
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of the doping level of PEDOT:PSS film by treatment with hydrazine to improve the Seebeck coefficient†
Temesgen Atnafu Yemataabd,
Yun Zhenga,
Aung Ko Ko Kyawac,
Xizu Wanga,
Jing Songa,
Wee Shong Chin*b and
Jianwei Xu *ab
*ab
aInstitute of Materials Research and Engineering, Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Singapore 138634, Republic of Singapore. E-mail: jw-xu@imre.a-star.edu.sg
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Republic of Singapore. E-mail: chmcws@nus.edu.sg
cDepartment of Electrical and Electronic Engineering, Southern University of Science and Technology, Shenzhen 518055, P. R. China
dChemical Engineering Department, Bahirdar University, P.O. Box 26, Bahirdar, Ethiopia
First published on 9th January 2020
Abstract
As the most popular conducting polymer, poly(3,4 ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) is widely used for a variety of applications, including thermoelectrics. This paper reports the modulation of the doping level by treatment with hydrazine to improve the Seebeck coefficient of PEDOT:PSS films. PEDOT:PSS films were first treated with formic acid followed by hydrazine, leading to a significant increase in the Seebeck coefficient from 17.5 to 42.7 μV K−1, about 2.5 times higher than that of the pristine film partially at the expense of electrical conductivity. An optimum power factor of 93.5 μW K−2 m−1, being 2.4 times that of the one treated with only formic acid, was achieved. The substantial improvement in the Seebeck coefficient and the power factor is collectively attributed to the removal of the PSS, and more importantly, the reduction of the doping level of PEDOT by the hydrazine treatment, which is evidenced clearly by UV-vis-NIR spectroscopy, XPS and Raman spectroscopy.
1. Introduction
Thermoelectric (TE) devices can directly convert thermal energy into electrical energy without requiring any moving parts.1,2 The effectiveness of TE materials is determined by the dimensionless TE figure-of-merit ZT = S2σT/κ, where S, T, σ, and κ stand for the Seebeck coefficient, absolute temperature, electrical conductivity, and thermal conductivity, respectively. The product of the square of S and the σ is termed as the power factor (PF), which is always used to evaluate the performance of TE materials. Until now, high ZT values have been reported for traditional inorganic TE materials such as Bi2Te3-based alloys3,4 and PbTe-based compounds.5,6 However, most high performance inorganic TE materials have several limiting factors such as scarcity and toxicity of elements, difficulty in materials processing, and poor mechanical flexibility.7,8 Alternatively, organic conducting polymer based thermoelectrics show great potential due to the cost-effectiveness, facile synthesis, large area processing, mass production as well as the high σ, and low κ.9–11 Among many organic conducting polymer TE materials studied,10,12–14 poly(3,4-ethyl-enedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) is a promising candidate for the practical organic based thermoelectric generators. Studies showed that the σ of PEDOT:PSS films could be greatly enhanced with various post-treatment methods.15–23 These post treatments enlarge the charge carrier concentration, and bipolaron are the main charge carriers. However, this high doping level results in a small S of PEDOT:PSS because of the extra charge carriers.24 Therefore, appropriate methods that enable to improve the S are desired as the ZT involves the square of the S.Previous studies already demonstrated that it was possible to tune the charge carrier concentration (normally by adjusting the oxidation level) in PEDOT:PSS and in turn affected both the S and the σ to achieve an optimized power factor. For instance, the precise control of the redox level of PEDOT:tosylate chains by simply using tetrakis(dimethylamino)ethylene as a de-doping agent resulted in a large PF of 324 μW K−2 m−1 and a ZT of 0.25 at room temperature.25 Besides, the addition of DMSO into PEDOT:PSS solution and then treatment with poly(ethylene oxide) led to a PF of 157 μW K−2 m−1.26 Also a multistep process was employed with ultrafiltration and dedoping of PEDOT:PSS by hydrazine and the highest PF of 115.5 μW K−2 m−1 was eventually recorded for the hydrazine treated PEDOT:PSS27 and an optimized power factor 112 μW K−2 m−1 was observed for PEDOT:PSS treated with a mixture of DMSO and hydrazine.24 A recent report has shown that the PEDOT:PSS thin film treated with sulfuric acid and different concentrations of sodium hydroxide resulted in a PF value of 334 μW K−2 m−1.28 Therefore, approaches that can tune the charge carrier concentration could be efficient for tuning the S and the σ of PEDOT:PSS films. An earlier report showed that PEDOT:PSS bulky films treated with formic acid exhibited a σ of 1900 S cm−1, an S 20.6 μV K−1 and a maximum PF of 80.6 μW K−2 m−1.29 However, the ZT value is still lower than those reported value28 which is mostly because of the low S. In this work, both the σ, and the S and thus PF of PEDOT:PSS films are improved by using multiple treatments with formic acid followed by hydrazine.
2. Experimental
2.1 Preparation and treatment of PEDOT:PSS films
PEDOT:PSS solution (Clevios PH 1000, PEDOT:PSS ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 and concentration by weight = 1.3%) was purchased from Heraeus. All other chemicals were purchased from Sigma-Aldrich and used without further purification. The glass substrates were cleaned with deionized (DI) water, detergent, acetone, and isopropanol in an ultrasonic bath consecutively and dried with nitrogen gas. The glass substrate was subjected to UV-ozone for 15 min before use. The PEDOT:PSS solution was filtered using a 0.45 μm poly (vinylene difluoride) PVDF syringe filter. Fig. 1 schematically presents the procedure for preparation and treatment PEDOT:PSS films. The PEDOT:PSS films were prepared by drop-casting 300 μL PEDOT:PSS solution on the pre-cleaned glass substrate and drying at 50 °C for 10 min, and 80 °C for another 10 min. Finally, the PEDOT:PSS films were annealed at 130 °C for 10 min to ensure complete evaporation of the solvent. For the post treatment, initially 140 μL formic acid or EG was dropped onto the PEDOT:PSS films on a hot plate at 140 °C and dried for about 5 min. Afterward, these films were rinsed with DI water and then dried again on a hot plate at 140 °C for 5 min.
:2.5 and concentration by weight = 1.3%) was purchased from Heraeus. All other chemicals were purchased from Sigma-Aldrich and used without further purification. The glass substrates were cleaned with deionized (DI) water, detergent, acetone, and isopropanol in an ultrasonic bath consecutively and dried with nitrogen gas. The glass substrate was subjected to UV-ozone for 15 min before use. The PEDOT:PSS solution was filtered using a 0.45 μm poly (vinylene difluoride) PVDF syringe filter. Fig. 1 schematically presents the procedure for preparation and treatment PEDOT:PSS films. The PEDOT:PSS films were prepared by drop-casting 300 μL PEDOT:PSS solution on the pre-cleaned glass substrate and drying at 50 °C for 10 min, and 80 °C for another 10 min. Finally, the PEDOT:PSS films were annealed at 130 °C for 10 min to ensure complete evaporation of the solvent. For the post treatment, initially 140 μL formic acid or EG was dropped onto the PEDOT:PSS films on a hot plate at 140 °C and dried for about 5 min. Afterward, these films were rinsed with DI water and then dried again on a hot plate at 140 °C for 5 min.
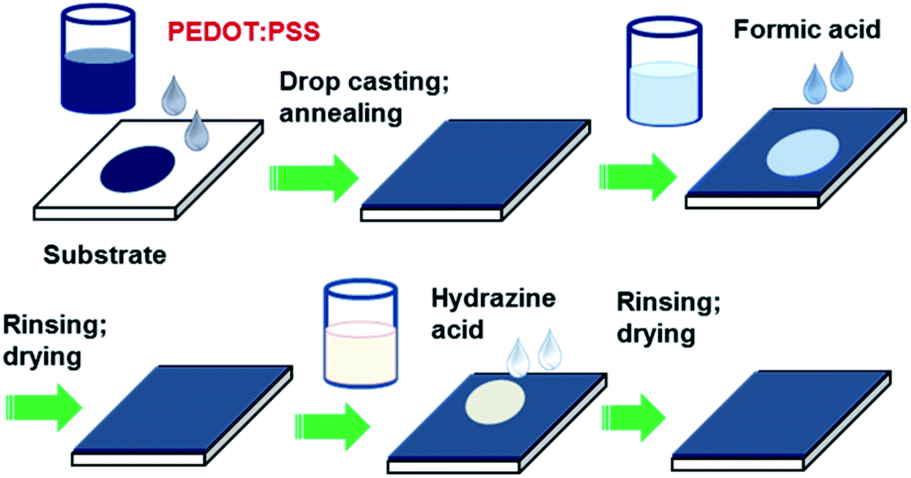 | ||
| Fig. 1 Schematic of preparation and sequential post-treatment of PEDOT:PSS films with formic acid and hydrazine. | ||
Next, the hydrazine treatment was performed by dropping 200 μL hydrazine aqueous solution with different concentrations (0.05%, 0.1%, 0.15%, 0.2%, 0.25%, 1.0% and 2% by weight) onto the previously formic acid- or EG-treated PEDOT:PSS films on a hot plate at 150 °C and dried for about 10 min. And then these films were washed thoroughly with DI water and dried again at 140 °C for 5 min.
2.2 Characterization
The sheet resistance (Rs) was measured using the four-point probe method (Laresta-GP MCP-T610 from Mitsubishi Chemical) at the room temperature and the corresponding thickness of the PEDOT:PSS films were obtained using a surface profiler (KLA-Tencor). The σ was calculated by using the relationship σ = 1/(sheet resistance × thickness). The S was measured with a homemade setup in a humidity-controlled room with relative humidity (RH) of 55% (Fig. 2). It consists of two stages, about 5 mm apart; one is integrated with a heater to generate a temperature gradient in the test sample. First, two long and narrow Au electrodes with 20 mm long, 1 mm wide and 2 mm apart were thermally evaporated on the film. To minimize the experimental error, the PEDOT:PSS film outside the area of electrodes was also removed. The Au electrodes were connected to a Keithley 2400 source meter through the probes to measure the voltage difference (ΔV). Simultaneously K-type thermocouples are connected to a data logger (Omron ZR-RX45) at the same time to collect the actual temperatures of the surfaces of the films. The voltage probes and thermocouples were placed at the same temperature zone on each side so that the measured voltage corresponds to the actual thermal gradient between the two voltage probes. The measured thermovoltage was corrected by the thermovoltage of Au wire to obtain the absolute S of the material. The S value was estimated from the slope of the linear relationship between thermoelectric voltage and the temperature difference of the two probes (i.e. S = −ΔV/ΔT). The absorption spectra measurement was performed on an UV-vis-NIR spectrophotometer (Shimadzu UV-vis spectrophotometer UV-3600). The films were deposited on a quartz substrate by using a spin coating method. The Raman spectroscopy measurements were obtained using Raman microscopy (Renishaw) with a laser wavelength of 785.5 nm, a laser beam spot size of 200 μm and an accumulation time of 30 s. X-ray Photoelectron Spectrometer (XPS) of the films were obtained by a Theta Probe Angle-Resolved X-ray Photoelectron Spectrometer (ARXPS) System (Thermo Scientific) using monochromated, micro-focused Al Kα X-ray photons (hυ = 1486.6 eV) at a base pressure of 1 × 10−9 Torr and a step size of 0.1 eV. The curve fitting and linear background subtraction were carried out using the Avantage software. The carrier concentration (n) and mobility (μ) were measured using a Hall effect measurement system (Ecopia HMS-5000) with a Van der Pauw method. First, the Ag electrodes were made-up by depositing Ag metal onto the film layer through a shadow mask. The HMS-5000 measurement system comprises software with an (I–V) curve for checking the ohmic contacts. Scanning electron microscopy (SEM) images were obtained using a JSM-6700F field emission SEM. Atomic Force Microscopy (AFM) photographs were taken on a Bruker Dimension Icon Atomic Force Microscope using the tapping mode.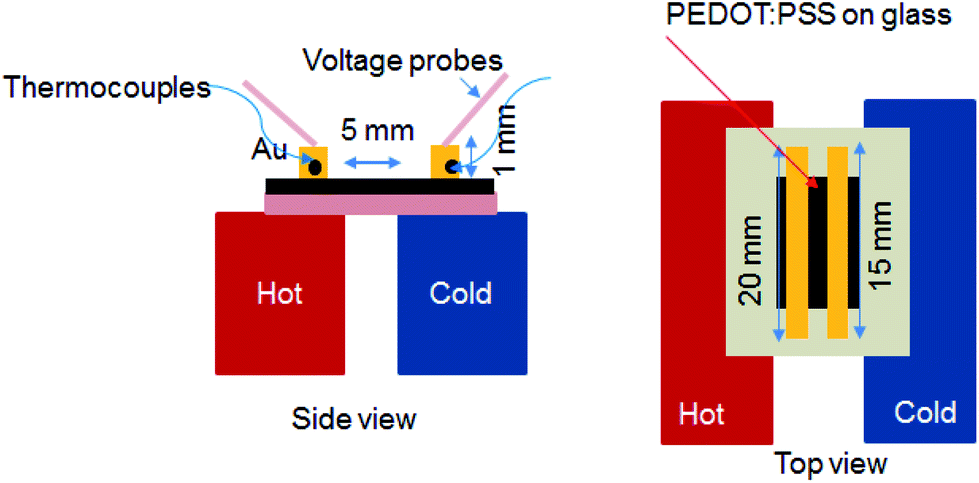 | ||
| Fig. 2 Schematic illustrations of the setup of S measurement and the electrode geometry. Side view (left) and top view (right). | ||
3. Results and discussion
The as-prepared PEDOT:PSS film from its aqueous solution has a very low σ of ∼0.3 S cm−1 and an S of about 17.5 μV K−1. After the treatment with formic acid, the σ of the treated PEDOT:PSS film was significantly enhanced to 1510 S cm−1 which is similar to previously reported value30,31 and the S is only slightly reduced to 16.1 μV K−1. Hence, the corresponding PF is 38.8 μW K−2 m−1 (Fig. S1a and b†). The improved TE property of the formic acid-treated films can be mainly attributed to the remarkably enhanced σ.Fig. 3a and b present the thermoelectric parameters (σ, S and PF) of the PEDOT:PSS films consecutively treated with formic acid followed by different concentrations of hydrazine. The S of the formic acid treated PEDOT:PSS films were further improved by dedoping with hydrazine in the sequential treatment. It was observed that as the concentration of hydrazine increased from 0 to 2 wt%, the S significantly increased from 16.1 to 43.9 μV K−1, while the σ reduced from 1510 to 103 S cm−1. The highest PF of 50.8 μW K−2 m−1 was achieved when the PEDOT:PSS films were treated with a concentration of 0.15 wt% hydrazine. The PF of the PEDOT:PSS can be further improved by a multiple-treatment of formic acid for three times followed by a single treatment of hydrazine. Fig. 3c and d show the S, σ, and PF of the multiple formic acid–hydrazine treated PEDOT:PSS films at different concentrations of hydrazine. The σ of the PEDOT:PSS films could be enhanced to 2139 S cm−1 after three times treatments with formic acid. After the subsequent treatment with hydrazine, the σ decreased from 2139 S cm−1 to 514 S cm−1, while the S increased from 16.2 to 42.7 μV K−1 as the concentration of hydrazine increased from 0 to 2 wt%. This results in a best PF value of 93.5 μW K−2 m−1, which is comparable to the PFs of some of PEDOT:PSS TE materials reported in the literature (Table S1†)24,29,32–36 when the film is treated with 0.15 wt% concentration of hydrazine.
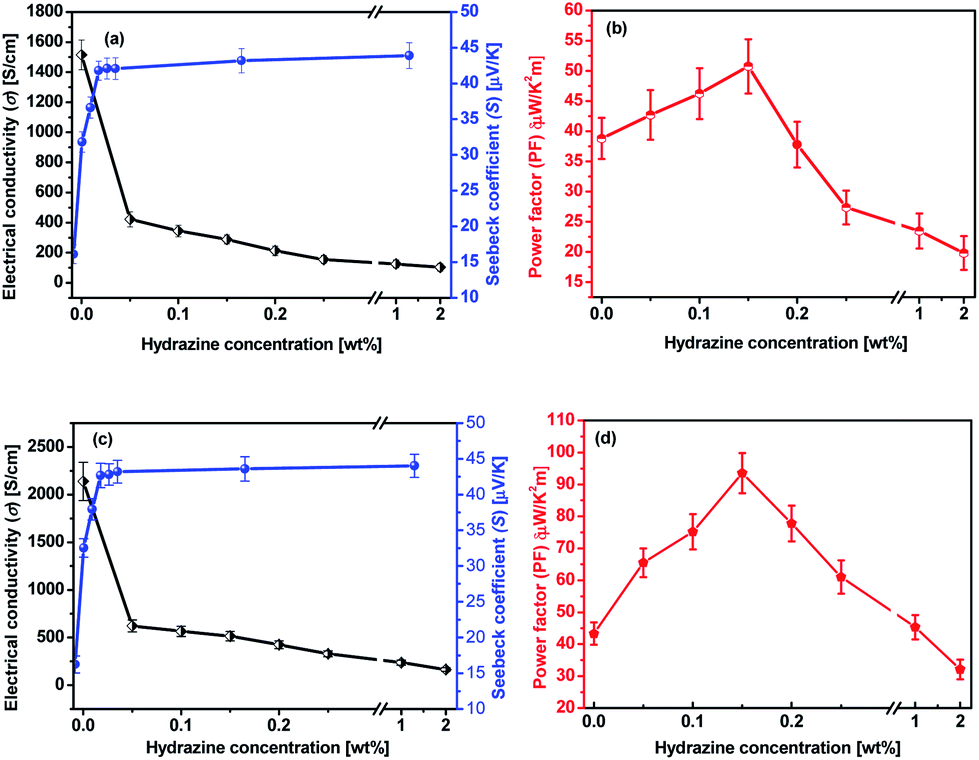 | ||
| Fig. 3 TE properties of PEDOT:PSS films as a function of the hydrazine concentration. (a and b) Formic acid × 1 time and hydrazine × 1 time; (c and d) formic acid × 3 times and hydrazine × 1 time. | ||
The mechanism for the TE properties enhancement of the films through the multiple formic acid–hydrazine treatments was investigated by different techniques. In the following discussions, the treatments are based on multiple treatments of formic acid unless otherwise stated.
Fig. 4 shows UV-vis-NIR absorption spectra of the untreated, formic acid-treated, and formic acid–hydrazine treated PEDOT:PSS films using different concentrations of hydrazine. The absorption band located at 225 nm was assigned to the aromatic rings of the PSS. The drop in the intensity of the absorption band after treatments indicates the removal of poly(4-styrenesulfonic acid) (PSSH) from the PEDOT:PSS films which agrees well with previously reported.37 The hydrazine treatment changes the optical spectral absorption due to dedoping. The previous studies have demonstrated that the chemical states of PEDOT chains play a vital role in affecting the magnitude of the S value.16,25,38 The neutral, polaron and bipolaron species of the PEDOT chains precisely correspond to different absorption wavelengths in the UV-vis-NIR spectra (Fig. 5). While the absorption of the PEDOT chain in the bipolaron state lies in the NIR region of above 1200 nm as a abroad band, the absorption for polaron and neutral states occurs at 900 and 600 nm, respectively.39,40 The absorption spectra of the formic acid-treated film appear similar to that of the pristine film, but the absorption is higher in the formic acid-treated film than in the pristine film, especially in the NIR region, suggesting that the formic acid treatment increases the charge carriers and hence bipolaron are the primary charge carriers. The lack of absorption peak at 600 nm also implies that neutral states are not dominant in the formic acid-treated film. This result is in good agreement with the increase in the σ and the decrease in the S. In contrast, a reduction in charge carrier concentration (vide infra) and dedoping in PEDOT:PSS by the formic acid–hydrazine treatment is noticeable through a switch in the PEDOT chemical state from bipolaron (∼1200 nm) to polaron (∼900 nm) and finally neutral (∼600 nm). Therefore, the appearance of the polaron and neutral state is consistent with the decrease in the σ and increase in the S illustrated in Fig. 3.
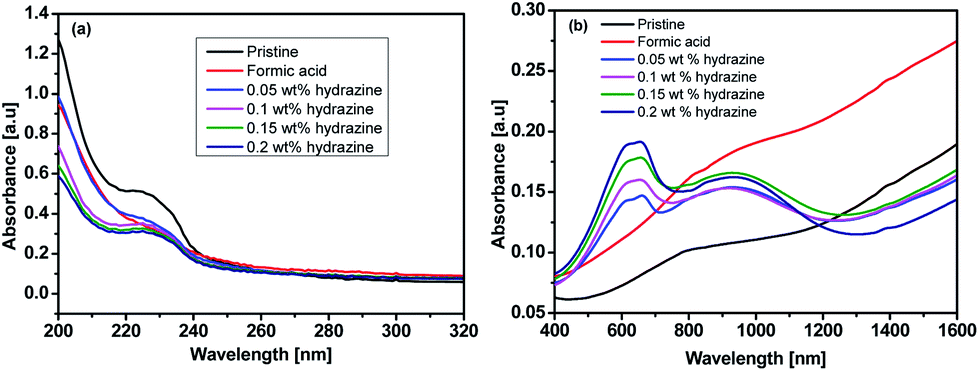 | ||
| Fig. 4 UV-vis-NIR absorption spectra of the untreated, formic acid-treated, and formic acid–hydrazine treated PEDOT:PSS films using different concentrations of hydrazine: (a) 200–320 nm (b) 400–1600 nm. | ||
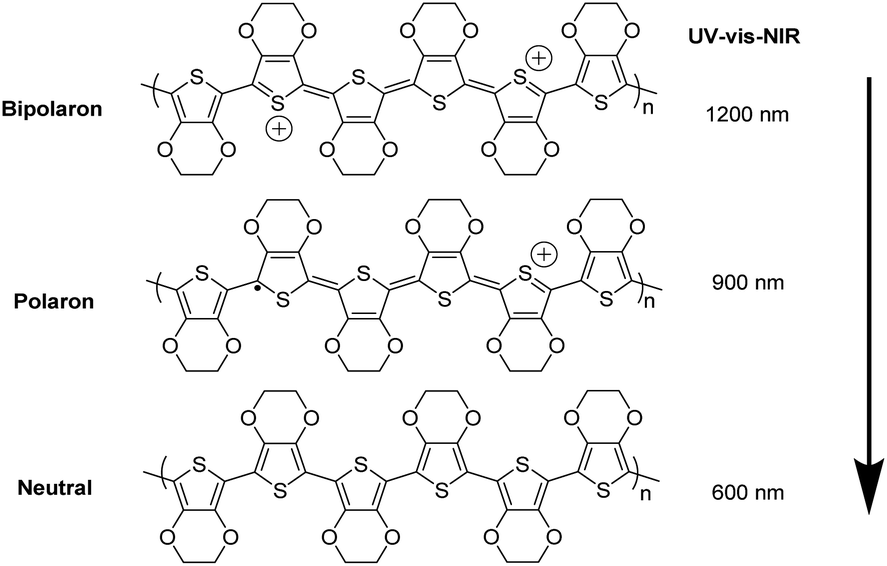 | ||
| Fig. 5 Schematic diagram of neutralization of chains of PEDOT:PSS by dedoping, i.e., bi-polaron (a di-cation charge carrier), polaron (a radical cation charge carrier) and neutral chain. | ||
The X-ray photoemission spectroscopy (XPS) spectra of the PEDOT and the PSS (Fig. 6) further demonstrated the loss of PSS from the PEDOT:PSS films after post-treatment. The XPS bands with binding energies between 166 and 172 eV are assigned to the S 2p band of the sulfur atoms in PSS, while the doublet XPS peaks with binding energies between 162 and 166 eV are assigned to the S 2p band of the sulfur atoms in the PEDOT.41,42 After the formic acid treatment or formic acid–hydrazine treatment, the S 2p intensity of the PEDOT relative to the PSS was significantly increased due to the removal of the PSS.43 Fig. 6a–c and S3† show the deconvoluted components of the S 2p XPS spectra. The binding energy of 165.3 & 164.1 eV, corresponding to S 2p1/2 and S 2p3/2 of formic acid treated PEDOT, shifted to the low binding energy at 164.9 & 163.7 eV, corresponding to S 2p1/2 and S 2p3/2 of 0.15 wt% hydrazine treated PEDOT, respectively, suggesting that the decrease in doping level after hydrazine treatment led to the decrease in binding energy, which concurred with the observation of UV-vis-NIR spectra. The lower energy shift of sulfur atoms in the PEDOT after the hydrazine treatment verifies the formation of dedoped PEDOT.
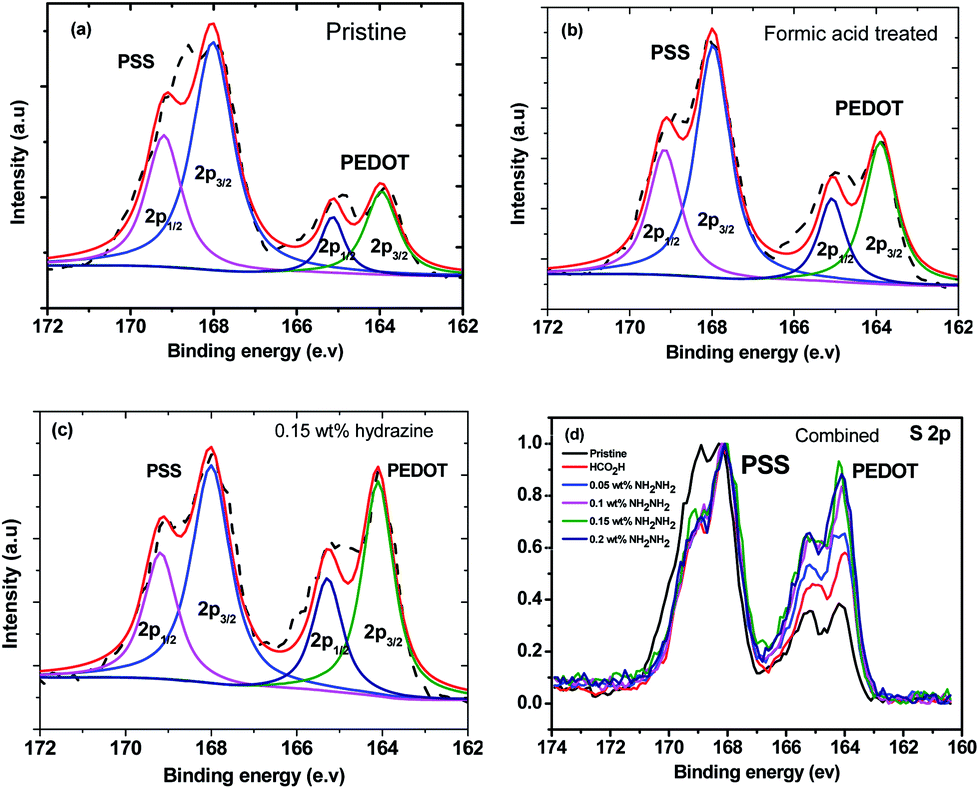 | ||
| Fig. 6 XPS spectra of (a) pristine PEDOT:PSS, (b) formic acid treated PEDOT:PSS, (c) 0.15 wt% hydrazine treated PEDOT:PSS, and (d) combination of normalized pristine PEDOT:PSS, formic acid treated PEDOT:PSS, and PEDOT:PSS films treated with different hydrazine concentrations. | ||
The neutralization reaction between free sulfonic acid (–SO3H) groups in the PSS units and hydrazine molecules might deactivate the interaction between the PEDOT and the PSS,44,45 and hence the PEDOT is dedoped.
Raman spectroscopy was performed to understand the enhanced TE properties of PEDOT:PSS films and to investigate the bipolaron, polaron and neutral states of the films as these state changes are accountable for TE properties of doped PEDOT:PSS films.46 Fig. 7 reveals the vibrational mode of the pristine and treated PEDOT:PSS films. The vibrational mode at 987 cm−1 was assigned to the deformation of the oxyethylene rings, while the vibrational modes at 1130 cm−1 and 1257 cm−1 were attributed to the PSS component and Cα–Cα′ interring stretching vibrations, respectively.46–49 However, the bands at 987, 1130 and 1257 cm−1 in the formic acid–hydrazine treated samples were weaker. The decrease in the intensity of formic acid–hydrazine treated films at 1130 cm−1 showed further loss of PSS by the dedoping process. Moreover, the vibrational mode of the formic acid–hydrazine films shifted from 1424 to 1417 cm−1 and became narrower than these of the untreated and formic acid treated films, suggesting that the change in the doping level from bipolaron to polaron and neutral state which is in agreement with UV-vis-NIR spectra. Together with UV-vis-NIR absorption spectra, it is evident that the hydrazine treatment dedoped the PEDOT chains to enhance the polaron concentration, leading to the decrease in the σ and the increment in the S.44 In general, the pristine PEDOT:PSS may contain both benzoid and quinoid structures. This band shift suggests a change from a predominated coil conformation of benzoid structure to a mixed linear-coil conformation of quinoid structure in the PEDOT chain after treatment,46,50 resulting in a quinoid dominant structure. The PEDOT chains are connected to the PSS chains through coulombic interactions and have core–shell or coiled structure because of repulsion between the long PSS chains.51 The formic acid–hydrazine treatment could weaken the ionic interaction between the PSS and the PEDOT, resulting in phase separation between PSS and PEDOT and a linear conformation of PEDOT chains. As a consequence, the σ is enhanced through a strong chain interaction. The same observations were seen on the Raman analysis of ethylene glycol treated PEDOT:PSS films as reported elsewhere.52 The peak around 1369 and 1509 cm−1 were observed for the Cβ–Cβ stretching and Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) =Cβ asymmetrical vibration of PEDOT, respectively. On the other hand, the partial removal of the PSS was observed by the reduction in the intensity of the Raman fingerprints of treated films compared to untreated films which are in agreement with the results obtained from UV and XPS.
=Cβ asymmetrical vibration of PEDOT, respectively. On the other hand, the partial removal of the PSS was observed by the reduction in the intensity of the Raman fingerprints of treated films compared to untreated films which are in agreement with the results obtained from UV and XPS.
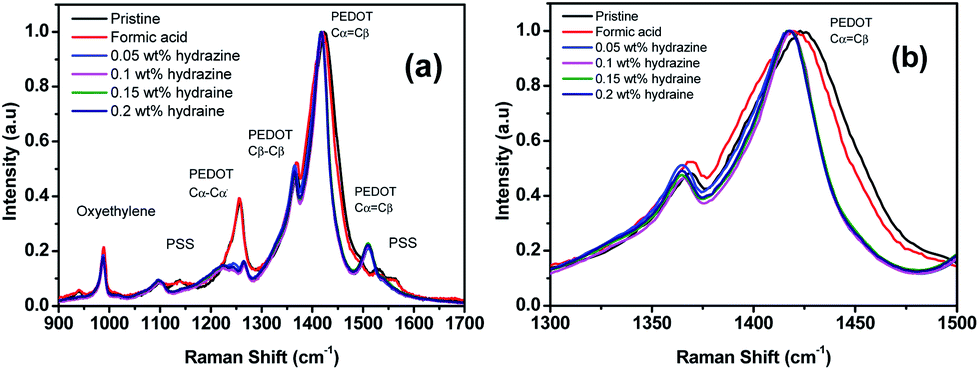 | ||
| Fig. 7 (a) Raman spectra of the PEDOT:PSS films before and after treatment with formic acid and formic acid–hydrazine. (b) The zoom-in spectra for the wavenumbers ranging from 1300 to 1500 cm−1. | ||
The AFM image of PEDOT:PSS films for formic acid and formic acid–hydrazine treated films was measured to study the possible alteration in the surface morphology and correlation between the σ and morphology (Fig. 8 and S2†). The height images of the pristine PEDOT:PSS films were smooth with a root-mean-square roughness (Rq) of 1.43 nm (Fig. 8a). The roughness increased from 1.43 to 1.60 nm after formic acid treatment (Fig. 8b) and to 1.82 nm after further dedoping with hydrazine (Fig. 8c). The formic acid and hydrazine treated PEDOT:PSS films showed PEDOT-rich regions. These results show that the PEDOT:PSS films treated by formic acid have improved the σ as a result of the reduced charge transport barriers and enlarged conducting PEDOT-rich core. After dedoping with hydrazine, some portions of the excess PSS-rich regions were further removed. The phase image of the pristine film does not show any apparent grains in the pristine film (Fig. 8d), implying that the PEDOT chains are well-intermixed with the PSS chains and the film is mostly covered by the PSS-rich domains. The interconnected large grains appeared in the phase images after formic acid and hydrazine treatment (Fig. 8e and f). These large grains were attributed to the interconnection of PEDOT-rich grains that were resulted from the strong phase separation between PSS-rich shell and PEDOT-rich core besides the depletion of PSS chain.53–55 AFM images of 0.2 wt% and 1 wt% hydrazine dedoped PEDOT:PSS films are shown in Fig. S2.† As compared to 0.15 wt% dedoped hydrazine the roughness further increases to 2.68 and 2.88 nm for 0.2 wt% and 1 wt% dedoped hydrazine, respectively, and interconnected large grains appeared in their phase images. Besides, the SEM images were studied to support the PSS chains aggregation. The white patches can be found in the formic acid–hydrazine treated PEDOT:PSS films (Fig. S4†). These patches have been formed from the PSSH that could be related to the dissociation of the acids in water. This explains that the formic acid–hydrazine can promote the phase separation and aggregation of the chains of PSSH from the film of PEDOT:PSS which is consistent with the previous report.56
 | ||
| Fig. 8 AFM height images: (a) pristine, (b) formic acid treated and (c) formic acid–0.15 wt% hydrazine treated. AFM phase images: (d) pristine, (e) formic acid treated and (f) formic acid–0.15 wt% hydrazine treated. All images captured with an area of 1 × 1 μm2. | ||
Hall effect measurement was carried out to determine the changes in n and μ quantitatively as they correlate to the dedoping process (Fig. 9). Mobility and carrier concentration increased after formic acid treatment. The mobility value is increased from 0.42 cm2 V−1 s−1 for the pristine film to 0.6 cm2 V−1 s−1 for the formic acid treated PEDOT:PSS film, while the carrier concentration value is increased from 4.1 × 1018 for pristine to 9 × 1022 cm−3 for formic acid treated PEDOT:PSS films. However, when the PEDOT:PSS film was subsequently treated with hydrazine, the mobility further increased, while the carrier concentration decreased as the increase of hydrazine concentration. The lower carrier concentration is responsible for the increase in the S.57,58 The S value for the hydrazine treated PEDOT:PSS films is ∼2.4 times higher than that for pristine PEDOT:PSS film and 2.7 times higher than that of the formic acid treated PEDOT:PSS film, which is mainly due to the change in the doping level. On the other hand, the higher mobility obtained in formic acid–hydrazine treated film is likely partially contributed to the better polymer alignment as well as the loss of the insulating PSS compared to the untreated sample.
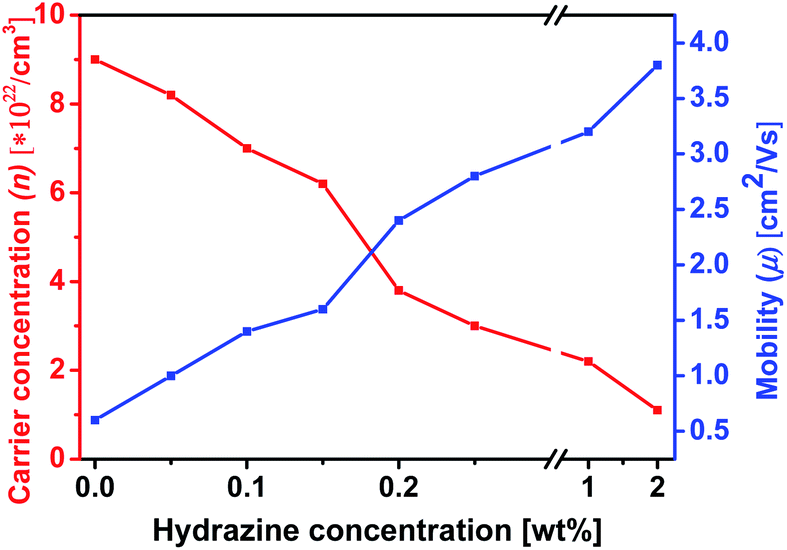 | ||
| Fig. 9 Variation of carrier concentration (n) and mobility (μ) of formic acid–hydrazine-treated PEDOT:PSS films with different weight concentration of proportional to hydrazine. The σ is proportional to n × μ and the S is inversely. | ||
Furthermore, the effects of the treatment on the stability of the σ and the S of PEDOT:PSS films were examined. It has been shown that the decrease of the σ and the humidity stability of PEDOT:PSS films is due to the hygroscopic and strongly acidic PSS that picks up the atmospheric water vapor.42,59,60 To examine the loss of PSS by the treatment, PEDOT:PSS films were exposed to the ambient atmosphere at a humidity of 75% and a temperature of 70 °C, and their σ and S were monitored over time. Fig. 10 shows the outcome of σ and S of PEDOT:PSS film treated with formic acid–0.15 wt% hydrazine as a function of exposure time in the air. The treated films maintain ∼90% of the original σ and S after 7 days exposure to air, implying the good stability of the PEDOT:PSS film after formic acid and hydrazine treatment. Reduction of water absorption and the removal of PSS resulted in a more compact structure and this also improves the stability of the treated film.59
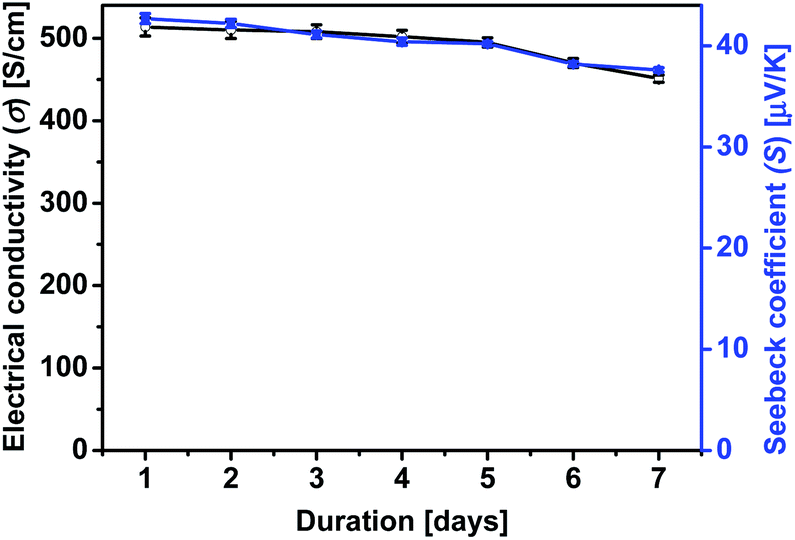 | ||
| Fig. 10 The effect of stability of σ and S at the optimum doping level with formic acid–0.15 wt% hydrazine treatment of PEDOT:PSS film as a function of exposure time (humidity: 75%; temperature: 70 °C). | ||
Lastly, a model for the mechanism of TE properties enhancement by the sequential formic acid and hydrazine treatment was suggested based on our observation (Fig. 11). The untreated PEDOT:PSS consists of a hydrophobic and conductive PEDOT-rich core and a hydrophilic and insulating PSS rich shell that exhibits a face-on packing and a layered structure. The solvent treatment interacts with the hydrophilic PSS and brings about a screening effect between the PEDOT and PSS chains, allowing the PSS chains to rearrange and the PEDOT chains to pack together with a higher order to form a layered structure.60,61 This layer structure could change chemical states of PEDOT chains that affect the carrier density in the PEDOT:PSS films.24
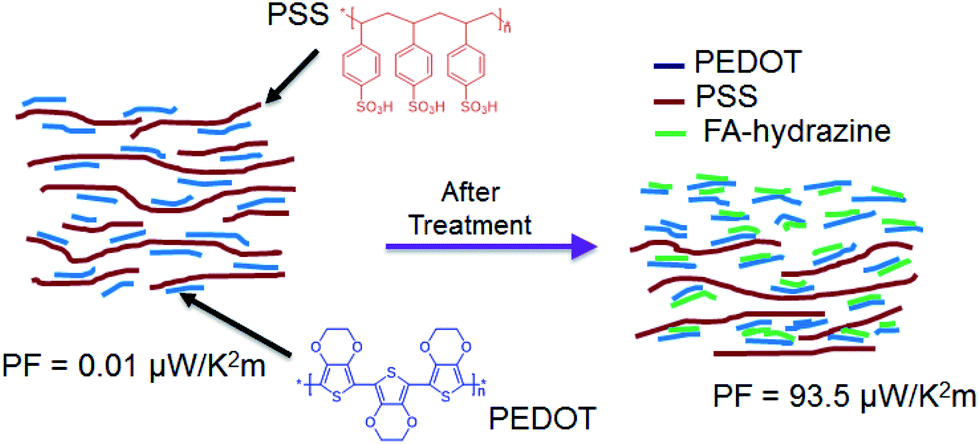 | ||
| Fig. 11 Chemical structures of PEDOT, PSS and schematic illustration of the mechanism of TE properties enhancement of PEDOT:PSS films caused by the sequential treatment of formic acid and hydrazine. | ||
4 Conclusions
In summary, we demonstrated that treatment with formic acid and hydrazine had improved the TE properties of PEDOT:PSS films. The formic acid treatment significantly enhanced the σ of the films, while modulation of the oxidation level of the PEDOT films through dedoping with hydrazine treatment resulted in an increase in the S from 17.5 μV K−1 for the pristine PEDOT:PSS to 42.7 μV K−1 for the treated PEDOT:PSS, respectively, to some extent, with the sacrifice of the σ. When the pristine films was treated with formic acid and then dedoped with different concentrations of hydrazine, the power factor reached its maximum value of 93.5 μW K−2 m−1 at the optimal condition, which was much higher than that in the pristine state due to a large increase in the S2 part of PF (σS2) to offset the decrease in the σ. The improved power factor is essentially due to the loss of insulating PSS and chemical dedoping as confirmed with UV-vis-NIR spectra, XPS and Raman spectra analysis. This work demonstrated that the Seebeck coefficient could be modulated by hydrazine to achieve a desired doping level, in other words, to make a balance between the three states of conducting PEDOT including the neutral, polaron and bipolaron, provide an effective way to attain a high PF of PEDOT:PSS films.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Agency for Science, Technology, and Research (A*STAR), Industry Alignment Fund, Pharos “Hybrid Thermoelectric Materials for Ambient Applications” Program (Grant No. 1527200019, 1527200021). T. A. Yemata would like to especially thank the SINGA scholarship from the A*STAR Graduate Academy.Notes and references
- L. E. Bell, Science, 2008, 321, 1457–1461 CrossRef CAS PubMed.
- S. B. Riffat and X. Ma, Appl. Therm. Eng., 2003, 23, 913–935 CrossRef.
- B. Poudel, Q. Hao, Y. Ma, Y. Lan, A. Minnich, B. Yu, X. Yan, D. Wang, A. Muto, D. Vashaee, X. Chen, J. Liu, M. S. Dresselhaus, G. Chen and Z. Ren, Science, 2008, 320, 634–638 CrossRef CAS PubMed.
- Y. Zheng, Q. Zhang, X. Su, H. Xie, S. Shu, T. Chen, G. Tan, Y. Yan, X. Tang, C. Uher and G. J. Snyder, Adv. Energy Mater., 2015, 5, 1401391 CrossRef.
- K. Biswas, J. He, I. D. Blum, C. I. Wu, T. P. Hogan, D. N. Seidman, V. P. Dravid and M. G. Kanatzidis, Nature, 2012, 489, 414–418 CrossRef CAS PubMed.
- Y. Pei, X. Shi, A. LaLonde, H. Wang, L. Chen and G. J. Snyder, Nature, 2011, 473, 66–69 CrossRef CAS PubMed.
- M. O'Neill and S. M. Kelly, Adv. Mater., 2011, 23, 566–584 CrossRef PubMed.
- E. Moulin, J. J. Cid and N. Giuseppone, Adv. Mater., 2013, 25, 477–487 CrossRef CAS PubMed.
- Y. Du, S. Z. Shen, K. Cai and P. S. Casey, Prog. Polym. Sci., 2012, 37, 820–841 CrossRef CAS.
- N. Dubey and M. Leclerc, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 467–475 CrossRef CAS.
- B. Zhang, J. Sun, H. Katz, F. Fang and R. Opila, ACS Appl. Mater. Interfaces, 2010, 2, 3170–3178 CrossRef CAS PubMed.
- B. Zhang, J. Sun, H. E. Katz, F. Fang and R. L. Opila, ACS Appl. Mater. Interfaces, 2010, 2, 3170–3178 CrossRef CAS PubMed.
- Y. Zheng, H. Zeng, Z. Qiang and J. Xu, J. Mater. Chem. C, 2018, 6, 8858–8873 RSC.
- T. A. Yemata, Q. Ye, H. Zhou, A. K. Kyaw, W. S. Chin and J. Xu, in Hybrid Polymer Composite Materials, Elsevier, 2017, pp. 169–195 Search PubMed.
- Q. Wei, M. Mukaida, K. Kirihara, Y. Naitoh and T. Ishida, Materials, 2015, 8, 732–750 CrossRef CAS PubMed.
- T. Park, C. Park, B. Kim, H. Shin and E. Kim, Energy Environ. Sci., 2013, 6, 788–792 RSC.
- Z. U. Khan, O. Bubnova, M. J. Jafari, R. Brooke, X. Liu, R. Gabrielsson, T. Ederth, D. R. Evans, J. W. Andreasen, M. Fahlman and X. Crispin, J. Mater. Chem. C, 2015, 3, 10616–10623 RSC.
- Y. Xia and J. Ouyang, ACS Appl. Mater. Interfaces, 2012, 4, 4131–4140 CrossRef CAS PubMed.
- A. K. K. Kyaw, T. A. Yemata, X. Wang, S. L. Lim, W. S. Chin, K. Hippalgaonkar and J. Xu, Macromol. Mater. Eng., 2018, 303, 1700429 CrossRef.
- T. A. Yemata, A. K. K. Kyaw, Y. Zheng, X. Wang, Q. Zhu, W. S. Chin and J. Xu, Polym. Int., 2020, 69, 84–92 CrossRef CAS.
- X. Wang, A. K. K. Kyaw, C. Yin, F. Wang, Q. Zhu, T. Tang, P. I. Yee and J. Xu, RSC Adv., 2018, 8, 18334–18340 RSC.
- A. K. K. Kyaw, G. D. H. Wong, T. A. Yemata and J. Xu, Org. Electron., 2019, 69, 7–12 CrossRef CAS.
- Q. Zhu, E. Yildirim, X. Wang, X. Y. D. S. Soo, Y. Zheng, T. L. Tan, G. Wu, S.-W. Yang and J. Xu, Front. Chem., 2019, 7, 783 CrossRef PubMed.
- H. Park, S. H. Lee, F. S. Kim, H. H. Choi, I. W. Cheong and J. H. Kim, J. Mater. Chem. A, 2014, 2, 6532–6539 RSC.
- O. Bubnova, Z. U. Khan, A. Malti, S. Braun, M. Fahlman, M. Berggren and X. Crispin, Nat. Mater., 2011, 10, 429–433 CrossRef CAS PubMed.
- C. Yi, A. Wilhite, L. Zhang, R. Hu, S. S. Chuang, J. Zheng and X. Gong, ACS Appl. Mater. Interfaces, 2015, 7, 8984–8989 CrossRef CAS PubMed.
- S. H. Lee, H. Park, W. Son, H. H. Choi and J. H. Kim, J. Mater. Chem. A, 2014, 2, 13380–13387 RSC.
- Z. Fan, P. Li, D. Du and J. Ouyang, Adv. Energy Mater., 2017, 7, 1602116 CrossRef.
- D. A. Mengistie, C.-H. Chen, K. M. Boopathi, F. W. Pranoto, L.-J. Li and C.-W. Chu, ACS Appl. Mater. Interfaces, 2014, 7, 94–100 CrossRef PubMed.
- J. E. McCarthy, C. A. Hanley, L. J. Brennan, V. G. Lambertini and Y. K. Gun'ko, J. Mater. Chem. C, 2014, 2, 764–770 RSC.
- D. A. Mengistie, M. A. Ibrahem, P. C. Wang and C. W. Chu, ACS Appl. Mater. Interfaces, 2014, 6, 2292–2299 CrossRef CAS PubMed.
- S. Liu, H. Deng, Y. Zhao, S. Ren and Q. Fu, RSC Adv., 2015, 5, 1910–1917 RSC.
- J. Kim, J. G. Jang, J.-I. Hong, S. H. Kim and J. Kwak, J. Mater. Sci.: Mater. Electron., 2016, 27, 6122–6127 CrossRef CAS.
- Z. Fan, D. Du, Z. Yu, P. Li, Y. Xia and J. Ouyang, ACS Appl. Mater. Interfaces, 2016, 8, 23204–23211 CrossRef CAS PubMed.
- E. Yang, J. Kim, B. J. Jung and J. Kwak, J. Mater. Sci.: Mater. Electron., 2015, 26, 2838–2843 CrossRef CAS.
- K. Fang-Fang, L. Cong-Cong, X. Jing-Kun, J. Feng-Xing, L. Bao-Yang, Y. Rui-Rui, L. Guo-Dong and W. Jian-Min, Chin. Phys. Lett., 2011, 28, 037201 CrossRef.
- Y. Xia, K. Sun and J. Ouyang, Energy Environ. Sci., 2012, 5, 5325–5332 RSC.
- O. Bubnova, M. Berggren and X. Crispin, J. Am. Chem. Soc., 2012, 134, 16456–16459 CrossRef CAS.
- E. Nasybulin, S. Wei, M. Cox, I. Kymissis and K. Levon, J. Phys. Chem. C, 2011, 115, 4307–4314 CrossRef CAS.
- J. C. Gustafsson, B. Liedberg and O. Inganas, Solid State Ionics, 1994, 69, 145–152 CrossRef CAS.
- X. Crispin, S. Marciniak, W. Osikowicz, G. Zotti, A. van der Gon, F. Louwet, M. Fahlman, L. Groenendaal, F. De Schryver and W. R. Salaneck, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2561–2583 CrossRef CAS.
- Y. H. Kim, C. Sachse, M. L. Machala, C. May, L. Müller-Meskamp and K. Leo, Adv. Funct. Mater., 2011, 21, 1076–1081 CrossRef CAS.
- Y. J. Xia and J. Y. Ouyang, Macromolecules, 2009, 42, 4141–4147 CrossRef CAS.
- J. Luo, D. Billep, T. Waechtler, T. Otto, M. Toader, O. Gordan, E. Sheremet, J. Martin, M. Hietschold, D. R. T. Zahn and T. Gessner, J. Mater. Chem. A, 2013, 1, 7576 RSC.
- H. Kim, S. Nam, H. Lee, S. Woo, C.-S. Ha, M. Ree and Y. Kim, J. Phys. Chem. C, 2011, 115, 13502–13510 CrossRef CAS.
- S. Garreau, G. Louarn, J. P. Buisson, G. Froyer and S. Lefrant, Macromolecules, 1999, 32, 6807–6812 CrossRef CAS.
- S. Garreau, J. L. Duvail and G. Louarn, Synth. Met., 2001, 125, 325–329 CrossRef.
- A. A. Farah, S. A. Rutledge, A. Schaarschmidt, R. Lai, J. P. Freedman and A. S. Helmy, J. Appl. Phys., 2012, 112, 113709 CrossRef.
- Y. K. Han, M. Y. Chang, W. Y. Huang, H. Y. Pan, K. S. Ho, T. H. Hsieh and S. Y. Pan, J. Electrochem. Soc., 2011, 158, K88–K93 CrossRef CAS.
- M. Łapkowski and A. Proń, Synth. Met., 2000, 110, 79–83 CrossRef.
- U. Lang, E. Müller, N. Naujoks and J. Dual, Adv. Funct. Mater., 2009, 19, 1215–1220 CrossRef CAS.
- J. Ouyang, Q. Xu, C.-W. Chu, Y. Yang, G. Li and J. Shinar, Polymer, 2004, 45, 8443–8450 CrossRef CAS.
- S.-I. Na, G. Wang, S.-S. Kim, T.-W. Kim, S.-H. Oh, B.-K. Yu, T. Lee and D.-Y. Kim, J. Mater. Chem., 2009, 19, 9045–9053 RSC.
- J. Luo, D. Billep, T. Blaudeck, E. Sheremet, R. D. Rodriguez, D. R. Zahn, M. Toader, M. Hietschold, T. Otto and T. Gessner, J. Appl. Phys., 2014, 115, 054908 CrossRef.
- J. Luo, D. Billep, T. Waechtler, T. Otto, M. Toader, O. Gordan, E. Sheremet, J. Martin, M. Hietschold and D. R. Zahn, J. Mater. Chem. A, 2013, 1, 7576–7583 RSC.
- Y. Xia and J. Ouyang, J. Mater. Chem., 2011, 21, 4927–4936 RSC.
- Q. Wei, M. Mukaida, K. Kirihara, Y. Naitoh and T. Ishida, ACS Appl. Mater. Interfaces, 2016, 8, 2054–2060 CrossRef CAS PubMed.
- T.-C. Tsai, H.-C. Chang, C.-H. Chen, Y.-C. Huang and W.-T. Whang, Org. Electron., 2014, 15, 641–645 CrossRef CAS.
- A. M. Nardes, M. Kemerink, M. De Kok, E. Vinken, K. Maturova and R. Janssen, Org. Electron., 2008, 9, 727–734 CrossRef CAS.
- D. Alemu, H.-Y. Wei, K.-C. Ho and C.-W. Chu, Energy Environ. Sci., 2012, 5, 9662 RSC.
- Q. Wei, M. Mukaida, Y. Naitoh and T. Ishida, Adv. Mater., 2013, 25, 2831–2836 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07648d |
| This journal is © The Royal Society of Chemistry 2020 |