
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of metal–organic nanostructures and their structural transformations on metal surfaces
Rujia
Hou
,
Chi
Zhang
 *,
Lei
Xu
,
Yuanqi
Ding
and
Wei
Xu
*
*,
Lei
Xu
,
Yuanqi
Ding
and
Wei
Xu
*
Interdisciplinary Materials Research Center, School of Materials Science and Engineering, Tongji University, Shanghai 201804, People's Republic of China. E-mail: zhangchi11@tongji.edu.cn; xuwei@tongji.edu.cn
First published on 3rd April 2025
Abstract
Metal–organic nanostructures, composed of organic molecules as building blocks and metal atoms as linkers, exhibit high reversibility and flexibility and open up new vistas for the creation of novel metal–organic nanomaterials and the fabrication of functional molecule-based nanodevices. With the rapid development of emerging surface science and scanning probe microscopy, various metal–organic nanostructures, ranging from zero to two dimensions, have been prepared with atomic precision on well-defined metal surfaces in a bottom-up manner and further visualized at the submolecular (or even atomic) level. In such processes, the metal–organic interactions involved and the synergy and competition of multiple intermolecular interactions have been clearly discriminated as the cause of the diversity and preference of metal–organic nanostructures. Moreover, structural transformations can be controllably directed by subtly tuning such intermolecular interactions. In this perspective, we review recent exciting progress in the construction of metal–organic nanostructures on metal surfaces ranging from zero to two dimensions, which is mainly in terms of the selection of metal types (including sources), in other words, different metal–organic interactions formed. Subsequently, the corresponding structural transformations in response to internal or external conditions are discussed, providing mechanistic insights into precise structural control, e.g., by means of metal/molecule stoichiometric ratios (including through scanning probe microscopy (SPM) manipulations), thermodynamic control, introduction of extrinsic competing counterparts, etc. In addition, some other regulatory factors, such as the functionalization of organic molecules and the choice of substrates and lattices, which also crucially govern the structural transformations, are briefly mentioned in each part. Finally, some potential perspectives for metal–organic nanostructures are evoked.
 Rujia Hou | Rujia Hou received her bachelor degree in Engineering from Tongji University (School of Materials Science and Engineering), China in 2022. Since 2022, she has been working on a PhD degree at the School of Materials Science and Engineering, Tongji University, under the supervision of Prof. Chi Zhang and Prof. Wei Xu. Her research interests include molecular assemblies and chemical reactions on metal surfaces under ultrahigh vacuum conditions. |
 Chi Zhang | Chi Zhang received her bachelor and PhD degrees in Engineering from Tongji University (School of Materials Science and Engineering), China in 2012 and 2017 under the supervision of Prof. Wei Xu. Thereafter, she was engaged in postdoctoral research in RIKEN, Japan from 2018 to 2021 (supervisor: Dr Yousoo Kim, chief scientist). Since 2021, she has been working at the School of Materials Science and Engineering at Tongji University, China as a professor. Her main research interests are on-surface molecular assemblies and chemical reactions aiming at fundamental understanding of interfacial chemical processes. |
 Wei Xu | Wei Xu received his PhD degree in Science from Aarhus University, Denmark in 2008. Thereafter he was a postdoctoral fellow at Interdisciplinary Nanoscience Center (iNANO), Aarhus University, Denmark and at Departments of Chemistry and Physics, The Penn State University, USA. Since 2009 he has been a full professor at Tongji University, P. R. China. His main research interests are Scanning Tunneling Microscopy (STM) and Density Functional Theory (DFT) investigations of molecular self-assemblies and reactions on surfaces under ultrahigh vacuum conditions with the aim of controllably building functional surface nanostructures and gaining fundamental insights into physics and chemistry. |
1. Introduction
Metal–organic nanostructures, which are composed of tunable organic molecules and metals generally interconnected by intermediate-strength bonds, have attracted great interest due to their promising structural diversity and geometrical control, and have been widely applied in interdisciplinary fields including, but not limited to, chemistry, nanoscience and nanotechnology, materials science, and physics.1,2 Based on the fascinating design of organic molecules and the choice of embedded metals with infinite combinations, serving as building blocks and linkers, respectively, diverse nanostructures and nanomaterials have been fabricated ranging from zero to three dimensions (0D to 3D) with varied morphologies, sizes, and functions. Accordingly, such a supramolecular association of organic and inorganic components has shown great promise in energy storage and conversion,3,4 catalysis,4–6 gas sensing4 and storage or separation,7 drug delivery,1 and other related systems, with abundant properties including gas sorption, catalytic, magnetic, and optical properties.1 Therefore, the precise preparation of metal–organic nanostructures down to the (sub-)nanometer scale endows researchers with tremendous opportunities to develop a new class of appealing nanomaterials.In addition to conventional synthetic methodologies such as template synthesis, controlled precipitation, microemulsion techniques, and in situ crystallization,1 with the boom of surface science and technology, bottom-up strategies based on metal-molecule assembly on surfaces8 have emerged as a versatile method to finely regulate and control the metal–organic interactions involved and the resulting metal–organic nanostructures at the (sub-)nanometer or sub-molecular scale. Especially after the revolutionary invention of SPM in the early 1980s, typically including scanning tunnelling microscopy (STM)9 and atomic force microscopy (AFM)10 as prominent members of the family, surface science and technology has been extensively applied to the investigation of metal–organic nanostructures, allowing direct observation of on-surface assembly processes and related phenomena.
Specifically, STM achieves atomic-scale resolution (≤0.1 nm lateral, 0.01 nm vertical) by exploiting quantum tunneling current between a conductive tip and a substrate, enabling direct detection of electronic states and precise molecular manipulation on conductive surfaces.10–13 Despite these strengths, STM is inherently constrained by its reliance on conductive samples and susceptibility to environmental vibrations and contaminants. For example, thermal drift necessitates cryogenic conditions (i.e., liquid nitrogen or liquid helium conditions). Stringent ultrahigh vacuum (UHV) conditions and functionalized tips (e.g., CO-terminated metallic tips) have been generally used to enhance stability, minimize perturbations, and increase sub-molecular apparent contrast.14,15 In contrast, AFM operates across conductive, insulating, and biological substrates, and employs multimodal techniques (e.g., tapping mode, Kelvin probe force microscopy) to map topography, mechanical properties, and electrostatic interactions with sub-nanometer vertical resolution.15–17 Despite circumventing the conductivity limitations of STM, AFM has a comparatively lower lateral resolution of ∼1 nm, and tip-sample convolution artifacts require advanced solutions such as self-sensing qPlus sensors and machine learning-assisted image reconstruction.18–21 For additional insights into SPM, readers may consult the comprehensive review recently published by Jiang et al.15
In addition to SPM techniques as mentioned above, well-defined surfaces not only serve as ideal atomically flat platforms to align organic molecules and metal atoms with 2D confinement,22 but also provide atomic registry from substrate templates,23 as well as additional free surface adatoms.24,25 Moreover, apart from the solid/liquid interfaces under ambient conditions26–31 acting as prototypical systems in conjunction with more realistic situations, extreme conditions such as UHV conditions22 have also been applied to the SPM approaches as a prerequisite for achieving uncontaminated, clean experimental conditions without the interference of impurities or atmospheres for target model systems. Based on the UHV conditions, well-designed organic molecules with specific functional groups are then sublimated onto substrates, leading to the interactions both between adjacent molecules and between molecules and the underlying substrates, and thus to the potential molecular self-assembly. At the same time, metals originated from different sources (or in varied forms), e.g., intrinsic metal adatoms24,25,32 from substrates or extrinsic metal atoms,33–35 inorganic salts,36,37 and metal-based complexes,38 are also introduced into these molecular systems on substrates, further increasing the complexity and diversity of the intermolecular interactions involved via the addition of metal–organic interactions and metal–substrate interactions. Accordingly, the synergy and competition of these interactions play a subtle role in the stabilization of the consequent metal–organic nanostructures as well as in the induction of potential structural transformations.
In order to steer the intermolecular interactions involved and to construct and control the metal–organic nanostructures, several regulatory strategies concerning all the components involved in the metal–organic systems on metal surfaces (see Fig. 1 for more details) have been carefully implemented and proven effective, such as: (1) the functionalization of organic molecules (with functional groups related to oxygen, nitrogen, halogen elements, etc.), which provides potential sites for interaction with metals in certain molecular backbones; (2) the selection of metal types (including alkali metals, alkaline earth metals, transition metals, lanthanide and actinide metals, etc.), which determines the predominant metal–organic interactions that stabilize the systems (e.g., electrostatic interactions, coordination bonds, etc.) and their characteristics (directionality, saturation, strength, etc.); (3) the selection of metal sources (i.e., intrinsic adatoms, extrinsic atoms, salts, complexes, etc., as discussed above) that are associated with their preferred existing forms and influence the potential structural transformations;39 and (4) the choice of substrates and lattices (i.e., typical noble metal substrates such as Au, Ag, and Cu, and lattice planes such as the commonly used (111), (110), (100)), which anchor the target organic molecules and metals with specific atomic registries acting as 2D templates. Overall, these above parameters not only finely tune the intricate intermolecular interactions behind and thus determine the bottom-up construction of metal–organic nanostructures on metal surfaces, including dimensions and sizes, morphologies, and structural diversity, but also crucially govern the potential structural transformations. Additionally, some other internal or external conditions, typically like the stoichiometric ratios of metals to molecules and the thermodynamic control on the generated metal–organic nanostructures, also lead to the structural transformations, which generally drive the evolution towards the energetically more favorable structures. Thereby, fundamental understandings of these aspects would inspire precise structural control and stimulate further functionalization of metal–organic nanostructures and broader applications in multiple interdisciplinary fields.
 | ||
| Fig. 1 Schematic illustration of the construction of metal–organic nanostructures and their structural transformations on metal surfaces, which can be well tuned by several strategies, including functionalization of organic molecules, selection of metal types and sources, choice of substrates and lattices, thermodynamic control, etc. | ||
In this perspective, we first focus on the bottom-up construction of metal–organic nanostructures on metal surfaces mainly in terms of the selection of metal types (and sources) as briefly illustrated in Fig. 2, in other words, the different contributing metal–organic interactions involved. Several recent exciting research studies will be introduced and reviewed as representative examples, displaying the characteristic features of each type of metal–organic interaction. Next, the structural transformations of the corresponding series of metal–organic nanostructures in response to different internal or external conditions are further discussed with respect to the involvement of varied types of metals, for example, by regulating the stoichiometric ratio of metals to organic molecules (including through local SPM manipulations), thermodynamic control, introduction of extrinsic competing counterparts, etc., with the vision of providing mechanistic insights into their structural control and engineering. In addition, some other regulatory factors, such as the functionalization of organic molecules and the choice of substrates and lattices, which also crucially govern the structural transformations, are briefly mentioned in each section to enrich the strategies for the design and modification of metal–organic systems. Finally, some potential perspectives (including prospects and challenges) for metal–organic nanostructures on surfaces are also evoked.
 | ||
| Fig. 2 Some typical examples of metal–organic nanostructures constructed on metal surfaces and probed by SPM under UHV conditions, in terms of different types of metals embedded. Reproduced with permission.34,35,40–43 Copyright 2020 American Chemical Society; 2023 American Chemical Society; 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; 2013 National Academy of Sciences; 2021 American Chemical Society. | ||
2. Construction of metal–organic nanostructures
According to the types (and sources) of metals embedded in the metal–organic nanostructures, we will basically classify them into three parts and discuss each in the following sections, i.e., s-block alkali metals (and alkaline earth metals), d-block transition metals, and f-block lanthanide metals (and actinide metals) (Fig. 2). The integration of such metals generally leads to the variation in the predominant metal–organic interactions that stabilize the corresponding molecular systems, ranging from electrostatic interactions to coordination bonds with different bonding features. For instance, alkali (and alkaline earth) metals frequently form electrostatic ionic interactions with organic molecules36 on surfaces. Instead, d-block transition metals, with partially filled d orbitals, form highly directional coordination bonds with molecular ligands in predetermined coordination numbers and angles,22 resulting in the relatively rigid metal–organic coordination nanostructures. In addition, lanthanide (and actinide) metals, as f-block elements, generally form sets of metal–organic nanostructures with enhanced tunability and flexibility in geometries, featured by the mostly ionic nature44,45 of metal–organic interactions. Thanks to the complexity and diversity of such metal–organic interactions, a variety of nanostructures have been constructed on surfaces in different dimensions, generally ranging from 0D to 2D, and further experimentally detected by SPM techniques with submolecular or atomic resolution and theoretically elucidated by density functional theory (DFT) calculations.2.1 Alkali metals (and alkaline earth metals)
As one of the most potent reductive agents known, alkali metals are introduced into molecular systems on surfaces as extrinsic metals, in most cases in the form of pure metals and alkali metal salts (commonly, alkali halides). Notably, upon adsorption on metal surfaces, they are usually positively charged, no matter in the form of isolated atoms (due to the strong charge transfer between them and the underlying metal substrates)36,46 or interacting with adjacent organic molecules47–49 (owing to the strong charge redistribution) or matching counterpart anions (e.g., halogens such as Cl, Br, and I).36,46,50,51 Thus, various electrostatic ionic interactions are generally formed in the metal–organic nanostructures based on alkali metals, which will be shown in detail in the following cases.As early as 2010, Kern et al. introduced cesium (Cs) atoms to interact with a series of aromatic carboxylic acids on Cu(100), forming 2D networks with tunable dimensions.47 As an extension, they further compared this kind of “ionic assembly” with the traditional coordination by applying the same molecule, 7,7,8,8-tetracyanoquinodimethane (TCNQ), on Ag(100) to interact with Cs and manganese (Mn), respectively, forming Cs-TCNQ4 and Mn-TCNQ4.48 Almost at the same time, Besenbacher et al. employed potassium (K), known to play a significant role in the cellular environment, to direct the assembly of bioligand guanine (G) molecules, thereby resulting in the formation of K-stabilized G-quartets on Au(111), which offers further insights into the intermolecular interactions involved (i.e., a delicate balance between hydrogen bonding and electrostatic ionic interactions).52,53 In the wake of these pioneering efforts, alkali metals began to be used to interact with organic molecules on surfaces.
Similarly, based on the bio-related background of alkali metals and nucleobases, Xu and coworkers have conducted a comprehensive study concerning almost all kinds of alkali metals and nucleobase molecules (and their derivatives), including 9-ethylguanine (9eG) and NaCl, KBr, and CaCl2 (Fig. 3d),36 G and NaCl (Fig. 3c),54 9eG and Na,55 1-methylcytosine (1mC) and NaCl,56 cytosine (C) and NaCl,57 uracil (U) and Li, Na, K (Fig. 3a),40 and Cs (Fig. 3b),58 adenine (A) and NaCl, NaBr, and NaI,59etc. For example, as shown in Fig. 3a and b, U molecules can interact with different alkali metals (Li, Na, K, and Cs) by a combination of hydrogen bonds and electrostatic ionic interactions between metal centers and O sites to form various U clusters, i.e., U3Li trimer, U4Na tetramer, and U5K pentamer,40 and U5Cs1, U6Cs2, and U8Cs3 clusters58 on Au(111), respectively. In these structures, the size (or diameter) of the embedded metal cations (which are all positively charged) plays a critical role in determining both the aggregation numbers of the interacting molecules and the stabilization of the metal–organic motifs. In particular, linear arrays of Cs cations can be formed by stepwise addition of Cs atoms, which appeared as distinctly bright dots (see Fig. 3b), representing different properties with respect to the different types of alkali metals.
 | ||
| Fig. 3 Examples of metal–organic nanostructures constructed on metal surfaces based on alkali metals and alkaline earth metals from different sources. (a) Construction of trimeric, tetrameric, and pentameric uracil (U) clusters on Au(111), i.e., U3Li, U4Na, and U5K, from alkali metals (Li, Na, and K). Reproduced with permission.40 Copyright 2020 American Chemical Society. (b) A series of Cs-involved U clusters, U5Cs1, U6Cs2, and U8Cs3, formed on Au(111). Reproduced with permission.58 Copyright 2019 The Royal Society of Chemistry. (c) Solventless formation of G-quartet-Na (G4Na) complexes on Au(111) from NaCl. Reproduced with permission.54 Copyright 2015 American Chemical Society. (d) G4Ca complexes on Au(111) formed from CaCl2. Reproduced with permission.36 Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (e) NaCl–organic coordinated nanoarchitectures on Au(111). Reproduced with permission.60 Copyright 2015 The Royal Society of Chemistry. (f) Sierpiński triangle fractals formed from K on Cu(111). Reproduced with permission.41 Copyright 2023 American Chemical Society. (g) Na-interlinked metal–organic nanostructure on Au(111). Reproduced with permission.61 Copyright 2024 American Chemical Society. | ||
Among others, the alkali and alkaline earth halides, as alternative metal sources to pure metals, provide both metals and halogens to the molecular systems. In this process, the corresponding alkali and alkaline earth metals usually interact with the oxygen or nitrogen sites of the molecules, forming metal–organic interactions, while the remaining halogens either allow structural diversity by forming electrostatic interactions with hydrogen-rich sites instead or do not participate in the metal–organic nanostructures (probably diffusing on the bare substrates), depending on the availability of hydrogen-rich sites. Fig. 3c and d show two examples of how inorganic salts (NaCl and CaCl2 herein) interact with organic molecules (G and 9eG, respectively). Thus, the well-known G-quartet-M structures (M refers to Na/K/Ca) can be successfully achieved on Au(111) based on the introduction of the corresponding salts (NaCl, KBr, and CaCl2) in a solvent-free UHV environment via the cooperative electrostatic interactions and hydrogen bonds (Fig. 3d),36 which further allow the tautomeric recognition and separation of two G tautomers (G/9H and G/7H) by forming different assembled structures54 (Fig. 3c). The successful construction of the G4Na structure on Au(111) by introducing NaCl and G molecules thus enables the in situ identification and dynamic regulation of two G tautomers within molecular networks. This strategy establishes a dual-perspective framework bridging surface chemistry and biological systems to decipher metal–organic interactions, while creating methodological connections between biomolecular tautomerism studies and surface-confined system analyses.
In particular, alkali salts and pure alkali metals share some similarities in their interactions with organic molecules, while they also have certain differences in inducing structural evolution and in determining structural diversity.39 Based on the tetrapyridyl-porphyrin (H2TPyP) system on Au(111), pure Na and NaCl were dosed separately, leading to the distinct evolution of metal–organic structures, where the competition between halogens and organic molecules in interacting with alkali metals was revealed to be the key to such differences.39 These findings thus represent a feasible strategy for the construction and functionalization of various alkali-metal-based metal–organic nanostructures on metal surfaces.
Interestingly, in some special cases, the alkali halide salts (e.g., NaCl and KCl) tend to bind tightly together via strong ionic interactions and interact with organic molecules to form “salt (e.g., NaCl and KCl) – organic coordinated nanostructures”60,62,63 instead. As typically displayed in Fig. 3e, NaCl dimers (represented by purple circles) appear as bright protrusions in STM images and form various arrangements with 3,4,9,10-perylenetetracarboxylic diimide (PTCDI) molecules in different aggregation numbers, with similar apparent heights to those appearing in NaCl islands, which also verifies their dimer composition in the metal–organic structures.
More recently, organic molecules functionalized with halogen substitutions have also been brought into interaction with alkali metals on surfaces.41,61,64 Tunable 2D metal–organic frameworks (MOFs) were constructed on Au(111) based on flexible alkali–halogen bonding between alkali cations and ortho-bromo-substituted molecules by Zhao et al.64 Besides, Wu et al. further reported the sophisticated construction of molecular Sierpiński triangles (STs) via the Kδ+⋯Clδ− electrostatic interaction between K cations and 4,4′′-dichloro-1,1′:3′,1′′-terphenyl (DCTP) molecules on Cu(111) and Ag(111)41 as shown in Fig. 3f, which enriches the toolbox (besides halogen bonding, hydrogen bonding, coordination, and covalent bonding) for the bottom-up fabrication of functional supramolecular nanostructures. Interestingly, by introducing Na atoms into the halogen-functionalized molecular systems, a novel halogen separation strategy for the on-surface dehalogenative reactions was recently developed on Au(111)61 by our group, where Na not only isolates the dissociated halogens by forming salt islands, but also facilitates the C–Br activation under mild conditions. In such processes, a honeycomb Na-interlinked metal–organic intermediate nanostructure was also captured (Fig. 3g), with the electrostatic attraction between Na and surrounding Br substituents (similar to the Kδ+⋯Clδ− in the former case) determined to be the driving force for stabilizing the structures and further halogen isolation.
2.2 Transition metals
Metal–organic nanostructures based on transition metals, which are generally formed by the strong coordination bonds between transition metal (TM) ions or clusters and organic ligands, have long been promising candidates in various fields. In particular, TM-based MOFs, which are characterized by their porosity, high surface area, tunable architecture, magnetism, and optical properties,65 have significant value and substantial application potential in gas adsorption and storage,66,67 energy storage and conversion,68,69 and catalysis.70–72 In the field of surface science, transition metals are also frequently utilized in the construction of metal–organic nanostructures, including 0D clusters,73 1D chains,74 and 2D frameworks.32 These specific nano-architectures are largely determined by the coordination modes between metal ions and organic ligands. Gold (Au), silver (Ag), and copper (Cu) are among the most commonly employed metallic substrates, leading to a prevalence of metal–organic nanostructures incorporating these elements.75–77 Beyond these noble metals, transition metals like iron (Fe), nickel (Ni), and cobalt (Co) are also commonly utilized due to their unique properties that can significantly influence the electronic and magnetic behaviors of the resulting nanostructures.78–81Metal–organic nanostructures constructed by intrinsic metal adatoms from noble metal substrates such as Au, Ag, and Cu have been extensively applied to develop a wide range of diverse metal–organic nanostructures and will be discussed first. We will list several corresponding examples of the construction of metal–organic nanostructures by tuning various parameters, such as molecular coverages, adsorption configurations, annealing temperatures, types of underlying surfaces, etc.
In the construction of metal–organic nanostructures, organic ligands possessing specific hydrogen-containing functional groups, such as hydroxyl and amino groups, usually undergo dehydrogenation to coordinate with metal ions, thereby forming stable nanostructures.32,77,82 Similarly, the thiolate–copper coordination bond has also attracted considerable attention due to its electronic conjugation characteristics.83,84 In order to construct thiolate–metal complexes, Lackinger et al. selected the 1,3,5-tris(4-mercaptophenyl)benzene (TMB) molecule functionalized with thiol groups as a precursor molecule.85 Upon deposition on Cu(111) at room temperature, TMB molecules formed densely packed triangular structures after deprotonation. Further annealing at 160–200 °C led to the formation of thiol–copper coordination bonds between the dehydrogenated thiolate and Cu adatoms, constructing two distinct metal–organic nanostructures, i.e., the honeycomb networks and dimer row structures (Fig. 4a). The relative proportion of these two structures is influenced by the initial coverages of the precursors, with a higher coverage favoring the formation of dimer row structures. The fully conjugated electronic structure of thiolate–copper coordination bonds, along with their enhanced thermodynamic stability, renders them promising candidates for “solder” in molecular electronics.
 | ||
| Fig. 4 Examples of metal–organic nanostructures constructed on noble surfaces based on intrinsic transition metal adatoms derived from the substrates. (a) Two kinds of porous structures induced by Cu adatoms on Cu(111). Reproduced with permission.85 Copyright 2011 American Chemical Society. (b) Construction of metal–organic coordination nanostructure after deposition of TPDCN molecules on Cu(111) at room temperature. Reproduced with permission.86 Copyright 2017 American Chemical Society. (c) Formation of triangular metal–organic networks on Ag(111) through the coordination assembly of m-DICB at different coverages. Reproduced with permission.87 Copyright 2021 American Chemical Society. (d) Enantiomeric windmill-like tetramers based on DHBP molecules and Ag adatoms on Ag(100). Reproduced with permission.88 Copyright 2019 American Chemical Society. (e) A hexagonal coordinated ring on Cu(111) and a typical ring composed of coordination bonds and hydrogen bonds obtained on Au(111). Reproduced with permission.75 Copyright 2017 American Chemical Society. (f) Formation of linear assembled chains composed of N⋯Au coordination bonds and halogen bonds on Au(111). Reproduced with permission.89 Copyright 2024 SIOC, CAS, Shanghai, & WILEY-VCH GmbH. | ||
In addition to influencing different coordination patterns, molecular coverage even directly alters different types of intermolecular interactions at a submolecular level. For example, Xu et al. achieved a reversible interconversion of intermolecular bonds between metal–organic coordination bonds (Cu⋯N) and weak hydrogen bonds (CH⋯N) by regulating the surface molecular coverage.86 In this scenario, carbonitrile end groups of three (1,1′:3′,1′′-terphenyl)-4,4′′-dicarbonitrile (TPDCN) molecules formed the 3-fold coordination interaction with one Cu adatom from Cu(111) (Fig. 4b). Unexpectedly, as the molecular coverage increased, the nitrogen sites on the carbonitrile groups were prone to engage in hydrogen bonding interactions instead. This propensity ultimately resulted in the formation of a more compact self-assembled structure, wherein the structural integrity was maintained by an extensive network of hydrogen bonds. Interestingly, a similar influence of molecular coverage on the assembled structure was observed in the molecular system of chevron-shaped 1,3-phenylene diisocyanobenzene (m-DICB) on Ag(111).87 Under a low coverage condition, a variety of fractal structure STs were formed, while at a high coverage, new metal–organic networks were generated, with both structures involving coordination bonds between a single Ag adatom and three m-DICB molecules (Fig. 4c). Notably, the arc-shaped features typically indicated by a white arrow in the upper part of Fig. 4c are attributed to the “swinging” terminal m-DICB molecules, which lack coordination interactions between their terminal isocyano groups and Ag adatoms. Besides molecular coverage, the standing configuration of m-DICB on Cu(111) resulted in the formation of linear chains, illustrating that the molecular adsorption configuration on surfaces can also influence the construction of metal–organic nanostructures. Cu(111) is generally characterized by a higher adsorption energy and a smaller lattice constant. The reduced intermolecular spacing effectively enhanced the π–π stacking interactions between neighboring molecules, which thermodynamically stabilized the standing adsorption configuration over the flat one. In contrast, the larger lattice constant of Ag(111) resulted in an increased intermolecular distance, thereby significantly weakening the π–π stacking interactions. Moreover, the relatively lower adsorption energy also drove m-DICB molecules to adopt the planar adsorption configuration, synergistically resulting in the construction of metal–organic nanostructures, as shown in Fig. 4c.
Moreover, precise control of the annealing process facilitates the coordination interactions between molecules and intrinsic adatoms, leading to the formation of well-organized metal–organic nanostructures.77Fig. 4d illustrates two chiral structures formed by the direct interaction between dehydrogenated hydroxyl groups on 4,4′-dihydroxybiphenyl (DHBP) and Ag adatoms on Ag(100) at 490 K, with the central circular bright spots representing Ag adatoms.88 Interestingly, such metal–organic coordinated tetramers facilitated further dimerization reactions by subsequent annealing at 520 K, completing a crucial chiral transfer process that enhanced the selectivity of the reaction.90,91 This case indicates the important role of such metal–organic interactions in the further construction of covalent bonding.
Notably, substrate effects, including reactivity and lattice commensurability, can also significantly influence the construction of metal–organic nanostructures.74,92 The precursor, 1,3-bi(4-pyridyl)benzene (BPyB) with pyridyl functional groups, was chosen to investigate its interactions with adatoms from different metal surfaces.75 As illustrated in Fig. 4e, BPyB molecules tended to form closed-ring structures with Cu adatoms on Cu(111), where each node contains one Cu adatom coordinated with two BPyB molecules. Since Cu adatoms are not visible in the STM image, to exclude the influence of hydrogen bonding, STM lateral manipulations were performed and showed that the ring structure remained largely unbroken with only a negligible shift, further confirming the coordination bonds between pyridyl groups and Cu adatoms. In contrast, BPyB molecules only formed hydrogen-bonded structures on Ag(111), and even desorbed from the surface at approximately 330 K. Besides, similar ring structures were observed on Au(111), with a combination of hydrogen bonds and coordination bonds involved. After annealing at 350 K, the proportion of coordination bonds increased due to an increase in the density of Au adatoms at higher surface temperatures. Such a case clearly highlights the crucial role of metal surfaces in providing adatoms for the preparation of metal–organic nanostructures.
Similar to the above metal–organic structure obtained on Au(111) with the assistance of hydrogen bonds, coordination bonds and halogen bonds can also work together synergistically to form corresponding metal–organic nanostructures.89 As illustrated in Fig. 4f, well-assembled linear chains were constructed on Au(111) composed of triangular units through Br⋯Br halogen bonds, and each unit consisted of a central Au adatom interacting with three nitrogen sites on 2,7-dibromodibromo-carbazole (DBC).
The studies presented here demonstrate that a variety of metal–organic nanostructures can be obtained through the subtle interactions between organic molecules and substrate free adatoms, which are influenced by factors including precursor coverages, molecular adsorption configurations, temperatures, substrate effects, etc.
In addition to the utilization of surface metal adatoms as metal sources for the construction of metal–organic nanostructures, the direct deposition of extrinsic metal atoms, typically including Fe, Co, Zn, and Ni, can also lead to diverse metal–organic interactions with target organic molecules.33,93–97 Molecules with various functional groups (predominantly involving elements such as N and O) have been applied to interact with these metal atoms, which will be discussed below based on several representative studies.
For example, pyridyl groups generally serve as a functional group to interact with transition metals, as also shown in the case of Fig. 4e. Accordingly, a V-shaped 3-bis(4-pyridyl)-benzene (BPyB) molecule was chosen by Wang et al. The co-deposition of transition metal Fe atoms and BPyB molecules on Au(111), followed by annealing, allowed the construction of a series of perfect equilateral Sierpiński triangle structures through the 3-fold coordination between Fe and N sites on the pyridine rings (Fig. 5a).98 Besides, control experiments on Au(100) in conjunction with theoretical calculations elucidated the pivotal role of molecular free diffusion capability and structural matching between structures and surface lattices in the formation of the two-dimensional crystals of STs.
 | ||
| Fig. 5 Examples of metal–organic nanostructures constructed on metal surfaces based on extrinsic transition metals. (a) A series of BPyB-Fe-ST-n fractal structures constructed on Au(111), n = 0–5. Reproduced with permission.98 Copyright 2020 American Chemical Society. (b) TPPT–Fe coordination motifs on Ag(111) with multiple linearly arranged Fe atoms. Reproduced with permission.99 Copyright 2018 Springer Nature. (c) Co-HOTP network on Au(111). Reproduced with permission.100 Copyright 2022 American Chemical Society. (d) An isolated G-quartet-Fe complex on Au(111). Reproduced with permission.101 Copyright 2014 American Chemical Society. (e) Ni (derived from NiBr2)-incorporated metal–organic trimers on Au(111) and the corresponding Kondo effect induced by the on-top Br atom on a three-metal center. Reproduced with permission.37 Copyright 2019 American Chemical Society. | ||
In a more complicated system, an aromatic molecule based on terpyridine (tpy), namely tpy-phenyl-phenyl-tpy (denoted as TPPT), was selected to coordinate with extrinsic Fe atoms on Ag(111) to probe low-dimensional metal–organic systems with multinuclear coordination centers.99Fig. 5b depicts different stages of the coordination process, where the non-metalated left tpy component, encased within the blue square, retains its characteristic “V” shape. In contrast, the metalated right tpy within the green square appears brighter due to the coordination with a single Fe atom and displays a distinct protrusion at the center. Non-contact AFM (nc-AFM) images revealed that the pyridine rings at the extremities were tilted towards the center of the molecule, indicating rotation around the C–C bond to facilitate coordination between the N sites and Fe atoms. The red square highlights a metal–organic nanochain formed by two TPPT molecules coordinated with three Fe atoms, featuring two opposing tpy units and a bright center. Interestingly, the quasi-linear tri-iron cluster is in a cationic mixed-valence configuration, concomitant with a charge redistribution. Such an unusual tri-nuclear Fe complex was also proposed to have high chemical activity. In particular, the synthesis of tri-iron coordination complexes has been demonstrated to overcome the limitations of activity and selectivity inherent in mononuclear catalysts through the electronic synergistic effects of metal–metal bonds,102–104 which paves the way for the development of quantum transport devices based on multinuclear metal nodes.
Apart from the N-containing functional groups, O-containing ones (e.g., hydroxyl group) are also commonly involved in the coordination with transition metals. In a recent study by Écija et al., 2,3,6,7,10,11-hexahydroxytriphenylene (H6HOTP) molecule with six hydroxyl groups was applied to interact with Co on Au(111). The Co-HOTP metal–organic network was successfully prepared based on the complete deprotonation of the hydroxyl groups and 3-fold coordination between Co and O sites (Fig. 5c), which exhibited a substantial orbital magnetic moment.100 This finding is significant due to its potential for engineering antiferromagnetic materials via metal–organic coordination.
In some other cases, both N and O sites are involved in the coordination with transition metals. For example, 9-ethylguanine (9eG), a derivative of DNA bases, with both potential coordination sites for metal binding, has been extensively investigated to interact with Fe101,105,106 and Ni.34,37 The STM image and theoretical computational simulation presented in Fig. 5d provide an exquisite illustration of an isolated G-quartet-Fe (G4Fe1) complex formed on Au(111) at a low Fe/9eG stoichiometric ratio, which is facilitated by the synergistic effect of both preferential coordination bonds between Fe and O sites and intermolecular hydrogen bonds.101 It is noteworthy that at higher Fe/9eG stoichiometric ratios, continuous structural transformations from G4Fe1 to G3Fe1, G4Fe2, and G3Fe3 were also achieved, with further integration of N sites into the coordination with Fe, presenting coordination diversity. Such structural transformations were also demonstrated to be reversible in response to the additional deposition of Fe atoms or 9eG molecules at room temperature.
Similar to the case of alkali metals, transition metal halides (e.g., NiCl2, NiBr2, and FeBr2) can also serve as metal sources,34,37 with additional halogens located at the hydrogen-rich harbors. Xu et al. have detailed a strategy for the room-temperature co-deposition of 9eG molecules and NiBr2 on Au(111), forming close-packed islands with the elementary structural motif designated as G3Ni3Br4.37 Within this structure, three Ni metal centers were observed to interact simultaneously with the O and N sites, with the central bright feature attributed to the Br atom on the top of the Ni3 cluster (Fig. 5e). After annealing the sample at 390 K, the on-top Br atoms were effectively removed, as evidenced by the disappearance of bright dots, along with the absence of the Kondo effect recorded in the dI/dV spectra. This observation reveals a direct modification of the magnetic properties of the trimetallic center in the metal–organic nanostructure, which underscores the potential to fine-tune the electronic and magnetic properties of nanomaterials through precise control of their chemical architectures. Moreover, G3Ni3I3 motifs without on-top halogen atoms could also be obtained by the direct integration of pure Ni atoms and I atoms separately,34 indicating the difference between transition metal salts and the corresponding pure transition metals. Crucially, the reversible switch of the spin states within transition metal Ni clusters presents a novel avenue for the development of chemically sensitive spin valves, thereby offering innovative perspectives for advancing the field of spintronics.
2.3 Lanthanide metals (and actinide metals)
Lanthanide metals, a subset of rare earth elements, have garnered significant interest in the field of metal–organic nanostructures due to their unique properties, which are regarded as an invaluable treasure trove for the development of novel materials.107–110 In the preceding discourse, we have mainly delineated the construction and characterization of metal–organic nanostructures based on alkali and transition metals in research endeavours. The f-block elements typically exhibit high coordination numbers and malleable coordination environments, with typical values ranging between 6 and 12, but as low as 3 or 4 in the case of larger ligands, leading to the formation of architectures with unique pore sizes, shapes, and functionalities.45,111 Moreover, lanthanide-element-based nanostructures, beyond their remarkable structural diversity, have been demonstrated to possess a spectrum of distinctive physicochemical properties, including luminescence, magnetic properties, gas storage and separation capabilities, and multiphase catalytic activities.112–115Recently, lanthanide metals have been involved in the on-surface coordination chemistry to form complexes with organic ligands with various functional groups, including carboxylates, carbonyls, pyridyls, and nitriles, which engage in coordination through O and N sites.45,111 To gain insight into the comparative behaviour of different metals, Écija et al. have conducted a systematic investigation on the construction of metal–organic networks based on p-terphenyl-4,4′-dicarboxylic acid (TDA) and gadolinium (Gd), dysprosium (Dy), and erbium (Er), respectively, on Cu(111).116–118 It is noteworthy that, regardless of the specific metals involved, the resulting reticular network structures exhibited a high degree of similarity following the co-deposition of TDA molecules and metals, as displayed in Fig. 6a. Moreover, the carboxylic acid functional groups underwent a complete deprotonation process during annealing, allowing them to coordinate with the specific metal to form networks, as characterised by the 8-fold coordination between each metal atom and eight oxygen sites. Similarly, the formation of a reticular metal–organic network was also obtained by dosing Ho on Ag(100).119
 | ||
| Fig. 6 Examples of metal–organic nanostructures constructed on metal surfaces based on lanthanide metals. (a) Construction of reticular metal–organic networks induced by Gd, Dy, and Er on Cu(111), respectively. Reproduced with permission.116–118 Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; 2021 Wiley-VCH GmbH; 2023 The Royal Society of Chemistry. (b) Dy-directed metal–organic networks on Cu(111) upon thermal annealing. Reproduced with permission.120 Copyright 2016 The Royal Society of Chemistry. (c) Five-vertex Ce supramolecular architectures on Ag(111). Reproduced with permission.121,122 Copyright 2013 National Academy of Sciences; 2014 American Chemical Society. (d) Synthesis of Gd-SNPc after cyclopentamerization on Ag(111). Reproduced with permission.123 Copyright 2019 Springer Nature. | ||
In the case of the Dy-TDA reticular network (Fig. 6a), unexpectedly, further annealing to 450 K induced phase transition to another quasi-hexagonal dinuclear network as shown in Fig. 6b.120 Two small bright spots were observed at each node (the bottom part of Fig. 6b) and were attributed to two Dy atoms. Accordingly, within this network, the Dy metal no longer engages in the expected 8-fold Dy⋯O coordination interaction. Instead, the atomic structural model illustrates that each metal coordinates with six O sites from four different molecules, and the two opposite molecules in the middle of this motif interact with two Dy atoms simultaneously. Additionally, the O-containing carbonyl functional groups also have the ability to coordinate with lanthanide metals to form metal–organic nanostructures.124
Organonitrogen compounds are also recognized as effective molecule candidates for the construction of metal–organic nanostructures, providing N sites for potential coordination with metal ions. Among others, carbonitrile groups are widely applied to coordinate with lanthanide metal elements.
Specifically, dicarbonitrile-polyphenyl molecules, denoted as p-NC-(Ph)3-CN-p (see Fig. 6c), assembled into a quintessential pentameric unit structure upon dosing of Ce atoms, via the 5-fold coordination interactions between Ce and N sites on the carbonitriles.121,122 Furthermore, by precisely controlling the stoichiometric ratio of metals to organic molecules, phase transitions were induced, thereby allowing the stepwise preparation of a variety of intriguing structures, as shown in Fig. 6c. The axial coordination activity of Ce centers within these diverse structures demonstrates significant potential for applications in heterogeneous catalysis. Similar nitrogen-lanthanide coordination interactions are also discernible within molecules functionalized by pyridyl moieties.125
In addition to the aforementioned cases where carbonitrile terminal groups directly coordinate with lanthanide metals to form metal–organic nanostructures, these metal elements have also been applied in the on-surface synthesis based on such coordination interactions. For instance, the synthesis of pentacyclic Gd-SNPc (Gd-supernaphthalocyanine, see Fig. 6d) on Ag(111) was achieved using the lanthanide metal Gd, which activated the carbonitrile groups (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) of precursors (ADN) to facilitate the cyclopentamerization.123 In this process, more importantly, Gd acts as a key metal template to simultaneously coordinate with N sites on five neighboring molecules, where the metal atoms appear as bright protrusions (Fig. 6d). In contrast, the transition metal Fe provides 4-fold coordination, resulting in the formation of iron-naphthalocyanine (Fe-NPc) on Ag(111). This study reveals the influence of different types of metal elements in providing various template-controlled constructions of metal–organic nanostructures, which is pivotal for the rational design of extended naphthalocyanine-based multifunctional molecule-based nanomaterials.
N) of precursors (ADN) to facilitate the cyclopentamerization.123 In this process, more importantly, Gd acts as a key metal template to simultaneously coordinate with N sites on five neighboring molecules, where the metal atoms appear as bright protrusions (Fig. 6d). In contrast, the transition metal Fe provides 4-fold coordination, resulting in the formation of iron-naphthalocyanine (Fe-NPc) on Ag(111). This study reveals the influence of different types of metal elements in providing various template-controlled constructions of metal–organic nanostructures, which is pivotal for the rational design of extended naphthalocyanine-based multifunctional molecule-based nanomaterials.
In addition, actinide elements, which share similar chemical properties to the lanthanides, possess a greater number of valence electrons, allowing them to reach higher oxidation states.126–128 Although the actinides theoretically have the potential to form a wide variety of metal–organic nanostructures, current research on actinide-based nanostructures is predominantly conducted in solution or at solid–liquid interfaces, with comparatively fewer studies on their fabrication on metal substrates.129
Overall, this section encapsulates the intricate interplay between metals and organic ligands in the construction of well-defined metal–organic nanostructures, demonstrating the precision and predictability of such metal–organic interactions.
3. Structural transformations of metal–organic nanostructures
In this section, some typical cases regarding structural transformations of metal–organic nanostructures will be discussed based on the involvement of different types of metals, including alkali metals, transition metals, lanthanide metals, and bimetals (i.e., combination of two types of metals). Several common regulatory factors, including both internal and external conditions, represented by the variation of metal/molecule stoichiometric ratios (including through SPM manipulations), thermodynamic control, introduction of extrinsic competing counterparts, respectively, will be focused on. By analyzing the corresponding cases, we expect to provide some mechanistic insights into precise structural control and engineering at the submolecular level, which should shed light on the creation of responsive metal–organic nanomaterials and nanodevices.3.1 Alkali metals
In the case of alkali metals, due to the flexibility of alkali–molecule bonding in both interaction numbers and angles,64 the corresponding structural transformations are generally realized by regulating the stoichiometric ratios of alkali metals (in any form) to organic molecules as demonstrated in several related reports.39,55,58 This can be achieved either by directly controlling the metal dosage and/or the organic ligand coverage, which are originally associated with the metal–organic system, or even by introducing competing counterparts (i.e., extrinsic components other than the target molecules or metals, which will be discussed in the following two cases). For instance, by gradually dosing Na atoms to the 9eG-precovered Au(111) sample, the stepwise structural transformation from pure 9eG islands through 9eG4Na1 quartets to 9eG3Na2 triads was obtained, with the reverse process accessible by further addition of 9eG molecules to the former sample.55 Consequently, the interconversion between 9eG4Na1 quartets and 9eG3Na2 triads was achieved in a controllable manner, indicating the versatility of electrostatic ionic interactions between Na and O, N sites.Intriguingly, such structural interconversions can also be realized by adding the competing counterparts (e.g., iodine) to interact with Na cations instead of organic molecules.39 As shown in Fig. 7 from our previous work, H2TPyP molecules interacted with Na at a Na![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2TPyP stoichiometric ratio of ∼1:1, initially forming the close-packed phase I on Au(111), which transformed to more porous structures (mixture phase and phase II) with an increasing Na:H2TPyP ratio, and finally formed well-ordered porous phase III at a Na:H2TPyP stoichiometric ratio of ∼2:1. In this process, the calculated binding energy per H2TPyP molecule in the corresponding periodic nanostructures increases progressively with increasing Na dosage, indicating that such a structural transformation is thermodynamically driven. In contrast, when Na was supplied from NaCl instead, only phase I could be formed with the coexistence of excess NaCl islands, as the binding energy per Na decreases, indicating that the phase transition from I to III is thermodynamically unfavorable in the case of NaCl. It is also noteworthy that by introducing iodine (I2) into phase III, the additional Na tended to bind with I to form NaI islands, accompanied by a reverse structural transformation through the mixture phase to eventually phase I. Thereby, halogens and organic molecules are competitive counterparts to interact with alkali metals, which is the cause of the reversible phase transition. This study thus unambiguously displays the difference between pure alkali metal atoms and alkali halide salts as two types of metal sources in the construction of alkali–molecule interactions at the submolecular level.
:H2TPyP stoichiometric ratio of ∼1:1, initially forming the close-packed phase I on Au(111), which transformed to more porous structures (mixture phase and phase II) with an increasing Na:H2TPyP ratio, and finally formed well-ordered porous phase III at a Na:H2TPyP stoichiometric ratio of ∼2:1. In this process, the calculated binding energy per H2TPyP molecule in the corresponding periodic nanostructures increases progressively with increasing Na dosage, indicating that such a structural transformation is thermodynamically driven. In contrast, when Na was supplied from NaCl instead, only phase I could be formed with the coexistence of excess NaCl islands, as the binding energy per Na decreases, indicating that the phase transition from I to III is thermodynamically unfavorable in the case of NaCl. It is also noteworthy that by introducing iodine (I2) into phase III, the additional Na tended to bind with I to form NaI islands, accompanied by a reverse structural transformation through the mixture phase to eventually phase I. Thereby, halogens and organic molecules are competitive counterparts to interact with alkali metals, which is the cause of the reversible phase transition. This study thus unambiguously displays the difference between pure alkali metal atoms and alkali halide salts as two types of metal sources in the construction of alkali–molecule interactions at the submolecular level.
 | ||
| Fig. 7 Reversible structural transformation of alkali-metal-based metal–organic nanostructures on Au(111) in response to the dosage of (a)–(c) Na atoms and (d)–(f) iodine, respectively. Reproduced with permission.39 Copyright 2023 American Chemical Society. | ||
Accordingly, we further synthesized Na-porphyrins (i.e., Na-TPyP)130 on Au(111) with a high yield by the application of either Na atoms or NaCl as metal suppliers, followed by thermal treatment to induce the intramolecular dehydrogenation (that is, the N–H activation) within macrocycles. Such a metalation scenario implies the broad utility of metal sources not only in assembling organic molecules with possible structural transformations,39,131 but also in inducing molecular reactions.130
More interestingly, by the subtle integration or removal of such alkali–molecule interactions, covalent organometallic (OM) nanostructures with C–M–C connections have also been successfully steered,132 showing the structural transformation of covalent nanostructures mediated by non-covalent electrostatic interactions (Fig. 8). In our experimental endeavors, we exploited the inherent dynamics of C–M–C covalent bonds133 as well as the electrostatic interactions between the alkali metal Na and both bipyridyl and diketone moieties39,134 to fine-tune the ring-chain equilibrium (as detailed in Fig. 8). Upon the incorporation of Na atoms into the trans OM chains (obtained in response to O2 exposure135) based on 4,4′-diethynyl-2,2′-bipyridine (DEBPy) on Ag(111), the trans-to-cis isomerization of DEBPy-based molecular components was facilitated by the electrostatic interactions between Na and bipyridyl moieties. Simultaneously, the Csp–Ag–Csp bonds underwent spontaneous cleavage and reformation at room temperature, thereby effectively constructing Kagome networks composed of cis OM rings in the presence of Na. Considering the competitive preference of diketone moieties to interact with Na over cis-bipyridyl moieties, pyrene-4,5,9,10-tetraone (PT) molecules were introduced into the Kagome networks as the extrinsic competing counterparts. Intriguingly, the conversion from cis OM rings to trans OM chains was facilitated by the removal of Na (colored green), while Na interacted with PT molecules instead to form the PT + Na structure (colored brown).
 | ||
| Fig. 8 Ring-chain equilibrium between covalent organometallic chains and rings induced by the regulation of weak intermolecular interactions provided by Na. Reproduced with permission.132 Copyright 2024 American Chemical Society. | ||
In the whole process, by adding Na atoms to the trans OM chains, Na atoms initially interacted with two opposite N sites of the trans-molecules via twofold Na⋯N electrostatic interactions, forming the Na-interlinked trans-dimers, which are energetically more favorable than the situation without any interactions. Then, with the assistance of Na, trans-dimers underwent trans-to-cis isomerization, forming the energetically more stable cis Kagome networks via fourfold Na⋯N electrostatic interactions. Additionally, the competitive preference of PT to interact with Na over cis-DEBPy further resulted in the removal of Na and allowed the reverse cis-to-trans isomerization accompanied by the ring-to-chain conversion. The ring-chain equilibrium was thus controllably directed by subtly regulating the electrostatic interactions between Na and organic ligands. As an extension, such Na⋯N electrostatic interactions were further integrated into the on-surface reactions, which showed the potential in controlling the selectivity of covalent reaction products transformed from isomerically specific trans-chains (with intrinsic Ag adatoms) to cis-rings (in the presence of Na) on Ag(111).136 These two cases undoubtedly demonstrated the close connection between metal–organic interactions (and corresponding nanostructures) and covalent nanostructures.
3.2 Transition metals
In the construction of metal–organic nanostructures, the coordination diversity between transition metals and organic ligands is crucial for inducing structural transformations.106,137 By meticulously adjusting the co-deposition ratio of metals to organic molecules, variations in the coordination modes can generally be achieved, leading to structural phase transitions.33,105,106,138 For instance, by continuously depositing Cu atoms to ligand molecules containing dicarbonitrile groups, the corresponding stoichiometric ratio was altered, inducing a series of structural transformations from 1D self-assembled molecular wires to 2D hexagonal porous networks, culminating in the formation of 1D metal–organic nanochains.93Interestingly, Xu et al. uncovered a unique phenomenon that the phase transitions of metal–organic nanostructures could be triggered without alteration in the local stoichiometric ratio in the elementary coordinated motifs.139 It was observed that the co-deposition of thymine (T) and Ni atoms onto Au(111) resulted in the formation of 1D metal–organic nanochains, composed of coordinated trimers with T molecules of alternating chiralities (left part in Fig. 9). The central Ni atom coordinates with T molecules via O2 or O4 sites, and the adjacent trimers are connected by N–H⋯O hydrogen bonds. Notably, by introducing additional T molecules and subsequent annealing at 370 K, the molecular chains were observed to transform into 2D hybrid network structures, with T molecules in all coordinated trimers becoming homochiral and coordinating with Ni atoms only via specific O4 sites (right part in Fig. 9). This resulting network structure consists of alternating coordinated trimers and hydrogen-bonded dimers. Throughout this transformation, the local stoichiometric ratio of these two coordinated motifs remained constant at 1:3. Furthermore, the reverse transformation from the hybrid network structure to the metal–organic nanochains was accessible by the addition of more Ni atoms and annealing procedure, demonstrating the reversibility of the two metal–organic trimers. Thus, this study not only realized reversible transitions between two different metal–organic trimers, but also provided insights into dynamic coordination chemistry on surfaces.
 | ||
| Fig. 9 Reversible structural transformation of Ni-based metal–organic nanostructures on Au(111) in response to additional T molecules or Ni atoms. Reproduced with permission.139 Copyright 2016 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Moreover, STM manipulations have emerged as a potent tool for surface chemistry, with a wide range of applications in the creation of novel nanostructures. This technique can not only facilitate the translation, rotation, and chirality alteration of single molecules, but can also induce phase transitions in metal–organic nanostructures by the disruption or reformation of chemical bonds.99,101,140–143 As depicted in Fig. 10, the precise control of the cleavage and reformation of hydrogen bonds enabled the reversible transformation of metal–organic nanostructures.144 In this study, uracil (U) was selected for potential coordination with Ni atoms (Fig. 10a). The co-deposition of U molecules and Ni atoms on Au(111) resulted in the formation of triangular motifs (based on the Ni⋯O coordination) with different orientations (Fig. 10b). Subsequent thermal treatment at 400 K facilitated the growth of parallelogram-shaped clusters and the emergence of more complex clusters via the formation of N–H⋯O hydrogen bonds between triangular motifs. Fig. 10c illustrates the structural transformation induced by lateral manipulations (as indicated by the green arrows), which led to the disassembly of these advanced clusters, leaving the triangular motifs undisturbed. A reverse reassembly process could also be induced by seamlessly stitching them back together with the STM tip. This phenomenon also suggests that lateral manipulations can be applied to discriminate the hierarchy of intermolecular interactions.
 | ||
| Fig. 10 Structural transformation of metal–organic clusters on Au(111) induced by STM manipulation. Reproduced with permission.144 Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In this section, it has been established that the modulation of the stoichiometric ratios of metals to molecules, temperature control, and STM manipulation are critical in inducing structural transformations. Furthermore, the influence of small molecules, such as water (H2O), carbon dioxide (CO2), nitric oxide (NO), etc., on intermolecular interactions has also been demonstrated to be significant, with the potential to lead to the collapse of network structures.145–148 For example, in a recent study, Wu and coworkers observed that exposing 2D STs composed of Fe and tris(3-phenylphosphine) cyanide (C3PC) molecules to CO2 and CO atmospheres could induce structural transitions from 2D STs to 1D chain-like structures.149
3.3 Lanthanide metals
Lanthanide-based metal–organic nanostructures, similar to those involving alkali, alkaline earth, and transition metals as discussed above, exhibit a remarkable diversity and propensity for phase transitions, which are pivotal in the context of materials science and allow for the dynamic alteration of the structural and functional attributes of the nanostructures.45,111 The variability of the coordination environment and the unique electronic properties of the lanthanide ions contribute to the rich and complex behavior of these materials.150In addition to thermal treatment as a means of inducing phase transitions,120 another commonly used method is to modulate the stoichiometric ratios of molecules to metals,119,121,122 which leverages the delicate balance of coordination numbers and interactions between lanthanide metals and organic ligands within the metal–organic nanostructures. By modifying the ratios, it is possible to control the assembly of the constituents, resulting in the formation of distinct structural phases with varied properties. In Fig. 11a, p-NC-(Ph)4-CN-p (qdc), featuring terminal carbonitrile groups, was chosen to construct europium (Eu)-based metal–organic nanostructures with varying coordination numbers by precisely adjusting the stoichiometric ratios of metals to ligands.151 Through precise stoichiometric modulation, a continuum of nanostructures can be engineered, encompassing amorphous string-like networks based on 3-fold coordination nodes, advancing to grid-like configurations with 4-fold coordination arrangements, and further evolving into intricate tiling patterns anchored by 5-fold coordination nodes. This progression culminated in the formation of the ubiquitous hexagonal lattices characterized by 6-fold coordination nodes. Notably, this structural transformation was driven by the combination of the ionic nature of these metal–organic bonds, the modulation of the ratio between Eu and ligand molecules, and the intrinsic properties of lanthanide metals.116,152 Such Eu-based metal–organic bonds, which are predominantly ionic with minimal covalent contributions, facilitated the rearrangement of metal centers and ligands, thereby enhancing the flexibility of these coordination architectures. Moreover, the larger size of the Eu atom, in conjunction with the characteristic preference for higher coordination environments, ensured the capacity to accommodate an increased number of ligand molecules. This adaptability eventually led to an expansion in the number of coordination nodes, propelling the system towards the phase transformation to a more stable structure.
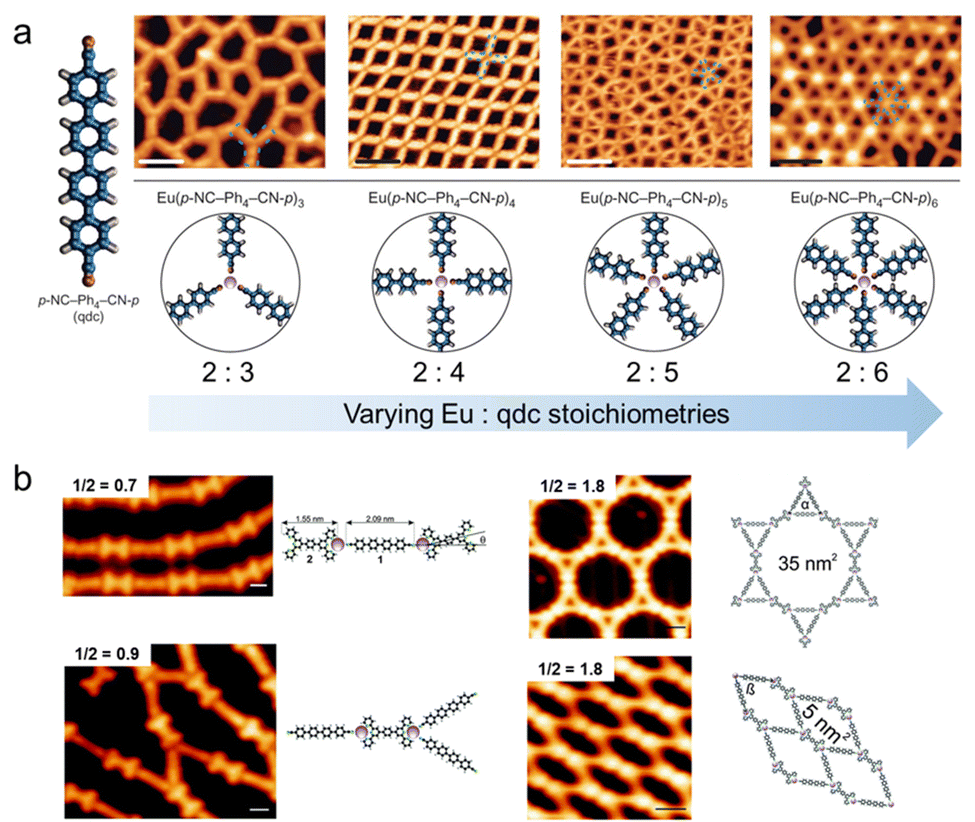 | ||
| Fig. 11 Structural transformation of lanthanide-based metal–organic nanostructures. (a) Various coordination networks obtained at increasing Eu:qdc stoichiometric ratios on Au(111). Reproduced with permission.151 Copyright 2016 Springer Nature Limited. (b) Supramolecular phases formed at different ligand stoichiometries (i.e., ligand 1 to ligand 2). Reproduced with permission.153 Copyright 2016 The Royal Society of Chemistry. | ||
In multimolecular systems, where two or more distinct molecular species coexist and interact, the regulation of their relative proportions also serves as a viable means of inducing phase transitions.153Fig. 11b illustrates the impact of altering the ratio of molecules 1 and 2, both functionalized with pyridyl groups, on the formation of distinct metal–organic nanostructures. At an initial molecular stoichiometric ratio of 0.7, alternating coordination of molecules 1 and 2 with Eu was observed, leading to the formation of 1D supramolecular chains. As the ratio was incrementally increased to 0.9, the structure evolved into a hybrid nanostructure, a mixture of 1D chains and networks inter-connected by the three-way junctions. Upon further increasing the ratio to 1.8, two distinct coexisting metallosupramolecular networks appeared, displaying hexagonal and fishnet assemblies, respectively. This structural transformation underscores the sensitivity of metal–organic nanostructures to the stoichiometry of their constituent ligands.
Another feasible way to control the phase transition of metal–organic nanostructures is to adjust the supramolecular pressure (i.e., change the amount of deposited molecular species and lanthanide atoms) while maintaining the stoichiometric ratio.125 Moreover, structural transformation can also be induced by lateral manipulations with a STM tip.154 In this scenario, the STM tip is used to nudge or move molecules laterally across the surface, effectively altering their relative positions and interactions with the central metal, leading to a direct change in the coordination number of the molecules. As shown in Fig. 12a, the successful construction of a pentagon composed of five pentamers (via the coordination between a central Gd atom and five adjacent p-NC-(Ph)4-CN-p molecules) was achieved by lateral manipulations, demonstrating the ability of STM manipulations to locally construct complex molecule-organic nanostructures with high precision. More interestingly, transformations between pentameric entity and tetramers were also achieved by directly manipulating the coordination nodes (Fig. 12b). Moreover, the newly generated pentamers and tetramers, once formed through STM manipulations, retained the stability necessary to be translated across the surface without significant structural disruption. This case thus nicely demonstrates an alternative strategy for the preparation and modification of specific metal–organic nanostructures in a localized manner.
 | ||
| Fig. 12 Structural transformation of Gd-supramolecules induced by STM lateral manipulations on Ag(111). (a) Formation of a pentagon by displacing five pentamers. (b) In situ tailoring and displacement of Gd–carbonitrile metal–organic supramolecules. Reproduced with permission.154 Copyright 2014 American Chemical Society. | ||
In addition, some other related studies have been nicely reviewed45,111 and are less discussed here to avoid repetition.
3.4 Bimetals
In this section, the construction of metal–organic nanostructures in composite systems that integrate multiple metal ions will be mainly discussed.155,156 Within this diverse landscape, bimetallic metal–organic nanostructures have emerged as a subject of considerable interest due to their unique structural and functional attributes.157–159 In particular, in comparison with single-metal systems, bimetallic MOFs have been shown to exhibit enhanced adsorption selectivity, augmented catalytic activity, and improved structural stability due to the synergistic interactions between metal nodes and the concomitant modulation of electronic structures.158,160–163Significant advances have been made in the synthesis of multimetallic metal–organic nanostructures in wet chemical environments.164 However, research on these structures in the field of surface chemistry remains relatively scarce.165,166 For example, organic ligands functionalized with pyridyl groups have been extensively explored by Lin and coworkers in the preparation of metal–organic nanostructures. Among others, 1,3,5-tris(pyridyl)benzene (TPyB) molecules interact with Cu atoms via N sites on the pyridyl moieties to form coordination bonds, leading to the construction of honeycomb-like network structures.33 Surprisingly, further introduction of Bi atoms into this structure resulted in the formation of two distinct binuclear metal–organic nanoclusters with multilayered interactions, including coordination bonds between pyridyl groups and Cu, as well as metal–metal bonds between Bi and Cu.167 This case indicates that the integration of another metal element would provide unexpected effects on the corresponding metal–organic nanostructures as well as additional intermolecular interactions involved. Consequently, it is crucial to investigate the synergistic and competitive relationships between different interactions involved in bimetallic nanostructures to gain a deeper understanding of the preparation rules and structural and property modifications.
Uracil (U) molecules, possessing potential sites for both ionic and coordination bonds, are considered ideal precursors for the construction of metal–organic nanostructures with both alkali and transition metals. In the study shown in Fig. 13,43 co-deposition of U and Na on Au(111), followed by annealing at 350 K, resulted in the formation of a metal–organic network structure based on U4Na1 tetramers via electrostatic interactions between Na and O sites (the upper panel of Fig. 13b). Interestingly, the introduction of Cs atoms into the structure and subsequent annealing at 370 K led to a structural transformation with the formation of 10-membered bimetallic motifs, which consisted of two U5Cs1 pentamers connected by a single Na atom and further organized into a different network structure. After further deposition of Na and annealing at 430 K, a U10Na4 network was formed, in which U molecules interacted with the central Na atoms, while Cs atoms stayed in the cavity and appeared as bright protrusions. This phase transition process reveals the synergistic and competitive electrostatic interactions between U and Cs and Na.
 | ||
| Fig. 13 Structural transformation of different 2D metal–organic networks on Au(111), including two bimetallic structures. (a) Illustration of the formation of U4Na1, U10Cs2Na1, U10Na4, U6Fe2, and U12Na4Fe2 structures. (b) Structural transformations starting from the U4Na1 structure induced by Cs and Fe, respectively. Reproduced with permission.43 Copyright 2021 American Chemical Society. | ||
Beyond alkali metals, transition metal Fe atoms were also introduced into the U4Na1 structure, followed by annealing at 800 K (the lower panel of Fig. 13b). This resulted in the formation of a highly porous U6Fe2 structure, with a central Fe atom coordinated to three U molecules, indicating the competition between electrostatic interactions and coordination bonds during this phase transition. Further deposition of Na led to the formation of a new U12Na4Fe2 bimetallic structure, with two U6Na2 hexamers connected by two Fe atoms, under the synergistic effects of electrostatic interactions and coordination bonds. Thus, it can be concluded that the synergy and competition between different electrostatic ionic interactions and coordination bonds, along with hydrogen bonds, enable the construction of various metal–organic nanostructures on metal surfaces and also facilitate phase transitions.
Apart from the synergy and competition among various types of intermolecular interactions, the different binding strengths and ground state electronic configurations of coordination centers on surfaces confer a degree of selectivity during coordination with different ligands.168 For instance, when both Cu and Fe atoms are involved in the assembly with tripyridyl ligands on a Au(111) surface, Fe–pyridyl coordination is thermodynamically and kinetically favored.169 Taking advantage of the higher binding energy between Cr atoms and 2,3,6,7,10,11-hexaiminotriphenylene (HITP) ligands compared to that between Ni and HITP, single-layer Cr3(HITP)2, a Cr-based 2D metal–organic framework, was successfully synthesized on Au(111) by metal substitution of Ni with Cr (Fig. 14).170 Notably, the Cr3(HITP)2 frameworks were inaccessible via direct deposition of 2,3,6,7,10,11-hexaaiminotriphenylene (HATP) molecules and Cr atoms. As shown in Fig. 14, the deprotonation of HATP molecules on the surface and their subsequent coordination with Ni led to the formation of the Ni3(HITP)2 hexagonal framework. Unexpectedly, upon the deposition of excess Cr atoms and subsequent annealing at 520 K, bright spots were observed at the joints, which were attributed to Cr atoms replacing Ni due to their higher apparent height, forming a bimetallic (NixCr3−x)(HITP)2 network. As the concentration of Cr increased, a complete substitution of Ni by Cr occurred with the complete transformation to a Cr3(HITP)2 network. This study illustrates a novel on-surface preparation strategy that exploits the coordination competition between different metals to facilitate the construction of specific metal–organic nanoarchitectures via metal substitution, which otherwise cannot be directly constructed.
 | ||
| Fig. 14 Structural transformation by metal substitution from Ni3(HITP)2, through (NixCr3−x)(HITP)2, eventually to Cr3(HITP)2 frameworks on Au(111) in response to the addition of Cr atoms. Reproduced with permission.170 Copyright 2023 Wiley-VCH GmbH. | ||
Overall, this section explores the critical factors that induce phase transitions in metal–organic nanostructures, including the balance of molecular-to-metal ratios, temperature, molecular coverage, introduction of competing counterparts, integration of other metals, and STM manipulations. Furthermore, to illustrate the impact of these factors more concretely, we have presented a series of case studies on metal–organic nanostructures based on various metal systems.
4. Conclusions and outlook
Through a combination of high-resolution SPM techniques and state-of-the-art theoretical calculations, various metal–organic nanostructures and their structural transformations on metal surfaces have been visualized in real space and rationalized at the single-bond level. Regarding the types of metals embedded in the target metal–organic nanostructures, s-block alkali (and alkaline earth) metals, d-block transition metals, and f-block lanthanide (and actinide) metals have been extensively introduced into molecular systems on surfaces, resulting in the different intermolecular bonding (e.g., electrostatic interactions and coordination bonds) and distinct characteristics in directionality, saturation, strength, etc., as discussed above in detail based on several typical examples. In addition, different sources of metals, such as intrinsic metal adatoms or extrinsic metal atoms and inorganic salts, have been applied, which may affect the structural transformations to some extent due to the potential competition between counterpart anions and organic molecules in grabbing the metal cations or the structural diversity further provided by the counterpart anions in interacting with the metal–organic motifs. Experimentally, these structural motifs have been identified in nanostructures on metal surfaces based on the submolecular morphologies and bond-resolved molecular skeletons, revealing different interaction modes including the aggregation numbers, bonding sites, and angles. Accordingly, the metal–organic interactions involved and their synergy and competition have been clearly discriminated with the assistance of theoretical calculations. Moreover, the corresponding structural evolutions have been controllably directed by the subtle tuning of such intermolecular interactions and have been evidenced to be responsive to some common regulatory factors (internal or external conditions), such as the stoichiometric ratios of metals to organic molecules (including situations realized by local SPM manipulations) and thermodynamic control predominantly, the introduction of extrinsic competing counterparts, the functionalization of organic molecules, and the choice of substrates and lattices, thereby providing mechanistic insights into precise structural and property control.Apart from the above aspects, it is also worth noting that although the metal–organic interactions are generally treated as relatively flexible and reversible, with a wide range of bond strengths generally weaker than carbon–carbon (C–C) covalent bonds, they can somehow determine or at least influence the construction of C–C bonded nanostructures. The seminal work of Lin et al. demonstrated the power of the metal-directed template (by virtue of the pyridyl–Cu–pyridyl coordination) in the efficient control of on-surface polymerization171 and further revealed the critical role of metal–organic preassembly in directing the construction of covalent bonds. It thus inspired a vast number of studies of on-surface reactions and synthesis based on molecular preassembly. Subsequently, such specific metal–organic interactions (e.g., between Fe/Cu/Ag/Na atoms and N sites of pyridyl groups, which control the molecular conformation of molecular precursors76,136,172,173 or the C–H activation site174) have been widely applied to provide coordination (or electrostatic interaction) templates,175 leading to the selective covalent coupling pathways or the formation of covalent nanostructures with specific molecular conformations. Such a reaction scenario is schematically illustrated in the upper panel of Fig. 15.
 | ||
| Fig. 15 Prospects for the application of metal–organic interactions and nanostructures in the regulation of on-surface reaction pathways, barriers, products, etc., which further influence the structures, processes, and properties. | ||
In addition, extrinsic metal atoms, including typical transition metals (e.g., Cu,171 Pd,176 Ni,177 and Au178) and lanthanide metals (e.g., Dy179), have been introduced into on-surface reactions, interacting with molecular precursors or embedding in corresponding intermediate structures, and have been proven to be efficient catalysts for lowering reaction temperatures or barriers. More recently, the alkali metal Na was further introduced into on-surface dehalogenative reactions by our group, where Na strongly interacts with dissociated intermolecular halogens and intramolecular halogens of molecular precursors,61 leading to the C–Br activation under mild conditions and halogen separation in the form of salt islands. Such studies show the potential of metal–organic interactions to lower reaction barriers and possibly also to stabilize previously unfavorable reaction products by regulating reaction equilibria (as shown in the right panel of Fig. 15). Aside from the catalytic effects provided by a single type of metal atoms, it is also noteworthy that cooperative catalysis involving the incorporation of two types of metals (e.g., Na and Ag herein) was also proposed in the previous report,134 indicating the great promise of bimetallic or even multimetallic catalysis in the on-surface synthesis stemming from the metal–organic interactions. It is also expected that the integration of metal atoms in combination with other extrinsic components would provide unpredictable opportunities, stimulating researchers to delve deeply into the related molecule-based catalytic systems on surfaces.180
Moreover, in our most recent work, metal–organic interactions have been further corroborated to provide additional intermolecular interactions not only to molecular precursors but also to reaction products, more specifically, covalent organometallic nanostructures (Fig. 8),132 which controllably direct the ring-chain equilibrium and realize the dynamic covalent chemistry on the surface. This kind of protocol is also displayed in the left panel of Fig. 15.
In summary, metal–organic interactions are pivotal not only for the construction of well-designed, sophisticated metal–organic nanostructures, but also for the fine-tuning of on-surface reactions, including but not limited to directing reaction pathways, lowering reaction barriers (regulating reaction equilibria), and determining reaction products (by favoring specific intermolecular interactions). This kind of strategy from metal–organic interactions to covalent bonding thus delicately bridges the gap between supramolecular and covalent chemistry and offers limitless possibilities for the fabrication of nanostructures with specific desired structures, processes, and properties or functionalities.
Significant progress has been achieved within the realm of surface science, yet a series of challenges and issues persist in the practical applications. Firstly, although STM and AFM technologies provide atomic-level imaging capabilities, various limitations are evident in sample preparation, operational complexity, and data analysis, which require further combination and development of other techniques. Secondly, the majority of current research has focused on the exploration of metal–organic nanostructures on metal surfaces, with comparatively less attention paid to those on insulating substrates, thus limiting the application in a wider range of material systems. Finally, the translation of fundamental research into practical applications still requires overcoming numerous technical obstacles.
Author contributions
Rujia Hou and Chi Zhang contributed to the preparation, creation and writing of the initial draft; Chi Zhang and Wei Xu contributed to the supervision and revising the draft; Lei Xu and Yuanqi Ding contributed to revising the draft.Data availability
Data availability is not applicable to this article as no new data were created or analyzed in this review article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (Grants No. 22202153, 22125203, and 22102117), and the Fundamental Research Funds for the Central Universities.References
- A. Carné, C. Carbonell, I. Imaz and D. Maspoch, Chem. Soc. Rev., 2011, 40, 291–305 RSC.
- L. Chen, R. Luque and Y. Li, Chem. Soc. Rev., 2017, 46, 4614–4630 RSC.
- B. Y. Guan, X. Y. Yu, H. B. Wu and X. W. Lou, Adv. Mater., 2017, 29, 1703614 CrossRef PubMed.
- D. Liu, J. Wan, G. Pang and Z. Tang, Adv. Mater., 2019, 31, 1803291 CrossRef.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 Search PubMed.
- Y. Peng, Y. Li, Y. Ban, H. Jin, W. Jiao, X. Liu and W. Yang, Science, 2014, 346, 1356–1359 CrossRef CAS PubMed.
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671–679 Search PubMed.
- G. Binnig and H. Rohrer, Rev. Mod. Phys., 1987, 59, 615–625 CrossRef CAS.
- G. Binnig, C. F. Quate and C. Gerber, Phys. Rev. Lett., 1986, 56, 930–933 Search PubMed.
- G. Binnig, H. Rohrer, C. Gerber and E. Weibel, Phys. Rev. Lett., 1982, 49, 57–61 Search PubMed.
- B. Flemming, Rep. Prog. Phys., 1996, 59, 1737 Search PubMed.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110–1114 CrossRef CAS PubMed.
- M. F. Crommie, C. P. Lutz and D. M. Eigler, Science, 1993, 262, 218–220 Search PubMed.
- K. Bian, C. Gerber, A. J. Heinrich, D. J. Müller, S. Scheuring and Y. Jiang, Nat. Rev. Methods Primers, 2021, 1, 36 CrossRef CAS.
- R. García and R. Pérez, Surf. Sci. Rep., 2002, 47, 197–301 CrossRef.
- M. Radmacher, M. Fritz, H. G. Hansma and P. K. Hansma, Science, 1994, 265, 1577–1579 CrossRef CAS PubMed.
- Q. Zhong, D. Inniss, K. Kjoller and V. B. Elings, Surf. Sci., 1993, 290, L688–L692 CrossRef CAS.
- F. J. Giessibl, Rev. Mod. Phys., 2003, 75, 949–983 CrossRef CAS.
- B. Alldritt, P. Hapala, N. Oinonen, F. Urtev, O. Krejci, F. Federici Canova, J. Kannala, F. Schulz, P. Liljeroth and A. S. Foster, Sci. Adv., 2020, 6, eaay6913 CrossRef CAS PubMed.
- F. J. Giessibl, Rev. Sci. Instrum., 2019, 90, 011101 CrossRef PubMed.
- N. Lin, S. Stepanow, M. Ruben and J. V. Barth, Surface-Confined Supramolecular Coordination Chemistry, Springer-Verlag, Berlin, Heidelberg, Germany, 2009 Search PubMed.
- R. Otero, F. Hümmelink, F. Sato, S. B. Legoas, P. Thostrup, E. Lægsgaard, I. Stensgaard, D. S. Galvão and F. Besenbacher, Nat. Mater., 2004, 3, 779–782 CrossRef CAS PubMed.
- P. Maksymovych, D. C. Sorescu, O. Voznyy and J. T. Yates, J. Am. Chem. Soc., 2013, 135, 4922–4925 CrossRef CAS PubMed.
- P. Maksymovych, O. Voznyy, D. B. Dougherty, D. C. Sorescu and J. T. Yates, Prog. Surf. Sci., 2010, 85, 206–240 CrossRef CAS.
- S. De Feyter and F. C. De Schryver, Chem. Soc. Rev., 2003, 32, 139–150 RSC.
- S.-S. Li, B. H. Northrop, Q.-H. Yuan, L.-J. Wan and P. J. Stang, Acc. Chem. Res., 2009, 42, 249–259 CrossRef CAS PubMed.
- D. Wang, Q. Chen and L.-J. Wan, Phys. Chem. Chem. Phys., 2008, 10, 6467–6478 RSC.
- Y. Yang and C. Wang, Chem. Soc. Rev., 2009, 38, 2576–2589 RSC.
- X.-H. Liu, C.-Z. Guan, D. Wang and L.-J. Wan, Adv. Mater., 2014, 26, 6912–6920 CrossRef CAS PubMed.
- K. S. Mali, J. Adisoejoso, E. Ghijsens, I. De Cat and S. De Feyter, Acc. Chem. Res., 2012, 45, 1309–1320 CrossRef CAS PubMed.
- F. Bebensee, K. Svane, C. Bombis, F. Masini, S. Klyatskaya, F. Besenbacher, M. Ruben, B. Hammer and T. R. Linderoth, Angew. Chem., Int. Ed., 2014, 53, 12955–12959 CrossRef CAS PubMed.
- J. Liu, T. Lin, Z. Shi, F. Xia, L. Dong, P. N. Liu and N. Lin, J. Am. Chem. Soc., 2011, 133, 18760–18766 CrossRef CAS PubMed.
- L. Xie, C. Zhang, Y. Ding and W. Xu, Angew. Chem., Int. Ed., 2017, 56, 5077–5081 CrossRef CAS PubMed.
- A. Langner, S. L. Tait, N. Lin, R. Chandrasekar, V. Meded, K. Fink, M. Ruben and K. Kern, Angew. Chem., Int. Ed., 2012, 51, 4327–4331 CrossRef CAS PubMed.
- C. Zhang, L. Wang, L. Xie, H. Kong, Q. Tan, L. Cai, Q. Sun and W. Xu, ChemPhysChem, 2015, 16, 2099–2105 CrossRef CAS PubMed.
- L. Xie, H. Lin, C. Zhang, J. Li, N. Merino-Díez, N. Friedrich, X. Bouju, Y. Li, J. I. Pascual and W. Xu, ACS Nano, 2019, 13, 9936–9943 CrossRef CAS PubMed.
- A. C. Papageorgiou, S. Fischer, S. C. Oh, Ö. Sağlam, J. Reichert, A. Wiengarten, K. Seufert, S. Vijayaraghavan, D. Écija, W. Auwärter, F. Allegretti, R. G. Acres, K. C. Prince, K. Diller, F. Klappenberger and J. V. Barth, ACS Nano, 2013, 7, 4520–4526 CrossRef CAS PubMed.
- R. Hou, Y. Guo, Z. Yi, Z. Zhang, C. Zhang and W. Xu, J. Phys. Chem. Lett., 2023, 14, 3636–3642 CrossRef CAS PubMed.
- Y. Ding, X. Wang, D. Li and W. Xu, J. Phys. Chem. C, 2020, 124, 5257–5262 Search PubMed.
- J. Dai, X. Zhao, Z. Peng, J. Li, Y. Lin, X. Wen, L. Xing, W. Zhao, J. Shang, Y. Wang, J. Liu and K. Wu, J. Am. Chem. Soc., 2023, 145, 13531–13536 CrossRef CAS PubMed.
- D. Ecija, J. I. Urgel, A. C. Papageorgiou, S. Joshi, W. Auwärter, A. P. Seitsonen, S. Klyatskaya, M. Ruben, S. Fischer, S. Vijayaraghavan, J. Reichert and J. V. Barth, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6678–6681 CrossRef CAS PubMed.
- D. Li, Y. Ding, X. Wang and W. Xu, J. Phys. Chem. Lett., 2021, 12, 5228–5232 CrossRef CAS.
- T. Vitova, P. W. Roesky and S. Dehnen, Commun. Chem., 2022, 5, 12 CrossRef CAS PubMed.
- D. Écija, J. I. Urgel, A. P. Seitsonen, W. Auwärter and J. V. Barth, Acc. Chem. Res., 2018, 51, 365–375 Search PubMed.
- C. Wäckerlin, C. Iacovita, D. Chylarecka, P. Fesser, T. A. Jung and N. Ballav, Chem. Commun., 2011, 47, 9146–9148 RSC.
- S. Stepanow, R. Ohmann, F. Leroy, N. Lin, T. Strunskus, C. Wöll and K. Kern, ACS Nano, 2010, 4, 1813–1820 CrossRef CAS PubMed.
- N. Abdurakhmanova, A. Floris, T.-C. Tseng, A. Comisso, S. Stepanow, A. De Vita and K. Kern, Nat. Commun., 2012, 3, 940 CrossRef PubMed.
- T. K. Shimizu, J. Jung, H. Imada and Y. Kim, Angew. Chem., Int. Ed., 2014, 53, 13729–13733 CrossRef CAS PubMed.
- D. Skomski, S. Abb and S. L. Tait, J. Am. Chem. Soc., 2012, 134, 14165–14171 CrossRef CAS PubMed.
- D. Skomski and S. L. Tait, J. Phys. Chem. C, 2013, 117, 2959–2965 CrossRef CAS.
- W. Xu, J.-G. Wang, M. Yu, E. Lægsgaard, I. Stensgaard, T. R. Linderoth, B. Hammer, C. Wang and F. Besenbacher, J. Am. Chem. Soc., 2010, 132, 15927–15929 CrossRef CAS PubMed.
- W. Xu, Q. Tan, M. Yu, Q. Sun, H. Kong, E. Laegsgaard, I. Stensgaard, J. Kjems, J.-G. Wang, C. Wang and F. Besenbacher, Chem. Commun., 2013, 49, 7210–7212 RSC.
- C. Zhang, L. Xie, L. Wang, H. Kong, Q. Tan and W. Xu, J. Am. Chem. Soc., 2015, 137, 11795–11800 CrossRef CAS PubMed.
- Y. Ding, L. Xie, D. Li, H. Shen, C. Li and W. Xu, Chem. Commun., 2022, 58, 3198–3201 RSC.
- L. Xie, C. Zhang, Y. Ding, W. E, C. Yuan and W. Xu, Chem. Commun., 2017, 53, 8767–8769 RSC.
- L. Xie, Y. Ding, X. Wang and W. Xu, Phys. Chem. Chem. Phys., 2019, 21, 9357–9361 Search PubMed.
- X. Wang, Y. Ding, D. Li, L. Xie and W. Xu, Chem. Commun., 2019, 55, 12064–12067 RSC.
- Y. Ding, X. Wang, D. Li, L. Xie and W. Xu, ACS Nano, 2019, 13, 6025–6032 CrossRef CAS PubMed.
- J. Hieulle, D. Peyrot, Z. Jiang and F. Silly, Chem. Commun., 2015, 51, 13162–13165 RSC.
- Z. Zhang, Y. Gao, Z. Yi, C. Zhang and W. Xu, ACS Nano, 2024, 18, 9082–9091 CrossRef CAS PubMed.
- Q. Guo, M. Huang, S. Lu and G. Cao, Nanotechnology, 2015, 26, 275603 CrossRef PubMed.
- D. Peyrot and F. Silly, J. Phys. Chem. C, 2017, 121, 20986–20993 CrossRef CAS.
- H. Shan, L. Zhou, W. Ji and A. Zhao, J. Phys. Chem. Lett., 2021, 12, 10808–10814 CrossRef CAS PubMed.
- D.-X. Yang, P.-F. Wang, H.-Y. Liu, Y.-H. Zhang, P.-P. Sun and F.-N. Shi, J. Solid State Chem., 2022, 309, 122947 CrossRef CAS.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- Y. He, Z. Yin, Z. Wang, H. Wang, W. Xiong, B. Song, H. Qin, P. Xu and G. Zeng, J. Mater. Chem. A, 2022, 10, 9788–9820 Search PubMed.
- S. Zheng, X. Li, B. Yan, Q. Hu, Y. Xu, X. Xiao, H. Xue and H. Pang, Adv. Energy Mater., 2017, 7, 1602733 CrossRef.
- G. Chen, Z. Li, Z. Huang, H. Lu, G. Long, J. S. Lezama Pacheco, J. B.-H. Tok, T. Z. Gao, Y. Lei, J. Zhou and Z. Bao, ACS Nano, 2023, 17, 9611–9621 CrossRef CAS PubMed.
- F. Sun, Q. Li, H. Xue and H. Pang, ChemElectroChem, 2019, 6, 1273–1299 CrossRef CAS.
- J. Panda, S. P. Tripathy, S. Dash, A. Ray, P. Behera, S. Subudhi and K. Parida, Nanoscale, 2023, 15, 7640–7675 RSC.
- H. Kong, L. Wang, Q. Tan, C. Zhang, Q. Sun and W. Xu, Chem. Commun., 2014, 50, 3242–3244 RSC.
- M. Knor, H.-Y. Gao, S. Amirjalayer, A. Studer, H. Gao, S. Du and H. Fuchs, Chem. Commun., 2015, 51, 10854–10857 Search PubMed.
- X. Zhang, N. Li, H. Wang, C. Yuan, G. Gu, Y. Zhang, D. Nieckarz, P. Szabelski, S. Hou, B. K. Teo and Y. Wang, ACS Nano, 2017, 11, 8511–8518 CrossRef CAS PubMed.
- J. Xiong, T. Qin, L. Hu, W. Yang, Z. Chen, H. Ding, J. Hu, Q. Xu and J. Zhu, J. Phys. Chem. Lett., 2023, 14, 9787–9792 CrossRef CAS PubMed.
- L. Song, J. Wang, H. Zhu, P. Huang, H. Lin, L. Chi and Q. Li, J. Phys. Chem. Lett., 2023, 14, 11286–11291 CrossRef CAS PubMed.
- Q. Sun, L. Cai, H. Ma, C. Yuan and W. Xu, Chem. Commun., 2015, 51, 14164–14166 RSC.
- M. Hua, B. Xia, M. Wang, E. Li, J. Liu, T. Wu, Y. Wang, R. Li, H. Ding, J. Hu, Y. Wang, J. Zhu, H. Xu, W. Zhao and N. Lin, J. Phys. Chem. Lett., 2021, 12, 3733–3739 CrossRef CAS PubMed.
- C. S. Kley, J. Čechal, T. Kumagai, F. Schramm, M. Ruben, S. Stepanow and K. Kern, J. Am. Chem. Soc., 2012, 134, 6072–6075 CrossRef CAS PubMed.
- S. Stepanow, N. Lin, D. Payer, U. Schlickum, F. Klappenberger, G. Zoppellaro, M. Ruben, H. Brune, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2007, 46, 710–713 CrossRef CAS PubMed.
- L. Song, B. Yang, F. Liu, K. Niu, Y. Han, J. Wang, Y. Zheng, H. Zhang, Q. Li and L. Chi, J. Phys. Chem. C, 2020, 124, 12390–12396 CrossRef CAS.
- J. He, C. Yang, Z. Xu, M. Zeller, A. D. Hunter and J. Lin, J. Solid State Chem., 2009, 182, 1821–1826 CrossRef CAS.
- X. Meng, E. Kolodzeiski, X. Huang, A. Timmer, B. Schulze Lammers, H.-Y. Gao, H. Mönig, L. Liu, W. Xu, S. Amirjalayer, D. Zhu and H. Fuchs, ChemNanoMat, 2020, 6, 1479–1484 Search PubMed.
- H. Walch, J. Dienstmaier, G. Eder, R. Gutzler, S. Schlögl, T. Sirtl, K. Das, M. Schmittel and M. Lackinger, J. Am. Chem. Soc., 2011, 133, 7909–7915 CrossRef CAS PubMed.
- L. Cai, Q. Sun, M. Bao, H. Ma, C. Yuan and W. Xu, ACS Nano, 2017, 11, 3727–3732 Search PubMed.
- S.-W. Li, R.-X. Zhang, L.-X. Kang, D.-Y. Li, Y.-L. Xie, C.-X. Wang and P.-N. Liu, ACS Nano, 2021, 15, 18014–18022 Search PubMed.
- B. Yang, N. Cao, H. Ju, H. Lin, Y. Li, H. Ding, J. Ding, J. Zhang, C. Peng, H. Zhang, J. Zhu, Q. Li and L. Chi, J. Am. Chem. Soc., 2019, 141, 168–174 CrossRef CAS PubMed.
- Y. Zhang, J. Lu, W. Gao, Y. Zhang, N. Li, S. Li, G. Niu, B. Fu, L. Gao and J. Cai, Chin. J. Chem., 2024, 42, 2717–2722 CrossRef CAS.
- H. Zhang, Z. Gong, K. Sun, R. Duan, P. Ji, L. Li, C. Li, K. Müllen and L. Chi, J. Am. Chem. Soc., 2016, 138, 11743–11748 CrossRef CAS PubMed.
- P. C. M. Grim, S. De Feyter, A. Gesquière, P. Vanoppen, M. Rüker, S. Valiyaveettil, G. Moessner, K. Müllen and F. C. De Schryver, Angew. Chem., Int. Ed. Engl., 1997, 36, 2601–2603 CrossRef CAS.
- C. Lyu, Y. Gao, K. Zhou, M. Hua, Z. Shi, P.-N. Liu, L. Huang and N. Lin, ACS Nano, 2024, 18, 19793–19801 CAS.
- J. Li, L. Solianyk, N. Schmidt, B. Baker, S. Gottardi, J. C. Moreno Lopez, M. Enache, L. Monjas, R. van der Vlag, R. W. A. Havenith, A. K. H. Hirsch and M. Stöhr, J. Phys. Chem. C, 2019, 123, 12730–12735 CrossRef CAS PubMed.
- F. Bischoff, Y. He, K. Seufert, D. Stassen, D. Bonifazi, J. V. Barth and W. Auwärter, Chem. – Eur. J., 2016, 22, 15298–15306 Search PubMed.
- A. Dmitriev, H. Spillmann, N. Lin, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2003, 42, 2670–2673 CrossRef CAS PubMed.
- P. Messina, A. Dmitriev, N. Lin, H. Spillmann, M. Abel, J. V. Barth and K. Kern, J. Am. Chem. Soc., 2002, 124, 14000–14001 CrossRef CAS PubMed.
- M. Yu, Y. Benjalal, C. Chen, N. Kalashnyk, W. Xu, R. Barattin, S. Nagarajan, E. Lægsgaard, I. Stensgaard, M. Hliwa, A. Gourdon, F. Besenbacher, X. Bouju and T. R. Linderoth, Chem. Commun., 2018, 54, 8845–8848 RSC.
- Y. Zhang, X. Zhang, Y. Li, S. Zhao, S. Hou, K. Wu and Y. Wang, J. Am. Chem. Soc., 2020, 142, 17928–17932 Search PubMed.
- C. Krull, M. Castelli, P. Hapala, D. Kumar, A. Tadich, M. Capsoni, M. T. Edmonds, J. Hellerstedt, S. A. Burke, P. Jelinek and A. Schiffrin, Nat. Commun., 2018, 9, 3211 CrossRef PubMed.
- C. Martín-Fuentes, S. O. Parreiras, J. I. Urgel, V. Rubio-Giménez, B. Muñiz Cano, D. Moreno, K. Lauwaet, M. Valvidares, M. A. Valbuena, P. Gargiani, W. Kuch, J. Camarero, J. M. Gallego, R. Miranda, J. I. Martínez, C. Martí-Gastaldo and D. Écija, J. Am. Chem. Soc., 2022, 144, 16034–16041 CrossRef PubMed.
- L. Wang, H. Kong, C. Zhang, Q. Sun, L. Cai, Q. Tan, F. Besenbacher and W. Xu, ACS Nano, 2014, 8, 11799–11805 CrossRef CAS PubMed.
- J. Klingele, S. Dechert and F. Meyer, Coord. Chem. Rev., 2009, 253, 2698–2741 CrossRef CAS.
- J. F. Berry and C. C. Lu, Inorg. Chem., 2017, 56, 7577–7581 CrossRef CAS PubMed.
- I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS.
- C. Zhang, L. Xie, Y. Ding, C. Yuan and W. Xu, Phys. Chem. Chem. Phys., 2018, 20, 3694–3698 Search PubMed.
- C. Zhang, L. Wang, L. Xie, Y. Ding and W. Xu, Chem. – Eur. J., 2017, 23, 2356–2362 Search PubMed.
- M. Gustafsson, A. Bartoszewicz, B. Martín-Matute, J. Sun, J. Grins, T. Zhao, Z. Li, G. Zhu and X. Zou, Chem. Mater., 2010, 22, 3316–3322 CrossRef CAS.
- G.-P. Li, G. Liu, Y.-Z. Li, L. Hou, Y.-Y. Wang and Z. Zhu, Inorg. Chem., 2016, 55, 3952–3959 Search PubMed.
- H. Xu, C.-S. Cao, X.-M. Kang and B. Zhao, Dalton Trans., 2016, 45, 18003–18017 Search PubMed.
- Q.-Y. Yang, K. Li, J. Luo, M. Pan and C.-Y. Su, Chem. Commun., 2011, 47, 4234–4236 Search PubMed.
- S. O. Parreiras, J. M. Gallego and D. Écija, Chem. Commun., 2023, 59, 8878–8893 Search PubMed.
- S.-N. Zhao, L.-J. Li, X.-Z. Song, M. Zhu, Z.-M. Hao, X. Meng, L.-L. Wu, J. Feng, S.-Y. Song, C. Wang and H.-J. Zhang, Adv. Funct. Mater., 2015, 25, 1463–1469 Search PubMed.
- J.-M. Zhou, W. Shi, N. Xu and P. Cheng, Inorg. Chem., 2013, 52, 8082–8090 CrossRef CAS PubMed.
- Y. Zhang, S. Liu, Z.-S. Zhao, Z. Wang, R. Zhang, L. Liu and Z.-B. Han, Inorg. Chem. Front., 2021, 8, 590–619 RSC.
- F. Saraci, V. Quezada-Novoa, P. R. Donnarumma and A. J. Howarth, Chem. Soc. Rev., 2020, 49, 7949–7977 Search PubMed.
- J. I. Urgel, B. Cirera, Y. Wang, W. Auwärter, R. Otero, J. M. Gallego, M. Alcamí, S. Klyatskaya, M. Ruben, F. Martín, R. Miranda, D. Ecija and J. V. Barth, Small, 2015, 11, 6358–6364 Search PubMed.
- S. O. Parreiras, D. Moreno, B. Cirera, M. A. Valbuena, J. I. Urgel, M. Paradinas, M. Panighel, F. Ajejas, M. A. Niño, J. M. Gallego, M. Valvidares, P. Gargiani, W. Kuch, J. I. Martínez, A. Mugarza, J. Camarero, R. Miranda, P. Perna and D. Écija, Small, 2021, 17, 2102753 Search PubMed.
- S. O. Parreiras, D. Moreno, S. K. Mathialagan, B. Muñiz-Cano, C. Martín-Fuentes, M. Tenorio, L. Černa, J. I. Urgel, K. Lauwaet, M. Valvidares, M. A. Valbuena, J. M. Gallego, J. I. Martínez, P. Gargiani, R. Miranda, J. Camarero and D. Écija, Nanoscale, 2023, 15, 7267–7271 Search PubMed.
- M. Uphoff, G. S. Michelitsch, R. Hellwig, K. Reuter, H. Brune, F. Klappenberger and J. V. Barth, ACS Nano, 2018, 12, 11552–11560 CrossRef CAS PubMed.
- B. Cirera, L. Đorđević, R. Otero, J. M. Gallego, D. Bonifazi, R. Miranda and D. Ecija, Chem. Commun., 2016, 52, 11227–11230 RSC.
- D. Écija, J. I. Urgel, A. C. Papageorgiou, S. Joshi, W. Auwärter, A. P. Seitsonen, S. Klyatskaya, M. Ruben, S. Fischer, S. Vijayaraghavan, J. Reichert and J. V. Barth, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6678–6681 CrossRef PubMed.
- J. I. Urgel, D. Ecija, W. Auwärter, A. C. Papageorgiou, A. P. Seitsonen, S. Vijayaraghavan, S. Joshi, S. Fischer, J. Reichert and J. V. Barth, J. Phys. Chem. C, 2014, 118, 12908–12915 CrossRef CAS.
- Q. Fan, J.-N. Luy, M. Liebold, K. Greulich, M. Zugermeier, J. Sundermeyer, R. Tonner and J. M. Gottfried, Nat. Commun., 2019, 10, 5049 Search PubMed.
- D. Moreno, J. Santos, S. O. Parreiras, C. Martín-Fuentes, K. Lauwaet, J. I. Urgel, R. Miranda, N. Martín, J. M. Gallego and D. Écija, Chem. – Eur. J., 2023, 29, e202300461 Search PubMed.
- D. Moreno, S. O. Parreiras, J. I. Urgel, B. Muñiz-Cano, C. Martín-Fuentes, K. Lauwaet, M. Valvidares, M. A. Valbuena, J. M. Gallego, J. I. Martínez, P. Gargiani, J. Camarero, R. Miranda and D. Écija, Small, 2022, 18, 2107073 Search PubMed.
- P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef CAS PubMed.
- M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 Search PubMed.
- F. T. Edelmann, J. H. Farnaby, F. Jaroschik and B. Wilson, Coord. Chem. Rev., 2019, 398, 113005 Search PubMed.
- E. Rheinfrank, M. Pörtner, M. D. C. Nuñez Beyerle, F. Haag, P. S. Deimel, F. Allegretti, K. Seufert, J. V. Barth, M.-L. Bocquet, P. Feulner and W. Auwärter, J. Am. Chem. Soc., 2021, 143, 14581–14591 CrossRef CAS PubMed.
- Z. Yi, C. Zhang, Z. Zhang, R. Hou, Y. Guo and W. Xu, Precis. Chem., 2023, 1, 226–232 CrossRef CAS.
- Z. Yi, Y. Guo, R. Hou, Z. Zhang, Y. Gao, C. Zhang and W. Xu, J. Am. Chem. Soc., 2023, 145, 22366–22373 CrossRef CAS PubMed.
- R. Hou, Y. Gao, Y. Guo, C. Zhang and W. Xu, ACS Nano, 2024, 18, 31478–31484 CrossRef CAS PubMed.
- Q. Fan, T. Wang, J. Dai, J. Kuttner, G. Hilt, J. M. Gottfried and J. Zhu, ACS Nano, 2017, 11, 5070–5079 CrossRef CAS PubMed.
- X. Liu, A. Matej, T. Kratky, J. I. Mendieta-Moreno, S. Günther, P. Mutombo, S. Decurtins, U. Aschauer, J. Repp, P. Jelinek, S.-X. Liu and L. L. Patera, Angew. Chem., Int. Ed., 2022, 61, e202112798 CrossRef CAS PubMed.
- C. Zhang, E. Kazuma and Y. Kim, J. Am. Chem. Soc., 2022, 144, 10282–10290 CrossRef CAS PubMed.
- L. Xu, C. Zhang, R. Hou, Y. Gao, Z. Zhang, Z. Yi, C. Zhang and W. Xu, J. Phys. Chem. Lett., 2024, 15, 11862–11868 CrossRef CAS PubMed.
- B. Li, X. Zhao, J. Guo, X. Shi and W. Wang, J. Phys. Chem. C, 2022, 126, 6662–6667 CrossRef CAS.
- C. Li, X. Zhang, N. Li, Y. Wang, J. Yang, G. Gu, Y. Zhang, S. Hou, L. Peng, K. Wu, D. Nieckarz, P. Szabelski, H. Tang and Y. Wang, J. Am. Chem. Soc., 2017, 139, 13749–13753 CrossRef CAS PubMed.
- H. Kong, C. Zhang, L. Xie, L. Wang and W. Xu, Angew. Chem., Int. Ed., 2016, 55, 7157–7160 CrossRef CAS PubMed.
- V. Simic-Milosevic, J. Meyer and K. Morgenstern, Angew. Chem., Int. Ed., 2009, 48, 4061–4064 CrossRef CAS PubMed.
- C. Dri, M. V. Peters, J. Schwarz, S. Hecht and L. Grill, Nat. Nanotechnol., 2008, 3, 649–653 CrossRef CAS PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Nat. Chem., 2014, 6, 41–46 CrossRef CAS PubMed.
- D. Zhong, J.-H. Franke, S. K. Podiyanachari, T. Blömker, H. Zhang, G. Kehr, G. Erker, H. Fuchs and L. Chi, Science, 2011, 334, 213–216 CrossRef CAS PubMed.
- H. Kong, L. Wang, Q. Sun, C. Zhang, Q. Tan and W. Xu, Angew. Chem., Int. Ed., 2015, 54, 6526–6530 CrossRef CAS PubMed.
- L. Xie, H. Jiang, D. Li, M. Liu, Y. Ding, Y. Liu, X. Li, X. Li, H. Zhang, Z. Hou, Y. Luo, L. Chi, X. Qiu and W. Xu, ACS Nano, 2020, 14, 10680–10687 CrossRef CAS PubMed.
- D. Li, L. Sun, Y. Ding, M. Liu, L. Xie, Y. Liu, L. Shang, Y. Wu, H.-J. Jiang, L. Chi, X. Qiu and W. Xu, ACS Nano, 2021, 15, 16896–16903 CrossRef CAS PubMed.
- F. Buchner, K. Seufert, W. Auwärter, D. Heim, J. V. Barth, K. Flechtner, J. M. Gottfried, H.-P. Steinrück and H. Marbach, ACS Nano, 2009, 3, 1789–1794 CrossRef CAS PubMed.
- J. Čechal, C. S. Kley, R. Pétuya, F. Schramm, M. Ruben, S. Stepanow, A. Arnau and K. Kern, J. Phys. Chem. C, 2016, 120, 18622–18630 CrossRef.
- C. Li, Z. Xu, Y. Zhang, J. Li, N. Xue, R. Li, M. Zhong, T. Wu, Y. Wang, N. Li, Z. Shen, S. Hou, R. Berndt, Y. Wang and S. Gao, Natl. Sci. Rev., 2023, 10, nwad088 CrossRef CAS PubMed.
- S. Subudhi, D. Rath and K. M. Parida, Catal. Sci. Technol., 2018, 8, 679–696 RSC.
- J. I. Urgel, D. Écija, G. Lyu, R. Zhang, C.-A. Palma, W. Auwärter, N. Lin and J. V. Barth, Nat. Chem., 2016, 8, 657–662 CrossRef CAS PubMed.
- J.-C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53–61 CrossRef PubMed.
- G. Lyu, Q. Zhang, J. I. Urgel, G. Kuang, W. Auwärter, D. Ecija, J. V. Barth and N. Lin, Chem. Commun., 2016, 52, 1618–1621 RSC.
- J. I. Urgel, D. Ecija, W. Auwärter and J. V. Barth, Nano Lett., 2014, 14, 1369–1373 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- L. Chen, H.-F. Wang, C. Li and Q. Xu, Chem. Sci., 2020, 11, 5369–5403 Search PubMed.
- S. Liu, Y. Qiu, Y. Liu, W. Zhang, Z. Dai, D. Srivastava, A. Kumar, Y. Pan and J. Liu, New J. Chem., 2022, 46, 13818–13837 RSC.
- M. Ahmed, Inorg. Chem. Front., 2022, 9, 3003–3033 Search PubMed.
- X. Yang and Q. Xu, Cryst. Growth Des., 2017, 17, 1450–1455 Search PubMed.
- A. M. Rice, G. A. Leith, O. A. Ejegbavwo, E. A. Dolgopolova and N. B. Shustova, ACS Energy Lett., 2019, 4, 1938–1946 CrossRef CAS.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Catal. Sci. Technol., 2016, 6, 5238–5261 RSC.
- S. Abednatanzi, P. Gohari Derakhshandeh, H. Depauw, F.-X. Coudert, H. Vrielinck, P. Van Der Voort and K. Leus, Chem. Soc. Rev., 2019, 48, 2535–2565 RSC.
- H. Yang, Y. Wang, J. Lei, L. Shi, X. Wu, V. Mäkinen, S. Lin, Z. Tang, J. He, H. Häkkinen, L. Zheng and N. Zheng, J. Am. Chem. Soc., 2013, 135, 9568–9571 CrossRef CAS PubMed.
- J. Liu, J. Li, Z. Xu, X. Zhou, Q. Xue, T. Wu, M. Zhong, R. Li, R. Sun, Z. Shen, H. Tang, S. Gao, B. Wang, S. Hou and Y. Wang, Nat. Commun., 2021, 12, 1619 CrossRef CAS PubMed.
- J. I. Urgel, D. Ecija, W. Auwärter, D. Stassen, D. Bonifazi and J. V. Barth, Angew. Chem., Int. Ed., 2015, 54, 6163–6167 CrossRef CAS PubMed.
- L. Yan, B. Xia, Q. Zhang, G. Kuang, H. Xu, J. Liu, P. N. Liu and N. Lin, Angew. Chem., Int. Ed., 2018, 57, 4707–4711 Search PubMed.
- M. Lischka, R. Dong, M. Wang, N. Martsinovich, M. Fritton, L. Grossmann, W. M. Heckl, X. Feng and M. Lackinger, Chem. – Eur. J., 2019, 25, 1975–1983 Search PubMed.
- Z. Shi, J. Liu, T. Lin, F. Xia, P. N. Liu and N. Lin, J. Am. Chem. Soc., 2011, 133, 6150–6153 CrossRef CAS PubMed.
- W. Zhong, T. Zhang, D. Chen, N. Su, G. Miao, J. Guo, L. Chen, Z. Wang and W. Wang, Small, 2023, 19, 2207877 Search PubMed.
- T. Lin, X. S. Shang, J. Adisoejoso, P. N. Liu and N. Lin, J. Am. Chem. Soc., 2013, 135, 3576–3582 CrossRef CAS PubMed.
- S. Xing, Z. Zhang, X. Fei, W. Zhao, R. Zhang, T. Lin, D. Zhao, H. Ju, H. Xu, J. Fan, J. Zhu, Y.-Q. Ma and Z. Shi, Nat. Commun., 2019, 10, 70 Search PubMed.
- H. Liang, S. Xing, Z. Shi, H. Zhang and L. Chi, ChemPhysChem, 2020, 21, 843–846 CrossRef CAS PubMed.
- X. Zhang, N. Xue, C. Li, N. Li, H. Wang, N. Kocić, S. Beniwal, K. Palotás, R. Li, Q. Xue, S. Maier, S. Hou and Y. Wang, ACS Nano, 2019, 13, 1385–1393 CAS.
- H. Xu, R. Chakraborty, A. K. Adak, A. Das, B. Yang, D. Meier, A. Riss, J. Reichert, S. Narasimhan, J. V. Barth and A. C. Papageorgiou, Angew. Chem., Int. Ed., 2024, 136, e202319162 Search PubMed.
- J. Adisoejoso, T. Lin, X. S. Shang, K. J. Shi, A. Gupta, P. N. Liu and N. Lin, Chem. – Eur. J., 2014, 20, 4111–4116 CrossRef CAS PubMed.
- S. Xing, B. Liu, W. Wang, J. Guo and W. Wang, Chem. – Asian J., 2018, 13, 2023–2026 CrossRef CAS PubMed.
- B. Lowe, J. Hellerstedt, A. Matěj, P. Mutombo, D. Kumar, M. Ondráček, P. Jelinek and A. Schiffrin, J. Am. Chem. Soc., 2022, 144, 21389–21397 Search PubMed.
- B. Cirera, J. Björk, R. Otero, J. M. Gallego, R. Miranda and D. Ecija, J. Phys. Chem. C, 2017, 121, 8033–8041 CrossRef CAS.
- C. Zhang, R. Hou and W. Xu, Small Methods, 2025, 2402118 CrossRef PubMed.
| This journal is © the Owner Societies 2025 |