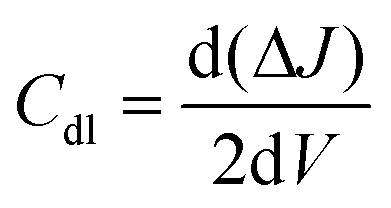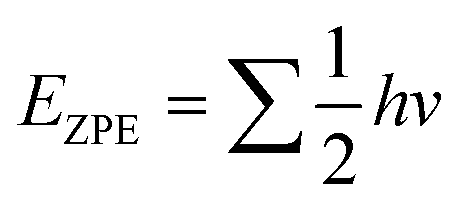DOI:
10.1039/D3NR05445D
(Paper)
Nanoscale, 2024,
16, 1758-1769
Insights of the efficient hydrogen evolution reaction performance in bimetallic Au4Cu2 nanoclusters†
Received
28th October 2023
, Accepted 24th November 2023
First published on 26th December 2023
Abstract
The design of efficient electrocatalysts for improving hydrogen evolution reaction (HER) performance using atomically precise metal nanoclusters (NCs) is an emerging area of research. Here, we have studied the HER electrocatalytic performance of monometallic Cu6 and Au6 nanoclusters and bimetallic Au4Cu2 nanoclusters. A bimetallic Au4Cu2/MoS2 composite exhibits excellent HER catalytic activity with an overpotential (η10) of 155 mV vs. reversible hydrogen electrode observed at 10 mA cm−2 current density. The improved HER performance in Au4Cu2 is due to the increased electrochemically active surface area (ECSA), and Au4Cu2 NCs exhibits better stability than Cu6 and Au6 systems and bare MoS2. This augmentation offers a greater number of active sites for the favorable adsorption of reaction intermediates. Furthermore, by employing X-ray photoelectron spectroscopy (XPS) and Raman analysis, the kinetics of HER in the Au4Cu2/MoS2 composite were elucidated, attributing the favorable performance to better electronic interactions occurring at the interface between Au4Cu2 NCs and the MoS2 substrate. Theoretical analysis reveals that the inherent catalytic enhancement in Au4Cu2/MoS2 is due to favorable H atom adsorption over it and the smallest ΔGH* value. The downshift in the d-band of the Au4Cu2/MoS2 composite influences the binding energy of intermediate catalytic species. This new catalyst sheds light on the structure–property relationship for improving electrocatalytic performance at the atomic level.
Introduction
The electrochemical process is an effective way to generate green hydrogen, which is considered an excellent renewable source of energy and alternative to fossil fuels.1–3 The major challenge has been to replace e the best-known efficient platinum (Pt) electrocatalyst by developing efficient catalysts with a lower hydrogen evolution reaction (HER) overpotential for electrocatalytic applications.4–13 Significant attention has been given to new strategies for improving the HER performance of catalysts by controlling the charge transfer process and lowering the H adsorption and desorption energies on the catalyst surface.14 The free energy of H adsorption at the edges of MoS2 is considered a viable HER catalyst to replace Pt. The defect-rich ultrathin MoS2 nanosheets (NSs) are found to be an exceptional electrocatalytic activity with a low onset overpotential.15–17 Nonetheless, their catalytic efficacy is significantly hampered by the restricted availability of catalytically active sites, primarily caused by the tendency for restacking and insufficient vertical (i.e., perpendicular to the plane) charge conductivity.18,19 Furthermore, it has been demonstrated that enhancing the surface of MoS2 through nanoparticle decoration is an efficient method for augmenting surface area and catalytically active sites, thus enhancing intrinsic activity.20–22 However, the lack of atomic-level precision in the size and alloy composition of traditional nanoparticles shows the difficulties in establishing an accurate relationship between structure and properties.14,23–25,26
Therefore, a defined structure of a catalyst would be required to tune the catalytic property at the atomic level.27–29 As a result, significant attention has been directed towards creating highly precise architectures and employing ligand-protected metal nanoclusters with atomic-level precision.30–32 Metal NCs, due to their ultrasmall small sizes of nearly 2 nm with fascinating properties and atomic precision, have gained significant attention towards their applications in catalysis.33–36 Atomically precise gold nanoclusters offer precise control over their geometric structure, electronic properties, and surface ligands, thus holding significant appeal as catalysts for electrochemical reactions due to their ultrasmall size and high surface-active sites.37–41 Zhao et al. investigated the performance of Au25 nanoclusters on MoS2 for the hydrogen evolution reaction, achieving an overpotential of 280 mV.42 The study emphasizes the potential of NCs to enhance the performance of HER via altering the interface of Au NCs with MoS2 NSs. Gratious et al. reported the enhancement of the HER activity by incorporating Au11 nanoclusters on MoS2 NSs, leading to an overpotential of 292 mV, and verified the stability of the H* intermediate using computational analysis.43 However, bimetallic nanoclusters could potentially offer improved catalytic activity and selectivity compared to their monometallic nanoclusters due to the synergistic effects of heteroatoms.44 A few investigations have explored the use of doped Au NCs with various dopants, including noble metals Pt, Pd, Ru, and Ag.39,45 A few studies have been reported on the doping of precious materials Pd and Pt into atomically precise Au25 and Au26 NCs.46 Zhu et al. investigated Au2Pd6 NCs over MoS2 showing significantly improved HER catalytic activity and achieved an overpotential of nearly 225 mV vs. RHE at a current density of 10 mA cm−2.45 Tang et al. compared the HER performance of two types of bimetallic Au24Ag20 and Au22Ag20 NCs protected by alkynyl and halogen groups with overpotentials of 260 mV and 335 mV, respectively.47 Further, DFT reveals the enhancement in HER activity based on the lower binding energy of hydrogen to form an H* intermediate. However, the dopant including non-noble metals Cu with Au NCs for electrocatalytic HER performance has limited exploration.
Here, we synthesized atomically precise bimetallic Au4Cu2 NCs and monometallic Au6 and Cu6 NCs for hydrogen evolution reaction (HER) catalytic activity. A comprehensive analysis involving experimental and theoretical approaches provides valuable insights into the mechanistic aspects responsible for the improved HER catalytic activity observed in the bimetallic Au4Cu2/MoS2 composite. XPS studies confirm better charge transfer between Au4Cu2 NCs and MoS2, indicating strong electronic interactions at their interface. Theoretical analysis reveals that the intrinsic catalytic improvement in the Au4Cu2/MoS2 composite is due to its favorable H atom adsorption and the smallest ΔGH* value.
Experimental details
Materials
Sodium molybdate dihydrate (Na2MoO4·2H2O, 99.99%), thiourea (CH4N2S, 99%), tetrachloroauric acid trihydrate (HAuCl4·3H2O), copper nitrate trihydrate (Cu(NO3)2·3H2O), tetrakis(hydroxymethyl)phosphonium chloride (THPC), 3-mercaptopropionic acid (MPA), sodium hydroxide (NaOH), Nafion (5 wt% in low aliphatic alcohols), Pt/C (20 wt% Pt loading on Vulcan XC-72) were obtained from Sigma-Aldrich. Sulfuric acid (H2SO4, 98%) was purchased from Thermo-Fisher Scientific India Ltd. All the chemicals were purchased in ultrapure form; therefore, no additional purification was required. The glassware was thoroughly cleaned with aqua regia and rinsed with ultrapure water and acetone before use.
Characterization
Scanning electron microscopy (SEM) images were obtained using a JEOL, JSMIT-300. Transmission electron microscopy (TEM) and high-resolution TEM images were taken using a JEOL-JEM-F200 instrument. Powder X-ray diffraction (PXRD) patterns were analyzed on a Bruker D8 ADVANCE powder diffractometer with Cu Kα radiation. Raman spectra were recorded in a WITEC Focus Innovations Alpha-300 Raman confocal microscope using a 532 nm excitation laser. XPS measurements were recorded using a Thermo-Fischer Scientific Kα instrument. Waters Q-TOF mass spectrometer equipped with a Z-spray source was used for the electrospray ionization (ESI) mass spectrometry measurements. The mass of as-synthesized NCs was examined using matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry on a Bruker Daltonics Autoflex II TOF/TOF system. Ultraviolet–Visible (UV-Vis) absorption spectra were collected from a UV-Vis spectrophotometer (Shimadzu). Fourier-transform infrared (FTIR) spectra were performed in a PerkinElmer FTIR-spectrometer using KBr pellets. The PL spectra were recorded using a Fluorolog 3-221 (HORIBA Jobin Yvon) fluorimeter. Inductively coupled plasma mass spectrometry (ICP-MS) analysis was performed using an Agilent 7900 instrument.
Synthesis of monometallic copper and gold nanoclusters
The synthesis of Cu NCs involves a one-pot strategy. In a beaker, 10 mL of high performance liquid chromatography (HPLC) water was mixed with 75 mL of 1 M NaOH and 3 μL of THPC (80% in water), with vigorous stirring. Then, 100 μL of copper salt was added, resulting in a color change to light brown, indicating the reduction of copper salt. After 30 minutes of stirring, an MPA ligand was introduced, and the reaction continued under stirring in the dark at room temperature. The change in the reaction mixture's color after 24 hours confirmed the formation of MPA-capped gold NCs. The synthesis of gold nanoclusters follows the same one-pot strategy by taking gold salt as a metal precursor in the case of Au NCs.
Synthesis of bimetallic copper doped gold nanoclusters
The synthesis of copper-doped gold nanoclusters was previously reported with the same methodology as for Au6 NCs with minor modifications. In a one-pot synthesis, two metal precursors (Au3+ and Cu2+) with a [Cu2+]/[Au3+] ratio of 0.047 were analyzed using ICP-MS. The as-synthesized Cu6, Au6, and Au4Cu2 NCs were washed multiple times with HPLC water to eliminate excess ligands and centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for further analysis.
000 rpm for further analysis.
Synthesis of MoS2 NSs
The synthesis involves previously reported procedures. Initially, 800 mg of Na2MoO4·2H2O was dissolved in 10 mL of distilled water with magnetic stirring to prepare 2D MoS2 NSs. Separately, 1.65 mg of thiourea was mixed with 16.6 mL of distilled water. The colorless thiourea solution was slowly added to Na2MoO4·2H2O solution until it became clear. The mixture was then placed in a Teflon-lined stainless-steel autoclave and heated at 200 °C for 24 hours. After cooling to room temperature, black residues were obtained by centrifuging the solution at 12000 rpm for 30 minutes. These residues were washed with HPLC water and ethanol and dried at 60 °C in an air oven for further analysis.
Preparation of Cu6 NCs, Au6 NCs and Au4Cu2 NCs composites with MoS2 NSs
Initially, 2 mg of MoS2 was mixed well with 0.35 mL of water through ultrasonication for 10 min. After that, different amounts of Cu6 NCs, Au6 NCs, and Au4Cu2 NCs were added into the above-dispersed mixture and were allowed to sonicate for 30 min for better interaction between as-prepared NCs and MoS2. The composites were then separated using centrifugation to confirm the successful loading of each type of NCs on MoS2, and the ICP-MS technique analyzed the loading of NCs present in a given system.
Preparation of working electrode for HER
Typically, 2 mg of MoS2 NSs was suspended in a solution containing a mixture of ethanol, water, and Nafion (in a ratio of 1:4:0.55) and sonicated to produce a well-dispersed suspension. Thereafter, 150 μL of NCs solution was added and sonicated for 30 minutes to obtain a homogeneous ink. 5 μL of ink was drop-casted onto a 3 mm diameter glassy carbon rotating disk electrode followed by drying under an IR lamp for 5 hours. The NCs loading on MoS2 NSs was regulated by varying the amount of NCs in the dispersion of MoS2 NSs. Similarly, a working electrode for bare MoS2 NSs was prepared without NCs. The working electrode of commercial Pt/C was fabricated by 50 μg cm−2 Pt loading on the electrode surface.
Electrochemical measurements
All electrochemical measurements were recorded using an Autolab multichannel M204 PGSTAT (Metrohm) electrochemical station by taking 0.5 M H2SO4 aqueous solution at 25 °C. A conventional three-electrode system was used to assess the performance of the HER. The working electrode was a catalyst-coated glassy carbon RDE, a graphite rod served as a counter electrode, and the reference electrode was a saturated calomel electrode (SCE). The formula ERHE = ESCE + (0.241 + 0.059 pH) was used to convert all the potential values in the study to the reversible hydrogen electrode (RHE) scale. HER linear sweep voltammetry (LSV) curves were obtained at a scan rate of 10 mV s−1. To calculate the double-layer capacitance, cyclic voltammograms with scan rates ranging from 10 to 50 mV s−1 were acquired in the non-faradaic potential ranging from 0.0 to 0.3 V vs. RHE (Cdl). The double-layer capacitance was determined by plotting the difference between the anodic and cathodic currents (ΔJ = Ja − Jc) at 0.15 V vs. RHE as a function of scan rate.48 The Cdl was measured using the formula:| | 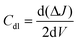 | (1) |
The electrochemically active surface area (ECSA) can be obtained by using the measured Cdl values of the samples, from the formula: ECSA = Cdl/Cs, where Cs indicates the specific capacitance of the flat standard electrode, i.e., Cs = 0.04 mF Cm−2.49 The electrochemical impedance spectra in the 100 kHz to 0.1 Hz frequency range were determined using an AC voltage having 10 mV amplitude. The measured impedance spectra were then fitted into an appropriate equivalent circuit model to ascertain the solution resistance and charge transfer resistance. A long-term durability test was done by collecting a chronoamperometric curve for 40 hours at a constant potential to attain a current density of 10 mA cm−2 at 25 °C. All the data are reported without iR-correction.
Computational details
The density functional theory (DFT) calculations were performed using the projected augmented wave (PAW) method implemented in the Vienna ab initio simulation package(VASP).50,51 The generalized gradient approximation with Perdew–Burke–Ernzerhof (GGA-PBE) was employed to describe the exchange–correlation functionals.52 Plane-wave basis sets were set at 550 eV to expand the electronic wave function. A Γ-centre (1 × 1 × 1) k-points grid was considered for sampling the Brillouin zone. A conjugate-gradient algorithm with an energy convergence criterion of 10–4 eV and Hellmann–Feynman force convergence criteria of <0.02 eV Å−1 was utilized during optimization. The DFT-D3 method was used to take into account the van der Waals interaction.52 The reaction free energies (ΔG) were calculated by considering the following equation:53,54where ΔE is the hydrogen binding energy, ΔEZPE is the change in zero-point energy, T is the temperature (300K), and ΔS is the entropy change for the reaction. ΔEZPE and entropies were calculated from the harmonic oscillator approximation.55 The zero-point energy (EZPE) was calculated using the following equation:| | 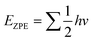 | (3) |
where h is the Planck constant and v is the vibrational frequency.
Results and discussion
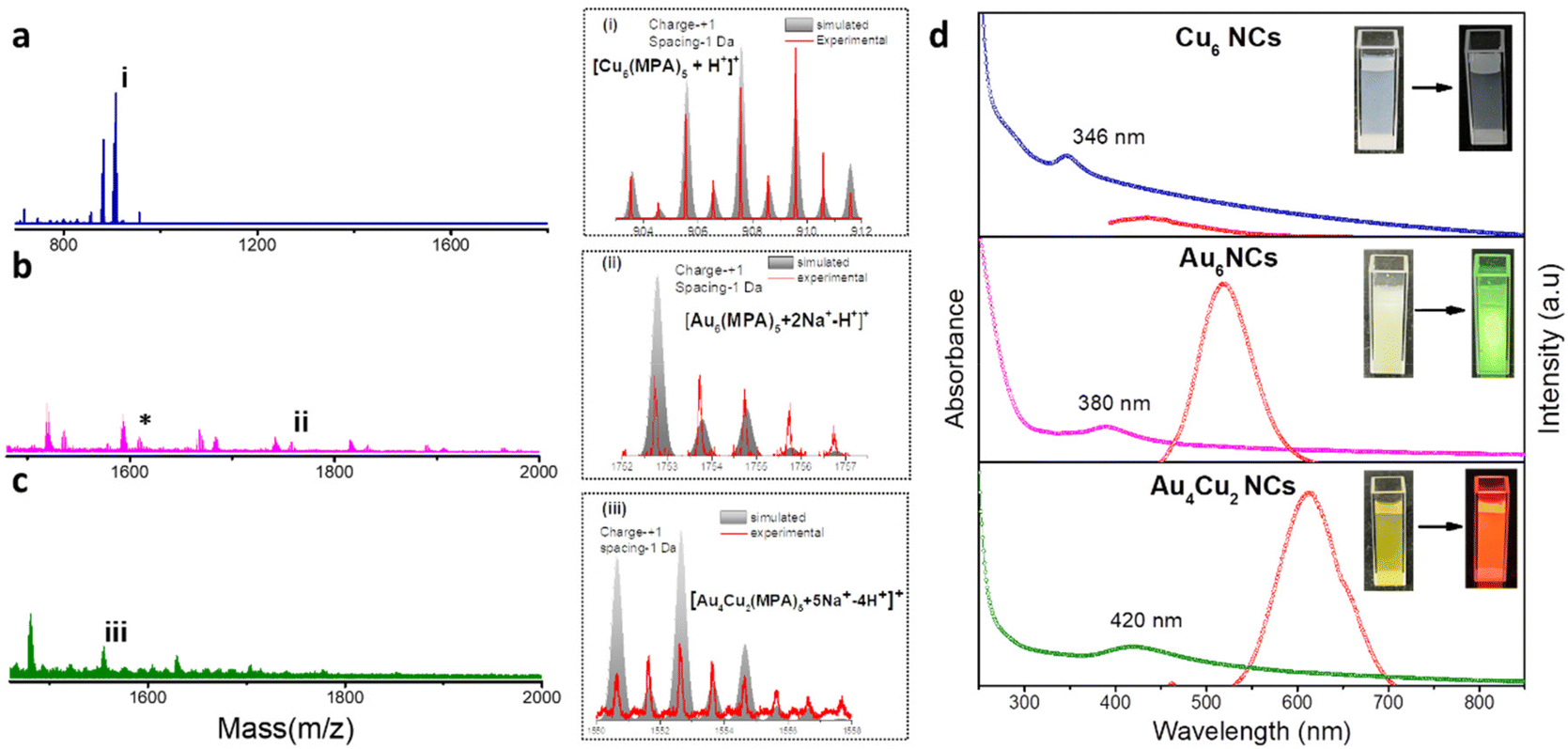
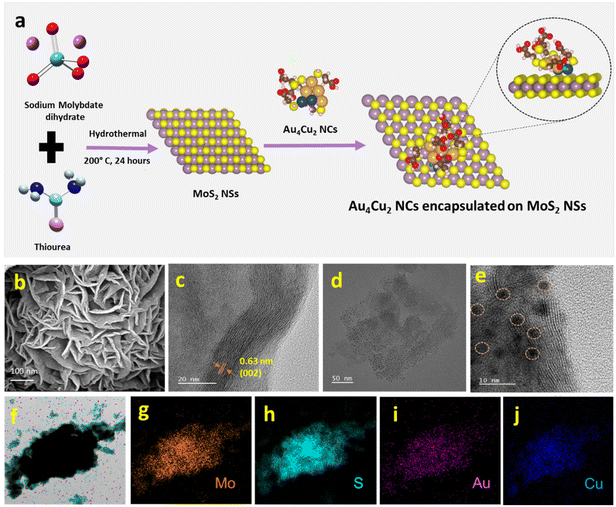
We have synthesized Au6 NCs, Cu6 NCs, and Au4Cu2 NCs utilizing a one-pot synthesis method in an aqueous medium with MPA as a capping ligand.56 The TEM images of as-prepared Cu6 NCs and Au6 NCs depict the formation of ultrasmall nanoclusters with spherical morphology and an acquired particle size distribution indicating a size of nearly 2 nm (Fig. S1†). The recorded TEM and HRTEM images of bimetallic Au4Cu2 NCs also depict the formation of ultrasmall nanoclusters with an average size of 2.03 ± 0.3 nm (Fig. S2a and b†). The elemental mapping by TEM of bimetallic Au4Cu2 NCs shows clear evidence of the presence of both Au and Cu in the prepared bimetallic NCs (Fig. S2c–f†). In-depth analysis through positive-mode ESI-mass spectrometry provided insights into the composition of the synthesized NCs. For the as-prepared Cu NCs, a prominent peak at m/z 903 Da corresponds to [Cu6(MPA)5 + H+]+, while for the Au6 NCs, the molecular ion peak observed at 1754 Da is attributed to [Au6(MPA)5 + 2Na+ − H+]+ (Fig. 1a and b). The additional peaks in mass spectrometry are attributed to the loss of fragments from the molecular ion.57 Notably, an additional peak at a separation of m/z 146 (marked with an asterisk) in the case of Au NCs is due to [Au6(MPA)4 + H+]+, suggesting the detachment of an MPA ligand fragment from Au6(MPA)5. This interpretation is supported by a close match between the isotopic experimental pattern and the simulated pattern, as depicted in Fig. S3.† Furthermore, in the case of the bimetallic AuCu NCs, the observed m/z peak at 1552 Da corresponds to the composition [Au4Cu2(MPA)5 + 5Na+ − 4H+]+ (Fig. 1c). Thus, the inclusion of Cu in Au NCs results in the replacement of a few Au atoms by Cu atoms. The assigned compositions in all as-prepared NCs are substantiated by the close match between the isotope distributions of the simulated and observed spectra, as seen in Fig. 1(i, ii, iii). The composition of as-prepared NCs was further confirmed with MALDI-MS analysis (Fig. S4†). The MALDI-MS analysis of Au NCs exhibits a molecular ion peak at m/z 1729 Da attributed to [Au6(MPA)5 + Na+], and other peaks at (a) m/z 1497 Da and (b) m/z 1207 Da can be assigned to [Au6(MPA)3 + H+] and [Au4(MPA)4 + H+], respectively, arising from the decomposition of (MPA)2 and Au2(MPA) fragments from Au6(MPA)5. Further, the MALDI-MS analysis of AuCu NCs includes a molecular ion peak at m/z 1507 Da that can be attributed to [Au4Cu2(MPA)5 + 3Na+], and other peaks at (a) m/z 1297 Da and (b) m/z 1073 Da can be assigned to [Au4Cu2(MPA)3 + 3Na+] and [Au4Cu2(MPA)2 + H+], respectively, arising from the fragmentation of (MPA)2 and (MPA)3 ligands from Au4Cu2(MPA)5. Investigation into the ligand binding characteristics of the NCs was conducted using FTIR spectra analysis (Fig. S5†). In the FTIR spectra of the MPA ligand, a peak is evident at 2567 cm−1, representing the S–H thiol group. However, upon the formation of as-prepared Cu6 NCs, Au6 NCs, and Au4Cu2 NCs, this peak disappears, providing evidence for the formation of covalent Au–S and Cu–S bonds in the NCs. The as-prepared NCs exhibit tunability in photophysical properties. The UV-Vis absorption spectra of the as-prepared NCs exhibit no SPR bands at 520 nm or 650 nm due to Au NPs or Cu NPs, suggesting no formation of nanoparticles.58,59 Interestingly, the UV-Vis absorption spectra of Cu6 NCs, Au6 NCs, and Au4Cu2 NCs exhibit prominent differences, as seen in Fig. 1d. The absorption peaks for monometallic Cu6 NCs and Au6 NCs were observed at 346 nm and 380 nm. After the inclusion of copper in Au6 NCs, the UV-Vis peak red-shifted to 420 nm in the case of Au4Cu2 NCs, revealing perturbation in the electronic structure of the Au6 NCs after Cu substitution. Similarly, the prepared NCs exhibit different photoluminescence (PL) properties. The Cu6 NCs are non-luminescent while Au6 NCs exhibit an emission peak at 520 nm which is red-shifted to 620 nm in the case of Au4Cu2 NCs, due to the tunability of the HOMO–LUMO gap after the inclusion of Cu atoms in Au NCs.56 Moreover, the recorded excitation spectra of as-prepared NCs can be seen in Fig. S6.† To explore the potential of as-prepared NCs in enhancing the electrocatalytic HER, we loaded them on MoS2, as seen in Fig. 2a. The MoS2 NSs were prepared by a previously reported strategy.60 The observed PXRD pattern of MoS2 exhibits distinctive peaks corresponding to planes (002), (100), (103), and (110), closely matching the MoS2 hexagonal structure (Fig. S7†).60
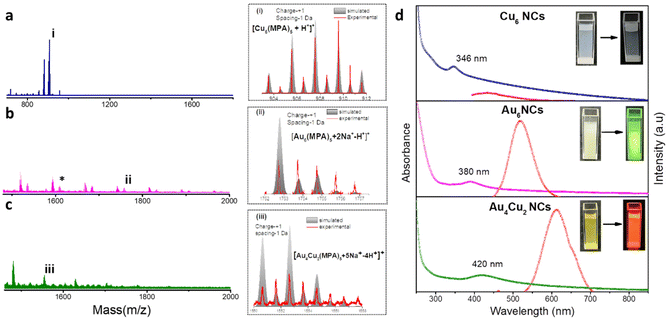 |
| | Fig. 1 ESI-mass spectra and corresponding isotopic patterns of (a) Cu6 NCs, (b)Au6 NCs, and (c) Au4Cu2NCs. (d) The corresponding UV-vis absorption and photoluminescence spectra. (Insets show the color of the NCs under visible and UV-light (λex = −365 nm).). | |
 |
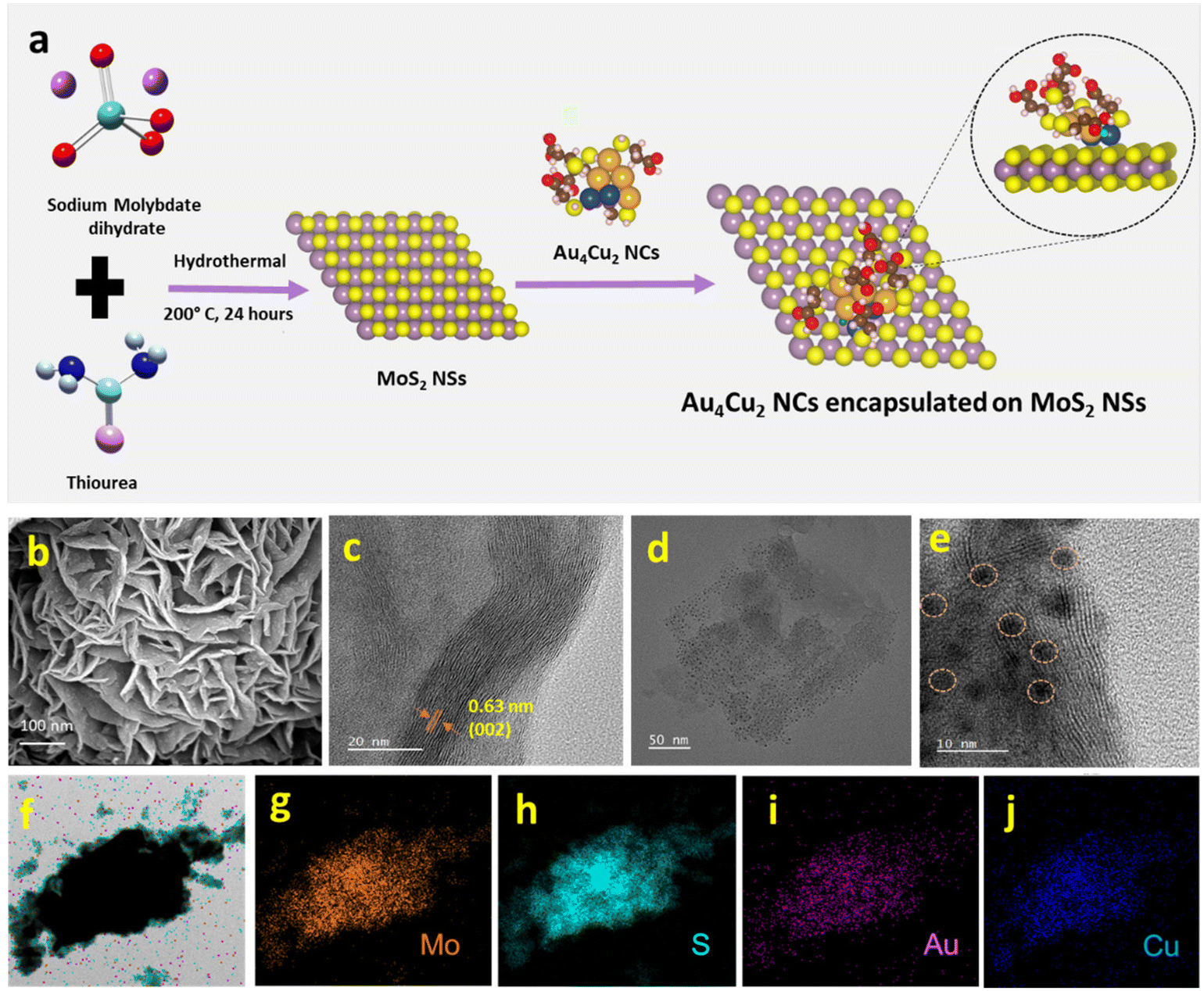
| | Fig. 2 (a) Schematic illustration of the synthesis of MoS2 and Au4Cu2/MoS2 heterostructures. (b) SEM and (c) HRTEM image of MoS2 NSs. (d) and (e) TEM and HRTEM images of Au4Cu2/MoS2. (f) STEM image of Au4Cu2/MoS2 with the corresponding elemental mappings of (g) Mo, (h) S, (i) Au, and (j) Cu. | |
The Au4Cu2 NCs/MoS2 composites were obtained by mixing as-prepared Au4Cu2 NCs in well-dispersed MoS2 NSs in an aqueous medium followed by 30 minutes of ultrasonication. To study the formation of Au4Cu2NCs/MoS2, further characterizations were performed involving SEM, TEM, and HRTEM. The SEM image of MoS2 clearly shows the sheet-like morphology, which is entangled to form a two-dimensional sheet-like configuration with many edges and corners (Fig. 2b). Further, the elemental mapping confirms the uniform distribution of Mo and S in MoS2 (Fig. S8†). The TEM images confirm the development of a sheet-like structure, aligning with the SEM analysis (Fig. S9†). Moreover, HRTEM images exhibit discernible lattice-fringes with spacings of 0.63 nm corresponding to the (002) plane of the MoS2 structure (Fig. 2c). Noticeably, TEM images of the composite reveal that the MoS2 surface is decorated by spherical Au4Cu2 NCs, and the NCs are uniformly encapsulated on the surface of MoS2 NSs (Fig. 2d–e†). The obtained EDX spectrum and elemental mapping confirm the presence and uniform distribution of Mo, S, Au, and Cu in the Au4Cu2/MoS2 heterostructure (Fig. S10,†Fig. 2f–j). The EDX spectrum quantifies the atomic ratio of Au/Cu of nearly 1.78, which is almost the same as that in Au4Cu2 NCs (inset to Fig. S10†). Thereafter, the TEM images of the Au6/MoS2 and Cu6/MoS2 composites show encapsulated NCs on the MoS2 NSs. Additionally, the elemental mapping suggests the presence of the considered NCs on MoS2 NSs (Fig. S11–12†).
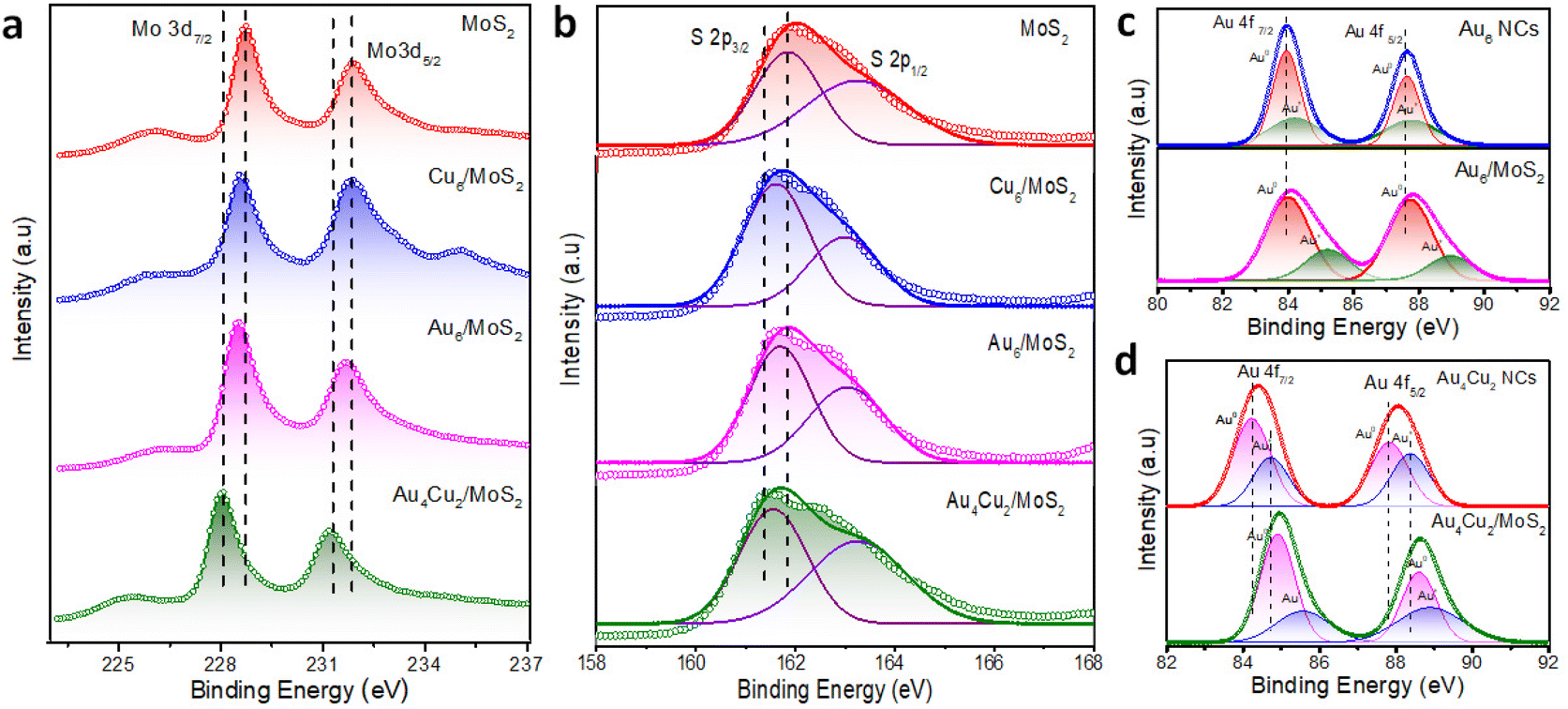
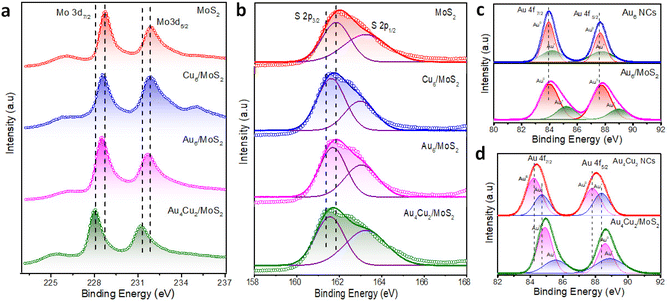
We carried out XPS and Raman investigations to look into the interactions between NCs and MoS2 NSs and study the inherent causes of the improvement in catalytic activity. The XPS survey spectra recorded for all considered catalysts can be seen in Fig. S13.† The Mo 3d XPS spectra of MoS2, Cu6/MoS2, Au6/MoS2, and Au4Cu2 /MoS2 are given in Fig. 3a. The peaks that appeared at 228.6 eV and 231.8 eV can be attributed to Mo 3d5/2 and Mo 3d3/2, respectively. Interestingly, Mo 3d in Au4Cu2/MoS2 nanocomposites shows a larger negative shift of 0.6 eV in binding energy (B.E) than that in Au6 NCs/MoS2 (0.3 eV) or Cu6 NCs/MoS2 (0.3 eV), in comparison with pristine MoS2, indicating better electronic interactions in case of bimetallic Au4Cu2NCs with MoS2. Similarly pronounced negative shifts (0.4 eV) are seen in the binding energies of S 2p in Au4Cu2/MoS2 compared to the other considered systems or bare MoS2 (Fig. 3b). Au 4f exhibits twin peaks between 80 and 90 eV for all considered catalysts, corresponding to Au 4f7/2 and Au 4f5/2.61 Further analysis involved the splitting of these two peaks revealing the presence of both Au(0) and Au(I) valence states, and these mixed oxidation states were well-matched with previous findings.62 Interestingly, the (B.E) of Au 4f in Au6NCs and Au4Cu2NCs shows positive shifts of 0.3 eV and 0.6 eV, respectively, with their heterostructures compared to bare Au6NCs and Au4Cu2NCs, which corroborates the effect of deposited NCs on the electronic properties of MoS2 (Fig. 3c–d). A similar kind of positive shift of 0.2 eV was observed in the Cu 2p of the pure nanocluster compared to its Cu6/MoS2 composite (Fig. S14†).63 These findings imply the considerable transfer of electron density between deposited NCs and the MoS2 interface. The Raman spectrum corresponding to MoS2 can be seen in Fig. S15.† The Raman peaks at 378 cm−1 and 405 cm−1 correspond to typical E2g1 and A1g1 vibration modes, respectively.64 Here the creation of multilayered MoS2 is indicated by the frequency difference (Δk ∼ 27 cm−1) between the phonon modes, E2g1 and A1g1. However, after Au4Cu2 NCs were encapsulated on MoS2, the in-plane E2g1 vibration frequency mode was shifted towards a higher wavenumber due to structural changes brought on by stacking or interfacial van der Waals interactions due to substantial electron density transfer between Au4Cu2 NCs and MoS2 NSs.
 |
| | Fig. 3 XPS analysis of MoS2, Cu6/MoS2, Au6/MoS2, and Au4Cu2NCs/MoS2: (a) Mo 3d, (b) S 2p, (c) XPS spectra of Au 4f in Au6 NCs and Au6/MoS2, and (d) XPS spectra of Au 4f in Au4Cu2 NCs and Au4Cu2/MoS2. | |
Electrocatalytic water reduction performance
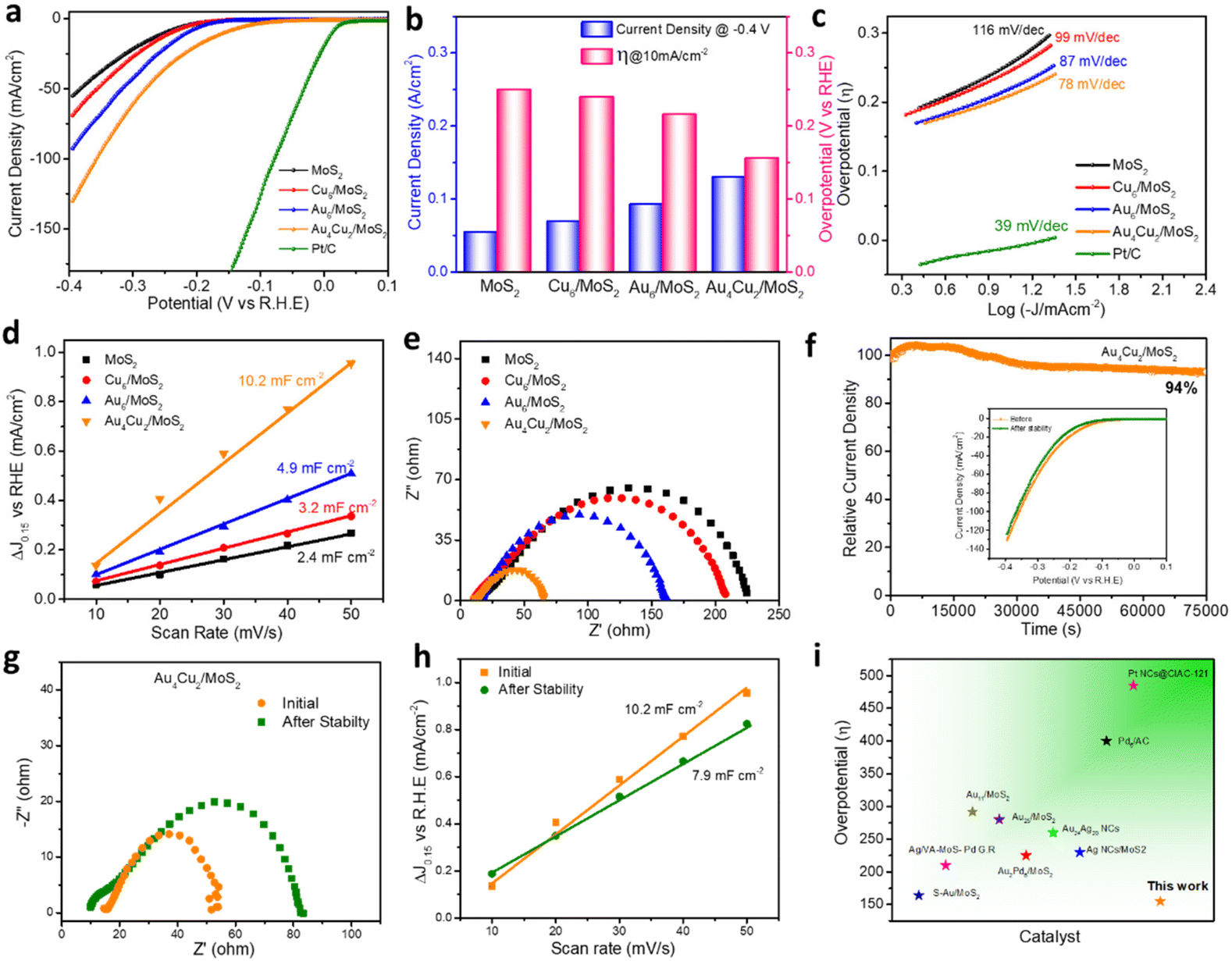
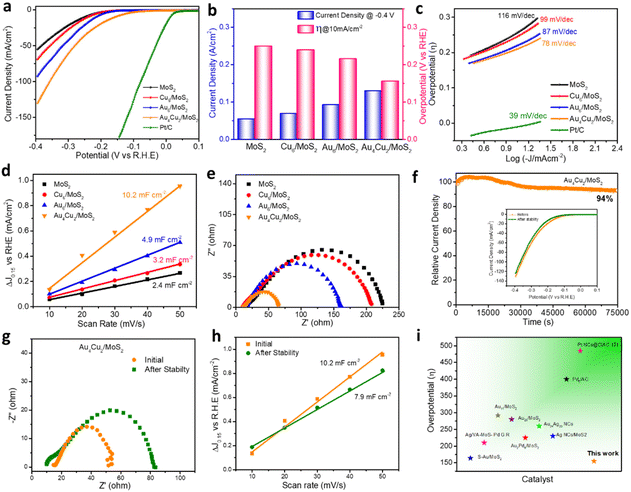
The electrocatalytic HER activity of bare MoS2 and considered nanocomposites was analyzed using a typical three-electrode system taking 0.5 M H2SO4 as an electrolyte. Commercial Pt/C was used as a benchmark catalyst using a metal loading of 50 μg cm−2 on the glassy carbon electrode. Pt/C displays the lowest overpotential of 13 mV vs. RHE (reversible hydrogen electrode) and a very high current density of nearly 500 mA cm−2 at a comparatively low overpotential, both of which highlight the catalyst's exceptional performance (Fig. S16†). Bare MoS2 shows a current density of 55 mA cm−2 at a potential of −0.4 V (Fig. 4a). The HER catalytic performance was found to depend on the different concentrations of NCs. For comparison, we investigated the best HER catalytic activity of all considered catalysts, such as Cu6/MoS2, Au6/MoS2, and Au4Cu2/MoS2, by taking different loading amounts of as-prepared NCs on the base material MoS2 (Fig. S17a–c†). The compared HER polarization curves show that the best HER activity is exhibited by a 2 wt% Au4Cu2/MoS2 nanocomposite, whereas the activity of 3 wt% Au6/MoS2 and 2 wt% Cu6/MoS2 composites is comparable to that of MoS2. Au4Cu2/MoS2 exhibits a prominent activity of 130 mA cm−2 which is 1.41, 1.91, and 2.36 times higher than that of Au6/MoS2, Cu6/MoS2, and bare MoS2, respectively (Fig. 4a). This implies that introducing Cu dopant into Au NCs can significantly enhance electrocatalytic performance. Fig. S18† emphasizes the charge transport region, wherein the current is governed by the rate of charge transfer at the electrode surface. Herein, the Au4Cu2/MoS2 catalyst shows an overpotential of 155 mV at a current density of 10 mA cm−2, which is notably lower than that of the other considered catalysts or bare MoS2 (Fig. 4b). Furthermore, other HER electrocatalytic factors, such as Tafel slope, charge-transfer resistance, and electric double-layer capacitance, were examined by varying the loading amounts of Au4Cu2 NCs on MoS2. The findings also indicate the most favorable outcomes with a loading of 2 wt% Au4Cu2 NCs on MoS2 (Fig. S19a–c†). The double-layer capacitance corresponding to variable loadings of Au4Cu2/MoS2 was analyzed by recording cyclic voltammograms at various scan rates (Fig. S20a–e†). The overall electrocatalytic parameters for Au4Cu2/MoS2 are tabulated in Table S1.† After that, to better comprehend the considered systems, Tafel slope analysis of various considered catalysts was used to determine the rate of electron transport at the electrode–electrolyte interface, as shown in Fig. 4c, of MoS2, Cu6/MoS2, Au6/MoS2, Au4Cu2/MoS2, and commercial Pt/C, created from the HER polarization curves. Commercial Pt/C exhibits the lowest Tafel slope of 39 mV dec−1, as reported in the literature.65 Furthermore, the Tafel slope of Au4Cu2/MoS2 is much smaller in comparison to the other considered catalysts, as seen in Fig. 4c. The Tafel slope obtained for the Au4Cu2/MoS2 bimetallic composite is 78 mV dec−1, which is considerably less than for the other considered catalysts Cu6/MoS2 (97 mV dec−1), Au6/MoS2 (87 mV dec−1), and bare MoS2 (116 mV dec−1), indicating greater enhancement in the HER kinetics of MoS2 in the presence of Au4Cu2 NCs. Moreover, the measured Tafel slope of 78 mV dec−1 for Au4Cu2/MoS2 suggests that HER may occur via a coupled Volmer–Heyrovsky-type mechanism, with the electrochemical adsorption–desorption process serving as the rate-limiting step.66 Further in-depth investigation of the electrochemically active surface area (ECSA) and charge transfer properties helped to clarify the cause of the increased electrocatalytic activity for Au4Cu2/MoS2 heterostructures. ECSA is also crucial because it influences the rate at which charge transfer reactions occur at the electrode–electrolyte interface. Fig. 4d shows a comparison plot of double layer capacitance (Cdl) as a function of scan rate. The Cdl, which directly affects the ECSA, was calculated using the cyclic voltammograms of MoS2 and other considered catalysts at various scan rates, as shown in Fig. S21.†67 The calculated Cdl for the Au4Cu2/MoS2 heterostructure is 10.2 mF cm−2, which is 2.0, 3.1, and 4.2 times higher than the estimated Cdl for Au6/MoS2, Cu6/MoS2, and bare MoS2. This suggests that the former has more active sites and ECSA due to the creation of an interfacial structure. The charge transfer kinetics at the electrode surface during the HER process were investigated using electrochemical impedance spectroscopy (EIS). The charge transfer process is represented by a semicircle at high frequencies, and the diameter of the semicircle equals the charge transfer resistance (Rct).68,69 The Nyquist curve presented in the inset of Fig. 4e shows faster charge transfer kinetics of the Au4Cu2/MoS2 composite as it exhibits the smallest diameter for the semicircle in comparison to the other catalysts and bare MoS2, due to the synergistic interaction between Au4Cu2 NCs and MoS2. This indicates a rapid electron transfer rate at the catalyst–electrolyte interface, leading to remarkable hydrogen evolution activity. The electrochemical performance of all the catalysts is shown in Table 1. Long-term durability is a crucial consideration when evaluating an electrocatalyst for improving catalytic performance. To examine the stability of the electrocatalyst, chronoamperometry measurements of MoS2, Cu6/MoS2, Au6/MoS2, and Au4Cu2/MoS2 were performed at a fixed potential to achieve an initial current density of 10 mA cm−2. The Cu6/MoS2 and Au6/MoS2 composites demonstrate initial current density stabilities of 55% and 60%, respectively, indicating slightly improved stability compared to bare MoS2 (52%) (Fig. S22†). However, the Au4Cu2/MoS2 composite shows better stability by maintaining nearly 94% of its initial current density even after 75000 seconds of stability testing. The inset of Fig. 4f shows the HER catalytic activity acquired before and after the chronoamperometry testing of Au4Cu2/MoS2, which demonstrates an increase in η10 of only 17 mV and a decrease in current density of 7 mA cm−2 after the stability test.
 |
| | Fig. 4 (a) Polarization curves of MoS2, NC-encapsulated MoS2, and Pt/C towards HER recorded at a scan rate of 10 mV s−1. (b) comparison of the current density at −0.4 V vs. RHE (blue columns) and overpotential at a current density of 10 mA cm−2 (pink columns). (c) Corresponding Tafel slopes obtained from the low-current density region. (d) Corresponding Cdl plot. (e) Nyquist plots of various catalysts. (f) Chronoamperometry plot of Au4Cu2/MoS2 (inset shows the HER polarization curve before and after catalysis). (g) Nyquist plot and (h) relative Cdl plot as a function of scan rate for Au4Cu2/MoS2 before and after catalysis. (i) Overpotential at a current density of 10 mA cm−2 of Au4Cu2/MoS2 in comparison with the overpotential of previously reported catalysts. | |
Table 1 Summary of HER overpotential, Tafel slope, double-layer capacitance, electrochemically active surface area, and charge-transfer resistance of MoS2, Cu6/MoS2, Au6/MoS2, Au4Cu2/MoS2, and commercial Pt/C
| Catalyst |
Overpotential at 10 mA cm−2 (mV vs. RHE) |
Tafel slope (mV dec−1) |
Double-layer capacitance Cdl (mF cm−2) |
Electrochemically active surface area ECSA (cm2) |
Charge-transfer resistance Rct (Ohm) |
| MoS2 |
252 |
116 |
2.4 |
60 |
210 |
| Cu6/MoS2 |
240 |
99 |
3.2 |
80 |
197 |
| Au6/MoS2 |
215 |
87 |
4.9 |
123 |
162 |
| Au4Cu2/MoS2 |
155 |
78 |
10.2 |
255 |
39 |
| Pt/C |
13 |
39 |
— |
— |
— |
Bare MoS2 shows a greater increase in η10 by 47 mV and a decrease in current density of nearly 2 times (Fig. S23a and b†). Moreover, the recorded EIS spectra before and after the durability test depict a greater increase in charge transfer resistance for bare MoS2 NSs in comparison to Au4Cu2/MoS2 (Fig. 4g). The Cdl of Au4Cu2/MoS2 is still three times greater than the original Cdl value of MoS2, indicating that the active sites are intact even after catalysis (Fig. 4h, Fig. S24†). We evaluated the stability of Au4Cu2/MoS2 by analyzing LSV for 2000 cycles at a scan rate of 50 mV s−1 to assess the change in overpotential at a constant current density. After the LSV cycling stability test, there is a decrease in overpotential by 9 mV at 10 mA cm−2 current density, which reveals the excellent stability of Au4Cu2/MoS2 (Fig. S25†). The findings imply that the loading of Au4Cu2 NCs enhances the stability of MoS2 by protecting the catalytically active sites during catalysis as a result of the robust electronic interactions between the two materials. There is no doubt that these results are better than previous findings so far (Fig. 4i and Table S2†). The nanosheet morphology of the catalyst (Au4Cu2/MoS2) remains unscratched after the stability test, as seen from its recorded TEM images (Fig. S26†).
Theoretical approach
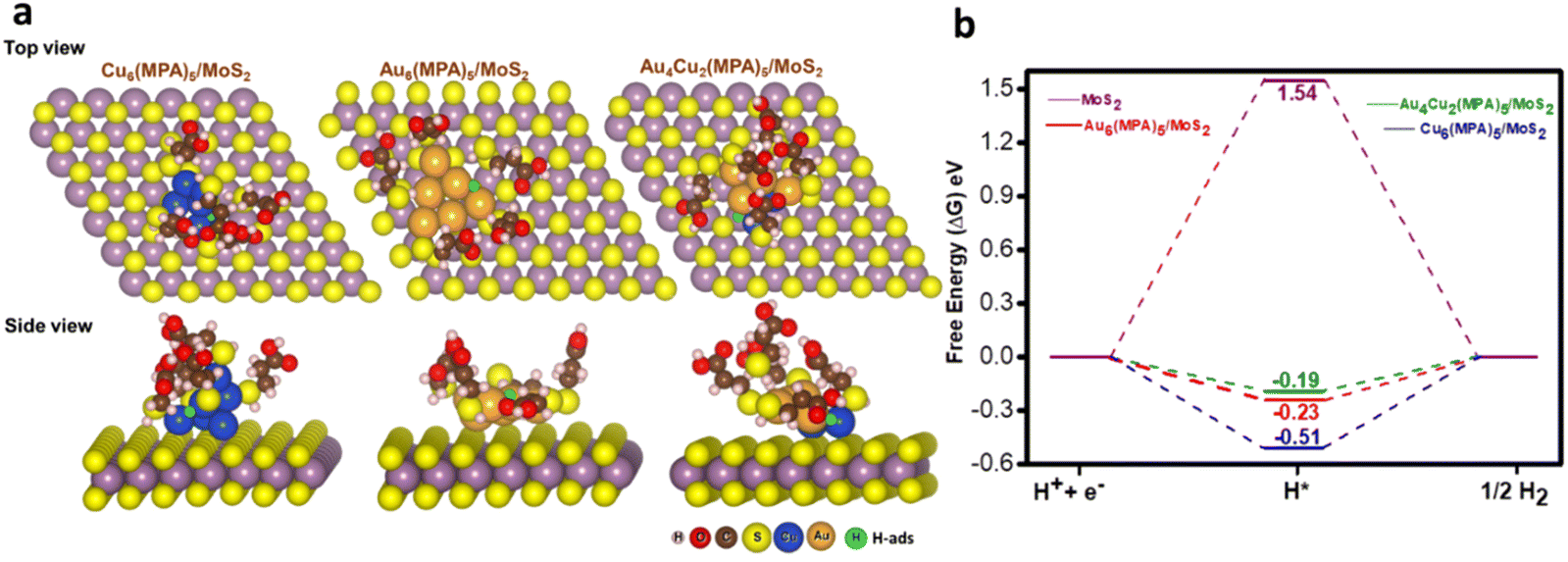
As we do not have the crystal structures of the nanoclusters, we modelled these clusters by considering a neutral ligand based on previous experimental and theoretical reports.56,70 We also followed a previous theoretical report where MPA is considered as a neutral ligand and the calculated results were very much in agreement with the experimental trend.56 On the other hand, the number of possible arrangements would be very large if we considered the possible oxidation states of the metal. Therefore, we modelled the clusters by considering the neutral ligand. DFT calculations were carried out to unveil the influence of pristine MoS2, Au6(MPA)5/MoS2, Cu6(MPA)5/MoS2, and Au4Cu2(MPA)5/MoS2 clusters on the HER. To mimic the experimentally synthesized cluster on an MoS2 support, initial optimization was performed for the Au6(MPA)5 and Cu6(MPA)5 clusters.55 In the Au6(MPA)5 cluster, two Au atoms were replaced with Cu atoms in all possible positions (A12, A13, A14, and A13′), as shown in Fig. S27.† Subsequently, Au6(MPA)5, Cu6(MPA)5, and Au4Cu2(MPA)5 clusters were deposited onto the MoS2 support, considering various possible orientations (Fig. S28–S30†). The final configuration with the most stable positions is shown in Fig. 5A.
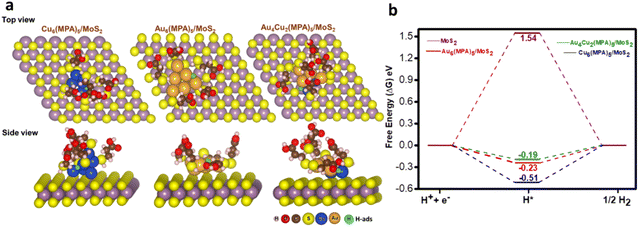 |
| | Fig. 5 (a) The top and side views of the most stable H-adsorption sites on the most stable orientation of clusters on the MoS2 support: Cu6(MPA)5/MoS2, Au6(MPA)5/MoS2, and Au4Cu2(MPA)5/MoS2. (b) Free energy diagrams of pristine MoS2, Cu6(MPA)5/MoS2, Au6(MPA)5/MoS2, and Au4Cu2(MPA)5/MoS2 for HER. | |
Here, all the possible sites in all the clusters deposited on the MoS2 surface for H adsorption are considered, as shown in Fig. 5a and Fig. S31–S34.† The Au–Au bridge, Cu–Cu bridge, Au–Cu bridge, on top of Au and top of S are found to be the most stable sites for H adsorption on Au6(MPA)5/MoS2, Cu6(MPA)5/MoS2, Au4Cu2(MPA)5/MoS2 and pristine MoS2 surface, respectively. It is the cooperative effect of Au–Cu that helped the better adsorption of H on the most active Au4Cu2(MPA)5/MoS2 catalyst. For the HER, an ideal catalyst would exhibit a nearly zero Gibbs free energy change (ΔG).71 Calculated ΔG values for all the considered catalysts are shown in Fig. 5b. Au4Cu2(MPA)5/MoS2 with a ΔG value of −0.19 eV is calculated to be the most efficient catalyst for HER, followed by Au6(MPA)5/MoS2 (−0.23 eV), Cu6(MPA)5/MoS2 (−0.51 eV), and MoS2 (1.54 eV), respectively. These findings are in excellent agreement with the experimental results. We further calculated the d-band center values for all the considered systems to understand their activity.72,73 The calculated d-band center values of Au4Cu2(MPA)5/MoS2, Au6(MPA)5/MoS2, Cu6(MPA)5/MoS2, and MoS2 with respect to the Fermi level (Ef) are −2.70, −1.81, −1.77, and −1.44 eV, respectively. These values illustrate that the Au4Cu2(MPA)5/MoS2 d-band center value is far from the Ef, followed by Au6(MPA)5/MoS2, Cu6(MPA)5/MoS2, and MoS2, respectively. The closer (farther) the d-band center value is to Ef, the stronger (weaker) the H bonding with the metal. Therefore, the d-band center values of the clusters follow the same trend in HER activity: Au4Cu2(MPA)5/MoS2 > Au6(MPA)5/MoS2 > Cu6(MPA)5/MoS2 > MoS2.
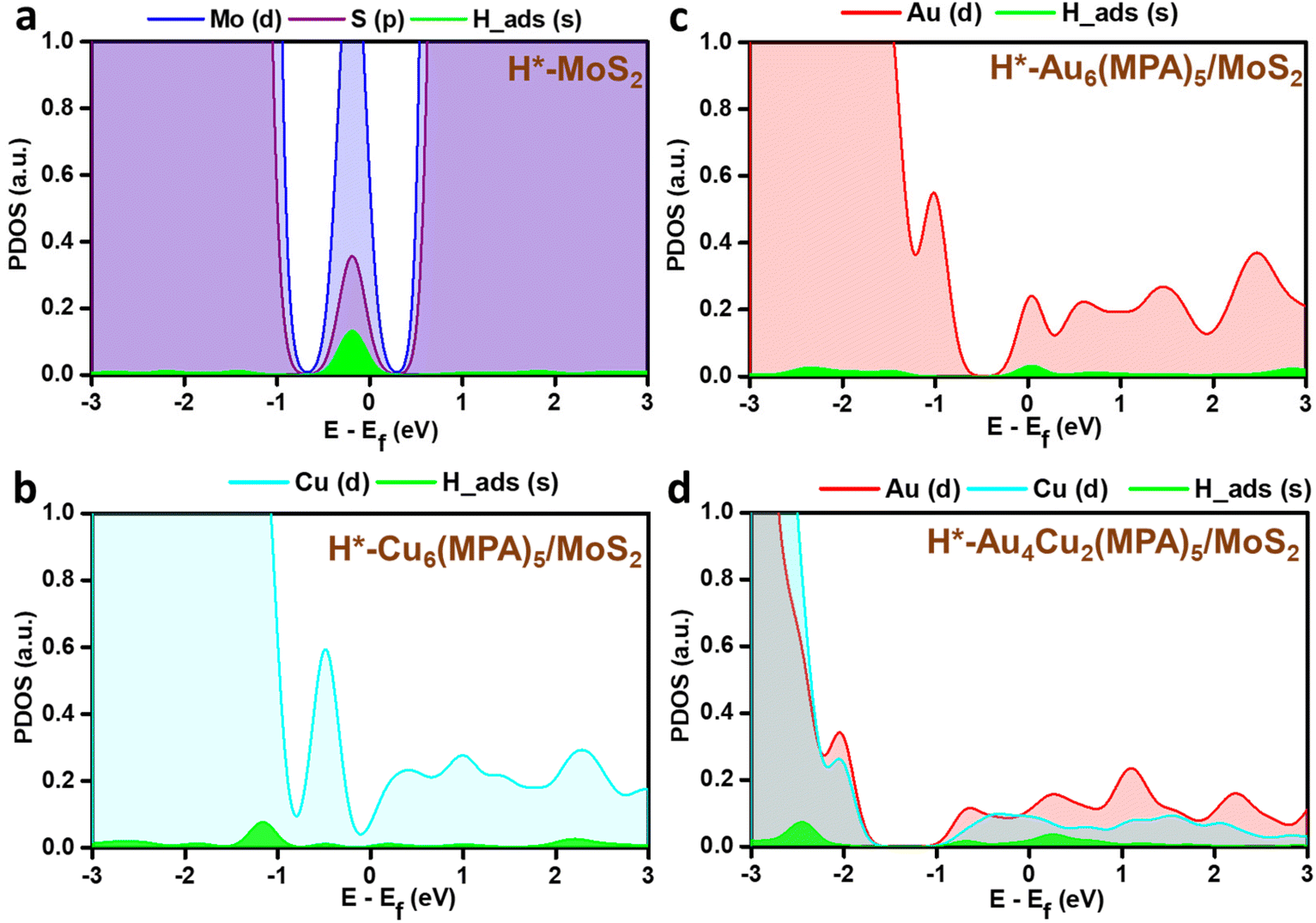
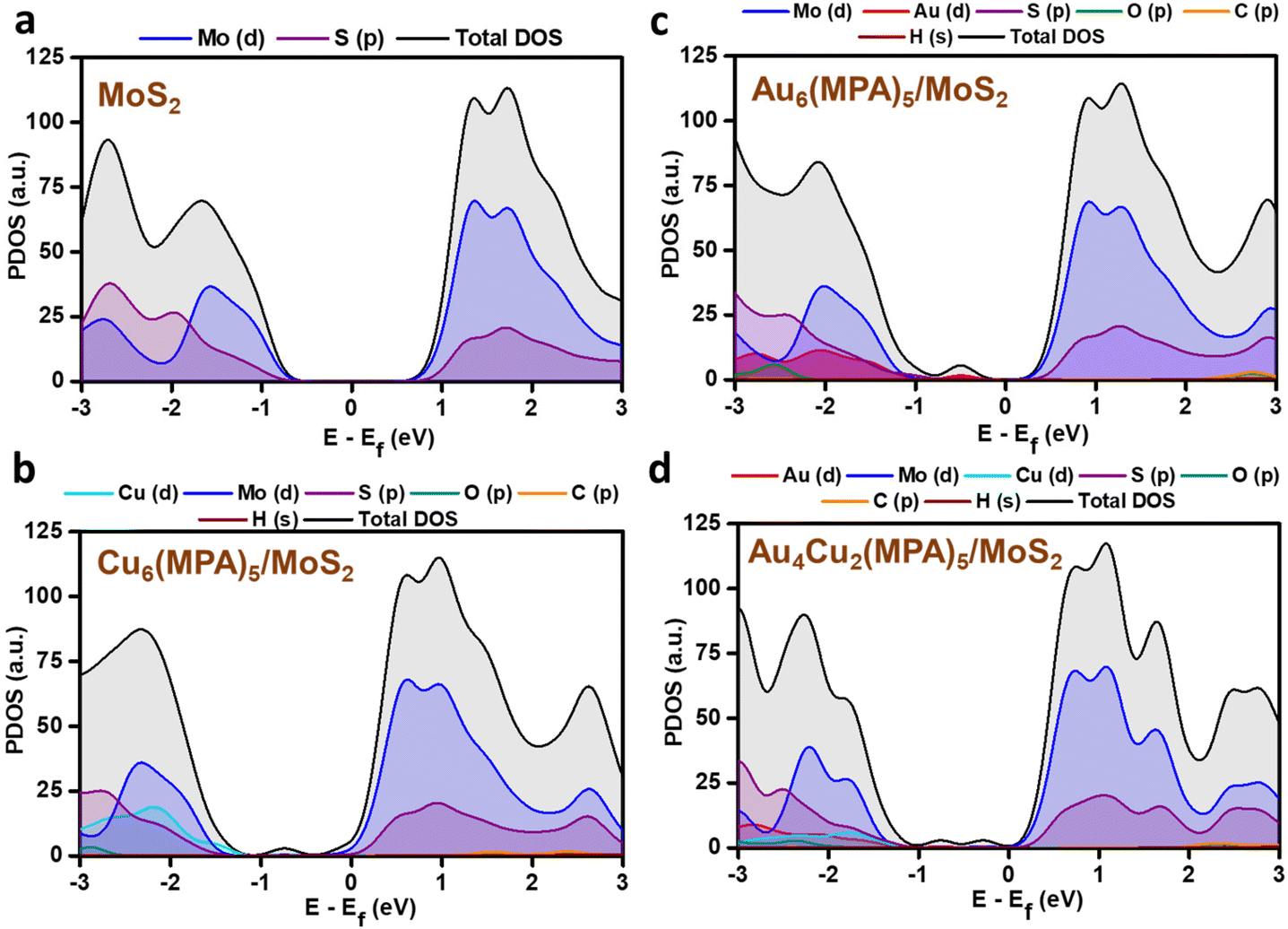
PDOS analysis of the H-adsorbed clusters (Fig. 6) reflects the interactions of H with different elements. The H states appear close to the Fermi level in the case of pure MoS2 surface and there is strong overlap with the Mo/S states, which could be due to strong interaction with the pure MoS2 surface. Similarly, the adsorbed H is found to be interacting with the Au/Cu, Au, and Cu states of Au4Cu2(MPA)5/MoS2, Au6(MPA)5/MoS2, and Cu6(MPA)5/MoS2 clusters, respectively. However, there is not much difference in the overlap between Au/Cu and H states in these clusters. Additionally, the Au6(MPA)5/MoS2 and MoS2 show metallic behavior after H adsorption. On the other hand, there is a shift in the valence band edge of Au6(MPA)5/MoS2 and Au4Cu2(MPA)5/MoS2 in the presence of an H atom (Fig. 6 and 7). This suggests that there is a strong interaction between the H atom and clusters and that the transition is maximum for MoS2, which is very much in agreement with the change in the Gibbs free energy.
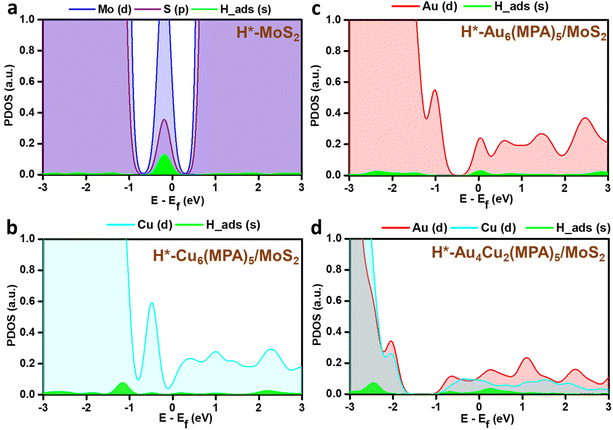 |
| | Fig. 6 Partial density of states (PDOS) of the 1s orbital of H in green and 4d orbitals of (a) Mo (blue) and S (purple), (b) Cu (cyan), (c) Au (red), and (d) Au and Cu after H adsorption. | |
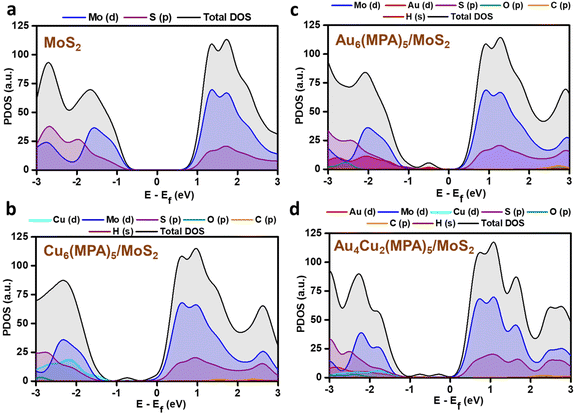 |
| | Fig. 7 Partial density of states (PDOS) of (a) pristine MoS2, (b) Cu6(MPA)5/MoS2, (c) Au6(MPA)5/MoS2, and (d) Au4Cu2(MPA)5/MoS2 before H adsorption. | |
Conclusions
In summary, Cu and Au are known to be poor HER catalysts, but here we found that combining them as bimetallic Au4Cu2 NCs over MoS2 made the combination more effective for electrocatalytic water splitting in comparison to monometallic Au6 NC and Cu6 NC composites with MoS2. The bimetallic Au4Cu2/MoS2 composite exhibits excellent current density at minimal overpotential among the other considered catalysts. Moreover, Au4Cu2/MoS2 shows a decrease in Tafel slope of 31 mV dec−1 and 4 times enhancement in Cdl in comparison to bare MoS2, indicating strong electronic interaction between the Au4Cu2 NCs and the MoS2 interface, as confirmed by XPS studies. Furthermore, due to the precisely structured Au4Cu2 NCs, theoretical calculations unveiled the better adsorption characteristics of H atoms in the Au4Cu2/MoS2 catalyst, leading to enhanced HER performance. These results further corroborated the observed downshift in the d-band of the Au4Cu2/MoS2 composite, directly impacting the binding energy of intermediate species. Hence, bimetallic nanoclusters are found to be promising for effective electrocatalytic water splitting in comparison to monometallic Au6 NC and Cu6 NC composites with MoS2.
Author contributions
AD contributed to the investigation, methodology, data curation, and writing of the original draft. HM and AD performed computational analysis. LS helped in the conceptualization and editing of the draft. Rashi helped with data curation and reviewing the draft. SG and SM helped in ESI-mass analysis. BP contributed to computational analysis. AP contributed to the supervision, validation, project administration, and final review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
SERB-DST is gratefully acknowledged for financial support. AD, Rashi, and L. S. acknowledge INST for doctoral fellowships and post-doctoral fellowships, respectively. We acknowledge IIT Indore for its laboratory and computational facilities. The work is supported by DST-SERB [Project No. CRG/2022/000836] and CSIR [Project No. 01(3046)/21/EMR-II]. H. M. thanks MHRD for PMRF and UGC for the research fellowship, respectively.
References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- P. Poizot and F. Dolhem, Energy Environ. Sci., 2011, 4, 2003–2019 RSC.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-e. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- T. T. Yang, T. L. Tan and W. A. Saidi, Chem. Mater, 2020, 32, 1315–1321 CrossRef CAS.
- T. Zeng, X. Meng, H. Huang, L. Zheng, H. Chen, Y. Zhang, W. Yuan and L. Y. Zhang, Small, 2022, 18, e2107623 CrossRef.
- S. L. Zhang, X. F. Lu, Z.-P. Wu, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 19068–19073 CrossRef CAS.
- W. Cheng, H. Zhang, D. Luan and X. W. Lou, Sci. Adv., 2021, 7, eabg2580 CrossRef CAS PubMed.
- L. Zeng, Z. Zhao, Q. Huang, C. Zhou, W. Chen, K. Wang, M. Li, F. Lin, H. Luo, Y. Gu, L. Li, S. Zhang, F. Lv, G. Lu, M. Luo and S. Guo, J. Am. Chem. Soc., 2023, 145, 21432–21441 CrossRef CAS PubMed.
- X.-K. Wan, H. B. Wu, B. Y. Guan, D. Luan and X. W. Lou, Adv. Mater., 2020, 32, 1901349 CrossRef CAS PubMed.
- J. W. Park, G. Park, M. Kim, M. Han, J. Jang, Y. Yamauchi, B. Yuliarto, P. Krüger, J. Kim, N. Park and H. Lim, Chem. Eng. J., 2023, 468, 143733 CrossRef CAS.
- H. Lim, K. Kani, J. Henzie, T. Nagaura, A. S. Nugraha, M. Iqbal, Y. S. Ok, M. S. A. Hossain, Y. Bando, K. C. W. Wu, H.-J. Kim, A. E. Rowan, J. Na and Y. Yamauchi, Nat. Protoc., 2020, 15, 2980–3008 CrossRef PubMed.
- H. Lim, J. Kim, K. Kani, M. K. Masud, H. Park, M. Kim, S. M. Alsheri, T. Ahamad, N. Alhokbany, J. Na, V. Malgras, Y. Bando and Y. Yamauchi, Small, 2020, 16, 1902934 CrossRef CAS PubMed.
- Q. Lu, G. S. Hutchings, W. Yu, Y. Zhou, R. V. Forest, R. Tao, J. Rosen, B. T. Yonemoto, Z. Cao, H. Zheng, J. Q. Xiao, F. Jiao and J. G. Chen, Nat. Commun., 2015, 6, 6567 CrossRef CAS PubMed.
- W. Li, X. Wang, D. Xiong and L. Liu, Int. J. Hydrogen Energy, 2016, 41, 9344–9354 CrossRef CAS.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- W. Jaegermann and H. Tributsch, Prog. Surf. Sci., 1988, 29, 1–167 CrossRef CAS.
- C. Tsai, K. Chan, J. K. Nørskov and F. Abild-Pedersen, Science, 2015, 640, 133–140 CAS.
- E. P. C. Higgins, A. A. Papaderakis, C. Byrne, R. Cai, A. Elgendy, S. J. Haigh, A. S. Walton, D. J. Lewis and R. A. W. Dryfe, J. Phys. Chem. C, 2021, 125, 20940–20951 CrossRef CAS.
- J. Wang, W. Fang, Y. Hu, Y. Zhang, J. Dang, Y. Wu, H. Zhao and Z. Li, Catal. Sci. Technol., 2020, 10, 154–163 RSC.
- Y. Li, S. Wang, Y. Hu, X. Zhou, M. Zhang, X. Jia, Y. Yang, B.-L. Lin and G. Chen, J. Mater. Chem. A, 2022, 10, 5273–5279 RSC.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- J. Choi, S. Seo, M. Kim, Y. Han, X. Shao and H. Lee, Small, 2023, 2304560 CrossRef PubMed.
- H. Yan, H. Xiang, J. Liu, R. Cheng, Y. Ye, Y. Han and C. Yao, Small, 2022, 18, 2200812 CrossRef CAS PubMed.
- S. Mondal, S. Dutta, S. Mal, S. K. Pati and S. Bhattacharyya, Angew. Chem., Int. Ed., 2023, 62, e202301269 CrossRef CAS PubMed.
- J. Ding, H. Yang, S. Zhang, Q. Liu, H. Cao, J. Luo and X. Liu, Small, 2022, 18, 2204524 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- Rashi, D. Bain, A. Devi, S. Chakraborty and A. Patra, ACS Sustainable Chem. Eng., 2023, 11, 1995–2004 CrossRef CAS.
- D. Bain, A. Devi, Rashi, S. Chakraborty, S. Kolay and A. Patra, J. Phys. Chem. C, 2023, 127, 18244–18251 CrossRef CAS.
- L. Sahoo, A. Devi and A. Patra, ACS Sustainable Chem. Eng., 2023, 11, 4187–4196 CrossRef CAS.
- M. Ghosal Chowdhury, L. Sahoo, S. Maity, D. Bain, U. K. Gautam and A. Patra, ACS Appl. Nano Mater., 2022, 5, 7132–7141 CrossRef CAS.
- Z. Cui, W. Jiao, Z. Huang, G. Chen, B. Zhang, Y. Han and W. Huang, Small, 2023, 19, 2301465 CrossRef CAS PubMed.
- T. Gao, X. Tang, X. Li, S. Wu, S. Yu, P. Li, D. Xiao and Z. Jin, ACS Catal., 2023, 13, 49–59 CrossRef CAS.
- T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 238 CrossRef CAS PubMed.
- Q. Zhu, X. Huang, Y. Zeng, K. Sun, L. Zhou, Y. Liu, L. Luo, S. Tian and X. Sun, Nanoscale Adv., 2021, 3, 6330–6341 RSC.
- X. Li, S. Takano and T. Tsukuda, J. Phys. Chem. C, 2021, 125, 23226–23230 CrossRef CAS.
- S. Zhao, R. Jin, H. Abroshan, C. Zeng, H. Zhang, S. D. House, E. Gottlieb, H. J. Kim, J. C. Yang and R. Jin, J. Am. Chem. Soc., 2017, 139, 1077–1080 CrossRef CAS PubMed.
- H. Mousavi, Y. Yin, S. K. Sharma, C. T. Gibson, V. Golovko, G. G. Andersson, C. J. Shearer and G. F. Metha, J. Phys. Chem. C, 2022, 126, 246–260 CrossRef CAS.
- S. Zhao, R. Jin, Y. Song, H. Zhang, S. D. House, J. C. Yang and R. Jin, Small, 2017, 13, 1701519 CrossRef PubMed.
- S. Gratious, A. Karmakar, D. Kumar, S. Kundu, S. Chakraborty and S. Mandal, Nanoscale, 2022, 14, 7919–7926 RSC.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- Y. Du, J. Xiang, K. Ni, Y. Yun, G. Sun, X. Yuan, H. Sheng, Y. Zhu and M. Zhu, Inorg. Chem. Front., 2018, 5, 2948–2954 RSC.
- W. Choi, G. Hu, K. Kwak, M. Kim, D.-e. Jiang, J.-P. Choi and D. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 44645–44653 CrossRef CAS PubMed.
- Y. Tang, F. Sun, X. Ma, L. Qin, G. Ma, Q. Tang and Z. Tang, Dalton Trans., 2022, 51, 7845–7850 RSC.
- J. Zhang, Y. Liu, C. Sun, P. Xi, S. Peng, D. Gao and D. Xue, ACS Energy Lett., 2018, 3, 779–786 CrossRef CAS.
- W. Li, D. Xiong, X. Gao, W.-G. Song, F. Xia and L. Liu, Catal. Today, 2017, 287, 122–129 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- A. Devi, H. Seksaria, D. Bain, S. Kolay, Rashi, A. De Sarkar and A. Patra, Phys. Chem. Chem. Phys., 2023, 25, 9513–9521 RSC.
- C. Comby-Zerbino, X. Dagany, F. Chirot, P. Dugourd and R. Antoine, Mater. Adv., 2021, 2, 4896–4913 RSC.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- S. Maity, D. Bain and A. Patra, J. Phys. Chem. C, 2019, 123, 2506–2515 CrossRef CAS.
- R. Gusain, N. Kumar, F. Opoku, P. P. Govender and S. S. Ray, ACS Appl. Nano Mater., 2021, 4, 4721–4734 CrossRef CAS.
- D. M. Chevrier, M. A. MacDonald, A. Chatt, P. Zhang, Z. Wu and R. Jin, J. Phys. Chem. C, 2012, 116, 26947–26947 CrossRef CAS.
- D. Bain, S. Maity, B. Paramanik and A. Patra, ACS Sustainable Chem. Eng., 2018, 6, 2334–2343 CrossRef CAS.
- X. Gao, Y. Lu, M. Liu, S. He and W. Chen, J. Mater. Chem. C, 2015, 3, 4050–4056 RSC.
- L. Liang and V. Meunier, Nanoscale, 2014, 6, 5394–5401 RSC.
- W. Zhou, M. Chen, M. Guo, A. Hong, T. Yu, X. Luo, C. Yuan, W. Lei and S. Wang, Nano Lett., 2020, 20, 2923–2930 CrossRef CAS PubMed.
- V. K. Singh, U. Gupta, B. Mukherjee, S. Chattopadhyay and S. Das, ACS Appl. Nano Mater., 2021, 4, 886–896 CrossRef CAS.
- A. Mondal, H. R. Inta, A. Roy, A. Kumar Mahato and V. Mahalingam, ACS Appl. Nano Mater., 2023, 6, 12040–12049 CrossRef CAS.
- J. Jin, J. Yin, H. Liu, B. Huang, Y. Hu, H. Zhang, M. Sun, Y. Peng, P. Xi and C.-H. Yan, Angew. Chem., Int. Ed., 2021, 60, 14117–14123 CrossRef CAS PubMed.
- L. Sahoo, S. Mondal, C. B. Nayana and U. K. Gautam, J. Colloid Interface Sci., 2021, 590, 175–185 CrossRef CAS PubMed.
- A. Posada-Amarillas, R. Pacheco-Contreras, S. Morales-Meza, M. Sánchez and J. C. Schön, Int. J. Quantum Chem., 2016, 116, 1006–1015 CrossRef CAS.
- N. K. Pandit, D. Roy, S. C. Mandal and B. Pathak, J. Phys. Chem. Lett., 2022, 13, 7583–7593 CrossRef CAS PubMed.
- H. Xin, A. Vojvodic, J. Voss, J. K. Nørskov and F. Abild-Pedersen, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 115114 CrossRef.
- Z. Chen, Y. Song, J. Cai, X. Zheng, D. Han, Y. Wu, Y. Zang, S. Niu, Y. Liu, J. Zhu, X. Liu and G. Wang, Angew. Chem., Int. Ed., 2018, 57, 5076–5080 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Lipipuspa
Sahoo
b,
Lipipuspa
Sahoo