
Intermediates involved in the reduction of SO2: insight into the mechanism of sulfite reductases†
Aishik
Bhattacharya
 ,
Soumya
Samanta
,
Arnab Kumar
Nath
,
Arnab
Ghatak
,
Somdatta Ghosh
Dey
* and
Abhishek
Dey
*
,
Soumya
Samanta
,
Arnab Kumar
Nath
,
Arnab
Ghatak
,
Somdatta Ghosh
Dey
* and
Abhishek
Dey
*
School of Chemical Sciences Indian Association for the Cultivation of Science, 2A & 2B, Raja SC Mullick Road, Kolkata, West Bengal PIN-700032, India. E-mail: somdattaghoshdey@gmail.com; abbeyde@gmail.com
First published on 21st June 2024
Abstract
Sulfite reductases (SiRs) catalyze the reduction of SO32− to H2S in biosynthetic sulfur assimilation and dissimilation of sulfate. The mechanism of the 6e−/6H+ reduction of SO32− at the siroheme cofactor is debated, and proposed intermediates involved in this 6e− reduction are yet to be spectroscopically characterized. The reaction of SO2 with a ferrous iron porphyrin is investigated, and two intermediates are trapped and characterized: an initial Fe(III)–SO22− species, which undergoes proton-assisted S–O bond cleavage to form an Fe(III)–SO species. These species are characterized using a combination of resonance Raman (with 34S-labelled SO2), EPR and DFT calculations. Results obtained help reconcile the different proposed mechanisms for the SiRs.
The reduction of sulfate to sulfide is a crucial step of the geochemical sulfur cycle, which controls biochemical sulfur assimilation and the respiration of sulfate-reducing bacteria.1,2 The reduction of sulfate to sulfide is catalyzed by two key metalloenzymes, ubiquitous in microorganisms, including sulfate-reducing bacteria and archaea as well as in methanogens.3,4 The reduction of sulfate (SO42−) requires its insertion into adenosine 5′-monophosphate to form adenosine 5′-phosposulfate, which is then reduced to release sulfite (SO32−). SO32− is then reduced in the active site of sulfite reductase (SiR). The reduction of sulfite is catalyzed by the siroheme cofactor (Fig. 1A) present in all the SiRs.5 The siroheme cofactor is bridged to an Fe4S4 cubane via one of its cysteine ligands.6–8 The mechanism of the 6e− reduction of SO32− to S2− is debated. In its active form siroheme and Fe4S4 clusters are reduced: i.e. the iron in the siroheme is in its Fe(II) state, and the Fe4S4 cluster is reduced. Although there are crystal structures of substrate-bound enzyme (Fig. 1A, left), there is no clarity on the oxidation states of the siroheme-Fe4S4 unit or sulfur in these structures, and two different mechanisms have been proposed (Fig. 1B).9,10 Initially, based on observation of 2e− and 4e− partially reduced species, trithionate and thiosulfate, during SO32− reduction, three consecutive 2e−/2H+ steps were proposed (Fig. 1B).11–17 Protons are provided by conserved arginine and lysine residues present in the active site.15 In addition, a sulfur monoxide (SO)-bound siroheme intermediate was proposed based on the crystal structure obtained by the oxidation of an S2−-bound siroheme-Fe4S4 site (Fig. 1A right).9 Recently, the direct 6e−/6H+ reduction of SO32− in the active site of an SiR (Fig. 1B) was called into question with the identification of a trisulfide formed between the two conserved cysteine residues of another dissimilatory sulfite reductase protein C (DsrC), which is encoded by all genomes that contain the genes of the catalytic A and B domains of an SiR and binds the DsrAB complex, as an end product of SO32− reduction. The rate of SO32− reduction by the SiR is first order with respect to the DsrC and about 15 times higher than the rate in the absence of the DsrC.18 The proposed mechanism invoked the attack on an Fe(III)–SO2−/Fe(III)–SO2H species (Fig. 1B), formed after the initial 2e− reduction of SO32−, by the two conserved cysteines of the DsrC to form a trisulfide.19 This trisulfide is then reduced to release sulfide, avoiding generation of trithionate and thiosulfate and resulting in sulfide as the only product of SO32− reduction.
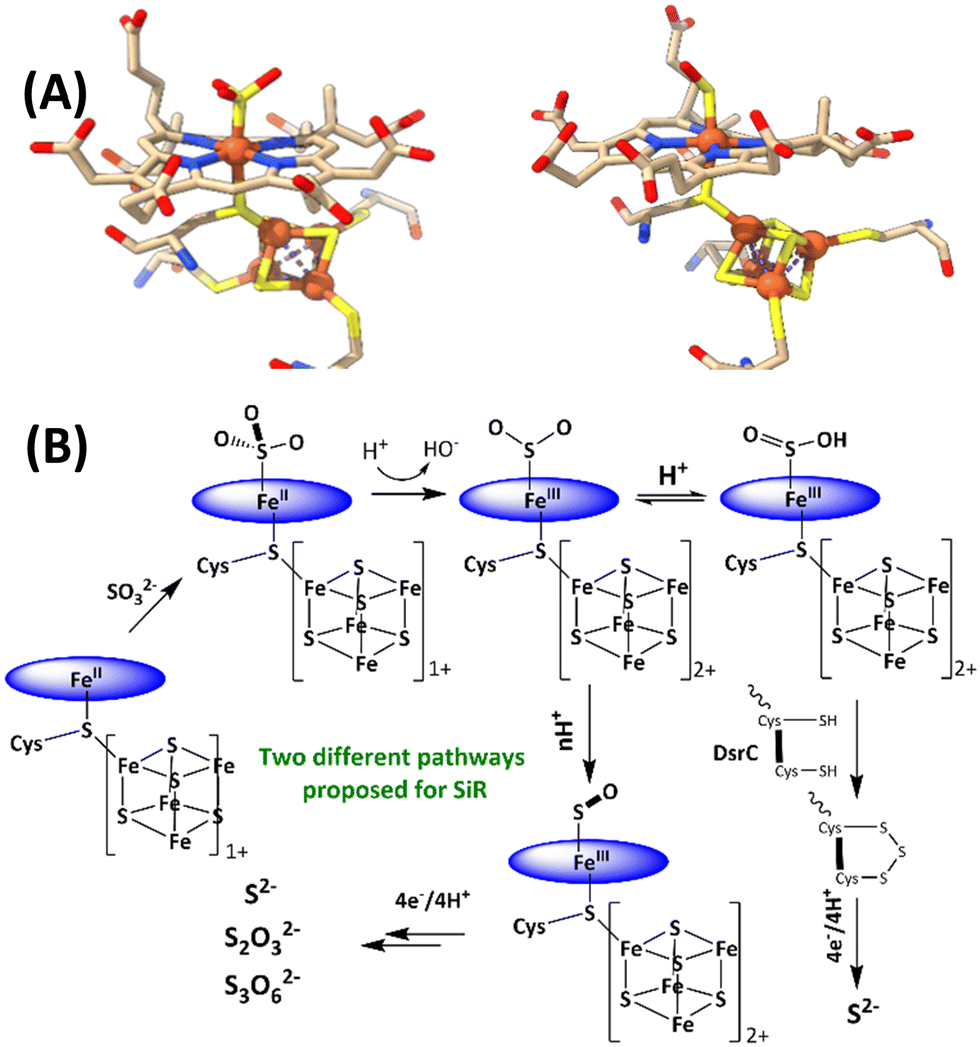 | ||
| Fig. 1 (A) active sites of SO32−-bound SiR and SO-bound SiR (pdb id: 7GEP) and (B) proposed mechanisms of the SiRs (direct 6e− reduction and trisulfide pathways are shown). The charge of the [Fe4S4] cluster (excluding the cysteines) is indicated next to the cubane. | ||
A synthetic analogue of the siroheme could be useful in understanding the mechanism.20 Unfortunately, the synthesis of this cofactor is not trivial and has not yet been achieved.21,22 Alternatively, simpler porphyrins such as iron tetraphenyl porphyrin (FeTPP), while not an exact model of a siroheme-Fe4S4 active site, has a reduction potential similar to that of the siroheme from different enzymes (which vary between −0.19 and −0.29 V at pH 7, and for FeTPP is −0.20 V at pH 7) and hence can be used to gain insight into this intriguing reaction.23,24 Initial attempts to investigate the reaction of SO2 with ferrous porphyrin in non-polar organic solvents inevitably resulted in the formation of sulfate-bound ferric porphyrin.21,25–27 Recently, the reaction of ferrous tetraphenyl porphyrin with SO2 was investigated in a protic organic solvent at room temperature (RT). The reaction proceeded to result in the 2e− reduction of SO2 to SO, and the released SO could be trapped using 2,3-dimethylbutadiene, resulting in the formation of a cyclic sulfoxide.28 An Fe(III)–SO intermediate was identified and was characterized using Mössbauer and EPR spectroscopy as a low-spin ferric porphyrin antiferromagnetically coupled to a triplet SO, resulting in an S = 1/2 species. Although the presence of the Fe–S bond could not be established experimentally, the observed S–O vibrations of this species were consistent with theoretically predicted vibrations for an Fe(III)–SO species.28 One of the 2e− needed is derived from iron porphyrin to which SO is bound, and the other comes from a free Fe(II)TPP. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of [Fe(III)TPP]+ and [Fe(III)TPP–SO]+ species was confirmed by Mössbauer spectroscopy.24,26 The Fe(III)–SO2−/Fe(III)–SO2H species, proposed in the SiR mechanism, has not however been observed either in the protein or in synthetic systems.
:1 ratio of [Fe(III)TPP]+ and [Fe(III)TPP–SO]+ species was confirmed by Mössbauer spectroscopy.24,26 The Fe(III)–SO2−/Fe(III)–SO2H species, proposed in the SiR mechanism, has not however been observed either in the protein or in synthetic systems.
To assess the involvement of any intermediate species prior to the [Fe(III)TPP–SO]+ species, the reaction of Fe(II)TPP with SO2 is allowed to proceed at −80 °C (MeOH-liq. N2 bath) for 10 min, and then, the reaction mixture is frozen in liq. N2. New EPR signals (Fig. 2A, green) are obtained with g = 2.36, 2.27, and 1.89, indicating the formation of another S = 1/2 low-spin intermediate species (henceforth referred to as Int-I) prior to the formation of [Fe(III)–SO]+ species (henceforth referred to as Int-II). In parallel to the Int-I signals, there is a g = 6.0 signal corresponding to a high-spin [Fe(III)TPP]+ species, suggesting that Int-I also results from the 2e− reduction of SO2, where one electron is derived from Fe(II)TPP to which SO2 to, and the other electron is derived from a free Fe(II)TPP, which gets oxidized to a high-spin [Fe(III)TPP]+ species with a g = 6.0 EPR signal. Mössbauer data suggests an equal population of these two species in the sample (Fig. S1, ESI†). When the solution is warmed up to −40 °C (10 min), the EPR signals originating from Int-I decrease, and the EPR signals from Int-II emerge (Fig. 2A, yellow), indicating that Int-I decays to produce Int-II (Fig. 2A, blue), i.e., Int-I is a species formed prior to Int-II in the reaction.
 | ||
| Fig. 2 (A) X-band EPR data collected at 77 K for a frozen solution of Fe(II)TPP (black), reaction mixture of Fe(II)TPP and SO2 in THF with 1%MeOH (v/v) kept at −80 °C for 10 min and later frozen in liq. N2 (green), kept at −40 °C for 10 min and later frozen in liq. N2 (yellow) and kept at RT for 10 min and further frozen in liq. N2 (blue, from reference 27). (B) Reaction mixture of Fe(II)TPP + SO2 in THF with 1%MeOH (v/v) kept at −80 °C for 10 min and later frozen in liq. N2 (green); reaction mixture of Fe(II)TPP + SO2 in THF with 1%MeOH (v/v) and 10 eq. of XylH+Cl− kept at −80 °C for 10 min and later frozen in liq. N2 (pink), kept at −40 °C for 10 min and further frozen in liq. N2 (yellow); and Fe(II)TPP + SO2 reaction mixture in THF with 1%MeOD (v/v) kept at −40 °C for 10 min and later frozen in liq. N2 (red). | ||
The transition of Int-I to Int-II depends on the availability of the protons. When the reaction of SO2 and Fe(II)TPP is performed at −80 °C (10 min) but in the presence of 10 eq. of xylidinium chloride (XylH+Cl−) as a proton source, the EPR signals from Int-I (Fig. 2B, green) are no longer observed and only those of Int-II are observed (Fig. 2B, pink). Similarly, when the MeOH proton source in the solution is deuterated (1% CD3OD instead of 1% MeOH, CD3OD represented as MeOD) and instead used as the proton source, the EPR signal of the reaction at −40 °C (Fig. 2B, red) shows Int-I and Int-II in a 5:1 ratio relative to MeOH where this ratio is 1:2, indicating that there is an H/D isotope effect in the conversion of Int-I to Int-II, consistent with a protonation step being involved in the reaction.
Int-I trapped at −80 °C is further characterized using resonance Raman spectroscopy of samples prepared with SO2 and comparing the vibrations with those observed when 34SO2 is used. In Int-I, vibration is observed at 984 cm−1, which shifts to 970 cm−1 on 34S substitution (Fig. 3A, top). This is consistent with an S–O stretching mode from an SO2-derived axial ligand.29,30 In the lower energy region, vibration is observed at 340 cm−1, which shifts to 337 cm−1 on 34S labelling (Fig. 3B, bottom). The energy of this vibration and isotope shift indicate that it is an Fe–S stretching mode.31,32
 | ||
| Fig. 3 Resonance Raman data on a frozen solution of Int-I (A) S–O region and (B) Fe–S region. 413.1 nm excitation and 10 mW laser power, prepared with 32SO2 (blue traces) and 34SO2 (red traces). The 32SO2 sample contains some unreacted Fe(II)TPP (green). *Indicates a solvent peak. | ||
In the past, Int-II had been tentatively assigned as a solvent-bound [Fe(III)TPP–SO]+ species with the S–O vibration at 1014 cm−1, which falls on the shoulder of a porphyrin band at 1004 cm−1. Accordingly, the 1014 cm−1 vibration shifts to 1005 cm−1 (Fig. 4A, top) on 34S substitution, confirming it to be an S–O stretching vibration, as previously assigned. The Fe–S vibration of Int-II is observed at 382 cm−1, which shifts to 377 cm−1 with 34S substitution (Fig. 4B, bottom). There is also another vibration at 206 cm−1, which shifts to 204 cm−1 with 34S (Fig. 4B, bottom) which may very well result from mixing of the Fe–S mode with a porphyrin vibration. There is some residual Fe–S vibration at 340 cm−1 from Int-I (Fig. 4B, bottom). Thus, the Fe–S and S–O vibrations, confirmed with 34S labelling, clearly indicate that Int-I and Int-II have Fe–S and S–O bonds. Samples prepared with MeOD did not show any shift in any of the S–O or Fe–S vibrations, suggesting that none of the species are likely to be protonated. Density functional theory (DFT) calculations are used to gain further insight.
 | ||
| Fig. 4 Resonance Raman data on a frozen solution of Int-II (A) S–O region and (B) Fe–S region. 413.1 nm excitation and 10 mW laser power, prepared with 32SO2 (blue traces) and 34SO2 (red traces). *Indicates a solvent peak and ** represents residual Int-I. | ||
DFT calculations are used to compute the hypothetical structure of possible intermediates in the reduction of SO2 to SO by Fe(II)TPP.33,34 The DFT method being used was reported to reproduce the Mössbauer and vibrational spectroscopy data of the [Fe(III)–SO]+ species quite well.28 EPR data indicates that Int-I and Int-II are S = 1/2 Fe(III) species, and the presence of a free [Fe(III)TPP]+ indicates that SO2 is reduced by 2e to its formal +2 oxidation state in both these species. Additionally, conversion of Int-I to Int-II requires a proton, as indicated by the EPR data obtained with MeOD and XylH+Cl−. The most likely description of Int-I is [Fe(III)TPP–SO2]− and Int-II has already been proposed to be [Fe(III)TPP–SO]+. The possibility of the formation of Fe(III)TPP–SO2H is removed by the lack of an H/D isotope effect in the S–O vibration. The DFT-calculated structure of an S = 1/2 [Fe(III)TPP–SO2]− with an axial MeOH (from the solvent) shows an Fe–S bond length of 2.33 Å and an S–O bond length of 1.50 Å (Table S1, ESI†). The computed S–O bond length shows a substantial increment from the reported 1.44 Å bond length of SO2 and is closer to that computed for free SO2− (one-electron-reduced SO2) consistent with the reduction of SO2.35,36 The Fe–S stretching vibration is computed to be at 339 cm−1, which shifts to 337 cm−1 on 34S substitution. The symmetric S–O vibration is calculated to be at 994 cm−1, which shifts by 7 cm−1 on 34S substitution to 987 cm−1. Computed Fe–S and S–O vibrations, and their shifts on 34S substitution are in excellent agreement with values obtained experimentally (Table S1, ESI†). Note that the S–O vibration of free SO2 is at 1156 cm−1, which, as expected, is higher than that of [Fe(III)TPP–SO2]− species, as reduction of SO2 is expected to weaken the S–O vibration. Analysis of the wavefunction of the [Fe(III)TPP–SO2]− species reveals very covalent interaction between Fe and SO2, and the dominant bonding interaction is the σ bond between the unoccupied dz2 orbital of the iron and occupied SO22− π* orbital (Fig. S2, ESI†).
The mechanism of SO2 reduction by Fe(II)TPP (Scheme 1A) thus involves a 2e− step, resulting in SO22− species bound to [Fe(III)TPP]+ to form [Fe(III)TPP–SO2]− Int-I, like the mechanism proposed for the SiR by Pereira and co-workers.18 No 1e−-reduced intermediate could be observed even at −80 °C, rather an additional electron is derived from a free Fe(II)TPP to result in a 2e reduction, which substitutes for the reduced [Fe4S4]+ cluster at the SiR active site. Protonation of this species leads to the cleavage of the S–O bond and formation of the [Fe(III)TPP–SO]+ species, Int-II. The reaction can proceed even at −40 °C with a weak proton source such as MeOH, albeit it is accelerated in the presence of XylH+Cl−. This indicates that the pKa of [Fe(III)TPP–SO2]− is higher than that of MeOH in THF. Thus, the 2e−-reduced intermediate in the active site of an SiR, which has several arginine and lysine residues, is most likely to be Fe(III)–SO2H. The reaction mechanism observed here suggests that the most likely mechanism of an SiR (Scheme 1B) involves the initial formation of the Fe(III)–SO2H species (Scheme 1B) after the 2e− reduction of SO2.18 The structure of the DsrC bound to the SiR shows that cysteinyl sulfur from the DsrC is less than 2 Å away from the siroheme, and the faster step is likely to be the attack of the cysteines of the DsrC protein resulting in the trisulfide.18,19
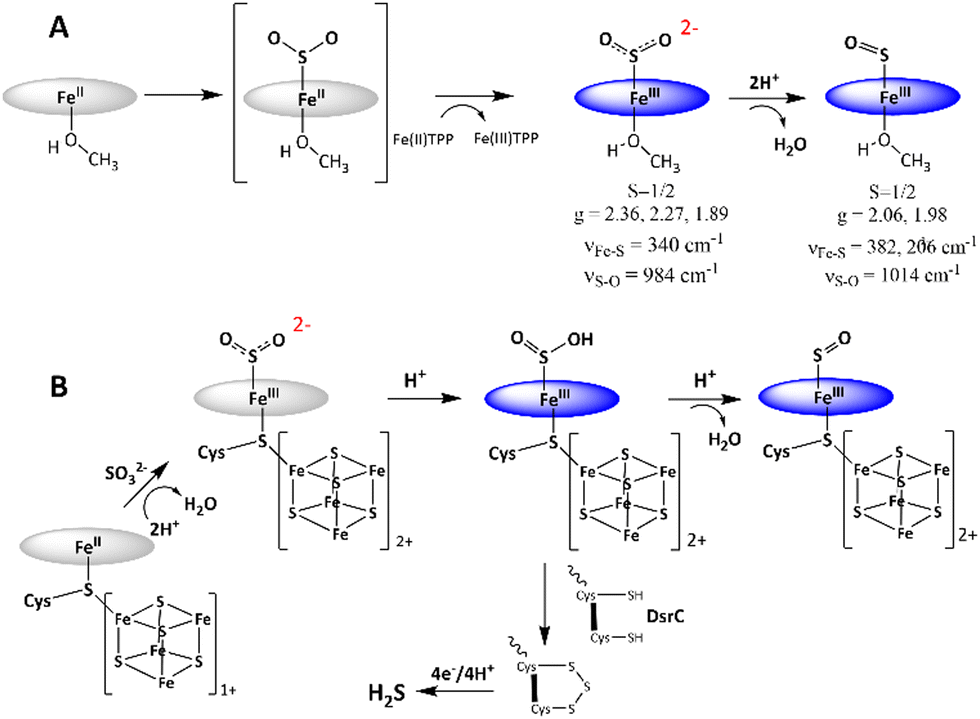 | ||
| Scheme 1 (A) Mechanism of SO2 reduction observed for Fe(II)TPP. Spectroscopic parameters are indicated below the proposed structures of the intermediates. (B) Mechanism of the SiR is refined based on the mechanism of SO2 reduction observed for Fe(II)TPP. The porphyrin ligand is indicated as a blue circle for clarity. | ||
The research is sponsored by the Department of Science and Technology, India SERB grant CRG/2021/000154.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- G. D. Fauque, in Ecology of Sulfate-Reducing Bacteria, ed. L. L. Barton, Springer US, Boston, MA, 1995, pp. 217–241 Search PubMed.
- J. Simon and P. M. H. Kroneck, in Advances in Microbial Physiology, ed. R. K. Poole, Academic Press, 2013, vol. 62, pp. 45–117 Search PubMed.
- M. Jespersen, A. J. Pierik and T. Wagner, Nat. Chem. Biol., 2023, 19, 695–702 CrossRef CAS PubMed.
- M. Jespersen and T. Wagner, Nat. Microbiol., 2023, 8, 1227–1239 CrossRef CAS PubMed.
- M. J. Murphy, L. M. Siegel, S. R. Tove and H. Kamin, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 612–616 CrossRef CAS PubMed.
- B. R. Crane, L. M. Siegel and E. D. Getzoff, Science, 1995, 270, 59–67 CrossRef CAS PubMed.
- J. A. Christner, E. Münck, P. A. Janick and L. M. Siegel, J. Biol. Chem., 1981, 256, 2098–2101 CrossRef CAS PubMed.
- I. Askenasy and M. E. Stroupe, in Transition Metals and Sulfur – A Strong Relationship for Life, ed. M. E. Sosa Torres and P. M. H. Kroneck, Metal Ions in Life Sciences, De Gruyter, Berlin, Boston, 2020, vol. 20, ch. 10, pp. 343–380.
- B. R. Crane, L. M. Siegel and E. D. Getzoff, Biochemistry, 1997, 36, 12120–12137 CrossRef CAS PubMed.
- P. A. Janick, D. C. Rueger, R. J. Krueger, M. J. Barber and L. M. Siegel, Biochemistry, 1983, 22, 396–408 CrossRef CAS PubMed.
- K. Parey, E. Warkentin, P. M. H. Kroneck and U. Ermler, Biochemistry, 2010, 49, 8912–8921 CrossRef CAS PubMed.
- J. M. Akagi, Biochem. Biophys. Res. Commun., 1983, 117, 530–535 CrossRef CAS PubMed.
- H. L. Drake and J. M. Akagi, J. Bacteriol., 1977, 132, 139–143 CrossRef CAS PubMed.
- H. L. Drake and J. M. Akagi, J. Bacteriol., 1976, 126, 733–738 CrossRef CAS PubMed.
- K. W. Smith and M. E. Stroupe, Biochemistry, 2012, 51, 9857–9868 CrossRef CAS PubMed.
- M. Surducan, A. M. V. Brânzanic and R. Silaghi-Dumitrescu, Int. J. Quantum Chem., 2018, 118, e25697 CrossRef.
- A. M. V. Brânzanic, U. Ryde and R. Silaghi-Dumitrescu, J. Inorg. Biochem., 2020, 203, 110928 CrossRef PubMed.
- A. A. Santos, S. S. Venceslau, F. Grein, W. D. Leavitt, C. Dahl, D. T. Johnston and I. A. C. Pereira, Science, 2015, 350, 1541–1545 CrossRef CAS PubMed.
- T. F. Oliveira, C. Vonrhein, P. M. Matias, S. S. Venceslau, I. A. C. Pereira and M. Archer, J. Biol. Chem., 2008, 283, 34141–34149 CrossRef CAS PubMed.
- A. M. V. Brânzanic, U. Ryde and R. Silaghi-Dumitrescu, Chem. Commun., 2019, 55, 14047–14049 RSC.
- L. Cai and R. H. Holm, J. Am. Chem. Soc., 1994, 116, 7177–7188 CrossRef CAS.
- C. Zhou, L. Cai and R. H. Holm, Inorg. Chem., 1996, 35, 2767–2772 CrossRef CAS.
- M. Hirasawa, M. Nakayama, T. Hase and D. B. Knaff, Biochim. Biophys. Acta Bioenerg., 1608, 2004, 140–148 Search PubMed.
- S. Bhunia, A. Ghatak, A. Rana and A. Dey, J. Am. Chem. Soc., 2023, 145, 3812–3825 CrossRef CAS PubMed.
- A. M. Stolzenberg, S. H. Strauss and R. H. Holm, J. Am. Chem. Soc., 1981, 103, 4763–4778 CrossRef CAS.
- W. R. Scheidt, Y. J. Lee and M. G. Finnegan, Inorg. Chem., 1988, 27, 4725–4730 CrossRef CAS.
- P. Cocolios, G. Lagrange, R. Guilard, H. Oumous and C. Lecomte, J. Chem. Soc., Dalton Trans., 1984, 567–574 RSC.
- A. Bhattacharya, A. Kumar Nath, A. Ghatak, A. Nayek, S. Dinda, R. Saha, S. Ghosh Dey and A. Dey, Angew. Chem., Int. Ed., 2023, 62, e202215235 CrossRef CAS PubMed.
- H. G. Houlton and H. V. Tartar, J. Am. Chem. Soc., 1938, 60, 544–548 CrossRef CAS.
- J. R. Durig, L. Zhou, T. Schwartz and T. Gounev, J. Raman Spectrosc., 2000, 31, 193–202 CrossRef CAS.
- A. C. Mot, C. Bischin, G. Damian, A. A. A. Attia, E. Gal, N. Dina, N. Leopold and R. Silaghi-Dumitrescu, J. Inorg. Biochem., 2018, 179, 32–39 CrossRef CAS PubMed.
- A. Rana, S. Amanullah, P. K. Das, A. B. McQuarters, N. Lehnert and A. Dey, J. Am. Chem. Soc., 2019, 141, 5073–5077 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- I. Jana, S. Naskar, M. Das and D. Nandi, Eur. Phys. J. D, 2019, 73, 233 CrossRef CAS.
- E. Krishnakumar, S. V. K. Kumar, S. A. Rangwala and S. K. Mitra, J. Phys. B: At., Mol. Opt. Phys., 1996, 29, L657 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, additional experimental and computation details are available free of charge in the supporting information. See DOI: https://doi.org/10.1039/d4cc02124j |
| This journal is © The Royal Society of Chemistry 2024 |