
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein ligation for the assembly and study of nonribosomal peptide synthetase megaenzymes†
Angelos
Pistofidis
and
T. Martin
Schmeing
 *
*
Department of Biochemistry and Centre de Recherche en Biologie Structurale, McGill University, Montréal, QC H3G 0B1, Canada. E-mail: martin.schmeing@mcgill.ca
First published on 7th February 2025
Abstract
Nonribosomal peptide synthetases (NRPSs) are biosynthetic enzymes found in bacteria and fungi, that synthesize a plethora of pharmaceutically relevant compounds. NRPSs consist of repeating sets of functional domains called modules, and each module is responsible for the incorporation of a single amino acid to the growing peptidyl intermediate. The synthetic logic of an NRPS resembles an assembly line, with growing biosynthesis intermediates covalently attached to the prosthetic 4′-phosphopantetheine (ppant) moieties of T (thiolation or transfer) domains for shuttling within and between modules. Therefore, NRPSs must have each T domain phosphopantetheinylated to be functional, and host organisms encode ppant transferases that affix ppant to T domains. Ppant transferases can be promiscuous with respect to the T domain substrate and with respect to chemical modifications of the ppant thiol, which has been a useful characteristic for study of megaenzymes and other systems. However, defined studies of multimodular megaenzymes, where different analogs are required to be affixed to different T domains within the same multimodular protein, are hindered by this promiscuity. Study of NRPS peptide bond formation, for which two T domains simultaneously deliver substrates to the condensation domain, is a prime example where one would want two T domains bearing different acyl/peptidyl groups. Here, we report a strategy where two NRPS modules that are normally part of the same protein are expressed as separate constructs, modified separately with different acyl-ppants, and then ligated together by sortase A of Staphylococcus aureus or asparaginyl endopeptidase 1 of Oldenlandia affinis (OaAEP1). We assessed various reaction conditions to optimize the ligation reactions and maximize the yield of the complex of interest. Finally, we apply this method in large scale and show it allows the complex built by OaAEP1-mediated ligation to be characterized by X-ray crystallography.
Introduction
Nonribosomal peptide synthetases (NRPSs) make natural product compounds that rank among the most widely used pharmaceutical agents, including the antibiotics linear gramicidin (Fig. 1(a)) and vancomycin,1 the immunosuppressant cyclosporin,2 and the anti-cancer drug actinomycin D.3 NRPSs are organized into sets of conserved domains, referred to as modules. Each module is responsible for the incorporation of a single monomer substrate into a growing peptide chain, through the combined function of its domains.4,5 NRPSs are typically multi-modular (∼2–18 modules), which can all be part of a single protein, or can be split into several, unusually multimodular, subunits that associate non-covalently. In the first step of a module's synthetic cycle, the adenylation (A) domain binds a cognate amino acid (or other acyl monomer) substrate from the cytoplasm, and activates it by adenylation (Fig. 1(b)).6 Then, the A domain catalyzes the thiolation of the aminoacyl moiety with the prosthetic 4′-phosphopantetheine (ppant) arm of the transfer (or thiolation; T) domain, covalently ligating the aminoacyl to this T domain.7 The T domain can then transport the aminoacyl moiety to other catalytic domains. In a canonical elongation module, the T domain's next stop is the acceptor site of the condensation (C) domain.8 The C domain catalyzes peptide bond formation between that aminoacyl moiety and the peptidyl or amino acyl group attached to the upstream module's T domain, elongating the peptide chain. The T domain then transports the elongated peptide downstream, to the next module, where it acts as the donor substrate at that module's C domain. Canonical initiation modules lack C domains, while initiation and elongation modules can have additional tailoring domains which the acyl-ppant-T domain visits before or after the C domain (for example, domain F1 in Fig. 1(b)).9 The nascent peptide is progressively passed downstream, being elongated and perhaps chemically modified at each module. It exists as an peptidyl-ppant-enzyme intermediate throughout synthesis, until it is released as a small molecule by the terminal (usually thioesterase) domain. Clearly, NRPS biosynthesis requires each T domain to bear a ppant moiety, and these are installed by a dedicated ppant transferase prior to the first round of synthesis, using coenzyme A as the source of ppant. | ||
| Fig. 1 Ligation of F1A1T1 and C2A2T2 by SaSrtA and OaAEP1 and the resulting ligation scars. (a) Chemical structure of linear gramicidin A, produced by the linear gramicidin synthetase. The first dipeptide highlighted by the dashed box is assembled by the first two modules of the pathway (F1A1T1C2A2T2). (b) The synthetic cycle of F1A1T1C2A2T2. A1 adenylates valine and ligates it to the ppant arm of T1. T1 transports the valinyl-ppant to F1, which catalyzes N-formylation using N10-formyltetrahydofolate. In the second module, A2 adenylates a glycine and ligates it to the ppant arm of T2. T1 and T2 both bind C2, which catalyzes amide bond formation to form fVal-Gly on the ppant of T2.fVal-GlyT2 is the donor substrate for the downstream C domain of the third module. (c) Ligation of F1A1T1AEP1/SrtA to AEP1/SrtAC2A2T2 by OaAEP1 and SaSrtA results in a ligation scar along the intermodular linker between T1 and C2. OaAEP1 leaves an Asn-Gly-Leu scar and SaSrtA leaves a Leu-Pro-Glu-Thr-Gly-Gly-Gly scar. Numbered residues Val770, Ser771, Leu772 are part of the native sequence of the intermodular linker between T1 and C2. | ||
There is great interest in understanding every step of canonical and non-canonical NRPS synthetic cycles, mechanistically and structurally. The most informative structural studies include all components of the NRPS and ligands (or faithful mimics of native ligands) that are relevant to a particular step of biosynthesis. Most catalytic steps feature intermediates linked to the T domain's ppant, so these, or analogs thereof, should be included for structural study. A classic approach is to create non-reactive analogs of the acyl-ppant moieties, for example replacing the thioester with an amide or thioether.10–14 Fortunately, as the Walsh and Marahiel groups discovered, ppant transferase enzymes have no discrimination against the thiol portion of coenzyme A, and little selectivity for T domain identity.15–17 Researchers have taken advantage of this promiscuity in the Bacillus subtilis ppant transferase Sfp, to place all manner of analogues on NRPS T domains, and onto the related polyketide and fatty acid synthetase acyl carrier protein domains.11–14,18–20 This has been fantastically useful for stages along the NRPS biosynthetic cycle and megaenzyme constructs that involve one T domain. However, NRPS condensation involves both donor and acceptor acyl-ppant-T domains, from adjacent modules, which are often in the same protein.12,21 Incubating Sfp with two different acyl-CoA analogues and an NRPS with two T domains would result in a heterogeneous mix of the NRPS complex loaded with different combinations of these analogs, unsuitable for further study (ESI,† Fig. S1). We sought a method to produce a homogeneous sample with different, appropriate analogs affixed at two T domains, to enable structural study of the condensation reaction.
Peptide cyclization and protein ligation are enzymatically catalyzed processes with great potential in ecology and biotechnology.22 Two prominent ligases are the peptidyl Asx-specific ligase asparaginyl endopeptidase 1 from Oldenlandia affinis (OaAEP1), whose natural function is to catalyze the cyclization of the kalata B1 to convert it into a potent insecticide,23 and sortase A from Staphylococcus aureus (SaSrtA) that uses calcium to ligate surface proteins to a pentaglycine motif on the bacterial cell wall.24 These enzymes recognize specific sequences at the C- and N-termini of proteins or peptides, called sorting sequences, and placement of sorting sequences on the same or different peptide chains results in intramolecular cyclization and intermolecular ligation, respectively. Bioengineering efforts have generated variants of OaAEP1 and SaSrtA with significantly increased rates of turnover.25–27 The wild type and bioengineered variants have been used in applications such as peptide cyclization,23,28 segmental isotopic labelling of proteins for nuclear magnetic resonance (NMR) analysis,29–33 ligation of proteins to solid support,34 protein assembly for atomic force microscopy-based single-molecule force spectroscopy (AFM-SMFS),35 ligation of peptide nucleic acids on epidermal growth factor receptors (EGFRs),36 modification of red blood cell surfaces with peptides and proteins,37 and immobilization of proteins on surfaces for single-molecule force spectroscopy.38
Here, we present the development of a strategy which combines promiscuous ppant transferase activity with protein ligation to create complexes of a megaenzyme with T domains each bearing different, appropriate non-hydrolysable analogs. We optimize protocols using OaAEP1 and SaSrtA ligation to “re-assemble” the dimodular linear gramicidin synthetase subunit A (LgrA)1,12 (Fig. 1(c) and ESI,† Fig. S2), and show effective covalent NRPS complex formation. These complexes can be purified to near homogeneity and can lead to co-complex structure determination by X-ray crystallography.
Results and discussion
Design of LgrA constructs for SaSrtA-mediated ligation
Linear gramicidin synthetase subunit A (LgrA) consists of module 1, F1A1T1, and module 2, (E* denotes an inactive epimerization domain) (Fig. 1(b)). Domain A1 selects and activates valine and ligates it to the ppant of T1 (T1-Val). Domain F1N-formylates T1-Val to T1-fVal, which is the donor substrate for condensation in C2. Meanwhile, A2 selects and activates glycine and ligates it to the ppant of T2 to make the acceptor T2-Gly substrate for condensation. Condensation transfers the fVal to T2-Gly, and thereafter T2-Gly-fVal travels to module 3 (in LgrB) to continue elongation.
(E* denotes an inactive epimerization domain) (Fig. 1(b)). Domain A1 selects and activates valine and ligates it to the ppant of T1 (T1-Val). Domain F1N-formylates T1-Val to T1-fVal, which is the donor substrate for condensation in C2. Meanwhile, A2 selects and activates glycine and ligates it to the ppant of T2 to make the acceptor T2-Gly substrate for condensation. Condensation transfers the fVal to T2-Gly, and thereafter T2-Gly-fVal travels to module 3 (in LgrB) to continue elongation.  is likely a remnant from an ancestorial NRPS where module 2 activated a chiral amino acid, but because it is nonfunctional1 we have omitted that domain from the experiments presented here. Therefore, we sought to make constructs of F1A1T1 and C2A2T2 that could be produced separately, modified with moieties fVal-NH-ppant and Gly-NH-ppant, and ligated together, to mimic the pre-condensation covalent LgrA complex (Fig. 1(c)).
is likely a remnant from an ancestorial NRPS where module 2 activated a chiral amino acid, but because it is nonfunctional1 we have omitted that domain from the experiments presented here. Therefore, we sought to make constructs of F1A1T1 and C2A2T2 that could be produced separately, modified with moieties fVal-NH-ppant and Gly-NH-ppant, and ligated together, to mimic the pre-condensation covalent LgrA complex (Fig. 1(c)).
We chose a split point for F1A1T1C2A2T2 to be between residues 770 and 771, in the middle of the flexible linker connecting T1 and C2 (Fig. 3(a)), giving each split construct one T domain.12 Mid-linker was chosen to eliminate potential steric occlusion of the sorting sequences, and to minimize the likelihood that a ligation scar would interfere with biosynthesis. We therefore used site directed mutagenesis to add codons encoding the sortase site Leu-Pro-Glu-Thr-Gly to the C-terminus of F1A1T1 (F1A1T1SrtA) and Gly-Gly-Gly to C2A2T2 (SrtAC2A2T2).27 Notably, all constructs contained octahistidine affinity tags and tobacco etch virus (TEV) protease cleavage sites. Fortunately, one efficient TEV cleavage site, ENLYFQ/G, leaves an N-terminal Gly after cleavage, so we used this Gly as part of the Gly-Gly-Gly of SrtAC2A2T2.29,35
Optimization of sortase A ligation
We expressed and purified F1A1T1SrtA and SrtAC2A2T2, and assessed SaSrtA mediated ligation with conditions adapted from Byrd and colleagues.27 Thus, 20 μM F1A1T1SrtA, 6 μM SrtAC2A2T2 and 5 μM SaSrtA were incubated for 30 minutes at room temperature and pH 7.5. Ligation reactions were quenched with EDTA and SDS-PAGE loading dye and analyzed by SDS-PAGE (Fig. 2(a)). A small band at the correct apparent molecular weight was visible on the gel, representing less than 1 μM product formation. We next varied the concentration of each LgrA module. At a constant concentration of SrtAC2A2T2, increasing the concentration F1A1T1SrtA did not increase the yield of desired product (Fig. 2(b)). Increasing the concentration of SrtAC2A2T2 at a constant concentration of F1A1T1SrtA, however, did increase yield (Fig. 2(c)). The ligation was more efficient when a higher concentration of SrtAC2A2T2 than F1A1T1SrtA was used, specifically a F1A1T1SrtA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SrtAC2A2T2 ratio of ∼1:1.9 (20 μM F1A1T1SrtA to 38 μM SrtAC2A2T2) (Fig. 2(b) and (c)).
:SrtAC2A2T2 ratio of ∼1:1.9 (20 μM F1A1T1SrtA to 38 μM SrtAC2A2T2) (Fig. 2(b) and (c)).
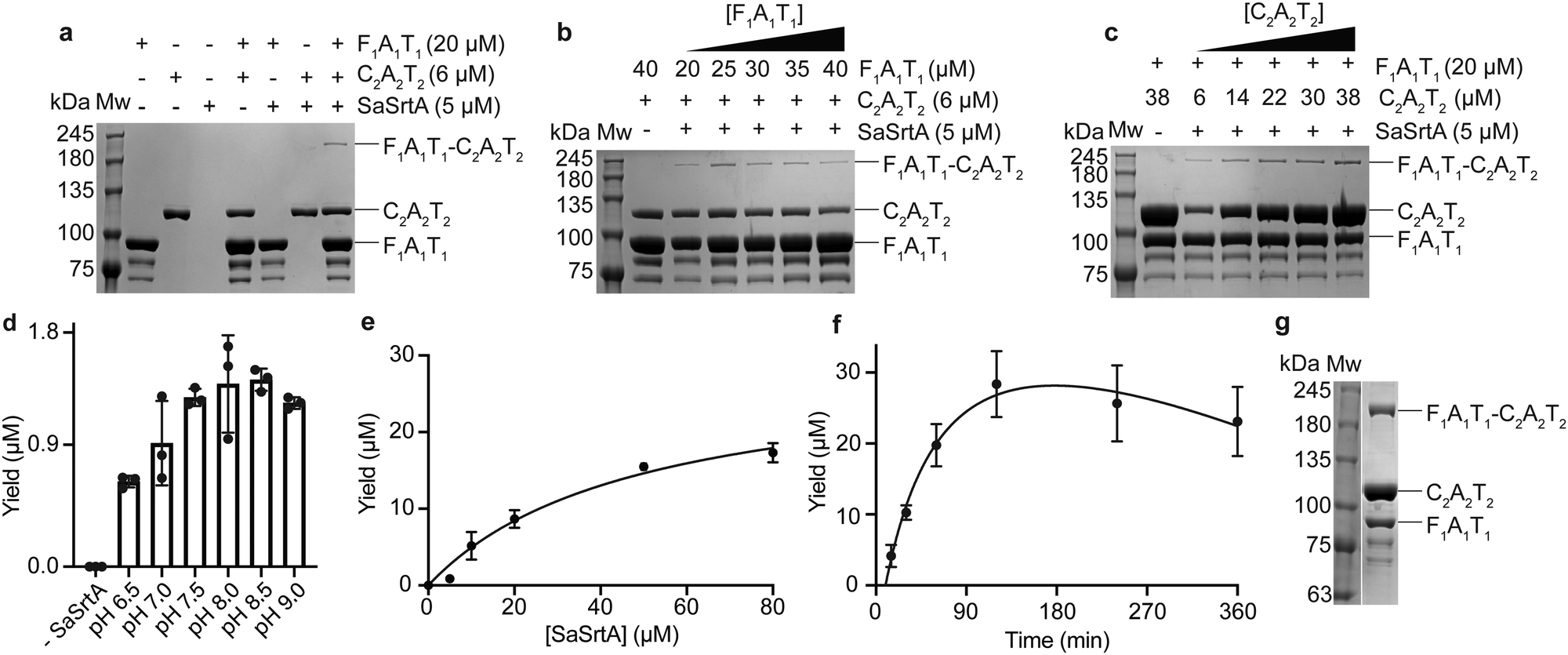 | ||
| Fig. 2 Optimization of SaSrtA-mediated ligation of F1A1T1 and C2A2T2. (a) Initial conditions for SaSrtA-mediated ligation, adapted from Byrd and colleagues.27 Lanes contain one, two or all three components (F1A1T1SrtA, SrtAC2A2T2 and SaSrtA) required for ligation. Extra bands in the F1A1T1SrtA-only lanes are likely C-terminal truncations of the F1A1T1SrtA, which do not possess the sorting sequence and are incompetent for ligation reaction. (b) Reactions with varied F1A1T1SrtA concentration. (c) Reactions with varied SrtAC2A2T2 concentration. For the optimizations shown in (d)–(f), bands corresponding to F1A1T1–C2A2T2 in the SDS-PAGE analysis were quantified and converted to yield in μM using a calibration curve of known F1A1T1C2A2T2 concentrations. These experiments were carried out in triplicate (n = 3) and bars or data points represent the mean yield between the three replicates and the error bars correspond to the standard deviation. (d) SaSrtA-ligation at pHs between 6.5–9.0. (e) Reactions with varied SaSrtA concentration. (f) Time trials of the SaSrtA ligation. (g) SaSrtA-mediated ligation using the optimized conditions: 120 μM F1A1T1SrtA, 210 μM SrtAC2A2T2, 80 μM SaSrtA at pH 7.5 for 120 minutes. | ||
Protein ligation with SaSrtA is typically performed at pH 7–8.27,29,31 To maximize the yield of ligated product we screened a broader pH range. At pHs lower than 6.5 there was significant precipitation of protein, so those reactions were discarded. SaSrtA displayed optimal activity at 7.5–8.5 and was less active below (pH 6.5–7.0) or above (pH 9.0) these values (Fig. 2(d)). We thus kept the pH at 7.5, the literature standard, for subsequent trials.27,29,31,39 We next ligated F1A1T1SrtA and SrtAC2A2T2 with higher concentrations of each LgrA construct, to maximize the amount of SaSrtA-ligated F1A1T1–C2A2T2 for downstream experiments. F1A1T1SrtA and SrtAC2A2T2 were increased to 120 μM and 210 μM and incubated with 5 to 80 μM SaSrtA. The yield of ligated F1A1T1–C2A2T2 increased with SaSrtA concentration up to the highest value of 80 μM, although there was only a ∼10% difference in yield from 60 and 80 μM SaSrtA (Fig. 2(e)). Finally, we performed time trials, with a reaction of 120 μM F1A1T1SrtA, 210 μM SrtAC2A2T2 and 80 μM SaSrtA, at pH 7.5, incubated between 15 minutes and 3 hours. The yield of F1A1T1–C2A2T2 increased rapidly up to 2 hours, with yield falling slowly thereafter (Fig. 2(f) and ESI,† Fig. S5a). Our final conditions for SaSrtA-mediated ligation (120 μM F1A1T1SrtA, 210 μM SrtAC2A2T2, 80 μM SaSrtA, pH 7.5, 2 hours) gave ∼28 μM F1A1T1–C2A2T2, a ∼24% yield with respect to F1A1T1SrtA incorporation into F1A1T1–C2A2T2 (Fig. 2(g) and Table 1).
| OaAEP1 | SaSrtA | |
|---|---|---|
| [F1A1T1] (μM) | 120 | 120 |
| [C2A2T2] (μM) | 210 | 210 |
| [Ligase] (μM) | 1 | 80 |
| pH | 6.5 | 7.5 |
| Reaction time (minutes) | 30 | 120 |
| Yield (mg) per mL of reaction | 10.23 ± 0.60 | 5.85 ± 0.96 |
| Yield [F1A1T1–C2A2T2] (μM) | 49.7 | 28.4 |
| % Molar yield with respect to F1A1T1% | 41.4 | 23.6 |
| Molar yield with respect to C2A2T2 | 23.6 | 13.5 |
SaSrtA-ligated condensation complex formation, crystallization, and influence of scar on activity
With optimised SaSrtA-ligation conditions, we proceeded to make the covalent F1A1T1–C2A2T2 pre-condensation complex, with fVal-NH-ppant on T1 and Gly-NH-ppant on T2. fVal-NH-CoA and Gly-NH-CoA were synthesized as described previously (ESI,† Fig. S6),11,12,40,41 and were incubated separately with F1A1T1SrtA and SrtAC2A2T2, respectively, in the presence of the promiscuous phosphopantetheine transferase Sfp.15,42,43 Intact protein mass spectrometry showed appropriate loading of amino acyl-NH-ppants onto each T domain (ESI,† Fig. S7a and b). A large scale SaSrtA ligation produced ∼12 mg of F1A1T1-fVal–C2A2T2-Gly, which was purified by anion exchange and size exclusion chromatography (Fig. 2(g) and ESI,† Fig. S8a, c). Ligated F1A1T1-fVal–C2A2T2-Gly has the same migration volume as wild-type F1A1T1C2A2T2 on size exclusion chromatography (ESI,† Fig. S9c). Sortase A-ligated F1A1T1-fVal–C2A2T2-Gly was subjected to broad sparse array crystallization screening, but no crystal hits were obtained. This result was somewhat surprising, since unliganded F1A1T1C2A2T2 had previously produced crystals in the condensation conformation,12 and substrate analogs would be expected to help rather than hinder the propensity to form a stable conformation.SaSrtA-ligated F1A1T1-fVal–C2A2T2-Gly differs from previously crystallized F1A1T1–C2A2T2 in two ways: the substrate analogs, and the scar introduced by SaSrtA. To assay whether this scar affected LgrA, we leveraged a multiple turnover tripeptide synthesis assay, where LgrA is fused with the terminal C domain of BmdB.12,41,44,45 The T1:C2 linker of F1A1T1–C2A2T2CT3 was modified to include the sequence of the SaSrtA ligation scar (Leu-Pro-Glu-Thr-Gly-Gly-Gly) (Fig. 3(a), linker 2). Compared to a construct with wild type T1:C2 linker sequence, F1A1T1–C2A2T2CT3 containing the SaSrtA ligation scar showed ∼25% less tripeptide synthesis (Fig. 3(b)). This activity would still have meant that a structure of SaSrtA-ligated F1A1T1-fValC2A2T2-Gly could provide insight into condensation in NRPSs, but hints that it not the ideal sample to study. Thereafter, we pivoted to another ligase, asparaginyl endopeptidase 1 from Oldenlandia affinis (OaAEP1), which is reported to have a higher rate of ligation and result in a smaller scar.26,27,29,31
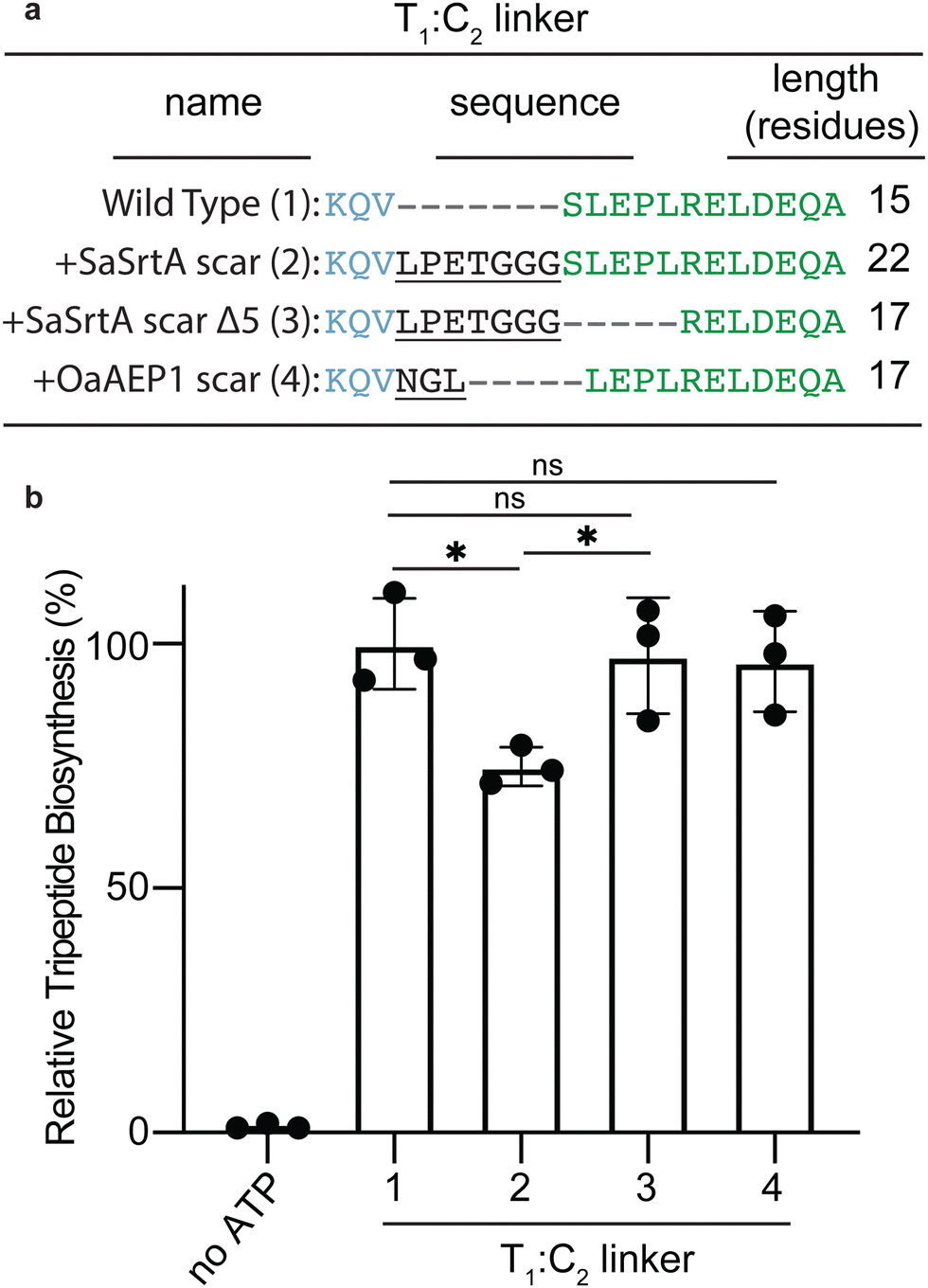 | ||
| Fig. 3 Impact of the SaSrtA and OaAEP1 ligation scars on the activity of F1A1T1–C2A2T2CT3. (a) Sequence and length LgrA T1:C2-linker modifications that were introduced by ligation, and into F1A1T1C2A2T2CT3, by site-directed mutategenesis, to investigate their effect on biosynthetic activity: (1) wild type linker; (2) wild type linker extended by the SaSrtA ligation scar; (3) linker 2 shortened by five amino acids; and (4) wild type linker extended by replacing Ser1171 with the OaAEP1 ligation scar. (b) Tripeptide biosynthesis by F1A1T1C2A2T2CT3 constructs with various linkers introduced by site-directed mutagenesis.12,41 Bars shown are the average of three replicates (n = 3) and the error bars are the standard deviation of the replicates. Statistical significance was determined using a Student's t-test. Introducing the SaSrtA ligation scar reduces activity by ∼25% (3; p = 0.0126; *). Removing five residues from downstream of the SaSrtA ligation scar allows the enzyme to regain wild type activity (3; compared to the activity of 2 p = 0.0346; *; compared to the activity of 1 p = 0.7933; ns). The OaAEP1 ligation scar does not affect the activity (4; p = 0.6736; ns). | ||
Optimization of AEP1 ligation
OaAEP1 ligates together constructs with a C-terminal Asn-Gly-Leu sequence and N-terminal Gly-Leu sequence and leaves a short Asn-Glu-Leu scar, via a mechanism similar to that of SaSrtA (Fig. 1 and ESI,† Fig. S2b). To test the effect of this shorter ligation scar on biosynthesis activity, this ligation scar was inserted into the T1:C2 linker in place of Ser771 in the LgrA-BmdB fusion used for peptide synthesis assays (F1A1T1–C2A2T2CT3; Fig. 3(a), linker 4). This construct displayed tripeptide synthesis activity indistinguishable from wildtype (Fig. 3(b)), indicating OaAEP1 could be useful for our purposes.The SaSrtA and OaAEP1 scars differ significantly in length and in sequence identity (Fig. 3(a)). It was not clear which of these features is detrimental to activity. To understand this question, we removed 5 residues in the linker of F1A1T1–C2A2T2CT3, downstream from the SaSrtA scar (Fig. 3(a)), which maintains the difference in sequence identity, but changes the total linker length to 17 residues, the same at that after OaAEP1 ligation, and performed tripeptide synthesis. Elimination of the five residues restored activity to wild type levels (Fig. 3(b)), suggesting that linker length was a culprit in the reduction of activity. Nonetheless, we did not re-design our SaSrtA strategy, but rather continued to OaAEP1 ligation, to investigate its utility in NRPS complex formation.
New constructs of F1A1T1 and C2A2T2 were created by site-directed mutagenesis to include the OaAEP1 ligation sites Asn-Gly-Leu at the C-terminus of F1A1T1 (F1A1T1AEP1), and Gly-Leu in C2A2T2 (AEP1C2A2T2) with the Gly being the remnant from the TEV scar after cleavage of the affinity purification tags by TEV protease. These proteins were expressed and purified for OaAEP1-ligation trials. Initial reaction conditions for OaAEP1-mediated ligation were adapted from Wu and colleagues.26 Under these conditions 8 μM F1A1T1AEP1, 5 μM AEP1C2A2T2 and 0.1 μM OaAEP1 were incubated at pH 7.5 and room temperature for 15 minutes. OaAEP1 ligation was quenched with SDS-PAGE loading dye and analyzed by gel electrophoresis (Fig. 4). Very little product was formed with these initial conditions, and a band that likely represents a reaction side-product appeared very close to that of F1A1T1–C2A2T2 (Fig. 4(a)). As we had done with SaSrtA, we performed optimizations to increase the yield of F1A1T1–C2A2T2, and also to reduce side-product formation. At a constant concentration of AEP1C2A2T2, increasing the concentration of F1A1T1AEP1 reduced the yield of the desired product and increased side product formation (Fig. 4(b)). Conversely and analogously to the sortase case, increasing the concentration of AEP1C2A2T2 at a constant concentration of F1A1T1AEP1 increased the yield of product, and it also reduced the amount of side product (Fig. 4(c)). The optimal ratio of F1A1T1AEP1:AEP1C2A2T2 was found to be 8 μM:14 μM (∼1:1.75).
 | ||
| Fig. 4 Optimization of OaAEP1-mediated ligation of F1A1T1 and C2A2T2. (a) Initial conditions for OaAEP1-mediated ligation adapted from Wu and colleagues.26 Lanes contain one, two or all three components (F1A1T1AEP1, AEP1C2A2T2 and OaAEP1) required for ligation. (b) Reactions with varied F1A1T1AEP1 concentration. (c) Reactions with varied AEP1C2A2T2 concentration. For the reactions shown in (d)–(f), bands corresponding to F1A1T1–C2A2T2 in the SDS-PAGE analysis were quantified and translated to yield in μM by comparison to a calibration curve of known concentration of wild type F1A1T1C2A2T2. These experiments were carried out in triplicate (n = 3), and bars (d) or data points (e) and (f) represent the mean yield of the three replicates. The error bars correspond to the standard deviation. (d) OaAEP1-ligation at pHs between 6.5–9.0. (e) Reactions with varied OaAEP1 concentration. (f) Time-trial of the OaAEP1 ligation. (g) OaAEP1-mediated ligation using the optimized conditions: 120 μM F1A1T1AEP1, 210 μM AEP1C2A2T2, 3 μM OaAEP1 at pH 6.5 for 30 minutes. | ||
We next sought to optimize the pH of the reaction. In nature, OaAEP1 functions inside the plant cell vacuole, where the pH is ∼5.5.46,47 Experiments on peptide modification and cyclization by OaAEP1 and the related enzyme butelase 1 showed optimal activity at around pH 6.0–7.0.26,48,49 Our pH trials with OaAEP1 were limited to a minimum pH of 6.5, as below pH 6.5, we observed precipitation in our samples. This minimum accessible pH showed maximum activity. Very similar activity was seen at pH 7.0, but activity became substantially lower at higher pHs (Fig. 4(d)). Subsequent OaAEP1 ligations were performed a pH 6.5. Optimization trials with high concentration of F1A1T1AEP1 (120 μM) and AEP1C2A2T2 (210 μM) and 0.1–3 μM OaAEP1 showed a markedly higher yield with increasing OaAEP1 concentration up to 1 μM, with no additional yield increase above 1 μM (Fig. 4(e)). Moreover, we observed higher amounts of side-product formation at concentrations of OaAEP1 above 1 μM (ESI,† Fig. S5b), so chose 1 μM OaAEP1 for further assays. The OaAEP1 time trial showed F1A1T1–C2A2T2 accumulating quickly for the first 30 minutes, followed by marked degradation (Fig. 4(e) and ESI,† Fig. S5b). Under these optimized conditions, 120 μM F1A1T1AEP1, 210 μM AEP1C2A2T2 and 1 μM OaAEP1, incubated at pH 6.5 for 30 minutes, yielded ∼50 μM product (incorporating ∼42% of F1A1T1AEP1 into F1A1T1–C2A2T2) (Fig. 4(g) and Table 1).
OaAEP1-ligated condensation complex formation, crystallization and structural characterization
F1A1T1AEP1 and AEP1C2A2T2 were loaded with fVal-NH-ppant and Gly-NH-ppant (ESI,† Fig. S7c and d), respectively, and joined using the optimized ligation conditions. The resulting F1A1T1-fVal–C2A2T2-Gly was purified by anion exchange chromatography followed by size-exclusion chromatography (ESI,† Fig. S8b and c), and migrated through the size exclusion column at the same volume as wild type F1A1T1C2A2T2. This OaAEP1-ligated F1A1T1-fVal–C2A2T2-Gly was put through a sparse matrix screen of crystallization conditions, and yielded a crystal hit that we iteratively optimized to produce large, single crystals. From these crystals, a structure could be determined at 3.6 Å resolution (ESI,† Table S1).The crystal structure of F1A1T1-fVal–C2A2T2-Gly contains one chain per asymmetric unit (Fig. 5(a) and (d)). F1–A1 and C2–A2 form typical “rigid cores” (also called “catalytic platforms”50) in each module, and are in very similar interdomain orientation to those seen in other structures of LgrA constructs (Fig. 5(a)).12 C2 displays the expected V-shaped pseudodimeric structure (ESI,† Fig. S9)51 of two lobes with chloramphenicol acetyl transferase (CAT) folds. The latch (residues 1124-1138) and floor (1044-1057) loops are ordered and observed in their expected positions, spanning the top and bottom of the gap between the lobes, respectively.12 T1 is bound to C2 at the expected T1:C2 binding site, in a very similar conformation to the high resolution structure of F1A1T1-fValC2 (ESI,† Fig. S10a and b).12 Strong density can be seen for the first few atoms of the ppant modification attached to Ser729 of T1 at the entrance of the donor substrate tunnel (Fig. 5(c)), but, not surprisingly for a thin, mobile moiety in moderate resolution electron density maps, the signal dissipates as the ppant reaches into the active site (Fig. 5(c) and ESI,† Fig. S10c).12
 | ||
| Fig. 5 Crystal structure of F1A1T1-fVal–C2A2T2-Gly. (a) Overall structure of the OaAEP1-ligated F1A1T1-fVal–C2A2T2-Gly complex. T2 is not at the expected binding site on C2. Two different views of the complex are shown. (b) Close up view of the OaAEP1 ligation scar and a composite omit map contoured at 0.9 σ. The three amino acids of the OaAEP1 scar (-Asn-Gly-Leu-; shown with black carbon atoms) do not interact with T1 or C2 and do not form a crystal contact with the Acore2 of the crystallographic symmetry mate. (c) Close-up view of the T1:C2 interface where fVal-NH-ppant is bound at the donor substrate tunnel. Density is observed for the first few atoms of the ppant moiety in this FO–FC difference map contoured at 3σ without carving. (d) T2 participates in a crystal contact with F1 of the adjacent symmetry mate, preventing it from engaging the T2 binding site on C2. (e) Close-up view of the crystal contact between T2 and F1 of the symmetry mate. | ||
Surprisingly and utterly disappointingly, T2 is not found to be engaging with the C domain acceptor site. Rather, it is ∼50 Å away, forming a crystal contact with the F1 of a symmetry mate (Fig. 5(d) and (e), ESI,† Fig. S10d). This conformation greatly limits the utility of this crystal form, as it cannot be used to observe the desired pre-condensation state and gain insight into peptide bond formation in NRPSs.
Yields of ligated complexes compared to the heterologous expression of F1A1T1C2A2T2
The protocols described here for native protein ligation of LgrA complexes were developed to obtain defined pre-condensation complexes, and clearly require more steps than direct expression and purification of dimodular LgrA F1A1T1C2A2T2. However, this is partially compensated for by the fact that the individual modules express more highly than the dimodule, which is not uncommon for multimodular systems.52 The yield of F1A1T1C2A2T2 from Escherichia coli BL21(DE3) entD-53 in Luria broth (LB) is typically ∼1.5 mg per L growth media, while F1A1T1 (with or without ligation sequences) yields ∼8 mg per L, and C2A2T2 constructs ∼6 mg per L. Large scale ligations were of 2 mL volume, with ∼21 mg of F1A1T1 and ∼47 mg of C2A2T2 and yielded ∼12 mg F1A1T1-fVal–C2A2T2-Gly or ∼20 mg F1A1T1-fVal–C2A2T2-Gly (Table 1). Thus, a larger scale reaction requires the amount of protein obtainable from <3 L of culture of cells expressing F1A1T1 plus <8 L of culture of cells expressing C2A2T2. To obtain comparable amounts of F1A1T1C2A2T2 by direct heterologous expression and purification of the dimodular construct, ∼8 L (for yield similar to SaSrtA ligation) or ∼13 L (for yield similar to OaAEP1 ligation) of culture is required. This comparison emphasizes that SaSrtA and OaAEP1 ligation can provide substantial amounts the ligated complexes, not very dissimilar from the amounts of dimodule obtained from expression of the large intact construct.Comparisons with alternative approaches
The “split-modify-combine” approach we performed here was successful in yielding the specifically double-labelled construct, but did not produce the desired structure. This was a known risk for this complex, as we could not crosslink acceptor and donor substrates at the C domain without greatly deviating from the natural substrates/intermediates. Other groups have cross-linked T domains at the active site of C domains,42,54,55 which provides insight for the overall condensation conformation, but cannot speak to chemistry of peptide bond formation. Other approaches to give faithful complexes besides “split-modify-combine” could be used: the simplest would be to perform studies with an NRPS that has a C domain with identical native acceptor and donor substrates. In this case, Sfp could be used to load both at once. Such NRPSs are quite rare and none have been crystallized.56,57 In addition, finding another dimodular NRPS that provides high resolution structures with near-native substate analogues would be challenging, explaining why we chose the LgrA system. Likewise, one could have found a C domain that can catalyze condensation between identical acyl donor and acceptor ppant moieties even if they are not the cognate substates, but the ideally, analogs are as close to native as possible.Other strategies for similar goals have also recently been reported.58 Cryle and colleagues used SaSrtA to link a T domain and a cytochrome P450 domain that are naturally in different pathways.39 This gave them control over the length and composition of their artificial linker, as well as the position of each domain (at the N- or C-terminus) in the ligated complex. In a different study, they also elegantly used a bioengineered SpyTag-SpyCatcher system to generate active NRPS chimeras for functional assays.13 A potential disadvantage of the SpyTag-SpyCatcher system is the sizable ligation scar (141 residues; ∼15 kDa) that could interfere with domain-domain interactions and/or be detrimental to crystallization of the complex, although we seem to have suffered from those drawbacks even with very small scars.59
It is notable that the SaSrtA ligation scar (Leu-Pro-Glu-Thr-Gly-Gly-Gly) in the F1A1T1–C2A2T2CT3 chimera lowered the activity by ∼25% (Fig. 3(b)). Linkers are important for tethering NRPS domains while providing sufficient flexibility for the large conformational changes.11,12,60 They typically display lower conservation than that seen within domains. Gulick and colleagues highlighted a portion of the A–T linker that displays preference for a Leu-Pro-Xxx-Pro sequence,61 although these residues interact with the A domain similarly in multiple structures,11,50,62–64 so may not be part of the flexible region. Similar conserved motifs have not been reported for T–C linkers. A–T and donor T–C linkers display ∼23% sequence identity (with other T–C and A–C linkers) and show a preference for alanine, proline, glycine, serine and glutamate.65,66 Three of these preferred residues (proline, glycine and glutamate) appear once or more in the ligation scar formed by SaSrtA. T–C linkers are ∼15–18 residues long and A–T linkers are ∼15 residues long.66 Introducing the seven residue-long SaSrtA-ligation scar in the T1–C2 linker increases its length by ∼1.5 fold. On the contrary, the OaAEP1 ligation scar had no effect on activity (Fig. 3) and only extended the T1–C2 linker by two residues. Interestingly, the OaAEP1 scar was ordered enough in the presented F1A1T1-fValC2A2T2-Gly structure that the full sequence could be modelled (Fig. 5(b)), but it does not make non-covalent interactions with T1 or C2, or alter the functional T1:C2 interaction at the donor site. Notably, crystal geometry places this OaAEP1 ligation scar sequence ∼8 Å from a symmetry-related molecule. The longer, presumably unstructured SaSrtA ligation scar sequence could prevent a F1A1T1-fVal–C2A2T2-Gly from assuming this position and dissuade crystallization, at least in this crystal form, consistent with the lack of crystals of F1A1T1-fVal–C2A2T2-Gly. The SaSrtA and OaAEP1 scars differ substantially in sequence length and identity. Our data showed that the extension of the T1:C2 linker could be an important cause of reduced biosynthetic activity, but it does not rule out the possibility of sequence having a significant affect.
Comparisons with ligation in other systems
Previous studies working with OaAEP1 and SaSrtA had optimized the concentration of N- and C-terminal fragments,26,27,31–33,67 concentration of OaAEP1 or SaSrtA,26,31,33,67 pH,35,67 temperature,33,67 reaction time,27,29–33,67 and the steric hindrance around the ligation sites.68 These studies report yields of 24–80% for SaSrtA27,29,31,33,67 and >90% for OaAEP1.32 Following our optimizations of OaAEP1 and SaSrtA ligation we obtained yields of ∼25% for SaSrtA and ∼40% for OaAEP1 (Table 1). Comparison of the optimized reactions shows OaAEP1 to perform better than SaSrtA in practically every way, making twice the amount of complex with 80-fold less ligase in a quarter of the time. Although our yields are lower than those reported in the literature, two milliliters of ligation reaction provided sufficient protein sample to carry out initial crystallographic screening and extensive crystal optimization trials. If increased yield (and reduced product breakdown) (Fig. 2(f), 3(f), ESI,† Fig. S5) is needed in future, published work on SaSrtA suggests that reaction in a dialysing membrane33 or in a spin concentrator with simultaneous concentration and dilution could be useful.29 Further optimizations, such as iteratively returning to the variables we assayed, preincubations of some reaction components, altering temperature, or changing the position of the split point could all be performed. Preincubation of ligation substrates (F1A1T1SrtA/AEP1 with SrtA/AEP1C2A2T2), or of individual substrates with SaSrtA/OaAEP1 could be assessed, as Ulrich and Cryle39 showed substrate preincubation has helpful in another system, but in our early experiments with SaSrtA preincubation did not increase yield and we did not yet return to it. We also did not alter temperature, because we saw significant protein precipitation at 30 °C and 37 °C. A split point could be chosen to maximize the affinity between the two ligation substrates to increase yield, for example splitting a domain or module, but dividing segment of protein that naturally is in a single, folded unit could be accompanied by defects in activity.52 Conversely, we placed our sorting sequences into the flexible intermodular NRPS linkers which should not interfere with protein folding and should offer excellent steric accessibly of the ligation sites.Outlook
Protein ligation has also been applied extensively to segmental isotopic labelling for NMR analysis.29–33,69–72 Our protocol could be used for the isotopic labelling of a specific NRPS domain(s) and subsequent ligation with non-labelled domains to assemble megaenzyme for NMR studies. Moreover, domains could be labelled with different fluorescent probes and ligated for fluorescence resonance energy transfer (FRET) experiments to interrogate the dynamics of NRPS modules.73–75 We are not aware of other systems that have leveraged this kind of protein ligation for X-ray crystallography or cryogenic-electron microscopy structure determination, but believe it could be useful in a variety of scenarios.The structure presented here is a case of “close but no cigar”, since the T2 is far from the T2:C2 binding site because of a crystal contact. Thus, the insight on peptide bond formation we desired could not be obtained. Fortunately, the broader story has a happier ending: in work that directly leveraged that presented here,41 we performed sample preparation which omitted the TEV cleavage step for F1A1T1AEP1, and so produced octahistidine-TEV-F1A1T1-fVal–C2A2T2-Gly, reasoning that the octahistidine-TEV sequence would break the F1:T2 crystal contact. Indeed, this led to a new crystal form, and high-resolution pre-and post-condensation structures provided the insight on condensation that we sought, proving the utility of protein ligation for megaenzyme complexes.
Materials and methods
Cloning and mutagenesis
All primer sequences are listed in ESI,† Table S2. Plasmids encoding F1A1T1AEP1, AEP1C2A2T2, F1A1T1SrtA and SrtAC2A2T2 (pBacTandem_F1A1T1AEP1, pBACTandem_AEP1C2A2T2, pBacTandem_F1A1T1SrtA and pBACTandem_SrtAC2A2T2, respectively) were amplified from pBACt_F1A1T1C2A2T212 and modified for sorting sequence inclusion using primers pBACt_FAT_NGL_Ins_For, pBACt_FAT_NGL_Ins_Rev, pBACt_GL_CAT_Ins_For, pBACt_GL_CAT_Ins_Rev, pBACt_SrtA_FAT_Ins_For, pBACt_SrtA_FAT_Ins_Rev, pBACt_SrtA_CAT_Ins_For and pBACt_SrtA_CAT_Ins_Rev. Parental plasmid was digested with DpnI and 2 μL of reactions were transformed in E. coli XL10gold (Agilent Technologies) for in-cell self-ligation of amplicons.Variants of F1A1T1C2A2T2CT3 used for the tripeptide biosynthesis assay were generated by site directed mutagenesis using the primer pairs LgrABmdB_ + NGL_For/Rev, LgrABmdB_ + LPETGGG_For/Rev and LgrABmdB_ + SrtA_Δ5_For/Rev with pBacTandem_FATCATC3 as template.12 Each primer was used in a separate amplification reaction for 15 cycles, then pairs of reactions containing complementary primers were mixed, prior to an additional 20 cycles of amplification. Reactions were all treated with DpnI to digest parental DNA, and 2 μL were transformed in E. coli XL10gold.
The plasmid encoding the pentamutant variant (Pro94Arg, Asp160Asn, Asp165Ala, Lys190Glu and Lys196Thr) of SaSrtA25 was obtained from Addgene (plasmid #75144) and the plasmid for the expression of OaAEP1 was kindly provided by the lab of Wu Bin at the Nanyang Technological University, Singapore.26
All constructs generated were verified by Sanger sequencing (Genome Quebec) and full-plasmid sequencing (Plasmidsaurus).
Protein expression and purification
For heterologous expression, pBacTandem_F1A1T1SrtA, pBACTandem_SrtAC2A2T2, pBacTandem_F1A1T1AEP1, and pBACTandem_AEP1C2A2T2 or pBACTandem_F1A1T1C2A2T2 were individually transformed in in E. coli BL21 (DE3) entD-cells.53 Separately, each of pBacTandem_FATCATC3, pBacTandem_FATCATC3_AEP1 and pBacTandem_FATCATC3_SrtA were transformed in E. coli BL21 (DE3) BapI.76 Starter cultures were prepared by adding one colony to Luria broth (LB) media, supplemented with 50 μg mL−1 of kanamycin and incubating overnight at 37 °C with shaking at 220 rpm. For large scale growth, 2.8 L shake flasks containing 1 L of LB media supplemented with 50 μg mL−1 kanamycin were inoculated with 10 mL of starter culture. Cultures were grown at 37 °C with shaking at 220 rpm until an OD600 of 0.5–0.7 was reached. Subsequently, cultures were transferred to 4 °C for 1 hour, then 0.5 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to the cultures to induce protein expression, which occurred for 16 hours at 16 °C while shaking at 220 rpm. Cells were harvested by centrifugation at 3993g for 20 minutes at 4 °C. Pellets were either used immediately for protein purification or stored at −20 °C for later use.For F1A1T1 construct purification, cells expressing F1A1T1SrtA and F1A1T1AEP1 were resuspended in 10 mL of IMAC A buffer (2 mM β-mercaptoethanol, 150 mM NaCl, 2 mM imidazole, 25 mM HEPES pH 7.5) per 1 L of cells grown. Resuspended cells were lysed by sonication and the lysate was clarified by centrifugation at 48000g for 30 minutes. Clarified supernatants were loaded on a 5 mL HiTrap IMAC FF column (Cytiva) equilibrated in IMAC A. The column was washed with 20 column volumes (CV) of IMAC A buffer and the protein was eluted with IMAC B (IMAC A with 250 mM imidazole). Resulting F1A1T1SrtA or F1A1T1AEP1 samples were incubated overnight with tobacco etch virus (TEV) protease to cleave the N-terminal octahistidine tag and simultaneously dialyzed against IMAC A buffer. Thereafter, protein was centrifuged at 3200g for 10 minutes and loaded back onto a 5 mL HiTrap IMAC FF column to remove cleaved octahistidine tags, remaining uncleaved protein and TEV protease. The flowthrough was pooled, concentrated and subjected to anion exchange chromatography. Protein was loaded onto a 20 mL MonoQ HR 16/10 (Cytiva) column equilibrated with a 90–10% mix of monoQ A buffer (0.5 mM tris(2-carboxyethyl)phosphine (TCEP), 25 mM HEPES pH 7.5) and monoQ B buffer (monoQ A with 1 M NaCl). The column was washed with 2 CV of 10% monoQ B buffer and 2 CV of 22% monoQ B buffer. The protein was eluted using a gradient from 22% to 40% monoQ B buffer (220–400 mM NaCl) over 9 CV. Fractions were evaluated by SDS-PAGE. Those containing purified protein were pooled and concentrated using a 10 kDa molecular weight cut off Amicon® Ultra-15 centrifugation concentrator (Millipore-Sigma). Unless used immediately, the protein was flash-frozen in liquid nitrogen and stored at −80 °C.
For purification of SrtAC2A2T2, AEP1C2A2T2 cell pellets were resuspended in 10 mL IC A buffer (IMAC A plus 3 mM CaCl2) per 1 L of cell culture. Cell lysis and metal affinity chromatography was performed as described above, except with IC A buffer and IC B buffer (IMAC B plus 3 mM CaCl2). SrtAC2A2T2 or AEP1C2A2T2 was then applied to a 30 mL calmodulin Sepharose 4B affinity (CBP) column (Cytiva) equilibrated in IC A buffer. The column was washed with 5 CV of IC A buffer and the protein was eluted with CBP B (2 mM β-mercaptoethanol, 3 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 25 mM HEPES, pH 7.5). Protein purity was evaluated by SDS-PAGE and purified fractions were pooled. To cleave the purification tags, the protein was incubated overnight with TEV protease and dialyzed against IC A buffer. Next, protein was loaded onto in-series HiTrap IMAC FF and CBP columns to remove cleaved tags, uncleaned protein and the TEV protease. The flowthrough was pooled and concentrated. Unless used immediately, the protein was flash-frozen in liquid nitrogen and stored at −80 °C.
Cell lysis, metal affinity purification and CBP affinity purification of F1A1T1C2A2T2 and F1A1T1C2A2T2CT3 constructs were performed as described for AEP1C2A2T2, except that a 10 mL CBP column was used. These proteins were further purified by size exclusion chromatography with a Superdex 200 Increase 10/300 GL (Cytiva) column and SEC buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 0.5 mM TCEP). Fractions containing pure protein were pooled, concentrated, flash frozen and stored at −80 °C.
OaAEP1 and SaSrtA were expressed in and purified from E. coli BL21 (DE3) following previously published protocols26,27 plus a gel filtration polishing step, initially with a HiLoad Superdex 75 16/60 (S75) (Cytiva) column equilibrated in SEC pH 7.5 (0.5 mM TCEP, 150 mM NaCl, 50 mM Tris pH 7.5). Once it was determined that OaAEP1 reactions would be performed at pH 6.5, its gel filtration polishing step was performed with SEC pH 6.5 buffer (same as SEC pH 7.5, but at pH 6.5). SDS-PAGE analysis of proteins used in this study are shown in ESI,† Fig. S11.
Optimization of the ligation reactions
For the optimization of both ligation strategies, the initial ligation reaction and the titration of F1A1T1 and C2A2T2 were carried out as single experiments. pH optimization, OaAEP1/SaSrtA titration and time-trials were carried out in triplicate for quantitative analysis. For quantitative analysis, known concentrations of wild-type F1A1T1C2A2T2 were diluted by the same factor and in the same buffer as reactions and run on the same gel. Band intensities on the gels were measured with the software ImageJ version 1.54k and a calibration curve was plotted in Microsoft Excel version 16.91 using the known concentrations of F1A1T1C2A2T2. Band intensities of the ligation products were thus used to calculate concentrations. All ligation reactions were carried out in a volume of 10 μL. All plots in Fig. 2 and 4 were made using GraphPad Prism version 10.4.0.All SaSrtA ligation reactions were quenched by the addition of 20 mM of EDTA. After the addition of EDTA different reactions were diluted differently for running on the SDS gels. For the initial ligation reaction, the F1A1T1SrtA: SrtAC2A2T2 titration and the pH trial, 1 μL of reaction was diluted in 9 μL of quenching buffer (0.05% bromophenol blue, 100 mM dithiothreitol (DTT), 10% (v/v) glycerol, 2% sodium dodecyl sulfate (SDS), 50 mM Tris–HCl, pH 6.8). For the SaSrtA titration and time trial, 1 μL was diluted in 49 μL of quenching buffer. The initial reaction, adapted from Byrd and colleagues,27 contained 20 μM, 6 μM and 5 μM SaSrtA in 1× SrtA ligation buffer pH 7.5 (50 mM Tris pH 7.5, 150 mM NaCl, 5 mM CaCl2, 0.5 mM TCEP) and was incubated for 30 minutes at room temperature. In the F1A1T1SrtA: SrtAC2A2T2 titration experiments, 1× SrtA ligation buffer pH 7.5 was mixed with 5 μM SaSrtA and 20–40 μM F1A1T1SrtA (at a constant concentration of SrtAC2A2T2) or 6–38 μM SrtAC2A2T2 (at a concentration of the F1A1T1SrtA) and incubated for 30 minutes at room temperature. For the pH trials, the pH of the 1× SrtA ligation buffer was varied, while keeping the concentration of all components constant: 20 μM F1A1T1SrtA, 38 μM SrtAC2A2T2, 5 μM SaSrtA, 30 minutes at room temperature. For the titration of SaSrtA, the F1A1T1SrtA and SrtAC2A2T2 were increased to 120 μM and 210 μM, respectively, in 1× SrtA ligation buffer pH 7.5 and SaSrtA was added at 5, 10, 20, 50 or 80 μM and incubated for 30 minutes at room temperature. In time course experiments, 120 μM F1A1T1SrtA, 210 μM SrtAC2A2T2 and 80 μM SaSrtA were mixed in 1× SrtA ligation buffer pH 7.5 at room temperature, and 1 μL aliquots were withdrawn and quenched at 15, 30, 60, 120, 240 and 360 minutes.
Initial ligation conditions for OaAEP1 were adapted from Wu and colleagues,26 and were: 1× AEP1 buffer pH 7.5 (1× SrtA buffer, without CaCl2), 8 μM F1A1T1AEP1, 5 μM AEP1C2A2T2 and 0.1 μM OaAEP1 at room temperature for 15 minutes. Module ratio optimization had those conditions, except that the concentrations of F1A1T1AEP1 and AEP1C2A2T2 were varied between 8–17 μM and 5–14 μM, respectively. In pH trials, the 1× AEP1 ligation buffer was altered to pH values between 6.5 and 9, with protein concentrations of 8 μM F1A1T1AEP1, 14 μM AEP1C2A2T2 and 0.1 μM OaAEP1, again at room temperature for 15 minutes. OaAEP1 concentration optimization reactions had 1× AEP1 buffer pH 6.5, 120 μM F1A1T1AEP1, 210 μM AEP1C2A2T2 and 0.1, 0.5, 1, 2 or 3 μM OaAEP1, with 15 minutes of room temperature incubation. Room temperature time course reactions of 120 μM F1A1T1AEP1, 210 μM AEP1C2A2T2, 1 μM OaAEP1 in 1× AEP1 buffer pH 6.5 were sampled with 1 μL withdrawals at 2, 5, 15, 30, 60 and 120 minutes. Quenching and preparation for SDS-PAGE were performed by adding 1 μL of the reaction mixtures to 9 μL (or 49 μL for OaAEP1 titration and time trials) of quenching buffer.
Synthesis and purification of substrate analogues
Non-hydrolyzable C domain substrate analogues, fVal-NH-CoA and Gly-NH-CoA were synthesized from amino coenzyme A (NH2-CoA) as previously published and characterized.11,12,41 Briefly, NH2-CoA was synthesized from amino-pantetheine (pant-NH; Wuxi Apptec)40 in a one-pot, three-enzyme reaction, and purified by preparatory HPLC with a YMC-Pack ODS-A preparatory column (250 × 20 mm) attached to a Waters Prep LCTM HPLC machine using a gradient of water to acetonitrile, with 0.1% trifluoroacetic acid (TFA) in each solution, with fractions content assessed by direct injection on an amaZon speed EDT (Bruker) ion trap mass spectrometer (ESI,† Fig. S6). Fractions containing NH2-CoA were pooled, frozen at −80 °C and lyophilized, and used in coupling reactions with protected glycine and formyl-valine compounds.
Tert-byloxycarbonyl (Boc)-protected glycyl-N-hydroxysuccinimide (Boc-Gly-NHS), NH2-CoA and N,N-diisopropylethylamine (DIPEA) at a ratio of 1:8:2 (NH2-CoA:Boc-Gly-NHS:DIPEA) in 80% dimethylformamide (DMF) was stirred overnight at room temperature. Boc-Gly-NH-CoA was dried by rotovap, deprotected with 95% TFA, 5% triisopropyl silane (TIPS) for 6 hours at room temperature and precipitated with pre-chilled diethyl ether, overnight at −20 °C. The sample was centrifuged at 3200g for 30 minutes, the diethyl ether was decanted, the compound was redissolved in 0.1% TFA, and purified by preparatory HPLC as above (using the same HPLC and column as for NH2-CoA). Resultant Gly-NH-CoA was resuspended in water at 50 mM and stored at −80 °C. fVal-NH-CoA was synthesized using NH2-CoA and formyl-valyl-NHS (fVal-NHS) using the same protocols as described for Gly-NH-CoA (and purified using the same HPLC and column), except that no Boc deprotection was performed.
Loading substrate analogues on F1A1T1 and C2A2T2
To load fVal-NH-ppant onto Ser729 of T1 in F1A1T1 constructs, one-pot reactions of 50 mM HEPES pH 7.5, 10 mM MgCl2, 40 μM F1A1T1AEP1 or F1A1T1SrtA, 200 μM fVal-NH-CoA and 10 μM Sfp were incubated overnight at room temperature.11,12 Ser1760 of T2 in C2A2T2 constructs was modified with Gly-NH-ppant using the same protocol, except with AEP1C2A2T2 or SrtAC2A2T2 in place of F1A1T1 constructs and Gly-NH-CoA in place of fVal-NH-CoA. Ppant loading was confirmed by LC–MS using a PLRP-S 1000A, 5 μm, 2.1 × 50 mm (Agilent) HPLC column attached to an Agilent 1260 series HPLC system and an amaZon speed EDT (Bruker) ion trap mass spectrometer (ESI,† Fig. S7). To purify F1A1T1-fValAEP1, F1A1T1-fValSrtA, AEP1C2A2T2-Gly or SrtAC2A2T2-Gly, the reactions were applied to a Superdex 200 16/60 column equilibrated in 1× AEP1 pH 6.5 or 1× SrtA buffer pH 7.5. Fractions were evaluated by SDS-PAGE. Those containing analog-loaded protein were pooled, concentrated with a 10 kDa molecular weight cut off Amicon® Ultra-15 centrifugation concentrator (Millipore-Sigma), flash frozen in liquid nitrogen and stored at −80 °C.Formation and purification of the F1A1T1-fVal–C2A2T2-Gly complexes for crystallization
As guided by our optimizations, reactions of 120 μM F1A1T1AEP1, 210 μM AEP1C2A2T2 and 1 μM OaAEP1 in 1× AEP1 buffer pH 6.5 were incubated for 30 minutes, or 120 μM F1A1T1SrtA, 210 μM SrtAC2A2T2 and 80 μM SaSrtA in 1× SrtA buffer pH 7.5 were incubated for 2 hours. F1A1T1-fVal–C2A2T2-Gly from each ligation was purified using a MonoQ HR 16/10 column (Cytiva) with monoQ A and monoQ B buffer. SaSrtA ligations were quenched by the addition of 20 mM EDTA prior to injection, while OaAEP1 ligation reactions were directly injected onto the MonoQ HR 16/10 column (Cytiva) equilibrated in 10% monoQ B buffer. The column was washed with 2 CVs of 10% monoQ B buffer and 2 CVs of 20% monoQ B buffer. Proteins were eluted from the column using a gradient from 20–36% monoQ B buffer over 9 CVs. Elution peaks were evaluated by SDS-PAGE and fractions containing F1A1T1-fVal–C2A2T2-Gly were pooled, concentrated and applied to a Superdex 200 Increase 10/300 GL (Cytiva) equilibrated in SEC buffer (0.5 mM TCEP, 150 mM NaCl, 25 mM HEPES pH 7.5). The purest elution fractions were concentrated and used for crystallization.Crystallization
F1A1T1-fVal–C2A2T2-Gly complexes were screened in a sparse matrix screen in a 96-well sitting-drop format, against five crystallization suites at three concentrations (2, 7 and 12 mg mL−1) and two temperatures (4 °C and 22 °C). Drops were set by adding 0.2 μL of crystallization solution and 0.2 μL of protein against a 50 μL reservoir. Crystal hits of OaAEP1-ligated F1A1T1-fVal–C2A2T2-Gly were optimized iteratively in a 24-well sitting-drop format with 2 μL crystallization solution and 2 μL of 7 mg mL−1 F1A1T1-fVal–C2A2T2-Gly, against a 500 μL reservoir at 4 °C. The crystal which yielded the structure, presented here, grew with a crystallization solution of 2% polyethylene glycol (PEG) 2000 monomethyl ether (MME), 0.9 M sodium succinate, 0.1 M HEPES pH 6.8, 0.1 M ammonium succinate and 0.5 mM 1-nonanoyl-2-hydroxy-sn-glycero-3-phosphocholine (09:0 lyso-PC), with streak seeding. Cryo-protection was performed by transfer into a solution of 2% PEG 2000 MME, 1.0 M sodium succinate, 0.1 M HEPES pH 6.8, 0.1 M ammonium succinate, 0.5 mM 09:0 lyso-PC, plus 10% PEG 400, then into a solution of the same composition but 20% PEG 400. Crystals were then looped and vitrified in liquid nitrogen. Diffraction data collection was carried out at the 24-ID-E-beamline of the Advanced Photon Source (APS) with a beam wavelength of 0.98 Å, and cooling to 200 K.Data indexing, integration and scaling was carried out using the software HKL2000 v721.77 Phases were calculated over two rounds of molecular replacement using the Phaser software v2.9.0,78 placing F1A1T1C2A2 as the first round and T2 the second.12 A restraint file for visible atoms of ppant attached on T1 was generated using eLBOW.79 Iterative refinement and modelling was performed with Coot v0.9.8.9280 and Phenix software suite v1.20-448781 to yield the final structure (ESI,† Table S1).
Tripeptide biosynthesis assay
Activity assays for F1A1T1C2A2T2CT3 and variants harbouring the -Leu-Pro-Glu-Th-Gly-Gly-Gly or -Asn-Gly-Leu-ligation scars in place of Ser1771 in the T1:C2 linker were carried out using the previously developed tripeptide biosynthesis assay.12,82 Briefly, solutions (40 μL) of 0.2 mM 5,10-mTHF, 2 mM valine, 1 mM glycine, 4 mM tryptamine, 5 mM ATP, 0.7 mM MgCl2, 1 mM TCEP, 150 mM NaCl and 50 mM HEPES pH 7.4 were pre-incubated for 10 minutes at 23 °C to allow 5,10-mTHF to convert to 10-fTHF. F1A1T1C2A2T2CT3 constructs were then added to the reactions and incubated for 5 hours at 23 °C. Negative controls lacked ATP. All reactions were carried out in triplicate. Reactions were quenched with 300 μL of 4:1 butanol:chloroform and vortexing, prior to incubation at −80 °C and lyophilization. Lyophylates were dissolved in 50 μL of methanol and vortexed thoroughly. Samples were transferred into HPLC vials and 40 μL aliquots were injected onto an Agilent 1260 series HPLC system with Eclipse XDB-C8 LC column (Agilent), and eluted with a gradient from water to acetonitrile in 0.1% TFA. Peak contents were confirmed by direct injection on an amaZon speed EDT (Bruker) ion trap mass spectrometer. Activity was quantified by integrating the area of the HPLC peak corresponding to the tripeptide product. Mean activity was calculated as the mean HPLC peak area of the three replicates and relative tripeptide biosynthesis was determined by calculating the HPLC peak area of the variants as a percentage of the wild type (F1A1T1C2A2T2–CT3). Error was determined as the standard deviation in the three replicates. The mean peak area, relative tripeptide biosynthesis and standard deviation were calculated using Microsoft Excel version 16.91. The bar chart of the relative tripeptide biosynthesis (Fig. 3) was plotted in GraphPad Prism v10.4.0.
Author contributions
AP: investigation, methodology, formal analysis, validation, data curation, visualization, writing – original draft. TMS: conceptualization, formal analysis, funding acquisition, resources, supervision, writing – review & editing.Data availability
Crystallographic data for F1A1T1-fVal–C2A2T2-Gly has been deposited at PBD under accession number 9MEH and can be obtained from https://www.rcsb.org/structure/9MEH.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This research was funded by grants to T. M. S. from Canadian Institutes for Health Research (grant PJT-178084). Infrastructure at the McGill University Centre de recherche en biologie structurale, used in this research, is supported by Fonds de Recherche du Québec (Health Sector) Research Centres grant #288558. We thank Yusuf Qutbuddin, Anirudh Mantri, Natalia Frota, Janice Reimer, Max Eivaskhani and the other members of the Schmeing laboratory, Karine Auclair, and Nancy Rogerson for advice and helpful discussion, and APS beamline staff for facilitating data collection. This work included data collected at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on the 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. We thank Christian Chalut and Christophe Guilhot for gift of Escherichia coli BL21 (DE3) entD-cells. AEP1-encoding plasmid was provided by Wu Bin (Nanyang Technological University).References
- N. Kessler, H. Schuhmann, S. Morneweg, U. Linne and M. A. Marahiel, J. Biol. Chem., 2004, 279, 7413–7419 CrossRef CAS PubMed.
- D. Colombo and E. Ammirati, J. Biol. Regul. Homeostatic Agents, 2011, 25, 493–504 CAS.
- T. M. Karpinski and A. Adamczak, Pharmaceutics, 2018, 10(2), 54 CrossRef PubMed.
- J. M. Reimer, A. S. Haque, M. J. Tarry and T. M. Schmeing, Curr. Opin. Struct. Biol., 2018, 49, 104–113 CrossRef CAS PubMed.
- K. D. Patel, M. R. MacDonald, S. F. Ahmed, J. Singh and A. M. Gulick, Nat. Prod. Rep., 2023, 40, 1550–1582 RSC.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- T. Stachelhaus, A. Huser and M. A. Marahiel, Chem. Biol., 1996, 3, 913–921 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz, V. Bergendahl and M. A. Marahiel, J. Biol. Chem., 1998, 273, 22773–22781 CrossRef CAS.
- J. Crosby and M. P. Crump, Nat. Prod. Rep., 2012, 29, 1111–1137 RSC.
- Y. Liu, T. Zheng and S. D. Bruner, Chem. Biol., 2011, 18, 1482–1488 CrossRef CAS PubMed.
- J. M. Reimer, M. N. Aloise, P. M. Harrison and T. M. Schmeing, Nature, 2016, 529, 239–242 CrossRef CAS PubMed.
- J. M. Reimer, M. Eivaskhani, I. Harb, A. Guarne, M. Weigt and T. M. Schmeing, Science, 2019, 366, 6466 CrossRef PubMed.
- T. Izore, Y. T. C. Ho, J. A. Kaczmarski, A. Gavriilidou, K. H. Chow, D. L. Steer, R. J. A. Goode, R. B. Schittenhelm, J. Tailhades, M. Tosin, G. L. Challis, E. H. Krenske, N. Ziemert, C. J. Jackson and M. J. Cryle, Nat. Commun., 2021, 12, 2511 CrossRef CAS PubMed.
- Y. T. C. Ho, T. Izoré, J. A. Kaczmarski, E. Marschall, M. S. Ratnayake, J. Tailhades, D. L. Steer, R. B. Schittenhelm, M. Tosin, C. J. Jackson and M. J. Cryle, Front. Catal., 2023, 3, 1184959 CrossRef.
- L. E. Quadri, P. H. Weinreb, M. Lei, M. M. Nakano, P. Zuber and C. T. Walsh, Biochemistry, 1998, 37, 1585–1595 CrossRef CAS PubMed.
- P. Tufar, S. Rahighi, F. I. Kraas, D. K. Kirchner, F. Lohr, E. Henrich, J. Kopke, I. Dikic, P. Guntert, M. A. Marahiel and V. Dotsch, Chem. Biol., 2014, 21, 552–562 CrossRef CAS PubMed.
- M. R. Mofid, R. Finking, L. O. Essens and M. A. Marahiel, Biochemistry, 2004, 43, 4128–4136 CrossRef CAS PubMed.
- J. L. Meier, R. W. Haushalter and M. D. Burkart, Bioorg. Med. Chem. Lett., 2010, 20, 4936–4939 CrossRef CAS PubMed.
- A. S. Worthington, H. Rivera, J. W. Torpey, M. D. Alexander and M. D. Burkart, ACS Chem. Biol., 2006, 1, 687–691 CrossRef CAS PubMed.
- S. Kapur, A. Worthington, Y. Tang, D. E. Cane, M. D. Burkart and C. Khosla, Bioorg. Med. Chem. Lett., 2008, 18, 3034–3038 CrossRef CAS PubMed.
- T. Stachelhaus and M. A. Marahiel, J. Biol. Chem., 1995, 270, 6163–6169 CrossRef CAS PubMed.
- H. E. Morgan, W. B. Turnbull and M. E. Webb, RSC Chem. Soc. Rev., 2022, 51, 4121–4145 RSC.
- K. S. Harris, T. Durek, Q. Kaas, A. G. Poth, E. K. Gilding, B. F. Conlan, I. Saska, N. L. Daly, N. L. Van Der Weerden, D. J. Craik and M. A. Anderson, Nat. Commun., 2015, 6, 10199 CrossRef CAS PubMed.
- L. A. Marraffini, A. C. DeDent and O. Schneewind, Microbiol. Mol. Biol. Rev., 2006, 70, 192–221 CrossRef CAS PubMed.
- I. Chen, B. M. Dorr and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11399–11404 CrossRef CAS.
- R. Yang, Y. H. Wong, G. K. T. Nguyen, J. P. Tam, J. Lescar and B. Wu, J. Am. Chem. Soc., 2017, 139, 5351–5358 CrossRef CAS PubMed.
- J. Li, Y. Zhang, O. Soubias, D. Khago, F.-A. Chao, Y. Li, K. Shaw and R. A. Byrd, J. Biol. Chem., 2020, 295, 2664–2675 CrossRef CAS PubMed.
- J. G. M. Bolscher, M. J. Oudhoff, K. Nazmi, J. M. Antos, C. P. Guimaraes, E. Spooner, E. F. Haney, J. J. G. Vallejo, H. J. Vogel, W. Hof, H. L. Ploegh and E. C. I. Veerman, FASEB J., 2011, 25, 2650–2658 CrossRef CAS PubMed.
- L. Freiburger, M. Sonntag, J. Hennig, J. Li, P. Zou and M. Sattler, J. Biomol. NMR, 2015, 63, 1–8 CrossRef CAS PubMed.
- K. M. Mikula, I. Tascón, J. J. Tommila and H. Iwaï, FEBS Lett., 2017, 591, 1285–1294 CrossRef CAS PubMed.
- S. B. Azatian, M. D. Canny and M. P. Latham, J. Biomol. NMR, 2022, 77, 25–37 CrossRef PubMed.
- K. M. Mikula, K. L., A. Plückthun and H. Iwaï, J. Biomol. NMR, 2018, 71, 225–235 CrossRef CAS PubMed.
- M. A. Refaei, A. Combs, D. J. Kojetin, J. Canagh, C. Caperelli, M. Rance, J. Sapitro and P. Tsang, J. Biomol. NMR, 2011, 49, 3–7 CrossRef CAS PubMed.
- L. Chan, H. F. Cross, J. K. She, G. Cavalli, H. F. P. Marting and C. Neylon, PLoS One, 2007, 2, e1164 CrossRef PubMed.
- Y. Deng, T. Wu, M. Wang, S. Shi, G. Yuan, X. Li, H. Chong, B. Wu and P. Zheng, Nat. Commun., 2019, 10, 2775 CrossRef.
- Z. Lu, Y. Liu, Y. Deng, B. Jia, X. Ding, P. Zheng and Z. Li, Chem. Commun., 2022, 58, 8448–8451 RSC.
- T. J. Harmand, N. Pishesha, F. B. H. Rehm, W. Ma, W. B. Pinney, Y. J. Xie and H. L. Ploegh, ACS Chem. Biol., 2021, 16, 1201–1207 CrossRef CAS PubMed.
- X. Ding, Z. Wang, B. Zheng, S. Shi, Y. Deng, H. Yu and P. Zheng, RSC Chem. Biol., 2022, 3, 1276–1281 RSC.
- V. Ulrich and M. J. Cryle, J. Pept. Sci., 2017, 23, 16–27 CrossRef CAS PubMed.
- I. Nazi, K. P. Koteva and G. D. Wright, Anal. Biochem., 2004, 324, 100–105 CrossRef CAS PubMed.
- A. Pistofidis, P. Ma, Z. Li, K. Munro, K. N. Houk and T. M. Schmeing, Nature, 2025, 638, 270–278 CrossRef CAS.
- M. J. Wheadon and C. A. Townsend, ACS Chem. Biol., 2022, 17, 2046–2053 CrossRef CAS.
- A. S. Worthington and M. D. Burkart, Org. Biomol. Chem., 2006, 4, 44–46 RSC.
- K. Bloudoff, C. D. Fage, M. A. Marahiel and T. M. Schmeing, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 95–100 CrossRef CAS PubMed.
- C. M. Fortinez, K. Bloudoff, C. Harrigan, I. Sharon, M. Strauss and T. M. Schmeing, Nat. Commun., 2022, 13, 548 CrossRef CAS PubMed.
- Y. Mathieu, J. Guern, A. Kurkdjian, P. Manigault, J. Manigault, T. Zielinska, B. Gillet, J.-C. Beloeil and J.-Y. Lallemand, Plant Physiol., 1989, 89, 19–26 CrossRef CAS.
- T. M. S. Tang and Y. P. L. Luk, RSC Org. Biomol. Chem., 2021, 19, 5048–5062 RSC.
- X. Hemu, A. E. Sahili, S. Hu, X. Zhang, A. Serra, B. C. Goh, D. A. Darwis, M. W. Chen, S. K. Sze, C.-F. Liu, J. Lescar and J. P. Tam, ACS Catal., 2020, 10, 8825–8834 CrossRef CAS.
- T. M. S. Tang, D. Cardella, A. J. Lander, X. Li, J. S. Escudero, Y.-H. Tsai and L. Y. P. Luk, Chem. Sci., 2020, 11, 5881–5888 RSC.
- A. Tanovic, S. A. Samel, L. O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 CrossRef CAS PubMed.
- T. A. Keating, C. G. Marshall, C. T. Walsh and A. E. Keating, Nat. Struct. Biol., 2002, 9, 522–526 CAS.
- M. Kaniusaite, J. Tailhades, E. A. Marschall, R. J. A. Goode, R. B. Schittenhelm and M. J. Cryle, Chem. Sci., 2019, 10, 9466–9482 RSC.
- C. Chalut, L. Botella, C. de Sousa-D’Auria, C. Houssin and C. Guilhot, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8511–8516 CrossRef CAS PubMed.
- G. W. Heberlig, J. J. La Clair and M. D. Burkart, Nature, 2024, 638, 261–269 CrossRef PubMed.
- C. Shi, B. R. Miller, E. M. Alexander, A. M. Gulick and C. C. Aldrich, ACS Chem. Biol., 2020, 15, 1813–1819 CrossRef CAS PubMed.
- D. J. Wilson, C. Shi, A. M. Teitelbaum, A. M. Gulick and C. C. Aldrich, Biochemistry, 2013, 52, 926–937 CrossRef CAS PubMed.
- Y. Yu and W. A. van der Donk, ACS Cent. Sci., 2024, 10, 1242–1250 CrossRef CAS PubMed.
- A. M. Gulick and C. C. Aldrich, Nat. Prod. Rep., 2018, 35, 1156–1184 RSC.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 690–697 CrossRef PubMed.
- S. Farag, R. M. Bleich, E. A. Shank, O. Isayev, A. A. Bowers and A. Tropsha, Bioinformatics, 2019, 35, 3584–3591 CrossRef CAS PubMed.
- B. R. Miller, J. A. Sundlov, E. J. Drake, T. A. Makin and A. M. Gulick, Proteins, 2014, 82, 2691–2702 CrossRef CAS PubMed.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. L. Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Nature, 2016, 529, 235–238 CrossRef CAS PubMed.
- D. F. Kreitler, E. M. Gemmell, J. E. Schaffer, T. A. Wencewicz and A. M. Gulick, Nat. Commun., 2019, 10, 3432 CrossRef PubMed.
- M. J. Tarry, A. S. Haque, K. H. Bui and T. M. Schmeing, Structure, 2017, 25, 783–793 CrossRef CAS PubMed.
- H. D. Mootz, D. Schwarzer and M. A. Marahiel, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5848–5853 CrossRef CAS PubMed.
- K. A. J. Bozhuyuk, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C. P. Niesert and H. B. Bode, Nat. Chem., 2018, 10, 275–281 CrossRef PubMed.
- D. A. Levary, R. Parthasarathy, E. T. Boder and M. E. Ackerman, PLoS One, 2011, 6, e18342 CrossRef CAS PubMed.
- A. Okuda, M. Shimizu, R. Urade, N. Fujii and M. Sugiyama, Angew. Chem., Int. Ed., 2022, 62, e202214412 CrossRef PubMed.
- L. Skrisovska and F. H. T. Allain, J. Mol. Biol., 2008, 375, 151–164 CrossRef CAS PubMed.
- M. Muona, A. S. Aranko and H. Iwaï, ChemBioChem, 2008, 9, 2958–2961 CrossRef CAS PubMed.
- A. E. L. Busche, A. S. Aranko, M. Talebzadeh-Farooji, F. Bernhard, V. Dotsch and H. Iwai, Angew. Chem., Int. Ed., 2009, 48, 6128–6131 CrossRef CAS PubMed.
- Y. Minato, T. Ueda, A. Machiyama, I. Shimada and H. Iwaï, J. Biomol. NMR, 2012, 53, 191–207 CrossRef CAS PubMed.
- J. Rüschenbaum, W. Steinchen, F. Mayerthaler, A.-L. Feldberg and H. D. Mootz, Angew. Chem., Int. Ed., 2022, 61, e202212994 CrossRef PubMed.
- X. Sun, J. Alfermann, H. Li, M. B. Watkins, Y.-T. Chen, T. E. Morrell, F. Mayerthaler, C.-Y. Wang, T. Komatsuzaki, J.-W. Chu, N. Ando, H. D. Mootz and H. Yang, Nat. Chem., 2024, 16, 259–268 CrossRef CAS PubMed.
- J. Alfermann, X. Sun, F. Mayerthaler, T. E. Morrell, E. Dehling, G. Volkmann, T. Komatsuzaki, H. Yang and H. D. Mootz, Nat. Chem. Biol., 2017, 13, 1009–1015 CrossRef CAS PubMed.
- B. A. Pfeifer, S. J. Admiraal, H. Gramajo, D. E. Cane and C. Khosla, Science, 2001, 291, 1790–1792 CrossRef CAS PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- N. W. Moriarty, R. W. Grosse-Kunstleve and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 1074–1080 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- P. D. Adams, R. W. Grosse-Kunstleve, L. W. Hung, T. R. Ioerger, A. J. McCoy, N. W. Moriarty, R. J. Read, J. C. Sacchettini, N. K. Sauter and T. C. Terwilliger, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 1948–1954 CrossRef PubMed.
- J. M. Reimer, I. Harb, O. G. Ovchinnikova, J. Jiang, C. Whitfield and T. M. Schmeing, ACS Chem. Biol., 2018, 13, 3161–3172 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00306c |
| This journal is © The Royal Society of Chemistry 2025 |