
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn activation-based high throughput screen identifies caspase-10 inhibitors†
José O.
Castellón
a,
Constance
Yuen
cd,
Brandon
Han
c,
Katrina H.
Andrews
a,
Samuel
Ofori
a,
Ashley R.
Julio
ab,
Lisa M.
Boatner
ab,
Maria F.
Palafox
abe,
Nithesh
Perumal
 ab,
Robert
Damoiseaux
cdfgh and
Keriann M.
Backus
*abcghi
ab,
Robert
Damoiseaux
cdfgh and
Keriann M.
Backus
*abcghi
aBiological Chemistry Department, David Geffen School of Medicine, UCLA, Los Angeles, CA 90095, USA. E-mail: kbackus@mednet.ucla.edu
bDepartment of Chemistry and Biochemistry, UCLA, CA 90095, USA
cCalifornia NanoSystems Institute (CNSI), UCLA, Los Angeles, CA 90095, USA
dDepartment of Molecular and Medical Pharmacology, UCLA, Los Angeles, CA 90095, USA
eDepartment of Human Genetics, David Geffen School of Medicine, UCLA, Los Angeles, CA 90095, USA
fDepartment of Bioengineering, Samueli School of Engineering, UCLA, Los Angeles, CA 90095, USA
gJonsson Comprehensive Cancer Center, UCLA, Los Angeles, CA 90095, USA
hEli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research, UCLA, Los Angeles, CA 90095, USA
iUCLA DOE Institute for Genomics and Proteomics, UCLA, Los Angeles, CA 90095, USA
First published on 4th February 2025
Abstract
Caspases are a family of highly homologous cysteine proteases that play critical roles in inflammation and apoptosis. Small molecule inhibitors are useful tools for studying caspase biology, complementary to genetic approaches. However, achieving inhibitor selectivity for individual members of this highly homologous enzyme family remains a major challenge in developing such tool compounds. Prior studies have revealed that one strategy to tackle this selectivity gap is to target the precursor or zymogen forms of individual caspases, which share reduced structural homology when compared to active proteases. To establish a screening assay that favors the discovery of zymogen-directed caspase-10 selective inhibitors, we engineered a low-background and high-activity tobacco etch virus (TEV)—activated caspase-10 protein. We then subjected this turn-on protease to a high-throughput screen of approximately 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds, with an average Z′ value of 0.58 across all plates analyzed. Counter screening, including against TEV protease, delineated bona fide procaspase-10 inhibitors. Confirmatory studies identified a class of thiadiazine-containing compounds that undergo isomerization and oxidation to generate cysteine-reactive compounds with caspase-10 inhibitory activity. In parallel, mode-of-action studies revealed that pifithrin-μ (PFTμ), a reported TP53 inhibitor, also functions as a promiscuous caspase inhibitor. Both inhibitor classes showed preferential zymogen inhibition. Given the generalized utility of activation assays, we expect our screening platform to have widespread applications in identifying state-specific protease inhibitors.
000 compounds, with an average Z′ value of 0.58 across all plates analyzed. Counter screening, including against TEV protease, delineated bona fide procaspase-10 inhibitors. Confirmatory studies identified a class of thiadiazine-containing compounds that undergo isomerization and oxidation to generate cysteine-reactive compounds with caspase-10 inhibitory activity. In parallel, mode-of-action studies revealed that pifithrin-μ (PFTμ), a reported TP53 inhibitor, also functions as a promiscuous caspase inhibitor. Both inhibitor classes showed preferential zymogen inhibition. Given the generalized utility of activation assays, we expect our screening platform to have widespread applications in identifying state-specific protease inhibitors.
Introduction
Human caspases are a family of 12 cysteine proteases, which are canonically associated with cell death,1–3 but have also been tied to nearly all cellular processes, spanning activation,4,5 proliferation,6,7 differentiation,8,9 and cell migration.10 Therefore, pharmacological manipulation of individual caspases represents an exciting opportunity to delineate caspase-specific functions and to intervene in human pathologies linked to dysregulated caspase activity, including metabolic11 and immune disorders,12–15 cancers,16–20 and neurodegenerative diseases.21 Selective inhibitors are available for caspase-1,22 caspase-2,23,24 caspase-6,25,26 and caspase-8.27,28 Despite these advances, obtaining highly selective inhibitors remains challenging, likely due in large part to the high sequence and structural homology of all caspases.Caspase-10 is one such caspase that, while particularly intriguing from a target perspective due to its important functions in immune cell apoptosis, still lacks selective inhibitors. In fact, caspase-10 is one of the only caspases that is not labeled by many conventional peptide-based caspase inhibitors.29,30 Caspase-10 is absent in rodents31 and shares high sequence homology with caspase-8. Efforts to delineate caspase-10's role in initiating apoptosis have been complicated by seemingly contradictory findings in immortalized cancer cells versus primary T cells.32,33 In cancer cells, caspase-8 rather than caspase-10 is the primary driver of extrinsic apoptosis.32–37 Caspase-10 has even been implicated as a dominant negative regulator of programmed cell death in some cancer types.1,38 In contrast, in primary T cells, caspase-10 contributes to the initiation of apoptosis;13 humans harboring inactivating mutations in caspase-10 experience autoimmunity and excessive T cell proliferation39,40 because of decreased apoptosis. These human phenotypes indicate non-functionally redundant roles for caspase-8 and -10 and highlight the likely value of selective inhibitors targeting each caspase, both for further characterizing the unique and overlapping activities of each protease and, more broadly, towards the production of new chemical tool compounds to manipulate adaptive immune cell function.
Type II kinase inhibitors, which target the inactive form of enzymes, exemplify one strategy to improve inhibitor selectivity.41–44 We took this approach in our prior work, which identified selective caspase-8 and caspase-2 inhibitors that function by targeting the zymogen, or uncleaved and inactive, precursor caspase proteoforms.24,27,45 However, in our previous study, developing selective inhibitors for caspase-10 that did not cross-react with caspase-8 proved elusive despite our best efforts.27 Consequently, there is an unmet need for new approaches to caspase-10 inhibitor discovery.
Here, we develop and apply a tobacco etch virus (TEV) activation-based screening platform to discover procaspase-10 inhibitors. To build this platform, we first generated an engineered caspase-10 protein (proCASP10TEV Linker) in which the caspase cleavage sites were replaced with TEV recognition sequences. This engineered protease showed low background, high stability, and robust TEV-dependent activity. After TEV activation, proCASP10TEV Linker protease showed comparable activity to recombinant active caspase-10. Enabled by this assay, we conducted a ∼100000 compound screen, which had a hit rate of ∼0.22% (calculated as compounds with a Z-score less than −3) and an average Z′-factor (a measure of how well positive and negative controls are separated)46 of 0.58 (Table S1, ESI†). Our subsequent re-screening and counter-screening efforts validated hits and delineated TEV inhibitors from those targeting proCASP10. Resynthesis of a thiadiazine-containing hit compound (SO265) unexpectedly revealed that compound rearrangement was driving the observed caspase-10 inhibition. Additionally, mode-of-action studies revealed that the hit compound pifithrin-μ (PFTμ), a previously reported inhibitor of TP53,47 is also a broadly reactive caspase inhibitor. Taken together, we expect that our hit compounds and innovative screening platform will advance efforts to discover potent and selective procaspase-10 inhibitors.
Establishing a TEV-protease activatable caspase-10
Guided by previous reports of TEV-activatable caspases,24,48,49 our first step was to engineer a TEV-activatable caspase-10 protein (Fig. 1(A)). For our first attempt at engineering a TEV-cleavable construct (proCASP10TEV), we replaced the aspartate cleavage site, D415, with the TEV recognition site (Fig. S1; ESI† D415ENLYFQG). While this protein did show substantial TEV-dependent increased activity, a high background in the absence of TEV protease was also observed (Fig. 1(B) and Fig. S2, ESI†). We ascribed this background activity to the formation of activated protease in our purified protein sample (Fig. S3, ESI†). The presence of TEV-independent protease activity, and therefore, cleaved caspase, while somewhat varied between recombinant protein batches, was consistently observed in our purified protein after labeling with Rho-DEVD-AOMK (Fig. S3, ESI†). The background activity was also observed to increase markedly over time, even at nanomolar protein concentrations and with increasing concentrations of the sodium citrate kosmotrope (Fig. S4, ESI†). Therefore, we deemed this construct incompatible with HTS, given the absolute requirement for a highly stable and consistently performing protein for large-scale screening applications. | ||
| Fig. 1 Robust TEV-cleavable functional caspase-10 is only cleaved in the presence of TEV protease and can be inhibited with a dual caspase-8/-10 inhibitor (KB7).27 (A) Design of engineered TEV cleavable caspase-10 containing TEV recognition site, ENLYFQG (pink). (B) Comparison of relative TEV-dependent and TEV-independent caspase activity for the indicated engineered proteins (333 nM) to cleave Ac-VDVAD-AFC fluorogenic substrate (10 μM) with/without TEV protease (667 nM). Sequences of engineered proteins can be found in Fig. S1 (ESI†). (C) Time course comparing activeCASP10 (blue) versus proCASP10TEV Linker with (red) and without (green) TEV protease (667 nM) in the presence of Ac-VDVAD-AFC fluorogenic substrate (10 μM). (D) Michaelis–Menten kinetics comparing proCASP10TEV Linker (333 nM) activity to activeCASP10 (333 nM) activity as measured by cleavage of the fluorogenic substrate over time. (E) Assessing the relative inhibition of proCASP10 Linker protein by dual procaspase-8/-10 inhibitor KB7 (10 μM) and negative control compound KB62 (10 μM).27 Assay conducted as in ‘B’. For B and E, data represents mean value ± standard deviation for two biological replicates. For C and D, data represent mean values ± standard deviation for three biological replicates. Statistical significance was calculated with unpaired Student's t-tests, *p < 0.05, **p < 0.001, ns, not significant p > 0.05. Relative fluorescence units, RFU. | ||
While caspase-10 is not known to harbor additional caspase cleavage sites, we postulated that D435 might also be recognized and cleaved, given the proximity to a likely caspase-recognition motif (PAED). Therefore, we inserted a second TEV motif to generate the proCASP2xTEV protein (Fig. S1, ESI†). Disappointingly, this enzyme showed low overall activity and negligible TEV-dependent activation, suggesting that sequence alterations at D435 are not well tolerated (Fig. 1(B)).
While caspases show high selectivity for aspartyl residues, we postulated that the glutamate residue in the TEV recognition sequence could be recognized and cleaved at a low level, thereby rationalizing the observed high TEV-independent background for the proCASP10TEV protein. To test this hypothesis, we next generated a caspase-10 construct (proCASP10TEV Linker) in which a two-alanine spacer was included to reposition this glutamate further from the remainder of the caspase recognition motif. We additionally optimized our expression and purification protocol to reduce self-activation, both by decreasing the induction time and by not freezing the cell pellets prior to purification (see methods). Pleasingly, the proCASP10TEV Linker protein exhibited both higher overall activity and reduced background compared to our initial construct (Fig. 1(B)). Further supporting the improved behavior of this protein, we detected no formation of cleaved caspase (∼20 kDa) by gel-based analysis (Fig. S5, ESI†).
Assessing the suitability of proCASP10TEV Linker for high throughput screening (HTS)
To test whether the proCASP10TEV Linker protein would faithfully recapitulate proCASP10 activity and thus prove suitable for HTS, we further assessed the activity of this protein. Gratifyingly, upon the addition of TEV protease, we observed marked TEV-dependent increased catalytic activity (Fig. 1(B) and (C)). TEV activation occurred rapidly and required only a modest (∼500 nM) concentration of TEV protease to achieve complete conversion of proCASP10TEV Linker (333 nM) to the cleaved proteoform (Fig. S6 and S7, ESI†). The proCASP10TEV Linker protein, once activated by TEV protease, showed near-comparable activity to activeCASP10 (Fig. 1(C)), with only a modest decrease in kcat and Km (Fig. 1(D)). We find that the protein labels robustly with our previously reported caspase-8/10 click probe KB61 (Fig. S6, ESI†),27 which corroborates that the proCASP10TEV Linker protein behaves similarly to proCASP10. The TEV cleavable construct was also inhibited by the dual caspase-8 and caspase-10 inhibitor KB7 (10 μM) and was not inhibited by the structurally matched inactive control, KB62 (10 μM) (Fig. 1(E)).27 These findings provided further evidence that our engineered protein behaves similarly to proCASP10 and, additionally, demonstrated that the protein is well suited to assess procaspase inhibition.We next vetted our assay conditions for HTS compatibility, with the goal of ensuring both high activity and high stability over longer assay periods. Additionally, we found that the proCASP10TEV Linker protein showed increased activity in the presence of the kosmotrope sodium citrate at increasing concentrations (37 mM, 111 mM, and 333 mM) (Fig. S8, ESI†). This added activity was restricted to the TEV-cleaved/activated protein; little TEV-independent activity was observed after chaotrope addition in the absence of TEV protease. Providing evidence of high enzyme stability, a favorable property for screening applications, proCASP10TEV Linker showed no substantial change in enzyme activity after prolonged (18 h) incubations times for both 4 °C and ambient temperature (Fig. S9, ESI†). Additionally, substrate turnover was observed to progress in a linear manner for the initial ∼2 h period, with increasing activity observed up to 6 h (Fig. S10, ESI†), which provides a wide time window for acquiring data. Thus, with high stability, low background, and high activity enzyme in hand, we turned to HTS implementation.
A small-scale screen of two pharmacologically active library compounds confirms assay compatibility with HTS
Toward large-scale library screening, our next step was to validate our TEV-mediated caspase activation assay and establish a miniaturized (384-well plate) semi-automated workflow. Following the workflow shown in Fig. 2(A), we first screened the ‘Library of Pharmacologically Active Compounds’ (LOPAC@1280), which is a widely utilized library of bioactive compounds that contain known promiscuous protease inhibitors such as E-64, along with a second library consisting of structurally diverse FDA approved drugs covering a broad spectrum of therapeutic areas.50 Key features of our screen include (1) the pre-incubation of compound library members with the proCASP10TEV Linker construct to favor the detection of procaspase inhibitors and (2) the automated dispensing of the premixed TEV protease and caspase substrate solution followed by automated plate reading. These two automation steps were designed to ensure the consistent timing of our assay to minimize both plate-to-plate and library-to-library variability. Confirming the robustness of our approach, this initial screen had a Z′-factor above 0.5 across 8 screened plates, with a total of 30 out of 2569 compounds affording a >50% reduction in caspase-10 activity relative to DMSO control (Fig. S11, ESI†). Illustrating the robust performance of our screen, the Z′-factor comparing our positive control KB7 inhibitor-treated wells to DMSO-treated wells27 was 0.90 (Fig. S12, ESI†)—values above 0.5 are considered an acceptable range for HTS Z′-factor.46 | ||
| Fig. 2 TEV cleavable proCASP10TEV Linker enables a >100000 compound screen. (A) Scheme of HTS setup with proCASP10TEV Linker (333 nM concentration) incubated with screening compounds (10 μM) for 1 h in a 384-well format followed by the addition of the fluorogenic substrate (Ac-VDAVAD-AFC, 10 μM). Endpoint reads were measured and collected after 1 h incubation with the TEV substrate solution. (B) Z-scores of >100000 compounds (arbitrarily numbered) screened against proCASP10TEV Linker. The red line indicates hits below a Z-score of −3. (C) Summary of filtering parameters for HTS screen. The first filtering step was to remove plates that were not within the desired range of Z′-factor (0.5–1.0), followed by a second filtering step that identified a total of 237 compounds with a Z-score of ≤−3 (D) SAR analysis of 237 hit compounds identified from HTS using the structure–activity landscape index (SALI) analysis as described in the DataWarrior SAR analysis methods section. The structure of recurring hit chemotypes identified by SAR cluster analysis (DataWarrior, v06.01.04.)51 are shown for cluster 1. All screening data is in Table S1 (ESI†). | ||
Large-scale screen identifies hit caspase inhibitors
Guided by the successful implementation of our pilot screen, we next deployed our platform to screen 118498 total compounds (all at 10 μM). In aggregate, this screen had Z′ factor ranging from 0.4–1.0 and a hit rate of 0.81% for a total of 963 compounds, affording a 50% decrease in proCASP10TEV Linker activity (Fig. S13, ESI†), with 237 unique hits showing a Z-score ≤−3 (Fig. 2(B)). The screen filtering steps are summarized in Fig. 2(C). Re-screen validation of the 237 compounds results in 38 compounds with <50% activity (Fig. S14A, ESI†). Z-Score analysis of the follow-up screen resulted in 97 (or approximately 41%) compounds below 3 standard deviations away from the mean of DMSO control samples (Fig. S14B, ESI†). Structural similarity clustering analysis using Datawarrior51 revealed that approximately 22% of the 237 hits (defined as Z-score ≤−3 or those values that are three standard deviations lower than the mean of the DMSO controls calculated per plate) clustered together, providing evidence of bona fide inhibitors. The 7 major hit clusters consisting of over 50 compounds are shown in Fig. 2(D). One prominent and intriguing chemotype was a class of thiadiazine-containing compounds identified in cluster 1 (Fig. 2(D)). Upon closer inspection of the hits, several compounds with clear electrophilic groups stood out, including several organometallic species such as cisplatin (Table S1, ESI†), several cyanoacryl sulfones, such as HTS-2 and the related HTS-6, which is a previously reported as non-specific κB kinase (IKK) inhibitor,52 and pifithrin-μ (PFTμ), a previously reported TP53 inhibitor47 (Scheme S1, ESI†). As caspase-10 is a cysteine protease, we anticipated that some of these electrophilic compounds might react with the catalytic nucleophile.
Hit prioritization by re-screening and counter-screening against activeCASP10
To further interrogate our hits and confirm on-target activity against procaspase-10, our next step was determining if our hits were selective for proCASP10 over the active proteoform. Using our previously reported fluorogenic caspase assay,24 we measured the relative inhibition of recombinant active CASP10 by each of our 237 prioritized hits. This screen had a Z′ factor of 0.73, resulting in 9 of 237 compounds having ≤50% activeCASP10 activity (Fig. S15A and Table S1, ESI†), with 78 compounds being 3 standard deviations below the DMSO control (Fig. S15B, ESI†). Notably, several of our aforementioned hits, including HTS-6, SO265, and PFTμ, showed substantially reduced inhibition of activeCASP10 compared to the proCASP10TEV Linker (Fig. S14A and S15A, ESI†). SAR clustering analysis of the counter screen showed ∼20 different clustering types for activeCASP10, but overall, less reactivity towards active compared to the pro-form (Fig. S16, ESI†), consistent with more favorable inhibition of the proenzyme. Together, our prioritized compounds (Scheme S1, ESI†) included 10 total inhibitors that showed preferential activity against proCASP10TEV when compared to activeCASP10.TEV assay identifies likely TEV inhibitors
As our proCASP10TEV linker screen requires the addition of TEV protease for caspase activation, we anticipated that some hits might be bona fide TEV protease inhibitors rather than caspase inhibitors. To test this hypothesis and to filter out such compounds, we established a TEV protease activity assay using a customized fluorogenic substrate (see methods) inspired by prior TEV protease assays.53 Following the workflow shown in Fig. 3(A), we first optimized both the substrate concentration and time points to ensure activity was within the enzyme's linear range (Fig. S17, ESI†). We find that TEV protease was highly active towards our fluorogenic substrate, with complete consumption of substrate within five minutes (Fig. 3(B)). Using automated dispensing to ensure the rapid and equal delivery of substrate across assay conditions, we observed comparable Michaelis–Menten kinetic parameters to those reported previously for TEV protease54 (Fig. 3(C)). | ||
| Fig. 3 Procaspase-10 HTS identified TEV protease inhibitors. (A) General scheme of TEV protease activity assay with our in-house TEV cleavable fluorogenic substrate, DABCYL-ENLYFQSGTK-5-FAM. (B) Shows relative fluorescent units (RFUs) for time-dependent increased fluorescence (λex = 495 nm λem = 550 nm) for TEV protease (100 nM) exposed to either vehicle (DMSO) or iodoacetamide (IA). (C) Michaelis–Menten kinetics of TEV protease (667 nM) with TEV substrate. (D)–(G) TEV protease (50 nM) activity assay treated with the indicated screening compounds at the indicated concentrations for 1 h in PBS buffer under ambient conditions. For D–G, data represent mean values ± standard deviation for three biological replicates. Statistical significance was calculated with unpaired Student's t-tests, *p < 0.05, **p < 0.01, ****p < 0.0001, ns, not significant p > 0.05. | ||
Using this optimized assay, we used freshly sourced compound stocks to evaluate the TEV protease inhibitory activity of ten prioritized hits (Fig. 3(D)–(G) and Fig. S18, S19, ESI†). Notably, while most compounds were sourced commercially, compound SO265 required in-house synthesis (Scheme S2, ESI† for the synthetic route for SO265). We observed substantial dose-dependent inhibition of TEV protease for HTS-1 (Fig. 3(D)), HTS-2 (Fig. S18A, ESI†), and HTS-3 (Fig. 3(E)). Some inhibition was also observed for HTS-6 (Fig. S18B, ESI†), HTS-4 (Fig. S18C, ESI†), and HTS-5 (Fig. S18D, ESI†). Therefore, we excluded these likely TEV inhibitors from further analysis. Encouragingly, several HTS hits stood out as having no appreciable TEV protease inhibitory activity (Fig. 3(F) and Fig. S19A, B, ESI†). Notably, PFTμ afforded a slight, but not significant, decrease in TEV protease activity at the highest tested concentration (100 μM) (Fig. 3(G)).
Caspase-10 inhibition is confirmed by activity assays
We next turned to confirm that our screening hits engage both recombinant and endogenous proCASP10 protein. We first rescreened a subset of the prioritized hits in our proCASP10TEV Linker assay. While we observed that most compounds only showed modest caspase-10 inhibition (Fig. S20, ESI†), several compounds stood out, including HTS-6 (Fig. S21, ESI†), the TEV protease inhibitor HTS-2 (Fig. S22, ESI†), and PFTμ (Fig. S23, ESI†). We then turned to gel-based activity-based protein profiling (ABPP) analysis to further corroborate our inhibition data. Having previously established ABPP gel-based assays for procaspase-8 and -10 using the KB61 click probe27 in HEK293T lysates, we first deployed this ABPP assay to assess compound engagement of recombinant procaspase-10 at the catalytic cysteine residue C401 by concentration-dependent competitive labeling of the initial screen compounds against the dual caspase-8/-10 click probe KB61 (Fig. S24, ESI†), focusing on the compounds that contained obvious electrophilic moieties, namely HTS-6 and HTS-2 and PFTμ. We find that PFTμ shows similar potency when compared to established caspase-8/10 dual inhibitor KB7.27 Both HTS-6 and HTS-2 exhibited high proteome-wide reactivity, indicating that their caspase engagement is likely driven by the high electrophilicity of the cyanoacryl sulfone moiety. Further illustrating the increased reactivity of these two compounds relative to PFTμ, we also observed increased competition of iodoacetamide-rhodamine (IA-Rho) (Fig. S25, ESI†), consistent with generalized cysteine reactivity rather than a caspase-10-specific effect. Due to their high reactivity, we excluded HTS-6 and HTS-2, from further analyses.Compound rearrangement drives caspase inhibition for SO265, which shows preferential inhibition of the pro-form of caspase-10
As the SO265 thiadiazine chemotype had shown pronounced procaspase-10 inhibition in our initial screen (approximately 80% proCASP10TEV Linker inhibition; Fig. 2(D) and Fig. S14, ESI†), we were surprised by the lack of activity using our resynthesized compound (20% proCASP10TEV Linker inhibition) (Fig. S26, ESI†). Substituted thiadiazine rings can isomerize to thiol imidazole moieties, gaining aromaticity.55 Therefore, we hypothesized that such a rearrangement of SO265, which could be favored by lower pH, could be driving its inhibitory activity (Fig. 4(A)). To test this hypothesis, we subjected SO265 to acidic conditions (10 mM HCl) prior to assessing caspase inhibitory activity. Consistent with our hypothesis, we observed increased inhibitory activity for stocks of SO265 pretreated with acid (Fig. S26, ESI†). This activity was generally restricted to caspase-10, with a more modest increase in inhibition observed for TEV protease (Fig. S27, ESI†).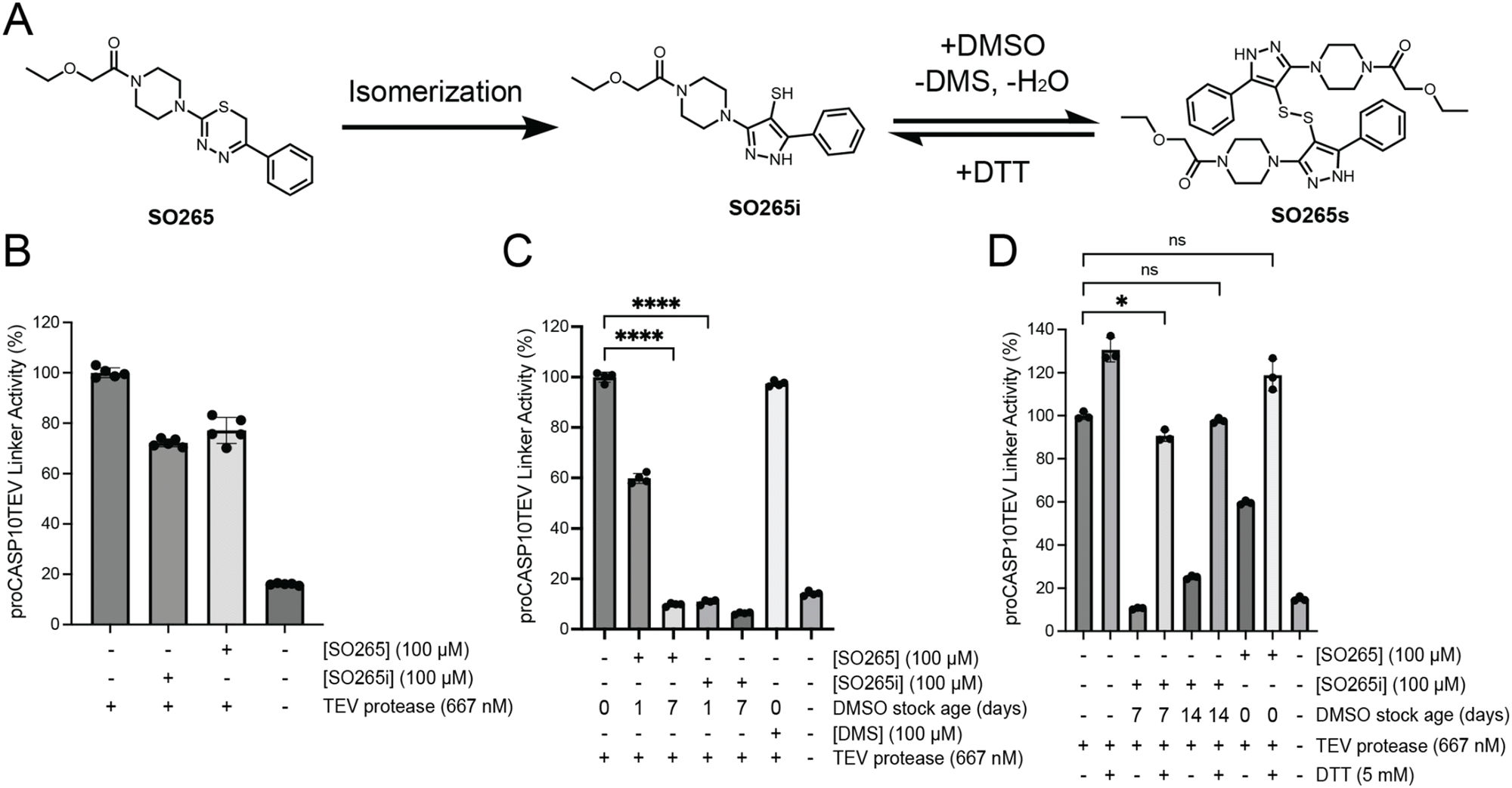 | ||
| Fig. 4 The rearranged product of SO265 inhibits proCASP10TEV linker activity. (A) Proposed scheme of SO265 isomerization and formation of the disulfide product (B) Relative activity of proCASP10 Linker protein (333 nM) treated with the indicated compounds for 1 h followed by addition of TEV protease (667 nM) and fluorogenic substrate (Ac-VDAVAD-AFC, 10 μM, and 333 mM sodium citrate) in PBS buffer. (C) The indicated compound stocks were subjected to either one- or seven-day incubation in DMSO at ambient conditions. Subsequently, compound-treated proCASP10 Linker protein was assayed as in ‘B’ with 1 h pre-exposure to either dimethyl sulfide (DMS) or the indicated compounds. (D) Compounds incubated for the indicated days in DMSO were further subjected to DTT (5 mM DTT) followed by evaluation for caspase inhibition as described in ‘B’. For B-D, data represent mean values ± standard deviation for four biological replicates. Statistical significance was calculated with unpaired Student's t-tests, *p < 0.05, ****p < 0.0001, ns, not significant p > 0.05. | ||
Guided by this increased activity, we next tested the putative isomer, SO265i (Fig. 4(A)), which we had isolated from the reaction mixture during the synthesis of SO265. While the masses of SO265 and SO265i are identical, the two compounds can be distinguished via H-NMR analysis (Fig. S28A, ESI†) and LC–MS analysis (Fig. S28B–F, ESI†). Notably, the thiadiazine methylene signal was only present in SO265, with loss in the SO265 spectra indicating that aromatization had indeed occurred. The SO265i spectra uniquely featured a signal for a thiol proton, consistent with the proposed isomerization. The presence of a thiol in SO265i was further corroborated by IR analysis (Fig. S29, ESI†). However, when tested in our activity assay, SO265i exhibited equivalent caspase-10 inhibition (30% inhibition) to SO265, indicating that the thiol imidazole was not the active species (Fig. 4(B)).
Following a 2-day incubation at room temperature in DMSO, repeated NMR analysis revealed that the SO265i stock had converted into a second species. This species was distinguished by the loss of the thiol proton and an increase in a signal matching the expected shift of dimethylsulfide (DMS) (Fig. S28A, ESI†). We hypothesized that DMSO-mediated oxidation of the SO265i thiol to a disulfide (SO265s) was occurring; this is a reported redox reaction that produces DMS as a byproduct and is promoted by low thiol pKa values.56,57 Therefore, to further assess whether SO265s was the active species responsible for caspase-10 inhibition, we incubated 50 mM DMSO stocks of SO265 and SO265i at room temperature for one, seven, and 14 days to promote the formation of SO265s. We then tested the activity of each of these compound stocks alongside DMS as a control (Fig. 4(C)). Consistent with SO265s as the active species, we observed a time-dependent increase in inhibition of proCASP10TEV Linker for both SO265 and SO265i; DMS had no effects on proCASP10TEV Linker activity. Providing further evidence of the likelihood that the rearranged disulfide structure is the active compound, the addition of 5 mM DTT to the compound stock completely abolished inhibitory activity (Fig. 4(D)). SO265s also exhibited increased reactivity with glutathione when compared to SO265 and SO265i (Fig. S30A, ESI†), indicative of the likelihood that SO265s functions as a cysteine-reactive covalent inhibitor. Gel-filtration of SO265s-modified protein failed to recover proCASP10TEV Linker activity, which further corroborates that SO265s acts as a covalent inhibitor (Fig. S30B, ESI†) that shows reduced potency compared to PFTμ (Fig. S30C, ESI†). Competitive gel-based ABPP analysis provided initial evidence that SO265s likely functions by labeling non-active site cysteine residue in caspase-10 (Fig. S30D, ESI†). Taken together, these data provide evidence that SO265s is a cysteine-reactive compound that blocks proCASP10TEV Linker activity with preferential inhibition of the zymogen (Fig. S31, ESI†). More broadly, we find that SO265s show some caspase selectivity, also inhibiting active caspase-8 (Fig. S32, ESI†), whereas only a slight decrease in activity was observed for active caspase-3 (Fig. S33, ESI†) and caspase-9 (Fig. S34, ESI†).
CETSA confirms caspase-10 labeling
As all our data, thus far, had been assessed for recombinant protein, we next opted to extend our analyses to assess the compound engagement of endogenous procaspase-8 and -10. For this, we turned to Cellular Thermal Shift Assay (CETSA)58 to measure binding-induced changes to protein thermal stability. To validate our assay, we subjected Jurkat lysates spiked with recombinant hexahistidine-tagged proCASP10 to CETSA analysis, comparing the thermal stability of the proCASP10 construct to that of the inactive catalytic cysteine mutant construct (proCASP10 C401A) with and without addition of KB7, as a positive control. We observe a marked decrease in thermal stability upon KB7 treatment for both the endogenous (anti-caspase-10 signal) and recombinant (anti-His signal) caspase-10 proteins (Fig. 5(A) and complete blots in Fig. S35, ESI†). The C401A mutant protein did not show a similar thermal shift (Fig. S36, ESI†), which is consistent with the covalent modification of the C401, which is the catalytic nucleophile, by KB7. Extension of this analysis revealed that, like KB7, PFTμ induced destabilization of the wildtype spiked proCASP10 and endogenous proCASP10 (Fig. 5(B) and complete blots in Fig. S37, ESI†) but not the C401A mutant protein (Fig. 5(C) and complete blots in Fig. S38, ESI†), which further confirms that PFTμ labels C401 Unexpectedly, the disulfide product (SO265s) caused some protein stabilization, suggesting an alternate mode of action compared to the two active site-directed inhibitors (Fig. 5(D) and complete blots in Fig. S39, ESI†). Corroborating an alternate mode of engagement by SO265s, we observed no competition in gel-based ABPP analysis using the KB61 click probe against procaspase-10 (Fig. S40A, ESI†), unlike PFTμ that labeled both procaspase-10 (Fig. S40A, ESI†) and procaspase-8 (Fig. S40B, ESI†). Disappointingly, we observed no similar compound-induced thermal stability shift for endogenous procaspase-8 upon KB7 treatment (Fig. S35, ESI†) nor PFTμ treatment (Fig. S37, ESI†), indicating that the CETSA assay was not suitable for evaluation of caspase-8 target engagement. Therefore, we returned to enzyme activity assays for broader target analysis.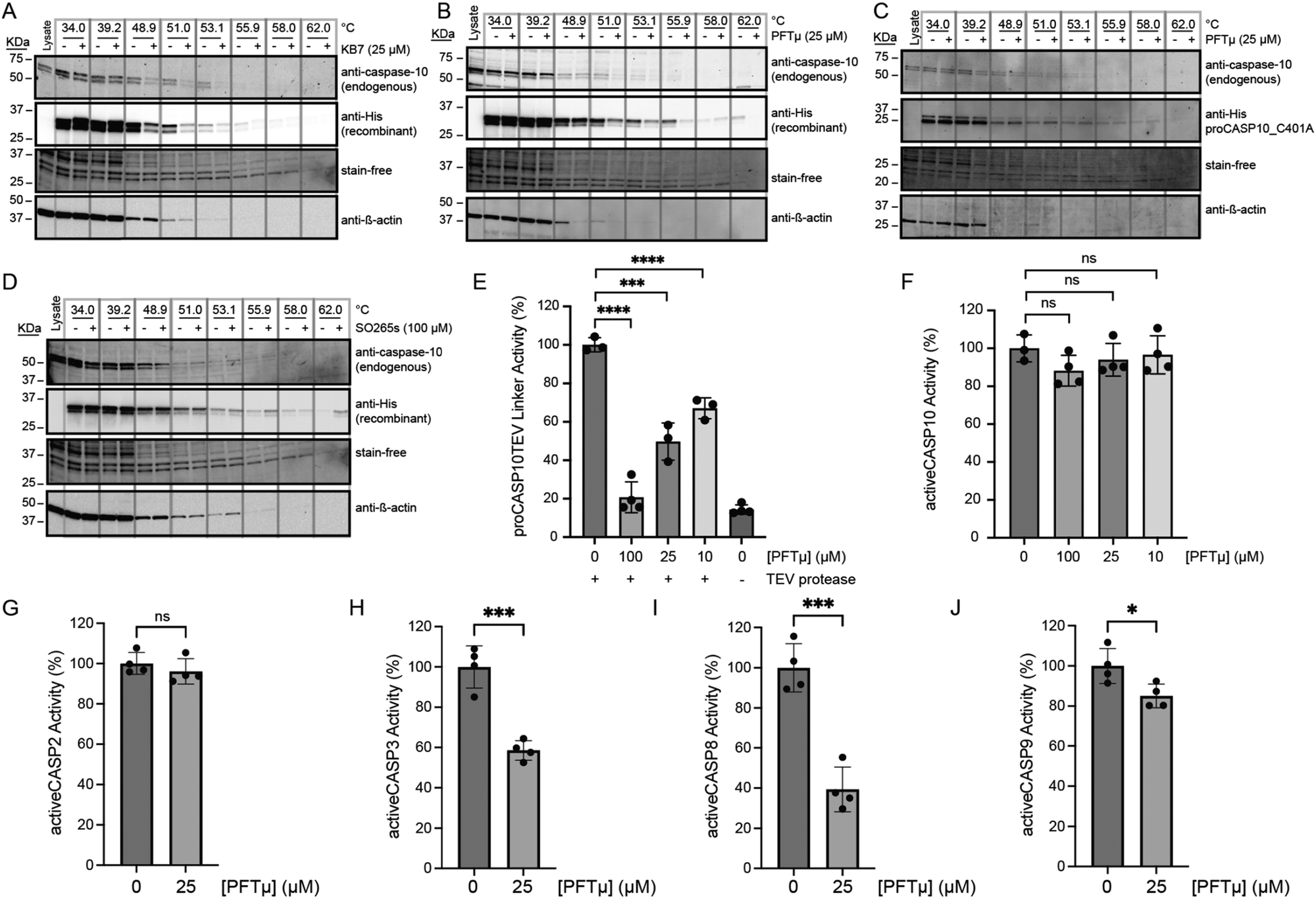 | ||
| Fig. 5 PFTμ and SO265s impact the stability of both recombinant and endogenous procaspase-10. (A)–(D) Cellular thermal shift assay (CETSA)58 analysis blot of Jurkat cell lysates spiked with the indicated recombinant caspase-10 proteins featuring C-terminal hexahistidine tag and treated with the indicated compounds at the indicated concentrations or vehicle (DMSO) and subjected to immunoblot analysis for using both anti-His and anti-caspase-10 antibodies to visualize recombinant and endogenous protein, respectively. Loading was visualized both with the BioRad Chemidoc Stain-Free imaging technology59 and with anti-β-actin. For ‘A, B, and D’, recombinant proCASP10 and for ‘C’, recombinant proCASP10_C401A were analyzed. (E) Relative activity of proCASP10TEV Linker protein (333 nM concentration in PBS buffer) exposed to PFTμ at the indicated concentrations for 1 h followed by TEV protease (667 nM) and analysis with Ac-VDVAD-AFC fluorogenic substrate (10 μM) in PBS supplemented with 333 mM sodium citrate. (F)–(J) Relative activity of recombinant active caspases in PBS buffer analyzed with Ac-VDVAD-AFC fluorogenic substrate (10 μM) after treatment with the indicated concentrations of PFTμ for 1 h. For activeCASP10, fluorogenic substrate (10 μM) in PBS was supplemented with 333 mM citrate. Recombinant proteins used in ‘F’ activeCASP10 (333 nM), ‘G’ activeCASP-2 (0.3 μM), ‘H’ activeCASP-3 (0.3 μM), and ‘I’ activeCASP9 (1 μM). For A–D, data is representative of two biological replicates. For E–J, data represent mean values ± standard deviation for four biological replicates. Statistical significance was calculated with unpaired Student's t-tests, *p < 0.05, ***p < 0.001, ****p < 0.0001, ns, not significant p > 0.05. | ||
Pifithrin-μ is a promiscuous caspase inhibitor that prevents FasL-mediated apoptosis
In contrast with the complex activation mechanism of SO265s, which likely could complicate assessing in-cell activity, we expected PFTμ to retain caspase inhibitory activity in complex cell environments. This expectation is further supported by PFTμ's well-documented anti-apoptotic activity, which has been ascribed to its function as a TP53 inhibitor.47 Therefore, we prioritized PFTμ for further analysis. Activity assay analysis revealed that PFTμ showed some preferential inhibition across a panel of analyzed caspases; consistent with our screening data, we observed preferential inhibition of procaspase-10 when compared to active caspase-10 (Fig. 5(E), (F) and Fig. S14, S15, ESI†). Similarly, PFTμ also preferentially labeled procaspase-2, as analyzed using our caspase-2 directed click probe24 and ABPP analysis (Fig. S41, ESI†). No detectable inhibition of active caspase-2 was observed, either by ABPP gel (Fig. S41, ESI†) or activity assay (Fig. 5(G)). PFTμ also significantly inhibited active caspase-3 and active caspase-8, with slight, albeit significant, inhibition observed for active caspase-9 (Fig. 5(H)–(J)). These data provided evidence that PFTμ is a promiscuous caspase inhibitor.We next turned to chemoproteomics to more broadly assess whether PFTμ labels endogenous caspases. We deployed our established cysteine chemoproteomic platform60 in which covalent labeling sites are identified in a competitive manner using the pan cysteine-reactive probe iodoacetamide alkyne (IAA) and isotopically enriched “light” and “heavy” biotin-azide capture reagents. Following the workflow shown in Fig. 6(A), out of 8925 total cysteines quantified, we find that 1070 total unique cysteines showed log2(H/L) values greater than 1.5, indicative of labeling by PFTμ. Included in this list were several known targets, including PARP1, HSP70 (HSPA1A), and PRDX proteins (PRDX1, PRDX2, PRDX5), which were previously identified via chemoproteomics using a clickable PFTμ analog,61 providing evidence of the robustness of our approach (Table S2, ESI†). Quite strikingly, several caspases stood out as having high log2(H/L) ratios (Fig. 6(B)), including caspase-8, which aligns with our aforementioned activity assay and gel-based ABPP analysis (Fig. 5(I) and Fig. S40, ESI†). Aligning with our prior discovery24 of structurally-related phenylpropiolate molecules that label caspase-2 at C370, we also observe that C370 is highly sensitive to PFTμ, which is also consistent with our gel-based analysis (Fig. S41, ESI†). The noncatalytic cysteine residue, C264, in caspase-6,26 was also observed to be labeled by PFTμ, as was the near-active site cysteine (C170) in caspase-3. We did not detect the catalytic cysteine nucleophile of caspase-3 (C163). Beyond these pro-apoptotic caspases, the catalytic cysteine in the pro-inflammatory caspase-4 (C258) was also labeled by PFTμ. The catalytic cysteine in caspase-10 was not identified, likely due to the long tryptic peptide that flanks this residue. Taken together, these data provide further evidence that PFTμ promiscuously labels many human caspases in addition to its previously reported targets.
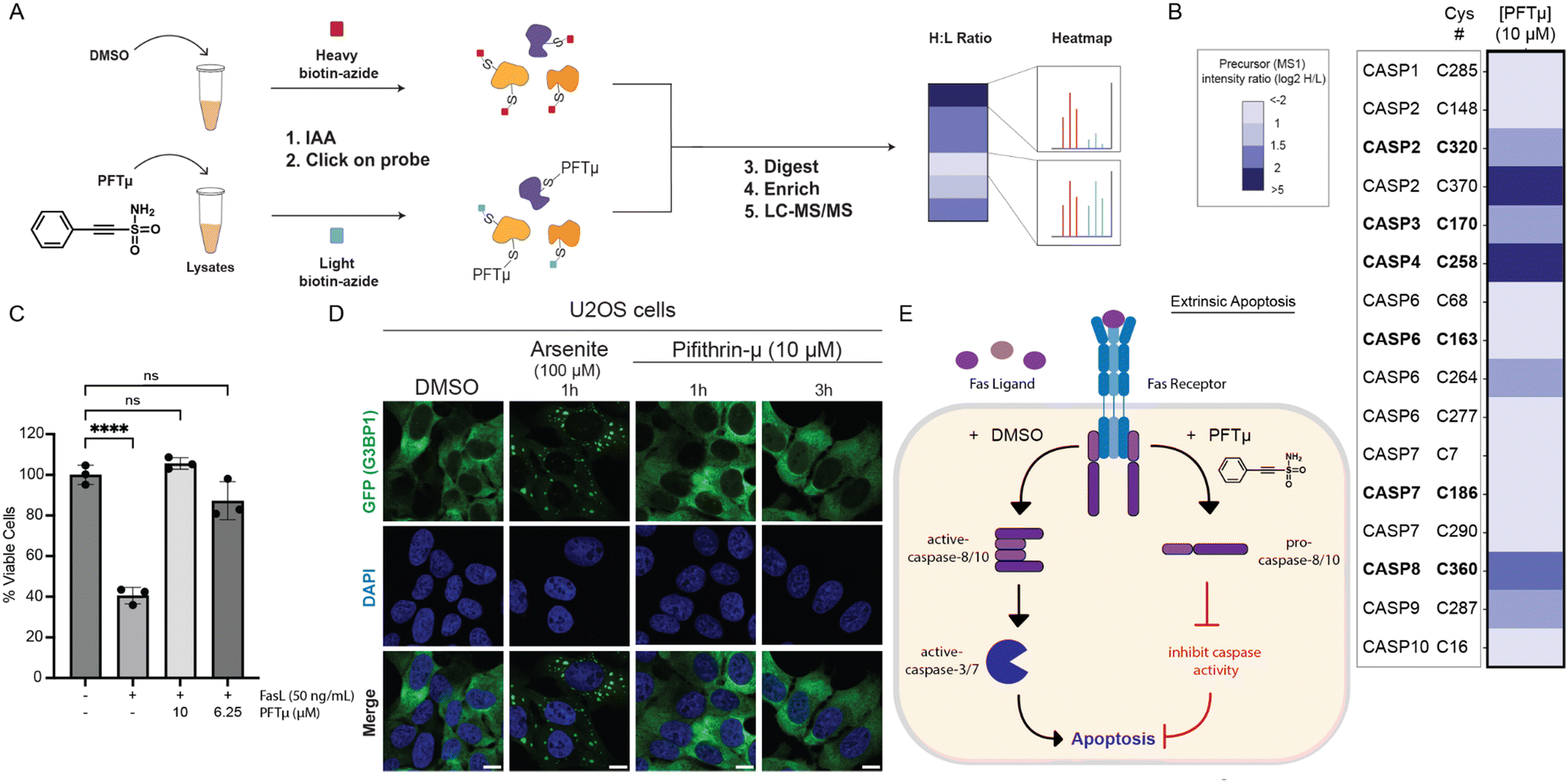 | ||
| Fig. 6 PFTμ labels initiator caspases and can protect Jurkat cells from FasL-mediated apoptosis. (A, B) Cysteine chemoproteomic analysis identifies protein targets of PFTμ. ‘A’ shows the general workflow in which Jurkat cell lysates were treated with either PFTμ (25 μM) or DMSO for 1 h, followed by IAA (200 μM) cysteine capping, conjugated via click chemistry to isotopically differentiated biotin-azide reagents,62,63 Single-pot solid-phase-enhanced sample-preparation (SP3)60,64,65 with on-resin tryptic digestion, enriched, and analyzed by LC–MS/MS. Heatmap in ‘B’ shows the mean quantified precursor intensity (log2H/L) for DMSO (H, heavy) versus compound treatment (L, light) for all caspase cysteines identified. Catalytic cysteine residues are annotated in bold. (C) CellTiter-Glo® measurement of relative viability of Jurkat cells subjected to PFTμ at the indicated concentrations for 1 h followed by FasL (50 ng mL−1, 3 h)-induced apoptosis. (D) Fluorescent microscopy of U2OS cells expressing GFP-G3BP1 and treated with PFTμ (10 μM) or positive control sodium arsenite. All scale bars = 10 μm. (E) Proposed model where PFTμ functions as a promiscuous initiator caspase inhibitor (labeling caspase-2, -8, and -10) and blocks FasL-mediated extrinsic apoptosis. For B, experiments were conducted in four biological replicates, with two samples additionally analyzed as technical replicates. For C, data represent mean values ± standard deviation for three biological replicates. All MS data can be found in Table S2 (ESI†). Statistical significance was calculated with unpaired Student's t-tests, ****p < 0.0001, ns, not significant p > 0.05. | ||
Guided by these findings, we next asked whether PFTμ could protect cells from extrinsic apoptosis induced by Fas ligand (FasL). While PFTμ's anti-apoptotic activity has been reported in other contexts,47,66–69 we selected FasL-mediated apoptosis as a model system due to the central role that caspase-8 and -10 play in this process12,70,71 and the less central role of TP53,72,73 with the goal of helping to define better the biologically active target(s) of PFTμ's anti-apoptotic activity. Using the CellTiter-Glo assay, we find that PFTμ affords near-complete protection from FasL (Fig. 6(C)) without inducing cytotoxic effects (Fig. S42, ESI†). These data align with caspase inhibition as contributing to PFTμ's activity.
As our recent work has shown that electrophile stress can lead to the formation of stress granules,74 we also opted to rule out PFTμ-induced stress granules as a potential confounding variable. Using a ΔΔG3BP1/2 KO cell line that stably expresses GFP-G3BP1,75,76 we find that PFTμ does not induce G3BP1 condensates using concentrations that are protective from apoptosis (Fig. 6(D)) and are consistent with prior studies using PFTμ as a putative TP53 inhibitor.47 Thus, we put forth a model (Fig. 6(E)) that caspases are likely biological targets of PFTμ that contribute to the reported anti-apoptotic activity.
Discussion
To enable high throughput procaspase-10 inhibitor discovery, here we developed and applied an engineered TEV-activatable caspase-10 protein to high throughput screening. Our high quality (an average Z′ of 0.58) ∼100000 compound semi-automated screen yielded 237 total hits with Z-score <−3. Subsequent rescreening and counter-screening delineated bona fide procaspase-10 inhibitors from those with activity against active caspase-10 or TEV protease. For screening hit PFTμ, orthogonal mode of action studies confirmed target engagement for both recombinant and endogenous procaspase-10. More broadly, we also expect that our and related activation screening platforms should prove broadly useful in the discovery of inhibitors targeting precursor proteases.
For such future activation-based caspase-10 high throughput screening campaigns, our work highlights intriguing opportunities. As TEV protease is a cysteine protease, we found that several of our hits functioned via engaging TEV rather than procaspase-10. We expect that these newly identified TEV protease inhibitors could serve as useful tools for synthetic biology studies that rely on TEV protease activity to control engineered circuits. To favor caspase rather than TEV inhibitor discovery, future studies could also consider using alternative non-cysteine protease enzymes for activation, which we expect would prove more substantially orthogonal to caspase cysteine protease activity. In addition to uncovering candidate TEV inhibitors, our screen also revealed that the thiadiazine chemotype is likely prone to disulfide rearrangement, as exemplified by our characterization of SO265s, which we found was the active species. Thus, our work both highlights the value of resynthesis in confirming screening hits and hints at the likely amenability of procaspase-10 to a disulfide trapping/tethering-style inhibitor discovery77–79 using additional disulfide-containing compounds.
Beyond the SO265 thiadiazine chemotype, PFTμ, a reported TP53 inhibitor, proved to be a key hit from our screen. Comparing active and procaspase-10 inhibitor activity, we found that PFTμ inhibited both proteoforms to some degree, with increased inhibition observed for the proenzyme. Looking beyond caspase-10, the in vitro, cell-based, and proteomic analysis together revealed that PFTμ also engages caspase-8, -3, -10, -5, and -6. These data allow us to put forth a model whereby PFTμ's reported activity as an anti-apoptotic agent is likely driven by caspase inhibition in addition to the previously reported TP53 inhibitory activity.47 Therefore, we anticipate that the cysteine-reactive ethynesulfonamide chemotype could prove broadly useful as a starting point for caspase inhibitor development campaigns. Further medicinal chemistry efforts will be needed to determine whether the reactivity and potency of ethynesulfonamide-containing compounds can be tuned to a level suitable for a selective chemical probe. We also expect our work to aid in phenotype interpretation for studies that have employed PFTμ to block p53 activity.
We also recognize some limitations of our work. Despite our extensive screening efforts, we failed to obtain potent, selective caspase-10 inhibitors, and, as such, we were unable to further inform the still cryptic functions of caspase-10. Due to the comparatively modest potency and low selectivity of our initial screening hits, we opted to proceed conservatively and restrict our mode-of-action studies to lysate- rather than cell-based analyses, so as to exclude possible electrophilic stress-mediated confounding activity, as has recently been reported for other classes of cysteine-reactive electrophiles.74,80 Our ABPP and CETSA analysis of SO265s suggest that caspase-10 harbors additional non-catalytic and possibly unique, ligandable cysteine residues. However, we have yet to pinpoint the positions and tractability of these putative cysteines. Future efforts to more fully characterize these still cryptic cysteines will benefit from more in-depth chemoprotomic and mutational analysis of caspase-10, together with solving the atomic resolution structure of caspase-10, which remains unresolved in the PDB.
In sum, our study sheds important light on the activity of two classes of cysteine-reactive electrophiles and provides a useful screening platform for future caspase-10 inhibitor discovery campaigns. We remain optimistic about the feasibility of developing a caspase-10 selective chemical probe, and hope that our screening hits, particularly those that we have yet to subject to counter screening analysis, will enable the discovery of potent and selective caspase-10 inhibitors.
Author contributions
J. O. C. and K. M. B. conceived of the project. J. O. C, C. Y., B. H., K. H. A., and A. R. J. collected data. K. H. A. and S. O. provided reagents. J. O. C., R. D., and L. M. B. performed data analysis. L. M. B., M. F. P., and N. P. wrote software. K. M. B. and R. D. provided guidance and funding. J. O. C. and K. M. B. wrote the manuscript and all authors revised and edited the manuscript.Data availability
The MS data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD053315.Conflicts of interest
K. M. B. is a member of the advisory board at Matchpoint Therapeutics. All remaining authors declare no conflicts of interest.Acknowledgements
This study was supported by National Institutes of Health (DP2 GM146246-02 to K. M. B.), Beckman Young Investigator Award (to K.M.B.), UCLA DOE Institute (DE-FC02-02ER63421 to K. M. B.), NSF GRFP (1000235263 to J. O. C.), UCLA Chemistry Biology Interface Training Program (T32GM136614 to A. R. J. and M. F. P.), UCLA Cellular and Molecular Biology Training Program (T32GM145388-03 to N.P), Clinical and Translational Sciences Institute Award Core voucher (to K. M. B.) as part of the NIH/National Center for Advancing Translational Science (UL1TR001881 to UCLA). The UCLA Molecular Screening Shared Resource is supported by Jonsson Comprehensive Cancer Center, award number P30CA016042 by the National Cancer Institute of the National Institutes of Health. We thank all members of the Backus lab for their helpful suggestions, Michael Lenardo for providing plasmids encoding caspase-10 and Dennis Wolan for providing the Rho-DEVD-AOMK probe.References
- S. Horn, M. A. Hughes, R. Schilling, C. Sticht, T. Tenev, M. Ploesser, P. Meier, M. R. Sprick, M. Macfarlane and M. Leverkus, Caspase-10 Negatively Regulates Caspase-8-Mediated Cell Death, Switching the Response to CD95L in Favor of NF-κB Activation and Cell Survival, Cell Rep., 2017, 19(4), 785–797, DOI:10.1016/j.celrep.2017.04.010.
- C. Y. Yang, C. I. Lien, Y. C. Tseng, Y. F. Tu, A. W. Kulczyk, Y. C. Lu, Y. T. Wang, T. W. Su, L. C. Hsu and Y. C. Lo, et al., Deciphering DED assembly mechanisms in FADD-procaspase-8-cFLIP complexes regulating apoptosis, Nat. Commun., 2024, 15(1), 3791, DOI:10.1038/s41467-024-47990-2 From NLM Medline.
- J. Wang, H. J. Chun, W. Wong, D. M. Spencer and M. J. Lenardo, Caspase-10 is an initiator caspase in death receptor signaling, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(24), 13884–13888, DOI:10.1073/pnas.241358198.
- O. Julien and J. A. Wells, Caspases and their substrates, Cell Death Differ., 2017, 24(8), 1380–1389, DOI:10.1038/cdd.2017.44.
- L. Wang, K. Main, H. Wang, O. Julien and A. Dufour, Biochemical Tools for Tracking Proteolysis, J. Proteome Res., 2021, 20(12), 5264–5279, DOI:10.1021/acs.jproteome.1c00289 From NLM Medline.
- E. Eskandari, G. L. Negri, S. Tan, M. E. Macaldaz, S. Ding, J. Long, K. Nielsen, S. E. Spencer, G. B. Morin and C. J. Eaves, Dependence of human cell survival and proliferation on the CASP3 prodomain, Cell Death Discovery, 2024, 10(1), 63, DOI:10.1038/s41420-024-01826-6.
- A. Alam, L. Y. Cohen, S. Aouad and R.-P. Sékaly, Early Activation of Caspases during T Lymphocyte Stimulation Results in Selective Substrate Cleavage in Nonapoptotic Cells, J. Exp. Med., 1999, 190(12), 1879–1890, DOI:10.1084/jem.190.12.1879.
- G. Pistritto, M. Jost, S. M. Srinivasula, R. Baffa, J. L. Poyet, C. Kari, Y. Lazebnik, U. Rodeck and E. S. Alnemri, Expression and transcriptional regulation of caspase-14 in simple and complex epithelia, Cell Death Differ., 2002, 9(9), 995–1006, DOI:10.1038/sj.cdd.4401061.
- S. Lippens, M. Kockx, M. Knaapen, L. Mortier, R. Polakowska, A. Verheyen, M. Garmyn, A. Zwijsen, P. Formstecher and D. Huylebroeck, et al., Epidermal differentiation does not involve the pro-apoptotic executioner caspases, but is associated with caspase-14 induction and processing, Cell Death Differ., 2000, 7(12), 1218–1224, DOI:10.1038/sj.cdd.4400785.
- A. Gorelick-Ashkenazi, R. Weiss, L. Sapozhnikov, A. Florentin, L. Tarayrah-Ibraheim, D. Dweik, K. Yacobi-Sharon and E. Arama, Caspases maintain tissue integrity by an apoptosis-independent inhibition of cell migration and invasion, Nat. Commun., 2018, 9(1), 2806, DOI:10.1038/s41467-018-05204-6.
- W. Shao, G. Yeretssian, K. Doiron, S. N. Hussain and M. Saleh, The Caspase-1 Digestome Identifies the Glycolysis Pathway as a Target during Infection and Septic Shock, J. Biol. Chem., 2007, 282(50), 36321–36329, DOI:10.1074/jbc.m708182200.
- F. Consonni, S. Moreno, B. Vinuales Colell, M.-C. Stolzenberg, A. Fernandes, M. Parisot, C. Masson, N. Neveux, J. Rosain and S. Bamberger, et al., Study of the potential role of CASPASE-10 mutations in the development of autoimmune lymphoproliferative syndrome, Cell Death Dis., 2024, 15(5), 315, DOI:10.1038/s41419-024-06679-6.
- H. F. Krug, Caspase-10 is the key initiator caspase involved in tributyltin-mediated apoptosis in human immune cells, J. Toxicol., 2012, 2012, 395482, DOI:10.1155/2012/395482 From NLM PubMed-not-MEDLINE.
- N. H. Philip, C. P. Dillon, A. G. Snyder, P. Fitzgerald, M. A. Wynosky-Dolfi, E. E. Zwack, B. Hu, L. Fitzgerald, E. A. Mauldin and A. M. Copenhaver, et al., Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(20), 7385–7390, DOI:10.1073/pnas.1403252111.
- F. Rieux-Laucat, F. Le Deist and A. Fischer, Autoimmune lymphoproliferative syndromes: genetic defects of apoptosis pathways, Cell Death Differ., 2003, 10(1), 124–133, DOI:10.1038/sj.cdd.4401190.
- M. Olsson and B. Zhivotovsky, Caspases and cancer, Cell Death Differ., 2011, 18(9), 1441–1449, DOI:10.1038/cdd.2011.30.
- Z. Cui, H. Dabas, B. C. Leonard, J. V. Shiah, J. R. Grandis and D. E. Johnson, Caspase-8 mutations associated with head and neck cancer differentially retain functional properties related to TRAIL-induced apoptosis and cytokine induction, Cell Death Dis., 2021, 12(8), 775, DOI:10.1038/s41419-021-04066-z.
- M. Jiang, L. Qi, L. Li and Y. Li, The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer, Cell Death Discovery, 2020, 6, 112, DOI:10.1038/s41420-020-00349-0.
- K. M. Groborz, M. Kalinka, J. Grzymska, S. Kołt, S. J. Snipas and M. Poręba, Selective chemical reagents to investigate the role of caspase 6 in apoptosis in acute leukemia T cells, Chem. Sci., 2023, 14(9), 2289–2302, 10.1039/d2sc05827h.
- Y. Shaulov-Rotem, E. Merquiol, T. Weiss-Sadan, O. Moshel, S. Salpeter, D. Shabat, F. Kaschani, M. Kaiser and G. Blum, A novel quenched fluorescent activity-based probe reveals caspase-3 activity in the endoplasmic reticulum during apoptosis, Chem. Sci., 2016, 7(2), 1322–1337, 10.1039/c5sc03207e.
- A. De Calignon, L. M. Fox, R. Pitstick, G. A. Carlson, B. J. Bacskai, T. L. Spires-Jones and B. T. Hyman, Caspase activation precedes and leads to tangles, Nature, 2010, 464(7292), 1201–1204, DOI:10.1038/nature08890.
- M. B. Boxer, A. M. Quinn, M. Shen, A. Jadhav, W. Leister, A. Simeonov, D. S. Auld and C. J. Thomas, A Highly Potent and Selective Caspase1 Inhibitor that Utilizes a Key 3-Cyanopropanoic Acid Moiety, ChemMedChem, 2010, 5(5), 730–738, DOI:10.1002/cmdc.200900531.
- E. Bosc, J. Anastasie, F. Soualmia, P. Coric, J. Y. Kim, L. Q. Wang, G. Lacin, K. Zhao, R. Patel and E. Duplus, et al., Genuine selective caspase-2 inhibition with new irreversible small peptidomimetics, Cell Death Dis., 2022, 13(11), 959, DOI:10.1038/s41419-022-05396-2.
- J. O. Castellón, S. Ofori, N. R. Burton, A. R. Julio, A. C. Turmon, E. Armenta, C. Sandoval, L. M. Boatner, E. E. Takayoshi and M. Faragalla, et al., Chemoproteomics Identifies State-Dependent and Proteoform-Selective Caspase-2 Inhibitors, J. Am. Chem. Soc., 2024, 146(22), 14972–14988, DOI:10.1021/jacs.3c12240.
- A. Tubeleviciute-Aydin, A. Beautrait, J. Lynham, G. Sharma, A. Gorelik, L. J. Deny, N. Soya, G. L. Lukacs, B. Nagar and A. Marinier, et al., Identification of Allosteric Inhibitors against Active Caspase-6, Sci. Rep., 2019, 9(1), 5504, DOI:10.1038/s41598-019-41930-7.
- K. S. Van Horn, D. Wang, D. Medina-Cleghorn, P. S. Lee, C. Bryant, C. Altobelli, P. Jaishankar, K. K. Leung, R. A. Ng and A. J. Ambrose, et al., Engaging a Non-catalytic Cysteine Residue Drives Potent and Selective Inhibition of Caspase-6, J. Am. Chem. Soc., 2023, 145(18), 10015–10021, DOI:10.1021/jacs.2c12240.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro and A. J. Olson, et al., Proteome-wide covalent ligand discovery in native biological systems, Nature, 2016, 534(7608), 570–574, DOI:10.1038/nature18002.
- O. Bucur, G. Gaidos, A. Yatawara, B. Pennarun, C. Rupasinghe, J. Roux, S. Andrei, B. Guo, A. Panaitiu and M. Pellegrini, et al., A novel caspase 8 selective small molecule potentiates TRAIL-induced cell death, Sci. Rep., 2015, 5(1), 9893, DOI:10.1038/srep09893.
- S. Dhani, Y. Zhao and B. Zhivotovsky, A long way to go: caspase inhibitors in clinical use, Cell Death Dis., 2021, 12(10), 949, DOI:10.1038/s41419-021-04240-3.
- F. J. Lopez-Hernandez, M. A. Ortiz, Y. Bayon and F. J. Piedrafita, Z-FA-fmk inhibits effector caspases but not initiator caspases 8 and 10, and demonstrates that novel anticancer retinoid-related molecules induce apoptosis via the intrinsic pathway, Mol. Cancer Ther., 2003, 2(3), 255–263 CAS . From NLM.
- R. U. Jänicke, D. Sohn, G. Totzke and K. Schulze-Osthoff, Caspase-10 in mouse or not?, Science, 2006, 312(5782), 1874, DOI:10.1126/science.312.5782.1874a From NLM.
- M. R. Sprick, M. A. Weigand, E. Rieser, C. T. Rauch, P. Juo, J. Blenis, P. H. Krammer and H. Walczak, FADD/MORT1 and Caspase-8 Are Recruited to TRAIL Receptors 1 and 2 and Are Essential for Apoptosis Mediated by TRAIL Receptor 2, Immunity, 2000, 12(6), 599–609, DOI:10.1016/s1074-7613(00)80211-3.
- J.-L. Bodmer, N. Holler, S. Reynard, P. Vinciguerra, P. Schneider, P. Juo, J. Blenis and J. Tschopp, TRAIL receptor-2 signals apoptosis through FADD and caspase-8, Nat. Cell Biol., 2000, 2(4), 241–243, DOI:10.1038/35008667.
- P. Juo, C. J. Kuo, J. Yuan and J. Blenis, Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade, Curr. Biol., 1998, 8(18), 1001–1008, DOI:10.1016/s0960-9822(07)00420-4.
- A. Kawahara, Y. Ohsawa, H. Matsumura, Y. Uchiyama and S. Nagata, Caspase-independent Cell Killing by Fas-associated Protein with Death Domain, J. Cell Biol., 1998, 143(5), 1353–1360, DOI:10.1083/jcb.143.5.1353.
- M. A. Grotzer, A. Eggert, T. J. Zuzak, A. J. Janss, S. Marwaha, B. R. Wiewrodt, N. Ikegaki, G. M. Brodeur and P. C. Phillips, Resistance to TRAIL-induced apoptosis in primitive neuroectodermal brain tumor cells correlates with a loss of caspase-8 expression, Oncogene, 2000, 19(40), 4604–4610, DOI:10.1038/sj.onc.1203816.
- T. Teitz, T. Wei, M. B. Valentine, E. F. Vanin, J. Grenet, V. A. Valentine, F. G. Behm, A. T. Look, J. M. Lahti and V. J. Kidd, Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN, Nat. Med., 2000, 6(5), 529–535, DOI:10.1038/75007.
- A. Mühlethaler-Mottet, M. Flahaut, K. B. Bourloud, K. Nardou, A. Coulon, J. Liberman, M. Thome and N. Gross, Individual caspase-10 isoforms play distinct and opposing roles in the initiation of death receptor-mediated tumour cell apoptosis, Cell Death Dis., 2011, 2(3), e125–e125, DOI:10.1038/cddis.2011.8.
- H. J. Chun, L. Zheng, M. Ahmad, J. Wang, C. K. Speirs, R. M. Siegel, J. K. Dale, J. Puck, J. Davis and C. G. Hall, et al., Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency, Nature, 2002, 419(6905), 395–399, DOI:10.1038/nature01063.
- K. Grønbaek, T. Dalby, J. Zeuthen, E. Ralfkiaer and P. Guidberg, The V410I (G1228A) variant of the caspase-10 gene is a common polymorphism of the Danish population, Blood, 2000, 95(6), 2184–2185 CrossRef . From NLM.
- B. Nagar, W. G. Bornmann, P. Pellicena, T. Schindler, D. R. Veach, W. T. Miller, B. Clarkson and J. Kuriyan, Crystal Structures of the Kinase Domain of c-Abl in Complex with the Small Molecule Inhibitors PD173955 and Imatinib (STI-571)1, Cancer Res., 2002, 62(15), 4236–4243 CAS . (acccessed 7/26/2024).
- R. Capdeville, E. Buchdunger, J. Zimmermann and A. Matter, Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug, Nat. Rev. Drug Discovery, 2002, 1(7), 493–502, DOI:10.1038/nrd839.
- R. L. M. van Montfort and P. Workman, Structure-based design of molecular cancer therapeutics, Trends Biotechnol., 2009, 27(5), 315–328, DOI:10.1016/j.tibtech.2009.02.003.
- Y.-L. Lin, Y. Meng, W. Jiang and B. Roux, Explaining why Gleevec is a specific and potent inhibitor of Abl kinase, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(5), 1664–1669, DOI:10.1073/pnas.1214330110.
- J. H. Xu, J. Eberhardt, B. Hill-Payne, G. E. González-Páez, J. O. Castellón, B. F. Cravatt, S. Forli, D. W. Wolan and K. M. Backus, Integrative X-ray Structure and Molecular Modeling for the Rationalization of Procaspase-8 Inhibitor Potency and Selectivity, ACS Chem. Biol., 2020, 15(2), 575–586, DOI:10.1021/acschembio.0c00019.
- J. H. Zhang, T. D. Chung and K. R. Oldenburg, A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays, J. Biomol. Screening, 1999, 4(2), 67–73, DOI:10.1177/108705719900400206 From NLM Publisher.
- E. Strom, S. Sathe, P. G. Komarov, O. B. Chernova, I. Pavlovska, I. Shyshynova, D. A. Bosykh, L. G. Burdelya, R. M. Macklis and R. Skaliter, et al., Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation, Nat. Chem. Biol., 2006, 2(9), 474–479, DOI:10.1038/nchembio809 From NLM Medline.
- D. C. Gray, S. Mahrus and J. A. Wells, Activation of Specific Apoptotic Caspases with an Engineered Small-Molecule-Activated Protease, Cell, 2010, 142(4), 637–646, DOI:10.1016/j.cell.2010.07.014.
- A. Oberst, C. Pop, A. G. Tremblay, V. Blais, J.-B. Denault, G. S. Salvesen and D. R. Green, Inducible Dimerization and Inducible Cleavage Reveal a Requirement for Both Processes in Caspase-8 Activation, J. Biol. Chem., 2010, 285(22), 16632–16642, DOI:10.1074/jbc.m109.095083.
- K. Kim, R. Damoiseaux, A. J. Norris, L. Rivina, K. Bradley, M. E. Jung, R. A. Gatti, R. H. Schiestl and W. H. McBride, High throughput screening of small molecule libraries for modifiers of radiation responses, Int. J. Radiat. Biol., 2011, 87(8), 839–845, DOI:10.3109/09553002.2011.560994.
- T. Sander, J. Freyss, M. von Korff and C. Rufener, DataWarrior: an open-source program for chemistry aware data visualization and analysis, J. Chem. Inf. Model., 2015, 55(2), 460–473, DOI:10.1021/ci500588j From NLM.
- J. W. Pierce, R. Schoenleber, G. Jesmok, J. Best, S. A. Moore, T. Collins and M. E. Gerritsen, Novel Inhibitors of Cytokine-induced IκBα Phosphorylation and Endothelial Cell Adhesion Molecule Expression Show Anti-inflammatory Effects in Vivo, J. Biol. Chem., 1997, 272(34), 21096–21103, DOI:10.1074/jbc.272.34.21096.
- M. Kraft, D. Radke, G. D. Wieland, P. F. Zipfel and U. Horn, A fluorogenic substrate as quantitative in vivo reporter to determine protein expression and folding of tobacco etch virus protease in Escherichia coli, Protein Expression Purif., 2007, 52(2), 478–484, DOI:10.1016/j.pep.2006.10.019.
- H. Nam, B. J. Hwang, D. Y. Choi, S. Shin and M. Choi, Tobacco etch virus (TEV) protease with multiple mutations to improve solubility and reduce self-cleavage exhibits enhanced enzymatic activity, FEBS Open Bio, 2020, 10(4), 619–626, DOI:10.1002/2211-5463.12828.
- E. L. Gerasimova, E. G. Gazizullina, D. I. Igdisanova, L. P. Sidorova, T. A. Tseitler, V. V. Emelianov, O. N. Chupakhin and A. V. Ivanova, Antioxidant properties of 2,5-substituted 6H-1,3,4-thiadiazines promising for experimental therapy of diabetes mellitus, Russ. Chem. Bull., 2022, 71(12), 2730–2739, DOI:10.1007/s11172-022-3702-0.
- C. N. Yiannios and J. V. Karabinos, Oxidation of Thiols by Dimethyl Sulfoxide, J. Org. Chem., 1963, 28(11), 3246–3248, DOI:10.1021/jo01046a528.
- T. J. Wallace and J. J. Mahon, Reactions of Thiols with Sulfoxides. II. Kinetics and Mechanistic Implications, J. Am. Chem. Soc., 1964, 86(19), 4099–4103, DOI:10.1021/ja01073a039.
- R. Jafari, H. Almqvist, H. Axelsson, M. Ignatushchenko, T. Lundbäck, P. Nordlund and D. M. Molina, The cellular thermal shift assay for evaluating drug target interactions in cells, Nat. Protoc., 2014, 9(9), 2100–2122, DOI:10.1038/nprot.2014.138.
- R. Ghosh, J. E. Gilda and A. V. Gomes, The necessity of and strategies for improving confidence in the accuracy of western blots, Expert Rev. Proteomics, 2014, 11(5), 549–560, DOI:10.1586/14789450.2014.939635.
- T. Yan, H. S. Desai, L. M. Boatner, S. L. Yen, J. Cao, M. F. Palafox, Y. Jami-Alahmadi and K. M. Backus, SP3-FAIMS Chemoproteomics for High-Coverage Profiling of the Human Cysteinome, ChemBioChem, 2021, 22(10), 1841–1851, DOI:10.1002/cbic.202000870.
- J. Yang, Z. Liu, S. Perrett, H. Zhang and Z. Pan, PES derivative PESA is a potent tool to globally profile cellular targets of PES, Bioorg. Med. Chem. Lett., 2022, 60, 128553, DOI:10.1016/j.bmcl.2022.128553 From NLM Medline.
- F. Shikwana, B. Heydari, S. Ofori, C. Truong, A. Turmon, J. Darrouj, L. Holoidovsky, J. Gustafson and K. Backus, CySP3-96 enables scalable, streamlined, and low-cost sample preparation for cysteine chemoproteomic applications, Mol. Cell. Proteomics, 2024, 100898, DOI:10.1016/j.mcpro.2024.100898.
- T. Yan, A. B. Palmer, D. J. Geiszler, D. A. Polasky, L. M. Boatner, N. R. Burton, E. Armenta, A. I. Nesvizhskii and K. M. Backus, Enhancing Cysteine Chemoproteomic Coverage through Systematic Assessment of Click Chemistry Product Fragmentation, Anal. Chem., 2022, 94(9), 3800–3810, DOI:10.1021/acs.analchem.1c04402.
- C. S. Hughes, S. Foehr, D. A. Garfield, E. E. Furlong, L. M. Steinmetz and J. Krijgsveld, Ultrasensitive proteome analysis using paramagnetic bead technology, Mol. Syst. Biol., 2014, 10(10), 757, DOI:10.15252/msb.20145625.
- C. S. Hughes, S. Moggridge, T. Müller, P. H. Sorensen, G. B. Morin and J. Krijgsveld, Single-pot, solid-phase-enhanced sample preparation for proteomics experiments, Nat. Protoc., 2019, 14(1), 68–85, DOI:10.1038/s41596-018-0082-x.
- M. A. Maj, J. Ma, K. N. Krukowski, A. Kavelaars and C. J. Heijnen, Inhibition of Mitochondrial p53 Accumulation by PFT-mu Prevents Cisplatin-Induced Peripheral Neuropathy, Front. Mol. Neurosci., 2017, 10, 108, DOI:10.3389/fnmol.2017.00108 From NLM PubMed-not-MEDLINE.
- K. Sekihara, N. Harashima, M. Tongu, Y. Tamaki, N. Uchida, T. Inomata and M. Harada, Pifithrin-mu, an inhibitor of heat-shock protein 70, can increase the antitumor effects of hyperthermia against human prostate cancer cells, PLoS One, 2013, 8(11), e78772, DOI:10.1371/journal.pone.0078772 From NLM Medline.
- L. Y. Yang, N. H. Greig, D. Tweedie, Y. J. Jung, Y. H. Chiang, B. J. Hoffer, J. P. Miller, K. H. Chang and J. Y. Wang, The p53 inactivators pifithrin-mu and pifithrin-alpha mitigate TBI-induced neuronal damage through regulation of oxidative stress, neuroinflammation, autophagy and mitophagy, Exp. Neurol., 2020, 324, 113135, DOI:10.1016/j.expneurol.2019.113135 From NLM Medline.
- J. Zhu, M. Singh, G. Selivanova and S. Peuget, Pifithrin-alpha alters p53 post-translational modifications pattern and differentially inhibits p53 target genes, Sci. Rep., 2020, 10(1), 1049, DOI:10.1038/s41598-020-58051-1 From NLM Medline.
- G. L. Coe, P. S. Redd, A. V. Paschall, C. Lu, L. Gu, H. Cai, T. Albers, I. O. Lebedyeva and K. Liu, Ceramide mediates FasL-induced caspase 8 activation in colon carcinoma cells to enhance FasL-induced cytotoxicity by tumor-specific cytotoxic T lymphocytes, Sci. Rep., 2016, 6(1), 30816, DOI:10.1038/srep30816.
- D. Milhas, O. Cuvillier, N. Therville, P. Clavé, M. Thomsen, T. Levade, H. Benoist and B. Ségui, Caspase-10 Triggers Bid Cleavage and Caspase Cascade Activation in FasL-induced Apoptosis, J. Biol. Chem., 2005, 280(20), 19836–19842, DOI:10.1074/jbc.m414358200.
- S. M. Uriarte, S. Joshi-Barve, Z. Song, R. Sahoo, L. Gobejishvili, V. R. Jala, B. Haribabu, C. McClain and S. Barve, Akt inhibition upregulates FasL, downregulates c-FLIPs and induces caspase-8-dependent cell death in Jurkat T lymphocytes, Cell Death Differ., 2005, 12(3), 233–242, DOI:10.1038/sj.cdd.4401549.
- A. Mohr, L. Deedigan, S. Jencz, Y. Mehrabadi, L. Houlden, S.-M. Albarenque and R. M. Zwacka, Caspase-10: a molecular switch from cell-autonomous apoptosis to communal cell death in response to chemotherapeutic drug treatment, Cell Death Differ., 2018, 25(2), 340–352, DOI:10.1038/cdd.2017.164.
- A. R. Julio, F. Shikwana, C. Truong, N. R. Burton, E. R. Dominguez 3rd, A. C. Turmon, J. Cao and K. M. Backus, Delineating cysteine-reactive compound modulation of cellular proteostasis processes, Nat. Chem. Biol., 2024 DOI:10.1038/s41589-024-01760-9.
- N. Kedersha, M. D. Panas, C. A. Achorn, S. Lyons, S. Tisdale, T. Hickman, M. Thomas, J. Lieberman, G. M. McInerney and P. Ivanov, et al., G3BP–Caprin1–USP10 complexes mediate stress granule condensation and associate with 40S subunits, J. Cell Biol., 2016, 212(7), 845–860, DOI:10.1083/jcb.201508028.
- B. Götte, M. D. Panas, K. Hellström, L. Liu, B. Samreen, O. Larsson, T. Ahola and G. M. McInerney, Separate domains of G3BP promote efficient clustering of alphavirus replication complexes and recruitment of the translation initiation machinery, PLoS Pathog., 2019, 15(6), e1007842, DOI:10.1371/journal.ppat.1007842.
- K. K. Hallenbeck, J. L. Davies, C. Merron, P. Ogden, E. Sijbesma, C. Ottmann, A. R. Renslo, C. Wilson and M. R. Arkin, A Liquid Chromatography/Mass Spectrometry Method for Screening Disulfide Tethering Fragments, SLAS Discovery, 2018, 23(2), 183–192, DOI:10.1177/2472555217732072.
- J. D. Sadowsky, M. A. Burlingame, D. W. Wolan, C. L. McClendon, M. P. Jacobson and J. A. Wells, Turning a protein kinase on or off from a single allosteric site via disulfide trapping, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(15), 6056–6061, DOI:10.1073/pnas.1102376108.
- D. A. Erlanson, A. C. Braisted, D. R. Raphael, M. Randal, R. M. Stroud, E. M. Gordon and J. A. Wells, Site-directed ligand discovery, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(17), 9367–9372, DOI:10.1073/pnas.97.17.9367.
- J. Sun, M. Gu, L. Peng, J. Guo, P. Chen, Y. Wen, F. Feng, X. Chen, T. Liu and Y. Chen, et al., A Self-Assembled Nano-Molecular Glue (Nano-mGlu) Enables GSH/H2O2-Triggered Targeted Protein Degradation in Cancer Therapy, J. Am. Chem. Soc., 2025, 147(1), 372–383, DOI:10.1021/jacs.4c11003.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cb00017c |
| This journal is © The Royal Society of Chemistry 2025 |