 CrossRef CAS PubMed.
- B. Rajabifar, G. F. Meyers, R. Wagner and A. Raman, Macromolecules, 2022, 55, 8731–8740 CrossRef CAS.
- Y. Zhou, J. Wen and Y. Nie, Macromolecules, 2024, 57, 3258–3270 CrossRef CAS.
- A. Chandrasekaran, C. Kim, S. Venkatram and R. Ramprasad, Macromolecules, 2020, 53, 4764–4769 CrossRef CAS.
- A. Bera, T. S. Akash, R. Ishraaq, T. H. Pial and S. Das, Macromolecules, 2024, 57, 1581–1592 CrossRef CAS.
- L. A. Miccio and G. A. Schwartz, Macromolecules, 2021, 54, 1811–1817 CrossRef CAS.
- L. Schneider and J. J. de Pablo, Macromolecules, 2021, 54, 10074–10085 CrossRef CAS.
- Y. Zhang and X. J. Xu, Polym. Chem., 2021, 12, 843–851 RSC.
- N. H. Park, D. Y. Zubarev, J. L. Hedrick, V. Kiyek, C. Corbet and S. Lottier, Macromolecules, 2020, 53, 10847–10854 CrossRef CAS.
- B. K. Wheatle, E. F. Fuentes, N. A. Lynd and V. Ganesan, Macromolecules, 2020, 53, 9449–9459 CrossRef CAS.
- S. T. Knox, S. J. Parkinson, C. Y. P. Wilding, R. A. Bourne and N. J. Warren, Polym. Chem., 2022, 13, 1576–1585 RSC.
- J. M. Bone, C. M. Childs, A. Menon, B. Póczos, A. W. Feinberg, P. R. LeDuc and N. R. Washburn, ACS Biomater. Sci. Eng., 2020, 6, 7021–7031 CrossRef CAS.
- T. Zhou, D. Qiu, Z. Wu, S. A. N. Alberti, S. Bag, J. Schneider, J. Meyer, J. A. Gámez, M. Gieler, M. Reithmeier, A. Seidel and F. Müller-Plathe, Macromolecules, 2022, 55, 7893–7907 CrossRef CAS.
- M. Meleties, D. Britton, P. Katyal, B. Lin, R. L. Martineau, M. K. Gupta and J. K. Montclare, Macromolecules, 2022, 55, 1239–1247 CrossRef CAS.
- A. Khajeh, D. Schweigert, S. B. Torrisi, L. Hung, B. D. Storey and H.-K. Kwon, Macromolecules, 2023, 56, 4787–4799 CrossRef CAS.
- J. Shi, N. J. Rebello, D. Walsh, W. Zou, M. E. Deagen, B. S. Leao, D. J. Audus and B. D. Olsen, Macromolecules, 2023, 56, 7344–7357 CrossRef CAS.
- A. N. Wilson, P. C. St John, D. H. Marin, C. B. Hoyt, E. G. Rognerud, M. R. Nimlos, R. M. Cywar, N. A. Rorrer, K. M. Shebek, L. J. Broadbelt, G. T. Beckham and M. F. Crowley, Macromolecules, 2023, 56, 8547–8557 CrossRef CAS PubMed.
- R. M. Ziolek, P. Smith, D. L. Pink, C. A. Dreiss and C. D. Lorenz, Macromolecules, 2021, 54, 3755–3768 CrossRef CAS.
- A. Braghetto, S. Kundu, M. Baiesi and E. Orlandini, Macromolecules, 2023, 56, 2899–2909 CrossRef CAS.
- M. A. Webb, N. E. Jackson, P. S. Gil and J. J. de Pablo, Sci. Adv., 2020, 6, eabc6216 CrossRef PubMed.
- Z. Y. Ren, S. H. Wang and Y. D. Zhang, CAAI Trans. Intell. Technol., 2023, 8, 549–580 CrossRef.
- A. Glielmo, B. E. Husic, A. Rodriguez, C. Clementi, F. Noé and A. Laio, Chem. Rev., 2021, 121, 9722–9758 CrossRef CAS.
- M. Botvinick, S. Ritter, J. X. Wang, Z. Kurth-Nelson, C. Blundell and D. Hassabis, Trends Cognit. Sci., 2019, 23, 408–422 CrossRef PubMed.
- S. T. Cahill, J. E. B. Young, M. Howe, R. Clark, A. F. Worrall and M. I. Stewart, J. Chem. Educ., 2024, 101, 2925–2932 CrossRef CAS.
- D. Lafuente, B. Cohen, G. Fiorini, A. A. García, M. Bringas, E. Morzan and D. Onna, J. Chem. Educ., 2021, 98, 2892–2898 CrossRef CAS.
- R. Qiu, Z. Lin, Z. Yang and L. Gao, J. Chem. Educ., 2024, 101, 328–336 CrossRef CAS.
- E. S. Thrall, S. E. Lee, J. Schrier and Y. Zhao, J. Chem. Educ., 2021, 98, 3269–3276 CrossRef CAS.
- S. Jiang, J. McClure, H. Mao, J. Chen, Y. Liu and Y. Zhang, J. Chem. Educ., 2024, 101, 675–681 CrossRef CAS PubMed.
- E. S. Thrall, F. Martinez Lopez, T. J. Egg, S. E. Lee, J. Schrier and Y. Zhao, J. Chem. Educ., 2023, 100, 4933–4940 CrossRef CAS.
- A. M. Hupp, J. Chem. Educ., 2023, 100, 1377–1381 CrossRef CAS.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- R. G. Brereton and G. R. Lloyd, Analyst, 2010, 135, 230–267 RSC.
- H. Abdi and L. J. Williams, Wiley Interdiscip. Rev.:Comput. Stat., 2010, 2, 433–459 CrossRef.
- A. Likas, N. Vlassis and J. J. Verbeek, Pattern Recognit., 2003, 36, 451–461 CrossRef.
- A. Géron, Hands-on machine learning with Scikit-Learn, Keras, and TensorFlow, O'Reilly Media, Inc., 2022 Search PubMed.
- B. Mahesh, Int. J. Sci. Res., 2020, 9, 381–386 Search PubMed.
- F. Chollet, Deep learning with Python, Simon and Schuster, 2021 Search PubMed.
- F. Rosenblatt, Psychol. Rev., 1958, 65, 386 CAS.
- Y. Lecun, L. Bottou, Y. Bengio and P. Haffner, Proc. IEEE, 1998, 86, 2278–2324 CrossRef.
- J. L. Elman, Cognit. Sci., 1990, 14, 179–211 CrossRef.
- S. Hochreiter, Neural Computation, MIT-Press, 1997 Search PubMed.
- G. E. Hinton, S. Osindero and Y.-W. Teh, Neural Comput., 2006, 18, 1527–1554 CrossRef PubMed.
- M. E. Deagen, B. Dalle-Cort, N. J. Rebello, T.-S. Lin, D. J. Walsh and B. D. Olsen, Macromolecules, 2024, 57, 42–53 CrossRef.
- T. Jin, C. W. Coley and A. Alexander-Katz, Macromolecules, 2023, 56, 1798–1809 CrossRef CAS.
- E. Z. Qu, A. M. Jimenez, S. K. Kumar and K. Zhang, Macromolecules, 2021, 54, 3034–3040 CrossRef CAS.
- E. van de Reydt, N. Marom, J. Saunderson, M. Boley and T. Junkers, Polym. Chem., 2023, 14, 1622–1629 RSC.
- S. Solorio-Fernández, J. A. Carrasco-Ochoa and J. F. Martínez-Trinidad, Artif. Intell. Rev., 2020, 53, 907–948 CrossRef.
- A. Saxena, M. Prasad, A. Gupta, N. Bharill, O. P. Patel, A. Tiwari, M. J. Er, W. P. Ding and C. T. Lin, Neurocomputing, 2017, 267, 664–681 CrossRef.
- R. Ma, H. Zhang and T. Luo, ACS Appl. Mater. Interfaces, 2022, 14, 15587–15598 CrossRef CAS.
- H. Li, C. R. Collins, T. G. Ribelli, K. Matyjaszewski, G. J. Gordon, T. Kowalewski and D. J. Yaron, Mol. Syst. Des. Eng., 2018, 3, 496–508 RSC.
- E. Brochu, V. M. Cora and N. De Freitas, 2010, arXiv preprint arXiv:1012.2599, DOI:10.48550/arXiv.1012.2599.
- P. M. Pittaway, S. T. Knox, O. J. Cayre, N. Kapur, L. Golden, S. Drillieres and N. J. Warren, Chem. Eng. J., 2025, 507, 160700 CrossRef CAS.
- C. Yan, X. M. Feng, C. Wick, A. Peters and G. Q. Li, Polymer, 2021, 214, 12 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |