
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromaticity transfer in an annulated 1,4,2-diazaborole: facile access to Cs symmetric 1,4,2,5-diazadiborinines†
Vignesh
Pattathil
 and
Conor
Pranckevicius
*
and
Conor
Pranckevicius
*
Department of Chemistry, Charles E. Fipke Centre for Innovative Research, University of British Columbia, Okanagan Campus, 3247 University Way, Kelowna, BC, Canada. E-mail: conor.pranckevicius@ubc.ca
First published on 2nd July 2024
Abstract
A tricyclic annulated 1,4,2-diazaborole is readily accessed via reaction of a bidentate pyridyl-carbene ligand with MesBBr2 followed by reduction. Dearomatization of the flanking rings is shown to increase reactivity of this heterocycle in the form of a B-centred alkylation with MeI. Its reaction with hydrido-, fluoro-, and chloro-boranes reveal an unprecedented ring expansion reaction to form a diverse family of B2C2N2 heterocycles, reduction of which allows facile access to the first examples of Cs symmetric 1,4,2,5-diazadiborinines. DFT calculations have shed light on the electronic structures of the reduced species and provide insight into mechanistic aspects of the observed ring-expansion.
Boron-containing heterocyclic compounds have received rapidly increasing attention in recent years due to their ability to activate inert bonds,1–9 and their tuneable photophysical properties have led to applications in optoelectronic devices.10–19 In particular, diazadiborinines, – six-membered aromatic rings featuring two C, two B, and two N atoms – have demonstrated themselves to be a privileged subclass of B-heterocycles (Scheme 1a). They have demonstrated a diverse array of challenging bond activations including H–H, C–H, C–F, and C–C bonds,20–29 and have been very recently applied as organocatalysts in hydroboration and N-formylation reactions.30–33 Diazadiborinines are synthesized via the reaction of a 2-lithio-1N-heterocycle, frequently imidazole or oxazole, with a borane source. Careful control of the addition of the reagents can afford either the 1,4,2,5- or 1,3,2,5-diazadiborinine isomers.34 Due to the nature of this procedure, the scope of known diazadiborinines is fairly narrow, and all reported examples possess either C2 or C2v symmetry. Given the ambiphilic nature of diazadiborinine-based bond activations, possessing the tools to modify this reactive scaffold without this symmetry constraint would allow chemists to further refine their emerging bond-activation and catalytic properties. Moreover, diazadiborinines bearing various electron donor/acceptor groups at exocyclic positions are predicted to be singlet fission chromophores,35 and the development of synthetic methods to modify different parts of these molecules independently would significantly facilitate this exploration.
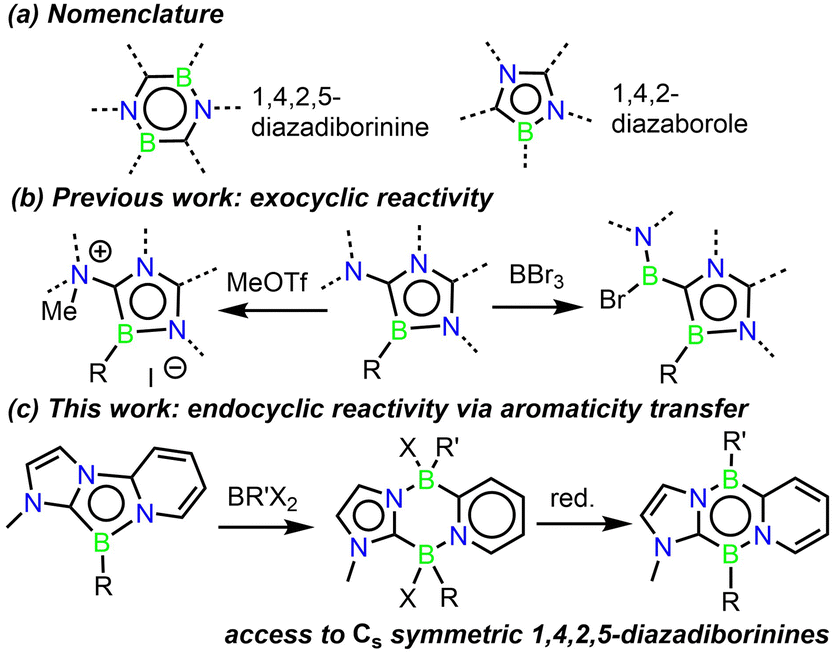 | ||
| Scheme 1 (a) B,N-heterocycles of interest to this work (b) Exocyclic reactivity reported with a monocyclic 1,4,2-diazaborole. (c) Endocyclic reactivity reported here with an annulated 1,4,2-diazaborole. | ||
1,4,2-Diazaboroles are another, albeit much less studied, class of aromatic B-heterocycles (Scheme 1a).36–38 They were first accessed by Kinjo and co-workers in 2015, and demonstrated exocyclic reactivity with methyl triflate, and formal borylene insertion at an exocyclic position upon reaction with boron tribromide (Scheme 1b). The use of the “F+” analogue Selectfluor was necessary to achieve endocyclic reactivity via the formation of a strong B–F bond.
The concept of aromaticity transfer has been utilized by Milstein and co-workers to achieve bond activations within otherwise unreactive heterocycles,39 where a de-aromatized fused ring induces reactivity through its thermodynamic drive to re-aromatize. Inspired by this, we began to explore the chemistry of 1,4,2-diazaboroles derived from a bidentate pyridyl-N-heterocyclic carbene framework (Scheme 1c). Our hypothesis being that the de-aromatized imidazole and pyridyl rings in this system would increase the reactivity of the central B-heterocycle. Herein, we report the isolation of a tricyclic 1,4,2-diazaborole and its unprecedented ring expansion reactions with chloro-, fluoro-, and hydrido-boranes to form 6-membered B2C2N2 diazadiborinine derivatives. Subsequent reduction has afforded the first examples of 1,4,2,5-diazadiborinines possessing Cs symmetry, establishing the first method for accessing these valuable heterocycles.
 | ||
Fig. 1 (a) Molecular structure of 3. Thermal ellipsoids are drawn at 50% probability. Mesityl groups are cropped, & hydrogen atoms are omitted for clarity. Selected bond lengths (Å): B1–N1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.466(2), B1–C1:1.493(3), C3–N1:1.404(2), C3–C4:1.392(2), C4–C5:1.373(3), C5–C6:1.424(3), C6–C7:1.358(3), C7–N1:1.391(2), C9–N2:1.402(2), C9–C8:1.346(3). (b) Frontier molecular orbitals of 3 (PBE0-D3(BJ)/6-311 + G(d,p)|SMD(benzene)). (c) Possible resonance forms of 3. :1.466(2), B1–C1:1.493(3), C3–N1:1.404(2), C3–C4:1.392(2), C4–C5:1.373(3), C5–C6:1.424(3), C6–C7:1.358(3), C7–N1:1.391(2), C9–N2:1.402(2), C9–C8:1.346(3). (b) Frontier molecular orbitals of 3 (PBE0-D3(BJ)/6-311 + G(d,p)|SMD(benzene)). (c) Possible resonance forms of 3. | ||
A 2-bromo-1,4,2-diazaborole derivative 2 could be readily isolated in high yield via the dropwise addition of a solution of N-mesityl-N′-(2-pyridyl)imidazol-2-ylidene 1 to MesBBr2 (Scheme 2). The product boronium salt 2 readily precipitates from hexane, can be isolated in 94% yield by simple filtration and was fully characterized by NMR spectroscopic methods. Selective reduction of 2 to the aromatic 1,4,2-diazaborole 3 was achieved via treatment with Collman's reagent, Na2[Fe(CO)4], and was isolated in 80% yield as an air-sensitive red crystalline solid which is stable for months under inert atmosphere. Interestingly, the use of KC8 as a reducing agent resulted in unselective reduction of 2, from which only trace quantities of 3 were isolated. Cyclic voltammetry of 3 in THF revealed an irreversible oxidation event at −0.78 V vs. Fc/Fc+ (see ESI†), suggesting the instability of the one-electron reduced intermediate, and the necessity of a two-electron reducing agent in the formation of 3. Tricyclic 1,4,2-diazaborole 3 displayed an 11B NMR signal at 12.3 ppm, similar to the derivative first reported by Kinjo and co-workers (18.4 ppm).371H NMR indicated the presence of a mirror plane in 3via the chemical equivalence of the ortho-Mes positions.
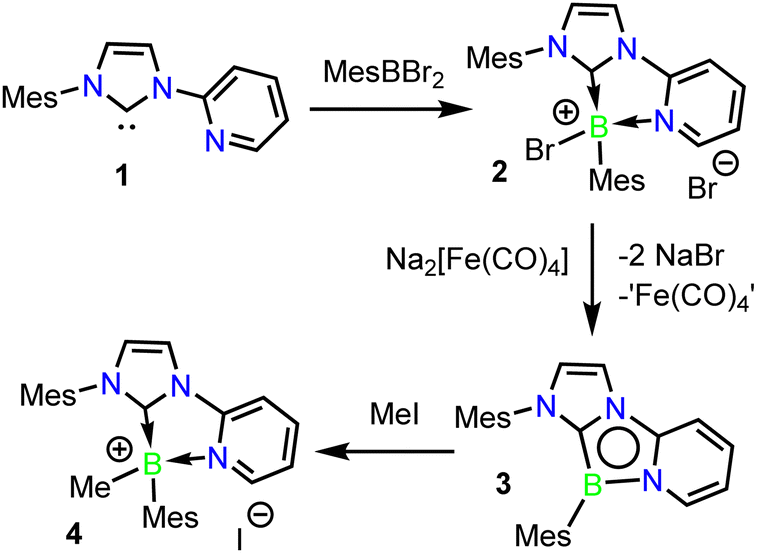 | ||
| Scheme 2 Synthesis of 1,4,2-diazaborole 3, and its methylation product 4. | ||
A single crystal X-ray diffraction study confirmed the formulation of 3 and revealed a B1–N1 distance of 1.466(2) Å and B1–C1 distance of 1.493(3) Å (Fig. 1a), suggesting partial double bond character between boron and both endocyclic atomic centres. The electronic structure of 3 was examined computationally (PBE0-D3(BJ)/6-311+G(d,p)|SMD(benzene)). The HOMO & LUMO of 3 are both significantly delocalized through the tricyclic conjugated π-system (Fig. 1b), although the greatest locus of electron density is centred on the boron atom, suggesting its nucleophilicity (Fig. 1c, B). Mayer bond order calculations revealed a B1–C1 bond order of 1.42, and a B1–N1 bond order of 1.13.
The molecular structure of 3 indicates somewhat localized single and double bonds in the flanking pyridyl ring (C–C bond range 1.358(3)–1.424(3) Å), suggesting its reduced aromaticity. This is further supported by the relatively upfield pyridyl 1H NMR shifts of 6.19 and 6.30 ppm for H–C5 and H–C6 (See Fig. 1a for numbering; 8.69 and 8.49 ppm in 2). Nucleus Independent Chemical Shift (NICS) calculations revealed a NICS(1)zz value of −35.0 ppm for the central diazaborole ring, revealing its significant aromatic character. Conversely, the flanking imidazole and pyridine rings have NICS(1)zz values of −19.8 and −15.6 ppm, respectively. The imidazole and pyridine rings in the unreduced 2 have NICS(1)zz values of −20.2 and −22.7 ppm, respectively, supporting the notion that the pyridine ring is comparatively de-aromatized in 3. NICS(0), NICS(1), and an ACID plot calculation are also consistent with this conclusion (see ESI†). This observation led us to anticipate an increased reactivity of 3 when compared with unconjugated 1,4,2-diazaboroles, as endocyclic reactivity is likely to be favoured by the re-aromatization of the flanking fused rings.39
Accordingly, combination of methyl iodide with 3 at 60 °C in benzene cleanly yielded the methylboronium iodide salt 4 (Scheme 2). Compound 4 was isolated in a 65% yield, and its structure was confirmed crystallographically (See ESI†). Compound 4 displays an 11B NMR signal at −0.2 ppm, similar to that observed for 2 (−1.1 ppm). This reactivity stands in contrast to that observed with a 1,4,2-diazaborole isolated by Kinjo and co-workers, where treatment with MeOTf led to methylation at an exocyclic nitrogen position (Scheme 1b),21 but is similar to B-centred alkylations reported with other reactive B-heterocycles.40,41
The reactivity of compound 3 with borane Lewis-acids was next explored. Their sterically larger profile was postulated to induce reactivity at the less-encumbered 5-position of the 1,4,2-diazaborole (Fig. 1c, C). In an initial experiment, 3 was combined with one equivalent of 9-borabicyclo[3.3.1]nonane (9-BBN) in C6D6. No initial reaction was observed, but upon warming to 60 °C overnight, a colour change from deep red to orange occurred along with the clean formation of a new product in the NMR spectra. 11B{1H} NMR revealed two boron environments at −5.6 and −8.5 ppm, supporting the presence of two 4-coordinate boron centres. 1H NMR indicated a C1 symmetric product with a 1:1 incorporation of 9-BBN and carbene moieties. A single crystal X-ray diffraction study revealed that the product is derived from insertion of the borabicyclo[3.3.1]nonane moiety into the 4,5-positions of the 1,4,2-diazaborole ring, forming the 1,4,2,5-diazadiborinine derivative 5 (Scheme 3; Fig. 2).
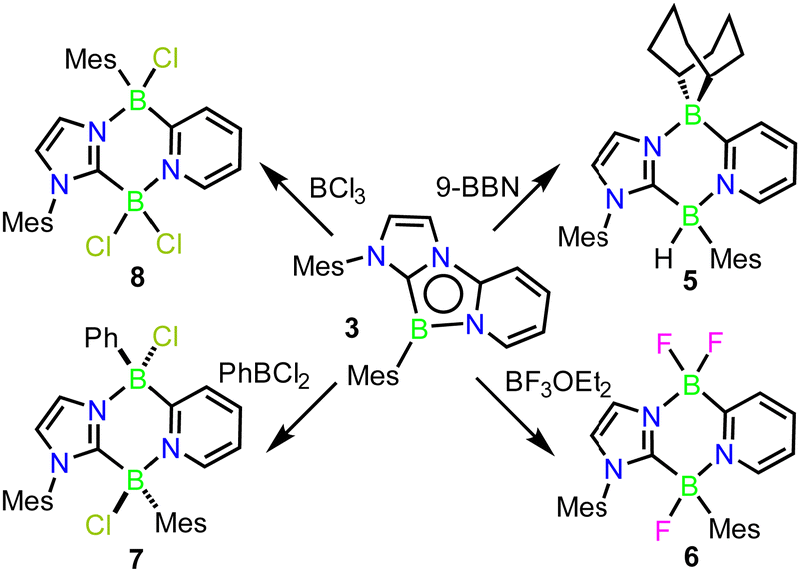 | ||
| Scheme 3 Ring expansion reactions of 3 with boranes. | ||
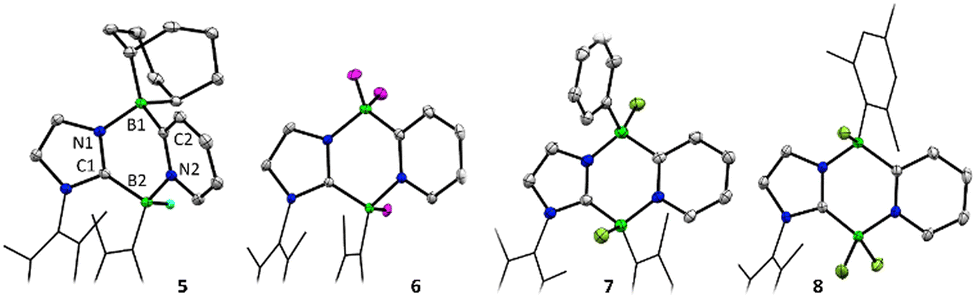 | ||
| Fig. 2 Molecular structures of 5–8. Thermal ellipsoids are drawn at 50% probability, mesityl groups are cropped, and selected hydrogen atoms are omitted for clarity. Selected bond lengths (Å) (Isostructural numbering in 5–8): (5) B1–N1:1.603(2), B1–C2:1.639(2), B2–C1:1.620(2), B2–N2:1.618(2), C1–N1:1.3434(17), C2–N2:1.3725(18). (6) B1–N1:1.561(2), B1–C2:1.627(3), B2–C1:1.637(2), B2–N2:1.626(2), C1–N1:1.3419(19), C2–N2:1.3622(19). (7) B1–N1:1.555(5), B1–C2:1.609(5), B2–C1:1.609(5), B2–N2:1.600(5), C1–N1:1.338(4), C2–N2:1.356(4). (8) B1–N1:1.554(3), B1–C2:1.625(3), B2–C1:1.593(2), B2–N2:1.575(2), C1–N1:1.338(3), C2–N2:1.354(3). | ||
To examine the scope of this transformation, reactions of 3 with other boranes were evaluated. While unselective reactions were observed with BBr3 and BI3, the addition of BF3·OEt2 or PhBCl2 resulted in the clean formation of new products 6 and 7 (Scheme 3), which were confirmed crystallographically to be the analogous ring expanded 1,4,2,5-diazadiborinine derivatives (Fig. 2). Both species were fully characterized by solutions NMR methods. These transformations constitute unique and unprecedented examples of Lewis-acid-induced ring expansion reactions of a 6π-aromatic B-heterocycle,42 and are a facile and high-yielding method to access hitherto-unknown non-symmetric 1,4,2,5-diazadiborinine derivatives.
The mechanism of the addition of BF3 to 3Ph (mesityl groups simplified as phenyl) was examined computationally (Fig. 3). The determined reaction profile indicates initial adduct formation with the nucleophilic 5-position of the central diazaborole ring (Int1) followed by fluoride migration to the Lewis acidic endocyclic boron centre. This reactivity supports the contribution of resonance structure C to the ground state of compound 3 (Fig. 1c). Disconnection of imidazole from the pyridyl group (TS2) is followed by migration of the Lewis acidic BF2 moiety to the nucleophilic imidazole nitrogen (TS3) to form compound 6Ph. Scission of the B–F and formation of the B–Nimidazole bonds were found to be barrierless processes, and the low-barrier TS3 is associated with reorientation of the BF2 moiety. The initial 1,3-dipolar B–F addition is distinct from the 1,3-cycloaddition reactions reported by Erker and co-workers in related dihydro-1,4,2-diazaboroles, which proceed across the 3,5-positions of the heterocycle.38
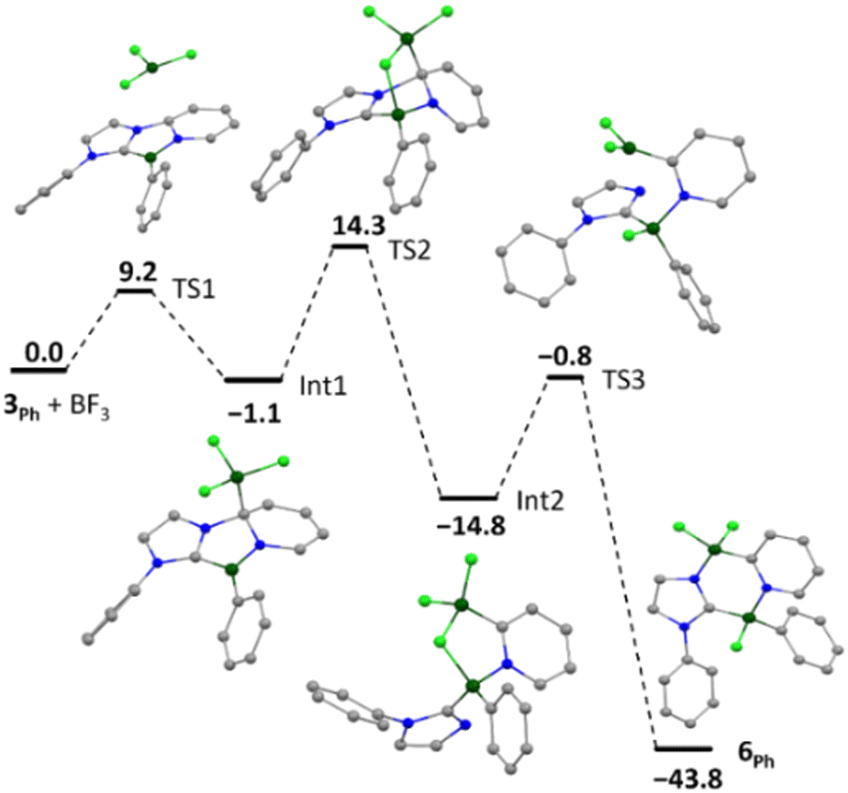 | ||
| Fig. 3 Computed pathway for the formation of the ring-expanded product 6Ph from 3Ph + BF3. Mesityl groups are simplified as phenyl. (PBE0-D3(BJ)/6-311 + G(d,p)|SMD(benzene)). Values given in kcal mol−1. | ||
Interestingly, the reaction of 3 with BCl3 resulted in a regioisomer of the expected product 8 (Scheme 3, Fig. 2), where the mesityl group had migrated to the boron centre bound to the imidazole N-moiety. We postulate that insertion in this case may be preceded by a fast initial exchange of the BCl3 with the B-mesityl group of 3. This would suggest that this ring expansion reaction may be amenable to a wider scope of 1,4,2-diazaboroles bearing different substituents at boron. The formation of 5–8 is curious in light of the selective B-methylation observed in 4. We postulate that only the relatively small methyl group can access the nucleophilic but sterically encumbered boron centre in 3.
Reduction of compounds 7 or 8 with an excess of KC8 in benzene cleanly afforded the Cs symmetric diazadiborinines 9 and 10 which were each isolated as deep red solids (Scheme 4). A molecular structure of 9 revealed delocalization of π-electrons through the central B2N2C2 ring (Fig. 4a), However, perturbation of the bonding is apparent. The endocyclic C–N and B–C bonds lengths differ notably (C1–N1:1.388(2); C2–N2:1.437(2); B2–C1:1.519(3); B1–C2:1.490(3) Å). This asymmetry is also reflected in the Mayer bond orders (C1–N1:1.22; C2–N2:1.08; B2–C1:1.18; B1–C2:1.36). Compound 9 displays two 11B NMR signals at 24.0 and 27.4 ppm, indicative of two inequivalent 3-coordinate boron centres. Similar to 3, the frontier molecular orbitals of 9 are delocalized over the tricyclic conjugated π-system (Fig. 4b), and a NICS(1)zz value of −24.9 ppm reveals significant aromaticity at the central ring. Compound 9 is highly fluorescent both in solution and the solid state and displays strong orange emission in C6H6 solution at 580 nm (excitation λ = 468 nm: Fig. 4c). TD-DFT calculations indicate that the major absorption band and corresponding emission result from a HOMO → LUMO transition (see ESI†). Compound 10 has similar spectroscopic and optical properties as 9.
 | ||
| Scheme 4 Reduction of ring-expanded products 7, 8 to the diazadiborinines 9, 10. | ||
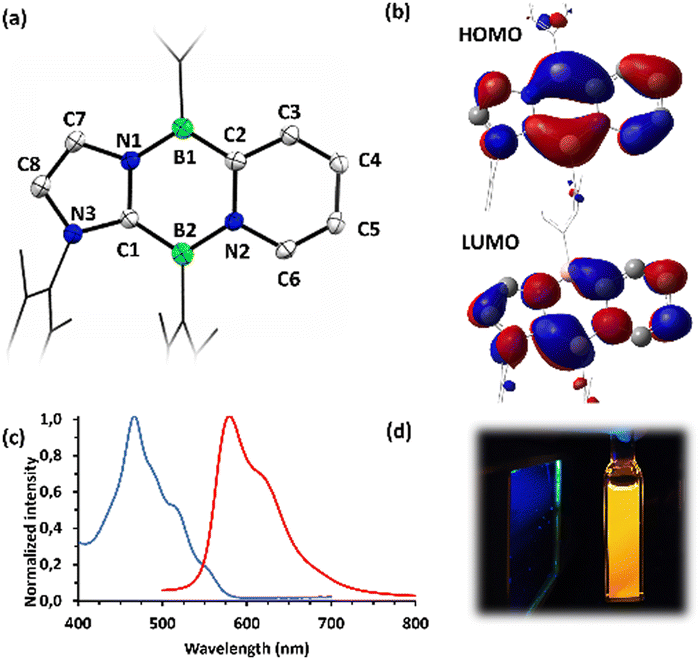 | ||
| Fig. 4 (a) Molecular structure of 9. Thermal ellipsoids are drawn at 50% probability, hydrogen atoms are omitted, and Mes/phenyl groups are cropped for clarity. Selected bond lengths (Å): B2–C1:1.519(3), B2–N2:1.441(3), B1–C2:1.490(3), B1–N1:1.452(3), C1–N1:1.388(2), C2–N2:1.437(2), C2–C3:1.421(3), C3–C4:1.352(2), C4–C5:1.427(3), C5–C6:1.342(3), C6–N2:1.407(2), C7–N1:1.397(2), C7–C8:1.345(3), C8–N3:1.372(3), C1–N3:1.384(2). (b) Frontier molecular orbitals of 9 (PBE0-D3(BJ)/6-311 + G(d,p)|SMD(benzene)). (c) Absorption and emission spectra in C6H6 solvent. (d) Compound 9 in C6H6 solution under broad spectrum UV irradiation. | ||
In summary, we have developed a novel annulated 1,4,2-diazaborole and demonstrated a unique ring expansion protocol with boranes to access the first examples of Cs symmetric 1,4,2,5-diazadiborinines. This protocol opens the door to fine tuning the optical and catalytic properties of the reactive diazadiborinine scaffold. Future studies will target expanding the scope of this reaction to include other substituted heterocycles, hetero-elements, and how we can use this reactivity to achieve reversible bond activations, catalysis, and desirable photophysical properties.
C. P. thanks NSERC Discovery Grant RGPIN-2021-03056 for supporting this work.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef PubMed
.
- Y. Su and R. Kinjo, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC
- J. He, F. Rauch, M. Finze and T. B. Marder, Chem. Sci., 2021, 12, 128–147 RSC
- E. von Grotthuss, M. Diefenbach, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2016, 55, 14067–14071 CrossRef CAS PubMed
- S. E. Prey, C. Herok, F. Fantuzzi, M. Bolte, H.-W. Lerner, B. Engels and M. Wagner, Chem. Sci., 2023, 14, 849–860 RSC
- K. Ota and R. Kinjo, Angew. Chem., Int. Ed., 2020, 59, 6572–6575 CrossRef CAS PubMed
- K. Ota and R. Kinjo, J. Am. Chem. Soc., 2021, 143, 11152–11159 CrossRef CAS PubMed
- M. M. Morgan, J. M. Rautiainen, W. E. Piers, H. M. Tuononen and C. Gendy, Dalton Trans., 2018, 47, 734–741 RSC
- J. W. Taylor, A. McSkimming, C. F. Guzman and W. H. Harman, J. Am. Chem. Soc., 2017, 139, 11032–11035 CrossRef CAS PubMed
-
B. J. Wang and M. P. Groziak, Advances in Heterocyclic Chemistry, Academic Press, 2016, vol. 118, pp. 47–90 Search PubMed
- S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77–89 CrossRef CAS
- Y. Guo, C. Chen and X. Wang, Chin. J. Chem., 2023, 41, 1355–1373 CrossRef CAS
- E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS
- J. Ruhl, N. Oberhof, A. Dreuw and H. A. Wegner, Angew. Chem., Int. Ed., 2023, 62, e202300785 CrossRef CAS PubMed
- M. M. Lorenzo-García, F. Fasano and D. Bonifazi, Patai's Chemistry of Functional Groups, pp. 1–93.
- V. M. Hertz, M. Bolte, H. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2015, 54, 8800–8804 CrossRef CAS PubMed
- P. G. Campbell, A. J. V. Marwitz and S. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed
- C. A. Jaska, D. J. H. Emslie, M. J. D. Bosdet, W. E. Piers, T. S. Sorensen and M. Parvez, J. Am. Chem. Soc., 2006, 128, 10885–10896 CrossRef CAS PubMed
- Z. Huang, S. Wang, R. D. Dewhurst, N. V. Ignat'ev, M. Finze and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 8800–8816 CrossRef CAS PubMed
- D. Wu, L. Kong, Y. Li, R. Ganguly and R. Kinjo, Nat. Commun., 2015, 6, 7340 CrossRef CAS PubMed
- B. Wang, Y. Li, R. Ganguly, H. Hirao and R. Kinjo, Nat. Commun., 2016, 7, 11871 CrossRef CAS PubMed
- B. Wang and R. Kinjo, Chem. Sci., 2019, 10, 2088–2092 RSC
- S. Portela and I. Fernández, Chem. – Eur. J., 2023, 29, e202300577 CrossRef CAS PubMed
- B. Wang, Y. Li, R. Ganguly, R. D. Webster and R. Kinjo, Angew. Chem., Int. Ed., 2018, 57, 7826–7829 CrossRef CAS PubMed
- Y. Su, Y. Li, R. Ganguly and R. Kinjo, Angew. Chem., Int. Ed., 2018, 57, 7846–7849 CrossRef CAS PubMed
- G. K. H. Goh, Y. Li and R. Kinjo, Dalton Trans., 2019, 48, 7514–7518 RSC
- D. Wu, R. Ganguly, Y. Li, S. N. Hoo, H. Hirao and R. Kinjo, Chem. Sci., 2015, 6, 7150–7155 RSC
- Y. Su, D. C. Huan Do, Y. Li and R. Kinjo, J. Am. Chem. Soc., 2019, 141, 13729–13733 CrossRef CAS PubMed
- B. Wang, K. Koshino and R. Kinjo, Chem. Commun., 2019, 55, 13012–13014 RSC
- D. Wu, R. Wang, Y. Li, R. Ganguly, H. Hirao and R. Kinjo, Chem, 2017, 3, 134–151 CAS
- Y. Shao, M. Huang, F. Gu, C. Zhao, L.-B. Qu and Z. Ke, Org. Chem. Front., 2021, 8, 1206–1215 RSC
- L. L. Liu, C. Chan, J. Zhu, C.-H. Cheng and Y. Zhao, J. Org. Chem., 2015, 80, 8790–8795 CrossRef CAS PubMed
- S. E. Prey and M. Wagner, Adv. Synth. Catal., 2021, 363, 2290–2309 CrossRef CAS
- R. Kinjo, J. Synth. Org. Chem., Jpn., 2021, 79, 1056–1064 CrossRef CAS
- E. Pradhan, J. N. Bentley, C. B. Caputo and T. Zeng, ChemPhotoChem, 2020, 4, 5279–5287 CrossRef CAS
- B. Su, Y. Li, R. Ganguly and R. Kinjo, Angew. Chem., Int. Ed., 2017, 56, 14572–14576 CrossRef CAS PubMed
- B. Su, Y. Li, R. Ganguly, J. Lim and R. Kinjo, J. Am. Chem. Soc., 2015, 137, 11274–11277 CrossRef CAS PubMed
- J. Li, C. G. Daniliuc, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2021, 60, 27053–27061 CrossRef CAS PubMed
- U. Gellrich, Y. Diskin-Posner, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2016, 138, 13307–13313 CrossRef CAS PubMed
- J. Gilmer, H. Budy, T. Kaese, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2020, 59, 5621–5625 CrossRef CAS PubMed
- J. Gilmer, M. Bolte, A. Virovets, H.-W. Lerner, F. Fantuzzi and M. Wagner, Chem. Eur. J., 2023, 29, e202203119 CrossRef CAS PubMed
- X. Su, T. A. Bartholome, J. R. Tidwell, A. Pujol, S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Rev., 2021, 121, 4147–4192 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2331652, 2331655, 2331653, 2331654, 2331660, 2331659, 2331656. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02414a |
| This journal is © The Royal Society of Chemistry 2024 |