
Simultaneous derivatization and exfoliation of a multilayered Ti3C2Tx MXene into amorphous TiO2 nanosheets for stable K-ion storage†
Yuan
Zhang‡
 ,
Zhuoheng
Bao‡
,
Rui
Wang
,
Yifan
Su
,
Yaping
Wang
*,
Xin
Cao
,
Rongxiang
Hu
,
Dawei
Sha
,
Long
Pan
* and
ZhengMing
Sun
*
,
Zhuoheng
Bao‡
,
Rui
Wang
,
Yifan
Su
,
Yaping
Wang
*,
Xin
Cao
,
Rongxiang
Hu
,
Dawei
Sha
,
Long
Pan
* and
ZhengMing
Sun
*
Key Laboratory of Advanced Metallic Materials of Jiangsu Province, School of Materials Science and Engineering, Southeast University, Nanjing 211189, P. R. China. E-mail: ypwang2011@seu.edu.cn; panlong@seu.edu.cn; zmsun@seu.edu.cn
First published on 10th January 2024
Abstract
Two-dimensional transition metal compounds (2D TMCs) have been widely reported in the fields of energy storage and conversion, especially in metal-ion storage. However, most of them are crystalline and lack active sites, and this brings about sluggish ion storage kinetics. In addition, TMCs are generally nonconductors or semiconductors, impeding fast electron transfer at high rates. Herein, we propose a facile one-step route to synthesize amorphous 2D TiO2 with a carbon coating (a-2D-TiO2@C) by simultaneous derivatization and exfoliation of a multilayered Ti3C2Tx MXene. The amorphous structure endows 2D TiO2 with abundant active sites for fast ion adsorption and diffusion, while the carbon coating can facilitate electron transport in an electrode. Owing to these intriguing structural and compositional synergies, a-2D-TiO2@C delivers good cycling stability with a long-term capacity retention of 86% after 2000 cycles at 1.0 A g−1 in K-ion storage. When paired with Prussian blue (KPB) cathodes, it exhibits a high full-cell capacity of 50.8 mA h g−1 at 100 mA g−1 after 140 cycles, which demonstrates its great potential in practical applications. This contribution exploits a new approach for the facile synthesis of a-2D-TMCs and their broad applications in energy storage and conversion.
 Long Pan | Long Pan earned his Ph.D. from Tsinghua University under the supervision of Prof. Xu-Ming Xie. He worked as a postdoc with Prof. Markus Niederberger in the Laboratory for Multifunctional Materials at ETH Zurich, Switzerland. He is now an associate professor at Southeast University. His current research interests are (1) the synthesis and assembly of 2D nanosheets, (2) energy storage materials and devices, and (3) solid-state electrolytes. |
Introduction
Owing to the unique low-dimensional effect, two-dimensional (2D) transition metal compounds (TMCs) possess high potential for applications in energy storage and conversion, including metal-ion batteries, supercapacitors, and catalysis.1 To be more specific, 2D morphologies endow TMCs with a large specific surface area,2,3 resulting in excellent ion transport, especially when used as electrochemical energy storage materials, for example, in the field of potassium (K)-ion storage.4–8 However, the ion storage performance of 2D TMCs is hindered by their sluggish ion storage kinetics because of insufficient active sites.9–11 In addition, most TMCs are insulators or semiconductors, leading to low electron conductivity and poor rate capability.12,13Recently, amorphous materials with a high degree of long-range disorder have attracted much attention due to their potential applications and prospects in the fields of energy storage and catalysis.8,14–17 Amorphous structures can modulate local atomic environments and electronic states, thereby not only offering ample active sites for ion storage but also facilitating ion transfer.18 With regard to this, the amorphization of 2D TMCs holds great potential for improving the ion storage kinetics, despite few attempts having been made.19 To address the electronic conductivity problem, a carbon coating has frequently been applied owing to its high conductivity and structural stability, which can not only accelerate the electron transfer in TMC electrodes but also accommodate the volume expansion during ion insertion and extraction.3,20 Therefore, the integration of amorphization with a carbon coating represents the most ideal solution to boosting the ion storage performance of 2D TMCs. However, this synergistic strategy has not been comprehensively employed in electrochemical energy storage, especially in K-ion batteries. Moreover, developing a facile one-step preparation methodology for carbon-coated amorphous 2D TMCs (labeled as a-2D-TMCs@C) remains a great technical challenge.
MXenes are a new family of novel 2D materials, with a general formula of Mn+1XnTx (where ‘M’ stands for a transition metal, ‘X’ stands for C or N, and ‘T’ stands for –OH, –O, –F…, n = 1, 2, 3, 4).21 MXenes possess many unique physical and chemical characteristics, and these characteristics render them promising for the energetic synthesis of a-2D-TMCs@C nanosheets. First, the ‘M’ atoms in MXenes can serve as the transition metal sources for the formation of TMCs, while the ‘X’ atoms can act as the carbon sources for the formation of carbon coatings. Second, the 2D morphology of MXenes can be preserved during its conversion, leading to the formation of 2D TMCs. For instance, our previous work has realized the partial transformation of Ti3C2Tx, Nb2CTx, and V2CTx MXenes into crystalline TiSe2, NbSe2, and VSe2. We also realized the in situ derivatization of pure Ti3C2Tx into TinO2n−1 quantum dots supported on carbon nanosheets.4,22,23 Although these progressive studies indicate the suitability of MXene precursors for the preparation of 2D TMCs and carbon coatings, it remains a great challenge to realize a one-step strategy for the conversion of MXenes into a-2D-TMCs@C.
In this contribution, we propose a facile approach for the simultaneous derivatization and exfoliation of a multilayered Ti3C2Tx MXene using a dilute (NH4)2S2O8 (denoted as APS) aqueous solution as a mild oxidant, resulting in the effective one-step preparation of carbon coated amorphous 2D TiO2 (labeled as a-2D-TiO2@C) for fast and stable K-ion storage. TiO2 is employed as a typical TMC because of its excellent structural stability during ion storage but it suffers from poor electronic conductivity compared to graphite.24–26 Multilayered Ti3C2Tx is used as the precursor based on two reasons: (i) Ti3C2Tx is the most symbolic MXene, and can act as a Ti and C source for a-2D-TiO2@C; and (ii) our strategy can simultaneously perform exfoliation of the multilayered Ti3C2Tx MXene and the derivatization process, thereby intentionally overcoming the conventional time-consuming delamination of the multilayered Ti3C2Tx MXene. Benefiting from the mild oxidation ability of the dilute (NH4)2S2O8 solution, the multilayered Ti3C2Tx MXene undergoes simultaneous derivatization and exfoliation, obtaining a-2D-TiO2@C with a large specific surface area and abundant active sites. Therefore, a-2D-TiO2@C delivers good rate capability (62.5 mA h g−1 at 3.0 A g−1) and excellent long-term capacity retention (86% at 1.0 A g−1 after 2000 cycles). This approach opens up a new avenue for the facile synthesis and broad applications of a-2D-TMCs@C.
Results and discussion
The preparation process is illustrated in Fig. 1. Multilayered Ti3C2Tx was obtained through the molten-salt method, in which CuBr2 was applied to etch the Al atomic layers of Ti3AlC2 under an argon atmosphere with a CuBr2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ti3AlC2 molar ratio of 6:1. During the etching, CuBr2 was reduced by Al, while metallic Cu was formed and remained as an impurity. Subsequently, the resultant multilayered Ti3C2Tx was dispersed in a 0.5M APS aqueous solution, which was then reacted and stirred for 8 h. In this crucial step, Ti3C2Tx underwent mild derivatization to give a-2D-TiO2@C. At the same time, the residual Cu impurities were slowly oxidized to soluble Cu2+, during which massive bubbles were generated (Fig. S1a†). The oxidation of Cu and the formation of bubbles in the interlayers provided the driving forces for interlayer expansion and exfoliation (see details discussed below).27,28
:Ti3AlC2 molar ratio of 6:1. During the etching, CuBr2 was reduced by Al, while metallic Cu was formed and remained as an impurity. Subsequently, the resultant multilayered Ti3C2Tx was dispersed in a 0.5M APS aqueous solution, which was then reacted and stirred for 8 h. In this crucial step, Ti3C2Tx underwent mild derivatization to give a-2D-TiO2@C. At the same time, the residual Cu impurities were slowly oxidized to soluble Cu2+, during which massive bubbles were generated (Fig. S1a†). The oxidation of Cu and the formation of bubbles in the interlayers provided the driving forces for interlayer expansion and exfoliation (see details discussed below).27,28
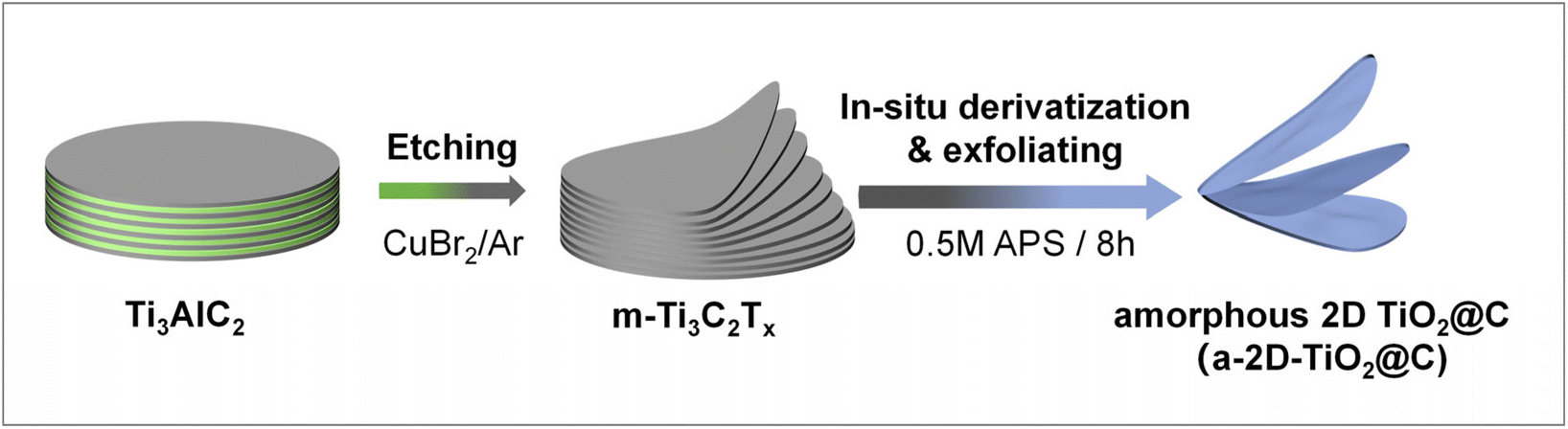 | ||
| Fig. 1 Schematic of simultaneous derivatization and exfoliation of multilayered Ti3C2Tx for the preparation of a-2D-TiO2@C. | ||
The derivatization mechanism of Ti3C2Tx into a-2D-TiO2@C can be explained using the redox potential criteria of reactants.29,30 Thermodynamically, the redox potential of S2O82−/SO42− (2.00 V vs. NHE) is much higher than those of Ti3+/Ti2+ (0.26 V vs. NHE), Ti4+/Ti3+ (0.052 V vs. NHE), and Cu2+/Cu (0.34 V vs. NHE). Note that the Ti valency in Ti3C2Tx is +2 and +3; thereby, they can be easily oxidized to Ti4+ by APS. In other words, the derivatization of Ti3C2Tx into TiO2@C is thermodynamically favorable, with a reaction equation 6S2O82− + Ti3C2 + 6H2O → 3TiO2 + 2C + 12SO42− + 12H+. The generation of H+ confirmed that the reaction solution altered from faint acid (pH = 3) to strong acid (pH = 1), as shown in Fig. S1b.† Similarly, the residual metallic Cu impurities can be oxidized to soluble Cu2+, with a reaction equation S2O82− + Cu → 2SO42− + Cu2+. In addition, the bubbles (Fig. S1a†) during the reaction can be ascribed to the decomposition of APS, with a reaction equation 2S2O82− + 2H2O → O2 + 4SO42− + 4H+.
To reveal the exfoliation process during the derivatization reactions, we conducted various ex situ investigations by analyzing samples at different reaction times, i.e., multilayered Ti3C2Tx (original state), intermediate product (react for 4 h), and a-2D-TiO2@C (final state). Fig. 2a–c show the SEM images of samples at different reaction times. In multilayered Ti3C2Tx, the layers are closely stacked (Fig. 2a), causing a low Brunauer–Emmett–Teller (BET) specific surface area of only 1.02 m2 g−1 (Fig. 2d). Note that metallic Cu impurities also contribute to the low specific area. The mass ratio of Cu was tested by performing inductively coupled plasma optical emission spectroscopy (ICP-OES) tests and Cu impurities contribute 57.2 wt% content in the original state product as shown in Fig. 2g. When it was stirred for 4 h, multilayered Ti3C2Tx underwent derivatization to give titanium oxide and slight exfoliation in the interlayer spacing was observed in the corresponding SEM image (Fig. S2† and Fig. 3b). The slightly exfoliated morphology of the intermediate product is also verified by the increased BET specific surface area of 43.83 m2 g−1 in Fig. 2e. Note that this change in morphology31,32 may be attributed to the oxidation of Cu in interlayers and the produced bubbles. In Fig. 2g, the mass ratio of Cu impurities decreased from 57.2 wt% (multilayered Ti3C2Tx) to only 0.5 wt% (intermediate product), indicating the removal and oxidation of Cu impurities. In a-2D-TiO2@C, the multilayered structure completely changed, and porous nanosheets were formed. The corresponding specific surface area increased to 137.41 m2 g−1 (Fig. 2c and f). The increase in the specific surface area was mainly caused by bubbles33–35 and the mass ratio of Cu impurities decreased from 0.5 wt% to 0.35 wt%.
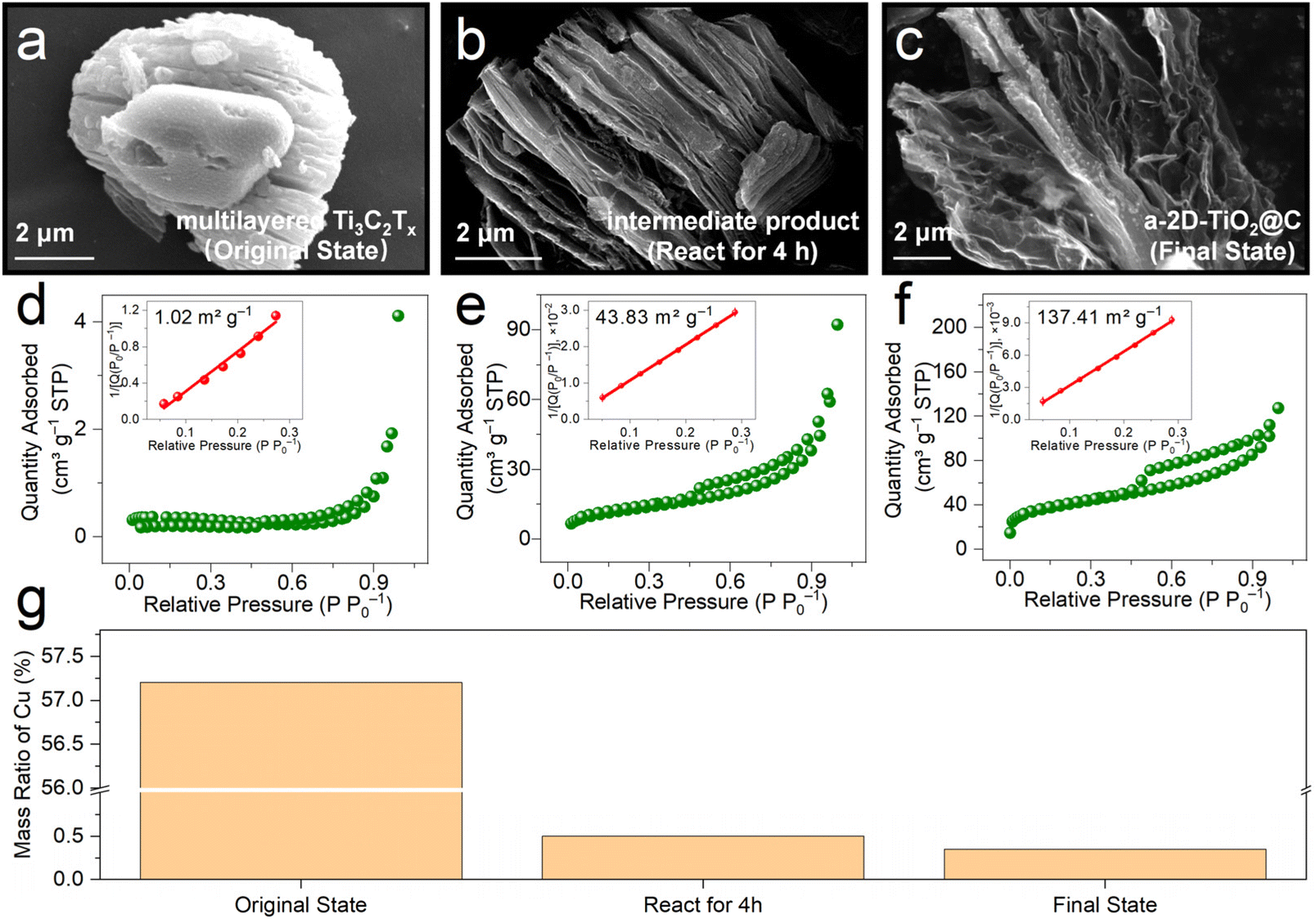 | ||
| Fig. 2 Ex situ investigations for analyzing samples at different reaction times. SEM images and nitrogen adsorption–desorption isotherms of (a and d) multilayered Ti3C2Tx (original state), (b and e) intermediate product (react for 4 h), and (c and f) a-2D-TiO2@C (final state). (g) Mass ratio of Cu in samples at different reaction times of the original state, sample after 4 h, and final state. | ||
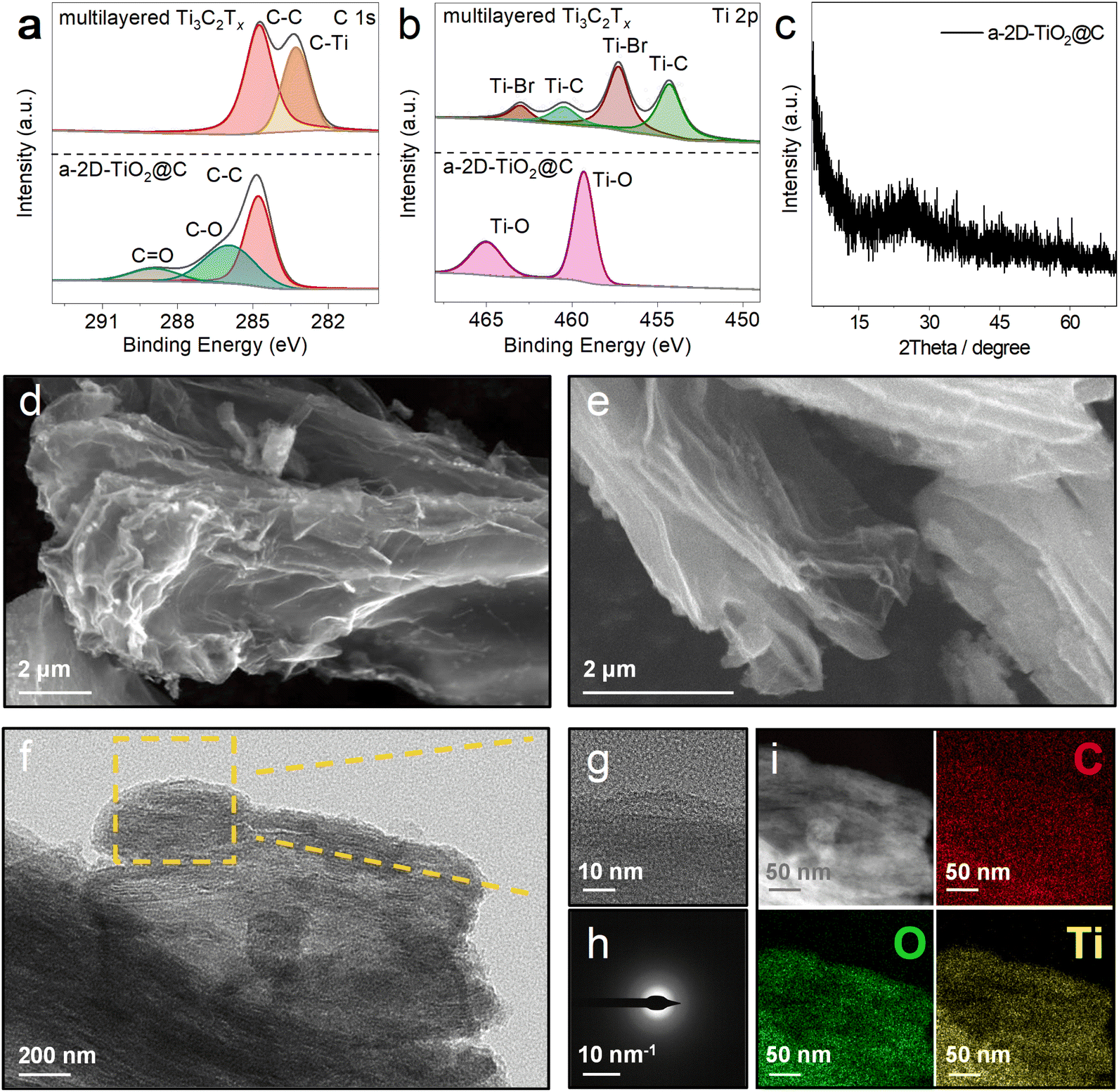 | ||
| Fig. 3 Phase and morphological characterization. (a and b) High-resolution XPS spectra of C 1s (a) and Ti 2p (b) for Ti3C2Tx and a-2D-TiO2@C. (c) XRD pattern of a-2D-TiO2@C. (d and e) SEM images, (f) TEM image, (g) HRTEM image, (h) SAED pattern, and (i) scanning TEM image and the corresponding element mappings of a-2D-TiO2@C. | ||
In order to verify the successful derivatization of multilayered Ti3C2Tx, X-ray photoelectron spectroscopy (XPS) was carried out to investigate the C 1s and Ti 2p chemical states of multilayered Ti3C2Tx and a-2D-TiO2@C, as shown in Fig. 3a and b. In the C 1s spectra (Fig. 3a), the characteristic C–Ti peak of multilayered Ti3C2Tx can be observed at 283.29 eV, which disappeared after the reaction. In addition, two new peaks at 285.93 and 288.93 eV appeared for a-2D-TiO2@C, which can be assigned to the C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively. Given that no other carbon-containing agents were added during the reaction, the C 1s XPS results indicate that the carbon element in multilayered Ti3C2Tx was successfully converted into carbon coatings with defects. Fig. S3† shows the Raman spectrum of a-2D-TiO2@C, in which the D and G bands at 1338 and 1575 cm−1 of carbon are noticed with a large ID/IG ratio of 0.98. The Raman results suggest that there is abundant sp3-hybridized carbon in the carbon coating, which is in line with the C 1s XPS results.36
O bonds, respectively. Given that no other carbon-containing agents were added during the reaction, the C 1s XPS results indicate that the carbon element in multilayered Ti3C2Tx was successfully converted into carbon coatings with defects. Fig. S3† shows the Raman spectrum of a-2D-TiO2@C, in which the D and G bands at 1338 and 1575 cm−1 of carbon are noticed with a large ID/IG ratio of 0.98. The Raman results suggest that there is abundant sp3-hybridized carbon in the carbon coating, which is in line with the C 1s XPS results.36
In the Ti 2p spectra (Fig. 3b), the characteristic peaks at 463.01, 457.27, 460.47, and 454.29 eV correspond to Ti–Br 2p1/2, Ti–Br 2p3/2, Ti–C 2p1/2, and Ti–C 2p3/2 respectively of multilayered Ti3C2Tx.37 These peaks all disappeared after the reaction. At the same time, peaks of Ti–O 2p1/2 at 465.01 eV and Ti–O 2p3/2 at 459.31 eV appeared for a-2D-TiO2@C, which can be assigned to the Ti–O bonds of TiO2.38 The formation of TiO2 was further confirmed by the Raman results (Fig. S3†), in which a peak at 154 cm−1 attributed to the Ti–O A1g vibration of TiO2 was detected. Fig. S4† presents the X-ray diffraction (XRD) pattern of multilayered Ti3C2Tx. The acute diffraction peaks at 43.3° and 50.4° are attributed to Cu (JCPDS 04-8036). After the reaction, the XRD pattern of a-2D-TiO2@C was recorded and it is displayed in Fig. 3c. No evident peaks were observed, except that a small broad peak existed at 20°–30°. This evidence implies that the derived TiO2 and carbon coatings were highly amorphous. To determine the weight contents in a-2D-TiO2@C, thermogravimetric analysis (TGA) was conducted under an air atmosphere, and the corresponding curve is presented in Fig. S5.† We could calculate that the carbon content of a-2D-TiO2@C is about 69.67 wt%.
To demonstrate the successful exfoliation upon the reaction, scanning electron microscopy (SEM) was employed. Fig. 3d and e show the SEM images of a-2D-TiO2@C and thin and porous nanosheets are observed, which are significantly different from the original multilayered Ti3C2Tx (as discussed above in Fig. 2a).39 The nanosheet morphology was further validated by the high-resolution transmission electron microscopy (HRTEM) result shown in Fig. 3f and g, in which layered structures are recognized. In addition, no lattice fringes are shown, once again verifying the amorphous structures of the obtained TiO2 and carbon coating. This finding is also confirmed by the diffuse diffraction rings in the selected area electron diffraction (SAED) image shown in Fig. 3h.40 As depicted in Fig. 3i, the distributions of O, C, and Ti elements are well overlapped, implying a uniform carbon coating on the TiO2 nanosheets.
The K-ion storage behavior and performance of a-2D-TiO2@C were investigated by assembling half cells using K metal as the counter electrode and potassium bis(fluorosulfonyl) imide (KFSI) as the electrolyte.41Fig. 4a shows the first three cyclic voltammetry (CV) curves at 0.1 mV s−1 of a-2D-TiO2@C. It can be observed that the first cathodic scan showed a wide peak at around 0.75 V, indicating the formation of the solid electrolyte interphase (SEI).42 The subsequent two cycles of the CV profiles overlap quite well, indicating the good K-ion storage reversibility of a-2D-TiO2@C. This can be further proved by the rate performance at different current densities shown in Fig. 4b. a-2D-TiO2@C delivered specific capacities of 183.4, 161.9, 149.5, 134.3, and 108.1 mA h g−1 at current densities of 0.1, 0.2, 0.3, 0.5 and 1.0 A g−1, respectively. When the current density was further increased to 2.0 A g−1 and 3.0 A g−1, the specific capacities remained at 79.5 and 62.5 mA h g−1. After the current density was returned to 0.5, 0.3, 0.2, and 0.1 A g−1, the specific capacity recovered to 131.8, 151.3, 164.9, and 184.8 mA h g−1, respectively, indicating the good K-ion storage reversibility of a-2D-TiO2@C.43–45
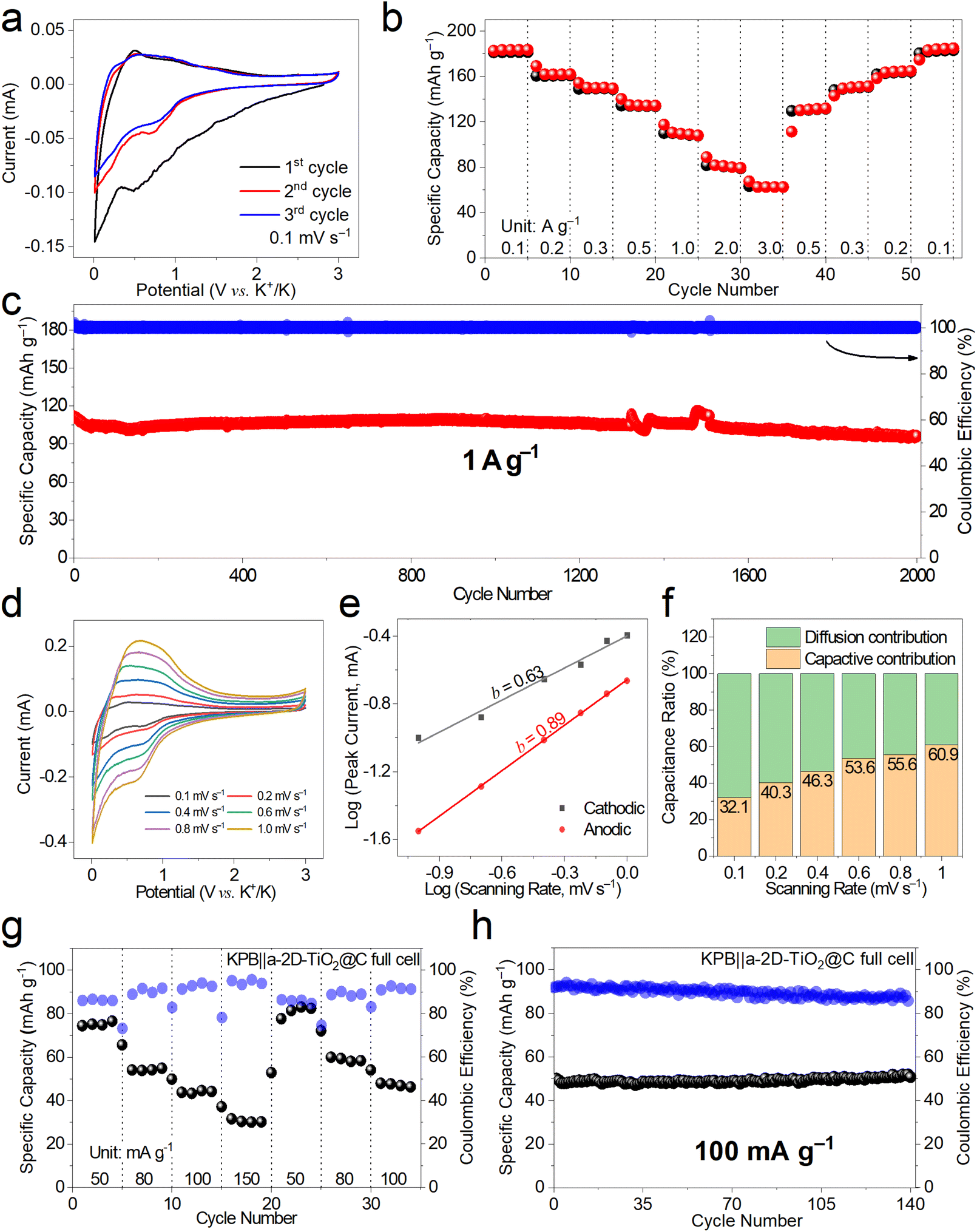 | ||
| Fig. 4 K+ storage behavior and performance. (a) The first three CV curves at 0.1 mV s−1, (b) rate performance at 0.1–3.0 A g−1, (c) long-term cyclability at 1 A g−1, (d) CV curves at 0.1–1.0 mV s−1, (e) b values of cathodic and anodic peaks, and (f) pseudocapacitance proportion of a-2D-TiO2@C. (g) Rate performance (50–100 mA g−1) and (h) cycling stability of KPB||a-2D-TiO2@C full cells. | ||
The cycling performance of a-2D-TiO2@C was assessed at a fixed current density of 1 A g−1, as shown in Fig. 4c and S6.† a-2D-TiO2@C delivered an initial specific capacity of 111.6 mA h g−1 and maintained a high specific capacity retention of 86% (96.1 mA h g−1) after long-term cycling for 2000 cycles. Note that a slight fluctuation at about the 1500th cycle was caused by an undesired power failure. The comparatively high retention of specific capacity indicates the tremendous potential of the a-2D-TiO2@C electrode compared to other TiO2-based anode materials (Table S1†).
CV measurements at different scanning rates of 0.1, 0.2, 0.4, 0.6, 0.8, and 1.0 mV s−1 were also performed as shown in Fig. 4d to figure out the b value derived from the slope of Log scanning rate versus Log peak current and the portion of diffusion contribution and capacitive contribution. The Log(I) − Log(v) curve is fitted to figure out the value of b. Cathodic b is calculated to be 0.63 while anodic b is calculated to be 0.89 (Fig. 4e). As the scanning rate increased, the contribution of pseudocapacitance increased (Fig. 4f), indicating that pseudocapacitance behavior is indispensable in the high-rate storage of K+.46
To further demonstrate the potential of a-2D-TiO2@C for practical applications, full cells were assembled using Prussian blue as the cathode (Fig. S8†). The working voltage was set at the range of 1.1–3.6 V according to Fig. S7.† In the first rate gradient at current densities of 50.0, 80.0, 100.0, and 150 mA g−1, the KPB||a-2D-TiO2@C full cells delivered specific capacities of 76.6, 54.9, 44.2, and 30.2 mA h g−1, respectively in Fig. 4g. When the current density was brought back to 50.0, 80.0, and 100.0 mA g−1, the KPB||a-2D-TiO2@C full cells delivered specific capacities of 82.5, 58.4, and 46.3 mA h g−1, which indicate a high retention of specific capacity. However, the rate performance of full cells is not as good as that of the cathode or anode in half cells, which may be due to electrodes. Note that the electrochemical performance of full cells not only depends on the electrode materials but also relies on the N/P ratios, electrolyte optimization, cell assembly, etc. In our future works, more effort will be devoted to optimizing the full cell performance.
Cycling performance (Fig. 4h) was assessed at 100 mA g−1. The KPB||a-2D-TiO2@C full cells delivered a specific capacity of 50.8 mA h g−1 after 140 cycles. The Ragone plot of KPB|| a-2D-TiO2@C full cells is depicted in Fig. S10† according to Fig. S9.† The full cell delivered power densities of 95.9, 148.5, 197.2, and 282.1 W kg−1 and the energy densities were 149.5, 107.6, 86.8, and 59.3 W h kg−1 at 50.0, 80.0, 100.0, and 150.0 mA g−1, respectively.
Conclusions
In this work, we exploit a new route to synthesize a-2D-TiO2@C using multilayered Ti3C2Tx as the precursor and a dilute (NH4)2S2O8 solution as a mild oxidant. The multilayered Ti3C2Tx MXene undergoes simultaneous derivatization and exfoliation, to give a-2D-TiO2@C with a large specific surface area and abundant active sites. Therefore, a-2D-TiO2@C shows a good long-term capacity retention of 86% after 2000 cycles at 1.0 A g−1 in K-ion storage. KPB||a-TiO2@C indicates its potential in practical applications (50.8 mA h g−1 at 100 mA g−1 after 140 cycles). Our work can be extended to a broader exploration of a-2D-TMCs@C, especially in MXene derivatization.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 52201242 and 52250010), the Young Elite Scientists Sponsorship Program by the CAST (No. 2021QNRC001), the Fundamental Research Funds for the Central Universities (No. 2242022R40018), and the Fund of Key Laboratory of Advanced Materials of Ministry of Education (AdvMat-2023-12) for financial support.References
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2(2), 17 CrossRef.
- J. B. Pang, R. G. Mendes, A. Bachmatiuk, L. Zhao, H. Q. Ta, T. Gemming, H. Liu, Z. F. Liu and M. H. Rummeli, Chem. Soc. Rev., 2019, 48(1), 72–133 RSC.
- Y. Q. Sun, Q. O. Wu and G. Q. Shi, Energy Environ. Sci., 2011, 4(4), 1113–1132 RSC.
- D. W. Sha, Y. R. You, R. X. Hu, X. Cao, Y. C. Wei, H. Zhang, L. Pan and Z. M. Sun, Adv. Mater., 2023, 35(15), 10 Search PubMed.
- J. F. Zheng, Y. J. Wu, Y. J. Sun, J. H. Rong, H. Y. Li and L. Niu, Nano-Micro Lett., 2021, 13(1), 37 CrossRef PubMed.
- Q. G. Pan, Z. P. Tong, Y. Q. Su, S. Qin and Y. B. Tang, Adv. Funct. Mater., 2021, 31(37), 27 CrossRef.
- H. Wang, F. Liu, R. H. Yu and J. S. Wu, Interdiscip. Mater., 2022, 1(2), 196–212 CrossRef.
- Z. Liu, X. Liu, B. Wang, X. Wang, D. Lu, D. Shen, Z. Sun, Y. Liu, W. Zhang and Q. Zhang, eScience, 2023, 100177, 2667–1417 Search PubMed.
- S. Anantharaj and S. Noda, Small, 2020, 16(2), 24 CrossRef PubMed.
- Y. Q. Zhang, L. Tao, C. Xie, D. D. Wang, Y. Q. Zou, R. Chen, Y. Y. Wang, C. K. Jia and S. Y. Wang, Adv. Mater., 2020, 32(7), 22 CrossRef PubMed.
- B. Chen, D. L. Chao, E. Z. Liu, M. Jaroniec, N. Q. Zhao and S. Z. Qiao, Energy Environ. Sci., 2020, 13(4), 1096–1131 RSC.
- J. X. Bi, Z. Z. Du, J. M. Sun, Y. H. Liu, K. Wang, H. F. Du, W. Ai and W. Huang, Adv. Mater., 2023, 35(16), 42 Search PubMed.
- A. M. Smith and S. M. Nie, Acc. Chem. Res., 2010, 43(2), 190–200 CrossRef CAS PubMed.
- Y. Z. Pei, H. Wang and G. J. Snyder, Adv. Mater., 2012, 24(46), 6125–6135 CrossRef CAS PubMed.
- C. G. Morales-Guio and X. L. Hu, Acc. Chem. Res., 2014, 47(8), 2671–2681 CrossRef CAS PubMed.
- Y. Li, Q. Zhang, Y. Yuan, H. Liu, C. Yang, Z. Lin and J. Lu, Adv. Energy Mater., 2020, 10, 2000717 CrossRef CAS.
- B. Xiao, Z. Sun, H. Zhang, Y. Wu, J. Li, J. Cui, J. Han, M. Li, H. Zheng, J. Chen, M. Cai, C. Ke, X. Wang, H. Liu, Z. Jiang, S. Zhang, D.-L. Peng, Z. Guo and Q. Zhang, Energy Environ. Sci., 2023, 16(5), 2153–2166 RSC.
- T. Q. Guo, L. D. Li and Z. C. Wang, Adv. Energy Mater., 2022, 12(24), 36 Search PubMed.
- J. W. Zhang, Y. Li, Z. Chen, Q. Liu, Q. G. Chen and M. H. Chen, Energy Environ. Mater., 2023, 6, e12573 CrossRef CAS.
- Y. M. He, W. J. Chen, C. T. Gao, J. Y. Zhou, X. D. Li and E. Q. Xie, Nanoscale, 2013, 5(19), 8799–8820 RSC.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23(37), 4248–4253 CrossRef CAS PubMed.
- H. Zhang, L. Yang, P. Zhang, C. Lu, D. Sha, B. Yan, W. He, M. Zhou, W. Zhang, L. Pan and Z. Sun, Adv. Mater., 2021, 33(21), e2008447 CrossRef PubMed.
- D. W. Sha, C. J. Lu, W. He, J. X. Ding, H. Zhang, Z. H. Bao, X. Cao, J. C. Fan, Y. Dou, L. Pan and Z. M. Sun, ACS Nano, 2022, 16(2), 2711–2720 CrossRef CAS PubMed.
- X. Y. Wang, L. Fan, D. C. Gong, J. Zhu, Q. F. Zhang and B. G. Lu, Adv. Funct. Mater., 2016, 26(7), 1104–1111 CrossRef CAS.
- W. C. Shi, J. S. Meng, Q. Li, Z. T. Xiao, X. M. Xu, M. S. Qin, X. Zhang and L. Q. Mai, Chem. Commun., 2020, 56(18), 2751–2754 RSC.
- P. Singh, M. Patel, A. Gupta, A. J. Bhattacharyya and M. S. Hegde, J. Electrochem. Soc., 2012, 159(8), A1189–A1197 CrossRef CAS.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, Chem. Mater., 2017, 29(18), 7633–7644 CrossRef CAS.
- M. K. Han, X. W. Yin, X. L. Li, B. Anasori, L. T. Zhang, L. F. Cheng and Y. Gogotsi, ACS Appl. Mater. Interfaces, 2017, 9(23), 20038–20045 CrossRef CAS PubMed.
- Y. Li, H. Shao, Z. Lin, J. Lu, L. Liu, B. Duployer, P. O. A. Persson, P. Eklund, L. Hultman, M. Li, K. Chen, X. H. Zha, S. Du, P. Rozier, Z. Chai, E. Raymundo-Pinero, P. L. Taberna, P. Simon and Q. Huang, Nat. Mater., 2020, 19(8), 894–899 CrossRef CAS PubMed.
- O. Mashtalir, M. Naguib, V. N. Mochalin, Y. Dall'Agnese, M. Heon, M. W. Barsoum and Y. Gogotsi, Nat. Commun., 2013, 4, 7 Search PubMed.
- Y. Wei, P. Zhang, R. A. Soomro, Q. Z. Zhu and B. Xu, Adv. Mater., 2021, 33(39), 30 CrossRef PubMed.
- M. Anayee, C. E. Shuck, M. Shekhirev, A. Goad, R. Wang and Y. Gogotsi, Chem. Mater., 2022, 34(21), 9589–9600 CrossRef CAS.
- L. Verger, C. Xu, V. Natu, H. M. Cheng, W. C. Ren and M. W. Barsoum, Curr. Opin. Solid State Mater. Sci., 2019, 23(3), 149–163 CrossRef CAS.
- P. Urbankowski, B. Anasori, T. Makaryan, D. Q. Er, S. Kota, P. L. Walsh, M. Q. Zhao, V. B. Shenoy, M. W. Barsoum and Y. Gogotsi, Nanoscale, 2016, 8(22), 11385–11391 RSC.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, Chem. Mater., 2017, 29(18), 7633–7644 CrossRef CAS.
- S. Wu, Y. Feng, K. Wu, W. Jiang, Z. Xue, D. Xiong, L. Chen, Z. Feng, K. Wen, Z. Li and M. He, J. Alloys Compd., 2023, 930, 167414 CrossRef CAS.
- V. Kamysbayev, A. S. Filatov, H. Hu, X. Rui, F. Lagunas, D. Wang, R. F. Klie and D. V. Talapin, Sci, 2020, 369(6506), 979–983 CrossRef CAS PubMed.
- W. L. Zhang, W. Wei, W. Liu, T. Guan, Y. Tian and H. B. Zeng, Chem. Eng. J., 2019, 378, 11 CrossRef.
- T. Zhang, L. M. Pan, H. Tang, F. Du, Y. H. Guo, T. Qiu and J. Yang, J. Alloys Compd., 2017, 695, 818–826 CrossRef CAS.
- R. Li, D. Rao, J. Zhou, G. Wu, G. Wang, Z. Zhu, X. Han, R. Sun, H. Li, C. Wang, W. Yan, X. Zheng, P. Cui, Y. Wu, G. Wang and X. Hong, Nat. Commun., 2021, 12(1), 3102 CrossRef CAS PubMed.
- J. Zheng, C. Hu, L. Nie, S. Zang, H. Chen, N. Chen, M. Ma and Q. Lai, Appl. Surf. Sci., 2023, 611, 155617 CrossRef CAS.
- G. Wang, Y. Li, Y. Liu, S. Jiao, B. Peng, J. Li, L. Yu and G. Zhang, Chem. Eng. J., 2021, 417, 127977 CrossRef CAS.
- X. J. Wang, S. Zhang, Y. Shan, L. Chen, G. Y. Gao, X. L. Zhu, B. Cao and X. M. He, Energy Storage Mater., 2021, 37, 55–66 CrossRef.
- H. J. Huang, R. Xu, Y. Z. Feng, S. F. Zeng, Y. Jiang, H. J. Wang, W. Luo and Y. Yu, Adv. Mater., 2020, 32(8), 11 CrossRef PubMed.
- N. N. Wang, C. X. Chu, X. Xu, Y. Du, J. Yang, Z. C. Bai and S. X. Dou, Adv. Energy Mater., 2018, 8(27), 27 Search PubMed.
- Z. L. Jian, W. Luo and X. L. Ji, J. Am. Chem. Soc., 2015, 137(36), 11566–11569 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr05723b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |