
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of phosphonoacetate analogues of the second messenger adenosine 5′-diphosphate ribose (ADPR)†
Ondřej Baszczyňski‡
b,
Joanna M. Watt ab,
Monika D. Rozewitzc,
Ralf Fliegertc,
Andreas H. Gusec and
Barry V. L. Potter*ab
ab,
Monika D. Rozewitzc,
Ralf Fliegertc,
Andreas H. Gusec and
Barry V. L. Potter*ab
aMedicinal Chemistry and Drug Discovery, Department of Pharmacology, University of Oxford, Mansfield Road, Oxford, OX1 3QT, UK
bWolfson Laboratory of Medicinal Chemistry, Department of Pharmacy and Pharmacology, University of Bath, Bath, BA2 7AY, UK. E-mail: barry.potter@pharm.ox.ac.uk
cThe Calcium Signalling Group, Department of Biochemistry and Molecular Cell Biology, University Medical Center Hamburg-Eppendorf, Martinistrasse 52, 20246 Hamburg, Germany
First published on 9th January 2020
Abstract
Adenosine 5′-diphosphate ribose (ADPR) is an intracellular signalling molecule generated from nicotinamide adenine dinucleotide (NAD+). Synthetic ADPR analogues can shed light on the mechanism of activation of ADPR targets and their downstream effects. Such chemical biology studies, however, are often challenging due to the negatively charged pyrophosphate that is also sensitive to cellular pyrophosphatases. Prior work on an initial ADPR target, the transient receptor potential cation channel TRPM2, showed complete pyrophosphate group replacement to be a step too far in maintaining biological activity. Thus, we designed ADPR analogues with just one of the negatively charged phosphate groups removed, by employing a phosphonoacetate linker. Synthesis of two novel phosphonoacetate ADPR analogues is described via tandem N,N′-dicyclohexylcarbodiimide coupling to phosphonoacetic acid. Neither analogue, however, showed significant agonist or antagonist activity towards TRPM2, underlining the importance of a complete pyrophosphate motif in activation of this particular receptor.
Introduction
The linear diphosphate adenosine 5′-diphosphate ribose (ADPR, Fig. 1a) is formed from nicotinamide adenine dinucleotide (NAD+) by two routes; either hydrolysis of the nicotinamide ribose by NAD+ glycohydrolases such as CD38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 or via the sequential action of poly-ADPR polymerase (PARP) and poly-ADPR glycohydrolase (PARG).2 PARP inhibitors are currently highly topical as drugs in oncology.3 ADPR's cyclic incarnation as cyclic ADP-ribose (cADPR) is a second messenger in cellular calcium signalling.4,5 cADPR is also hydrolysed to ADPR as a deactivation mechanism. ADPR targets include widespread macro domains6 and the non-selective cation channel, transient receptor potential cation channel, subfamily M, member 2 (TRPM2).7,8 They are associated with a diverse range of physiological processes that are still not yet fully understood. However, chemical biological studies of ADPR-mediated processes in whole cells are often hampered by ADPR's negative charges and lack of membrane permeability. Studies of these processes often require technically demanding and time-consuming patch clamp or microinjection experiments on single cells to deliver charged species physically through the cell membrane. Furthermore, ADPR is subject to hydrolysis of the pyrophosphate by enzymes (intracellular enzymes, ectoenzymes in the plasma membrane and enzymes present in biological fluids), including hydrolases of the NUDIX family. This means that ADPR itself is unlikely to be stable for any length of time in a biological setting, complicating the direct study of its effects. Thus, synthetic ADPR analogues that are both membrane permeant and stable in the biological environment would be valuable tools to unravel the downstream mechanisms and function of this ligand. Stable ADPR analogues may also be useful when clarification of the precise second messenger that is acting under physiological conditions has proved challenging.9,10
1 or via the sequential action of poly-ADPR polymerase (PARP) and poly-ADPR glycohydrolase (PARG).2 PARP inhibitors are currently highly topical as drugs in oncology.3 ADPR's cyclic incarnation as cyclic ADP-ribose (cADPR) is a second messenger in cellular calcium signalling.4,5 cADPR is also hydrolysed to ADPR as a deactivation mechanism. ADPR targets include widespread macro domains6 and the non-selective cation channel, transient receptor potential cation channel, subfamily M, member 2 (TRPM2).7,8 They are associated with a diverse range of physiological processes that are still not yet fully understood. However, chemical biological studies of ADPR-mediated processes in whole cells are often hampered by ADPR's negative charges and lack of membrane permeability. Studies of these processes often require technically demanding and time-consuming patch clamp or microinjection experiments on single cells to deliver charged species physically through the cell membrane. Furthermore, ADPR is subject to hydrolysis of the pyrophosphate by enzymes (intracellular enzymes, ectoenzymes in the plasma membrane and enzymes present in biological fluids), including hydrolases of the NUDIX family. This means that ADPR itself is unlikely to be stable for any length of time in a biological setting, complicating the direct study of its effects. Thus, synthetic ADPR analogues that are both membrane permeant and stable in the biological environment would be valuable tools to unravel the downstream mechanisms and function of this ligand. Stable ADPR analogues may also be useful when clarification of the precise second messenger that is acting under physiological conditions has proved challenging.9,10
 | ||
| Fig. 1 (a) ADPR; (b–e) ADPR analogues modified in the pyrophosphate motif and (f) the targets 1 and 2 of the present work. | ||
Small changes to the structure of ADPR have demonstrated profound effects on activity and, as a result, synthetic analogues closely based on the ADPR structure have been very informative.11–13 Analogues with modifications in the adenosine11,13 and terminal ribose14–17 motifs have been prepared via coupling of a modified 5′-O-phosphoryl adenosine or ribose to its 5′-O-phosphoryl counterpart using morpholidate, diphenylphosphonate or imidazolide methods to activate one phosphate towards nucleophilic attack and thus generate the pyrophosphate. Notwithstanding their utility for studying ADPR processes using single cell techniques such as patch clamping, further studies even with many of these designed analogues are limited by the negative charge and potential biological instability of the pyrophosphate moiety. Such pyrophosphate-containing ADPR analogues are thus not suitable for use in whole cell studies or for in vivo applications. The cell permeable peptide tat-M2NX has been shown to inhibit TRPM2 and reduce ischemic injury in male mice,18 suggesting that membrane permeable analogues closely related to ADPR in structure would find similar utility.
The negative charge of ADPR may be reduced by masking the charged phosphate groups. Indeed, ADPR that was acetylated and bioreversibly masked with a lipophilic group on the adenosine phosphate has been reported.19 However, pyrophosphates masked in this way can be more unstable towards hydrolysis by nucleophiles than their negatively charged counterparts, and this does not address the issue of degradation due to cellular pyrophosphatases. The use of phosphate bioisosteres in medicinal chemistry is an alternative method to improve cellular stability and potentially also improve membrane permeability.20 Previous studies using pyrophosphate replacement in ADPR have provided some new insights into ADPR binding. The methylene bis-phosphonate analogue of ADPR (α-β methylene ADPR, AMPCPR, Fig. 1b) contains a non-hydrolysable P–C–P pyrophosphate substitution that is stable towards pyrophosphatases and has therefore generated a number of crystal structures of ADPR targets bound to AMPCPR.21 Tóth et al. showed that AMPCPR still supports TRPM2 channel gating, albeit as a lower affinity partial agonist.22 AMPCPR has been used to uncouple the TRPM2 channel gating mechanism from its enzymatic activity by virtue of its pyrophosphatase resistant methylene bis-phosphonate linker.22,23 Two further ADPR analogues with similarly modified pyrophosphates (one methylene bisphosphate and one difluoromethylene bisphosphate, see Fig. 1e) that were also protected on some or all of the ADPR hydroxyl groups selectively inhibited TRPM2.24 These hydrolysis resistant analogues demonstrate that modification of the pyrophosphate can impart valuable attributes to the ADPR analogue. However, such methylene bis-phosphonate analogues still carry two negative charges at physiological pH, and thus are not membrane permeable.
Our previous highly ambitious complete replacements of the pyrophosphate group in ADPR (Fig. 1c and d), that are unlikely to be charged at physiological pH, unfortunately did not show TRPM2 agonist or antagonist activity.11,13 We therefore pursued a more conservative approach in this study, designing two ADPR analogues 1 and 2 with only one phosphorus centre replaced (Fig. 1f). We chose the isosteric phosphonoacetate group as a pyrophosphate mimic between the adenosine and the terminal ribose of ADPR, as this analogue would only carry a single negative charge at physiological pH. A phosphonoacetate motif should also be resistant to the intrinsic cellular pyrophosphatase activity of some ADPR targets as the P–C bond is more chemically stable than the pyrophosphate P–O bond.25 Therefore, such analogues could potentially be used to probe ADPR-mediated activities in isolation from pyrophosphatase activity. The first phosphonoacetate analogues of the 5-O-pyrophosphate in diphosphoinositol polyphosphates (PP-InsPs) were recognised and phosphorylated by the kinase domain of diphosphoinositol pentakisphosphate kinase 2 (PPIP5K2), showing that such substitution of a pyrophosphate did not prevent recognition of the analogue by its target enzyme and allowing downstream effects unrelated to dephosphorylation to be studied.26
We therefore report here the synthesis of phosphonoacetate ADPR analogues 1 and 2, studies on the stability of both analogues and their initial biological evaluation at TRPM2.
Synthesis of ADPR analogues
Synthesis of adenosine-5′-O-(2-phosphoryl) acetate ribose (Fig. 1f, 1) started with commercially available triethylphosphonoacetate 3 (Scheme 1). Selective deprotection of the carboxylic ester in the presence of the phosphate esters27 led to the corresponding carboxylic acid 4 (97%). Carboxylic acid 4 was esterified, by N,N′-dicyclohexylcarbodiimide (DCC) mediated coupling, with 2′,3′-O-isopropylidene adenosine 11 to obtain compound 5 (82%). Diethylphosphonate ester 5 was then trans-silylated by bromotrimethylsilane (TMSBr), followed by hydrolysis to obtain phosphonic acid 6 (best yield 46%). Selective removal of the phosphonate esters in the presence of the isopropylidene group was achieved by carrying out the reaction in pyridine, as the basic conditions buffer the HBr generated. Carrying out the same conversion with DCM as a solvent generated a complex mixture due to concomitant deprotection of the isopropylidene protecting group. Purification of the free phosphate 6 was carried out using normal phase flash chromatography on silica gel. Impurities were eluted first with an isocratic solvent system of ethyl acetate, methanol and water (7:2:1 v/v/v) containing 0.1% triethylamine. The desired product 6 was then eluted by switching to a second isocratic solvent system of isopropyl alcohol, water and aqueous ammonia (7:2:1 v/v/v) containing 0.1% triethylamine. First attempts to obtain compound 7 via DCC mediated coupling to 1′-O-methyl-2′, 3′-O-isopropylidene-β-D-ribofuranoside 12 were unsuccessful. However, after a short optimization, we found that at elevated temperature (50–70 °C) compound 6 forms a mixture of anhydrides, including most likely adenosine phosphonoacetate anhydride28 which only reacts with 12 at temperatures above 100 °C. Thus, the higher temperature of 120 °C (compared to the DCC mediated reaction of 4 and 11 that occurred at rt) plays a crucial role in the conversion of 6 to 7. Finally, with the optimized conditions, the DCC mediated coupling of 6 with 1′-O-methyl-2′, 3′-O-isopropylidene-β-D-ribofuranoside 12 generated the desired ADPR analogue 7 (best yield 57%). Deprotection of 7 led to target compound 1, which was obtained after HPLC purification in 0.1 M TEAB buffer with a gradient of acetonitrile as the triethylammonium salt (36%). Until deprotection of analogue 7, all reactions are stereospecific. The final ADPR analogue 1 is a mixture of α and β anomers at the terminal ribose (ratio α:β = 2:3, as detailed in the Experimental section).
 | ||
| Scheme 1 Synthesis of phosphonoacetate ADPR analogue 1. Reagents and conditions: (a) NaOH, water, EtOH, 20 °C (97%); (b) DCC, DMAP (cat.), THF, 20 °C (82%); (c) TMSBr, pyridine, 20 °C, 16 h (46%); (d) DCC, pyridine, 120 °C, 16 h (57%); (e) aqueous TFA 75%, 2 h, 20 °C (36%). | ||
Synthesis of adenosine-5′-phosphonoacetyl ribose (Fig. 1f, 2) started from diethylphosphonoacetic acid 4 (Scheme 2), that was coupled to 1′-O-methyl-2′,3′-O-isopropylidene-β-D-ribofuranoside 12 using DCC, to give compound 8 (best yield 74%). Trans-silylation and hydrolysis of 8, followed by silica gel chromatography gave phosphonic acid 9 (68%) contaminated with trace impurities. Compound 9 was directly coupled to 2′,3′-O-isopropylidene adenosine 11 via previously optimized DCC coupling at elevated temperature 120 °C to give compound 10 (56%). Deprotection of 10 led to the final compound 2, which was purified several times using HPLC in 0.1 M TEAB buffer with a gradient of acetonitrile as the triethylammonium salt (best yield 67%). Until deprotection of analogue 10, all reactions are stereospecific. The final ADPR analogue 2 is a mixture of α and β anomers at the terminal ribose (ratio α:β = 2:3, as detailed in the Experimental section).
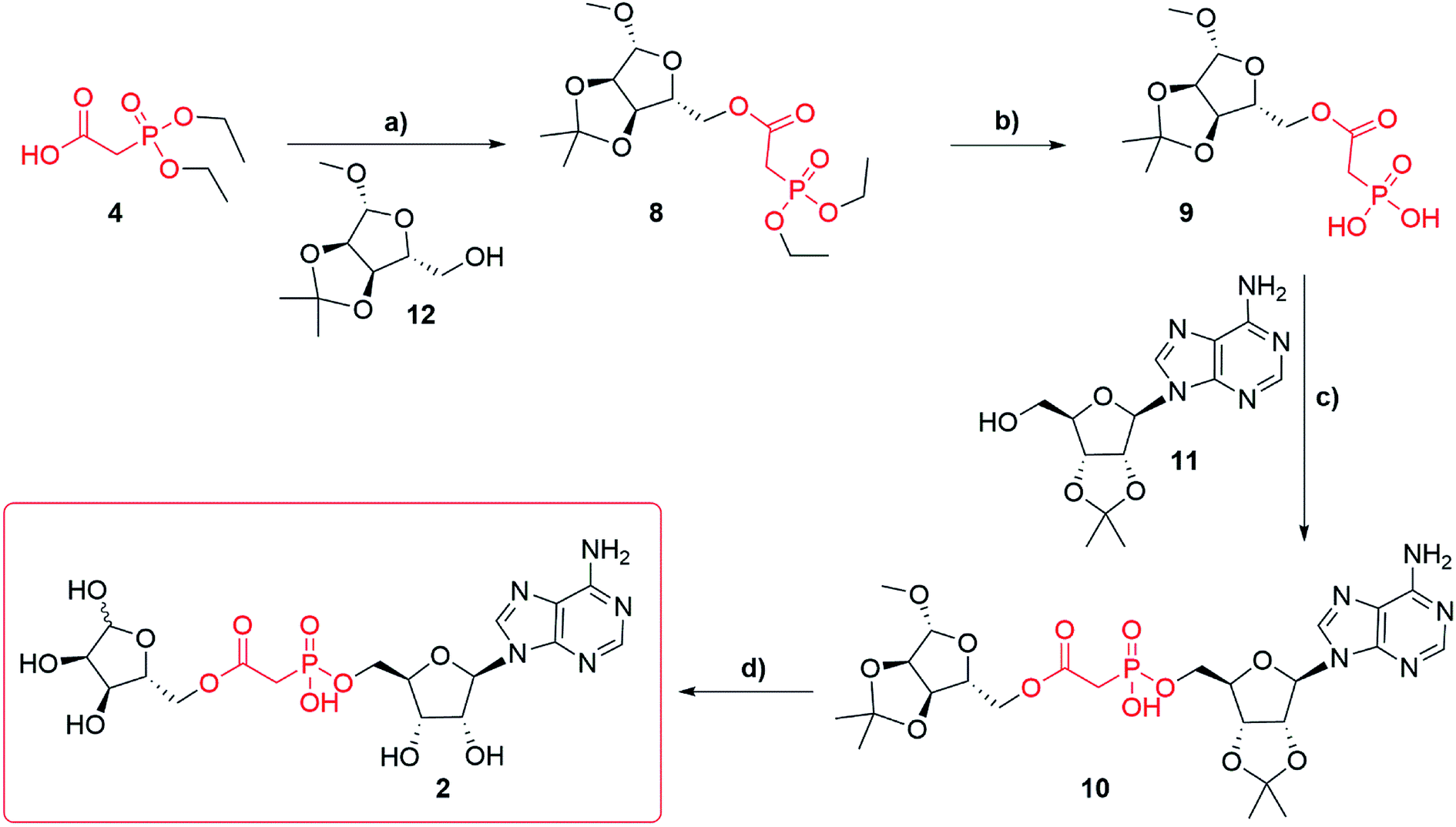 | ||
| Scheme 2 Synthesis of phosphonoacetate ADPR analogue 2. Reagents and conditions: (a) DCC, DMAP (cat.), THF, 20 °C (74%); (b) TMSBr, pyridine, 20 °C, 16 h (68%); (c) DCC, pyridine, 120 °C, 16 h (56%); (d) aqueous TFA 75%, 2 h, 20 °C (60%). | ||
Stability of target phosphonoacetates 1 and 2
The stability of 1 and 2 in solution was monitored by analytical HPLC, comparing freshly made samples from the solid compounds 1 and 2, stored at −20 °C. Both final analogues were synthesised as the corresponding triethylammonium salts (1.0 × Et3N and 1.2 × Et3N equivalents respectively, calculated from 1H-NMR spectra). While phosphonoacetate 1 is stable in this form in MilliQ water, TEAB (0.1 M) or MeCN solutions, we were surprised to observe that phosphonoacetate 2, degrades slowly in TEAB (pH 8–9, 0.1 M TEAB, ESI, Fig. S1†). HPLC of 2 (Rt = 3.9 minutes) after 16 h in 0.1 M TEAB buffer showed an additional peak at Rt = 2.7 minutes. This decomposition product showed a characteristic adenosine UV absorption (λmax = 259) and was analysed using mass spectrometry (ESI+ found [M − H]− 388.0676, C12H15N5O8P requires 388.0664) suggesting loss of the terminal ribose to generate adenosine 5′-phosphonoacetate (ESI, Fig. S1†). We suspect it is most likely that 2 undergoes basic hydrolysis of the terminal ribose from the phosphonoacetate ester mediated by triethylamine from the TEAB buffer.We then measured the stability of phosphonoacetate 2 under various aqueous conditions (Fig. 2). According to these findings, analogue 2 was stable in pure MilliQ water (pH 7) as the triethylammonium salt when stored in the fridge. Importantly, this should be sufficient for biological assays using the pure phosphonoacetate 2 when used immediately from a frozen sample. Increased pH 8–9 (in 0.1 M TEAB buffer) resulted in slow decomposition of 2 either at room temperature or when stored in the fridge. We prepared the sodium salt of analogue 2 in an attempt to overcome the possibility of triethylammonium counter ions generating a basic solution and thus causing the decomposition of 2. The triethylammonium salt of 2 was stirred with Chelex (Na+ form) resin, which was removed by filtration to generate the sodium salt of 2. However, we were surprised to find that the corresponding sodium salt of 2 was even more unstable and degraded completely either at room temperature or when stored in the fridge (Fig. 2).
 | ||
| Fig. 2 Stability of compound 2 under various aqueous conditions. | ||
It was rather unexpected that ADPR analogue 1 is stable under various aqueous conditions whereas ADPR analogue 2 is not. This led us to consider which feature of 2 might be responsible for the observed degradation. Our observations suggest that 2 is sensitive to basic hydrolysis of the phosphonoacetate ester in TEAB buffer but that, under the same conditions, the phosphonoacetate ester of 1 is stable. This suggests that the terminal ribose 5′′-O-ester may influence the stability in a way that is not observed with the adenosine ribose 5′-O-ester. The triethylamine base present in TEAB buffer may catalyse hydrolysis of the ester bond between the phosphonoacetate ester and the 5′′-O-terminal ribose. Indeed, we have previously observed that modifications to the terminal ribose can generate ADPR analogues that are particularly prone to instability.14 Analogue 1 is not susceptible to hydrolysis because the more stable phosphonate monoester is adjacent to the terminal ribose. Phosphate monoesters are stable under basic conditions due to the negative charge on the phosphonate oxygen preventing nucleophilic attack. Presumably, the 5′-O-ester in analogue 1 is more sterically hindered, due to its proximity to the less flexible adenosine, and thus is less susceptible to hydrolysis under mildly basic conditions. In future, synthesis of the attractive corresponding phosphonamide ADPR analogues in which the ester is replaced by an amide linker, may be one method to ensure stable analogues are generated, as exemplified in a recent synthesis of an inositol polyphosphate pyrophosphate analogue.29
Biological evaluation of 1 and 2
Due to our stability studies, we were aware of the possibility of degradation of the unstable ester bond of compound 2. This would have been undesirable in our assay to determine the agonist and antagonist effects of 2. In order to prevent degradation, the compounds were reconstituted using MilliQ water (pH 7), which was shown to generate a stable solution of both compounds (Fig. 2). The assay was carried out at pH 7.2. Immediately before the assay for agonist or antagonist effects, the solution of the analogue to be tested was verified by HPLC. No degradation was detected during the entire sequence of testing, or even when the pH 7 solution of the analogue was stored at −80 °C for 1 year.ADPR analogues 1 and 2 were tested for their agonist and antagonist activity on human TRPM2 (hTRPM2) using patch clamp experiments in HEK293 clones with stable expression of hTRPM2.13 Because the compound is infused into the cytoplasm of the cell via the patch-pipette, this method allows evaluation of the activity of the compounds independently of their membrane permeability. Following the results of our stability testing, phosphonoacetate 2 was stored as a lyophilized dry solid at −20 °C and made up in fresh MilliQ water immediately before testing to ensure that decomposition did not occur (vide supra). When applied at 100 μM or 1000 μM, neither 1 nor 2 had significant agonist effects at hTRPM2 (Fig. 3a). When applied with ADPR in the patch pipette (100 μM of ADPR with 300 μM or 900 μM 1 or 2), neither analogue showed significant antagonist effects on the ADPR-induced TRPM2 current (Fig. 3b). If activity was observed, we would have determined the membrane permeability of 1 and 2 by applying them extracellularly to whole cells, however this was not possible.
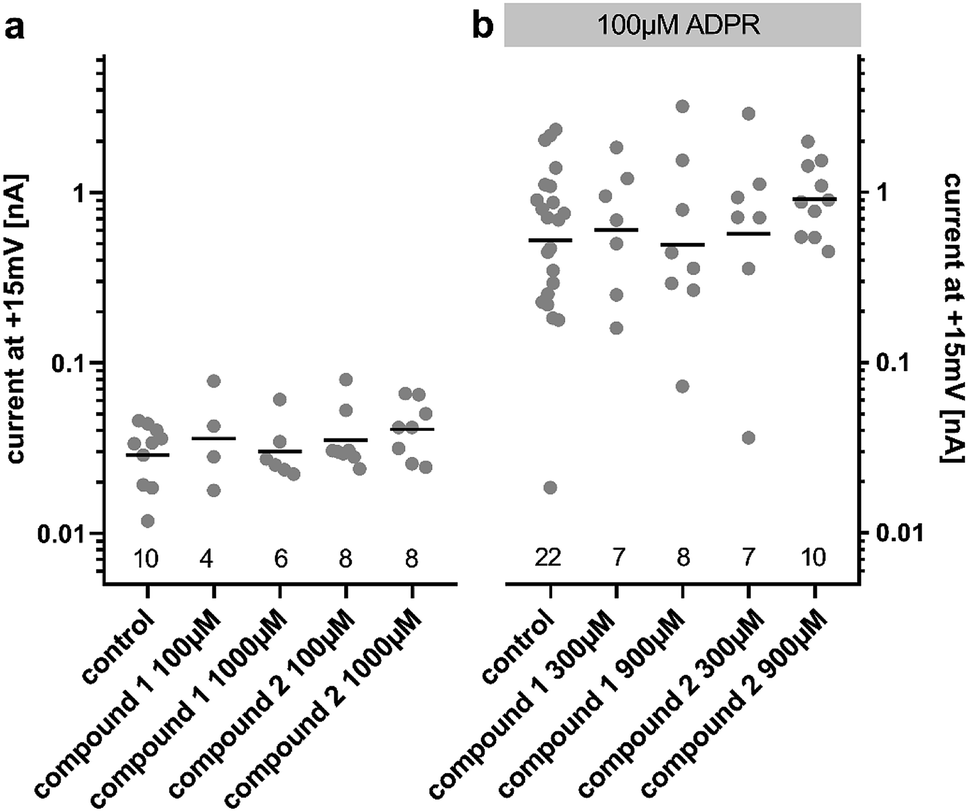 | ||
| Fig. 3 Effect of 1 and 2 on whole cell currents in hTRPM2 expressing HEK293 cells. Electrophysiological analysis of the impact of ADPR analogues on human TRPM2. (a) During whole cell patch clamp experiments using HEK293 cells with stable expression of the ion channel, the analogues were included in the pipette solution at 100 μmol L−1 or 1000 μmol L−1. The maximum outward current at +15 mV over a time of 450 s from individual cells is plotted at a logarithmic scale with a horizontal bar indicating the mean value. Neither compound did result in a significantly higher current than the buffer control. (b) To check for potential inhibition of TRPM2 currents from activation by ADPR, the compounds were added at 300 μmol L−1 or 900 μmol L−1 to a pipette solution already containing 100 μmol L−1 ADPR. Neither compound reduced the TRPM2 current significantly when compared to the control (pipette solution containing only ADPR). The numbers at the bottom indicate the number of cells analysed. | ||
These results highlight the sensitivity of hTRPM2 channel activation to modifications of the ADPR molecular structure. It is possible that both phosphate motifs of the pyrophosphate bridge are required for binding and activation or inhibition of the channel, so that even a subtle change in this part of the molecule may lead to the complete loss of activity. The single atom substitution in the methylene bis-phosphonate analogue AMPCPR corresponded to a roughly 40 fold reduction in potency at hTRPM2 and AMPCPR is also only a partial hTRPM2 agonist.22 Alternatively the substitution of the pyrophosphate with the phosphonoacetate bioisostere may alter the presentation of other critical binding partners in the hTRPM2 binding pocket. Our molecular modelling suggests the phosphonoacetate linker theoretically seems to be a very good spatial mimic of the pyrophosphate unit, with sufficient flexibility to allow the adenosine and terminal ribose binding partners to position themselves in the same orientation as ADPR (ESI, Fig. S2†), but this may not reflect the reality once solvent effects and binding interactions are taken into account.
Cryo-EM structures of hTRPM2 have recently improved understanding of the zebrafish hTRPM2 apo and ADPR-bound states30 importantly for hTRPM2 also in the presence of bound ligands.31,32 However, only recently has their resolution become detailed enough to place ADPR in the binding pocket and has revealed, for the first time, ADPR binding sites in hTRPM2 that may act synergistically.32 The requirement for an ADPR mimic to bind to both sites, which possess very distinct shapes,30 may explain why very few synthetic ADPR analogues have activated the channel.11,15 Despite this recent advance, the structure activity relationship for hTRPM2 is not fully understood, and ADPR analogues remain essential for ligand-based drug design.
The crystal structure of the macro domain of thermophilic protein Af1521 bound to ADPR was solved in high resolution (1.5 Å, PDB 2BFQ).6 With the exception of the adenosine ribose, each ADPR structural feature interacts closely with the macro domain structure. The adenosine 5′-O-phosphate has two hydrogen bonds to the backbone N–H of valine 43 and glycine 143. The ribose 5′-O-phosphate has hydrogen bonds to the backbone N–H of serine 141 and tyrosine 145. Furthermore, the pyrophosphate is stabilized by the helix dipole. Thus, the demonstrated highly specific binding interactions of macro domains and apparent similar selectivity of hTRPM2 binding require very specific ADPR binding partners. Our initial results with hTRPM2 suggest that phosphonoacetate analogues 1 and 2 may not be able to fulfil all these demands, but 1 and 2 may potentially find applicability in other systems that bind ADPR.
Conclusion
We have synthesised two phosphonoacetate analogues of ADPR, in which the pyrophosphate is substituted by a phosphonoacetate linker. These analogues retain all other ADPR features but would be predicted to carry only a single negative charge at physiological pH. Suitably protected ribose and adenosine building blocks were sequentially coupled to the phosphonoacetate core by carbodiimide-mediated coupling, followed by a global deprotection to give phosphonoacetates 1 and 2. Analogue 2, in which the phosphonoacetate ester is attached to the terminal ribose 5′′-hydroxyl, has unexpected instability in TEAB buffer. Future efforts would seek to avoid using ester links in such analogues, but likely employ amide linkages. Neither analogue showed any agonist activity at hTRPM2, nor antagonist effects on the ADPR-induced current at TRPM2, suggesting the full pyrophosphate motif is required for activity and adding to the growing picture of a highly specific binding interaction with many requisite ADPR features.Experimental section
General
Reagents and solvents were purchased from commercial sources and used without further purification, unless described otherwise. Triethylamine was dried with potassium hydroxide, purified by distillation, and stored over potassium hydroxide. MilliQ quality water was used for all aqueous experiments and purifications. All reactions were performed under an inert atmosphere of argon unless otherwise stated. For NMR experiments; 1H, 13C, and 31P NMR spectra were collected using either a Varian Mercury 400 MHz or Bruker Avance III 500 MHz Spectrometer. All 1H and 13C NMR assignments are based on COSY, HSQC, HMBC, and DEPT experiments. Chemical shifts (δ) are reported in parts per million (ppm) and splitting patterns are abbreviated as follows: br, broad; s, singlet; d, doublet; t, triplet; m, multiplet etc. High resolution mass spectra (HRMS) were obtained on a Bruker Daltonics micrOTOF mass spectrometer with electrospray ionisation (ESI). Analytical HPLC was performed on a Waters 2695 Alliance module coupled to a Waters 2996 PDA Detector (210–350 nm) equipped with Hichrom Guard Column for HPLC and a Phenomenex Synergi column (4u, MAX-RP, 80 Å, 150 × 4.60 mm), eluted at 1 mL min−1 with a gradient of MeCN in 0.05 M TEAB. Semi-preparative HPLC was performed on a Waters 2525 pump with manual FlexInject using a Phenomenex Gemini column (5u, C18, 110 Å, 250 × 10.00 mm), eluted at 5 mL min−1.:4 v/v) and a gradient of MeOH (0–10%). This procedure afforded the title compound 5 as colourless glass (0.98 g, 62%). 1H NMR (400 MHz, d6-DMSO) δ 8.34 (s, H-8), 8.16 (s, H-2), 7.34 (s, 2H, NH2), 6.19 (d, 1H, J = 2.68 Hz, H-1′), 5.47 (dd, 1H, J = 6.0 Hz, J = 2.96 Hz, H-2′), 5.04 (dd, 1H, J = 6.4 Hz, J = 3.09 Hz, H-3′), 4.39–4.35 (m, 1H, H-4′), 4.31–4.27 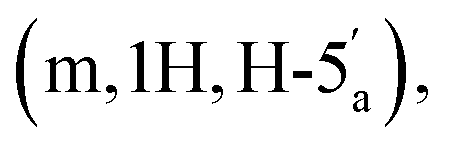 4.23–4.18
4.23–4.18  4.04–3.95 (m, 4H, CH2CH3), 3.11 (d, 2H, JH–P = 21.36 Hz, CH2P), 1.54 (s, 3H, C(CH3)2), 1.33 (s, 3H, C(CH3)2), 1.18 (m, 6H, J = 3.16 Hz, CH2CH3). 31P NMR (161 MHz, d6-DMSO) δ 19.64 (s). 13C NMR (100 MHz, d6-DMSO) δ 165.44 (COO), 156.14 (C-6), 152.75 (C-2), 148.81 (C-4), 139.83 (C-8), 119.08 (C-5), 113.49 (C(CH3)2), 89.00 (C-1′), 83.31 (C-4′), 83.08 (C-2′), 81.02 (C-3′), 64.56 (C-5′), 62.05 and 61.99 (CH2CH3), 33.22 (d, JC–P = 130.57 Hz, C–P), 26.99 and 25.19 (C(CH3)2), 16.06 and 16.01 (CH2CH3). HRMS (ES+) calcd for C19H29N5O8P 486.1748, found [M + H]+ 486.1789.:2:1 v/v/v + 0.1% Et3N) and the product was eluted using iPrOH–H2O–aq. ammonia (7:2:1 v/v/v + 0.1% Et3N). This procedure afforded the title compound 6 as a white foam, as the mono-triethylammonium salt (179 mg, 30%). 1H NMR (400 MHz, d6-DMSO) δ 8.59 (s, 1H, H-8), 8.16 (s, 1H, H-2), 7.33 (s, 2H, NH2), 6.13 (d, 1H, J = 3.16 Hz, H-1′), 5.54 (dd, 1H, J = 6.4 Hz, J = 3.2 Hz, H-2′), 5.10 (dd, 1H, J = 6.0 Hz, J = 3.0 Hz, H-3′), 4.39 (m, 1H, H-4′), 4.22
4.04–3.95 (m, 4H, CH2CH3), 3.11 (d, 2H, JH–P = 21.36 Hz, CH2P), 1.54 (s, 3H, C(CH3)2), 1.33 (s, 3H, C(CH3)2), 1.18 (m, 6H, J = 3.16 Hz, CH2CH3). 31P NMR (161 MHz, d6-DMSO) δ 19.64 (s). 13C NMR (100 MHz, d6-DMSO) δ 165.44 (COO), 156.14 (C-6), 152.75 (C-2), 148.81 (C-4), 139.83 (C-8), 119.08 (C-5), 113.49 (C(CH3)2), 89.00 (C-1′), 83.31 (C-4′), 83.08 (C-2′), 81.02 (C-3′), 64.56 (C-5′), 62.05 and 61.99 (CH2CH3), 33.22 (d, JC–P = 130.57 Hz, C–P), 26.99 and 25.19 (C(CH3)2), 16.06 and 16.01 (CH2CH3). HRMS (ES+) calcd for C19H29N5O8P 486.1748, found [M + H]+ 486.1789.:2:1 v/v/v + 0.1% Et3N) and the product was eluted using iPrOH–H2O–aq. ammonia (7:2:1 v/v/v + 0.1% Et3N). This procedure afforded the title compound 6 as a white foam, as the mono-triethylammonium salt (179 mg, 30%). 1H NMR (400 MHz, d6-DMSO) δ 8.59 (s, 1H, H-8), 8.16 (s, 1H, H-2), 7.33 (s, 2H, NH2), 6.13 (d, 1H, J = 3.16 Hz, H-1′), 5.54 (dd, 1H, J = 6.4 Hz, J = 3.2 Hz, H-2′), 5.10 (dd, 1H, J = 6.0 Hz, J = 3.0 Hz, H-3′), 4.39 (m, 1H, H-4′), 4.22 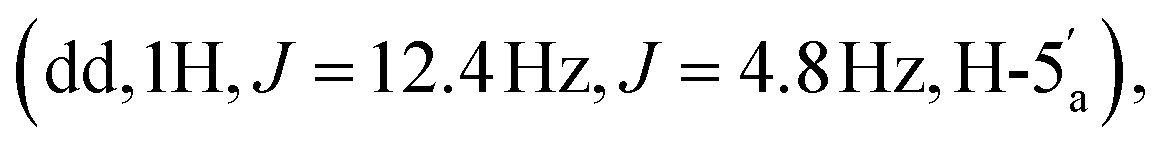 4.06
4.06  2.89 (q, 6H, J = 7.25, NCH2CH3), 2.53 (dq, 2H, JH–P = 22.4 Hz, Jgem = 8.0 Hz, CH2P), 1.53 (s, 3H, C(CH3)2), 1.18 (s, 3H, C(CH3)2), 1.11 (t, 6H, J = 7.26 Hz, NCH2CH3). 31P NMR (161 MHz, d6-DMSO) δ 8.14 (s). 13C NMR (100 MHz, d6-DMSO) δ 176.66 (COO) HMBC, 156.06 (C-6), 152.76 (C-2), 149.13 (C-4), 139.84 (C-8), 118.79 (C-5), 113.27 (C(CH3)2), 88.69 (C-1′), 83.40 (C-4′), 83.16 (C-2′), 81.01 (C-3′), 63.52 (C-5′), 45.05 (NCH2CH3), (d, 38.52 and 38.02, very low intensity, CH2P signal partially hidden by the DMSO signal, estimated from HSQC), 27.02 and 25.20 (C(CH3)2), 8.70 (NCH2CH3). HRMS (ES−) calcd for C15H19N5O8P 428.0977, found [M − H]− 428.0985.
2.89 (q, 6H, J = 7.25, NCH2CH3), 2.53 (dq, 2H, JH–P = 22.4 Hz, Jgem = 8.0 Hz, CH2P), 1.53 (s, 3H, C(CH3)2), 1.18 (s, 3H, C(CH3)2), 1.11 (t, 6H, J = 7.26 Hz, NCH2CH3). 31P NMR (161 MHz, d6-DMSO) δ 8.14 (s). 13C NMR (100 MHz, d6-DMSO) δ 176.66 (COO) HMBC, 156.06 (C-6), 152.76 (C-2), 149.13 (C-4), 139.84 (C-8), 118.79 (C-5), 113.27 (C(CH3)2), 88.69 (C-1′), 83.40 (C-4′), 83.16 (C-2′), 81.01 (C-3′), 63.52 (C-5′), 45.05 (NCH2CH3), (d, 38.52 and 38.02, very low intensity, CH2P signal partially hidden by the DMSO signal, estimated from HSQC), 27.02 and 25.20 (C(CH3)2), 8.70 (NCH2CH3). HRMS (ES−) calcd for C15H19N5O8P 428.0977, found [M − H]− 428.0985. 4.15–4.11 (m, 1H, H-4′′), 4.06–4.02
4.15–4.11 (m, 1H, H-4′′), 4.06–4.02 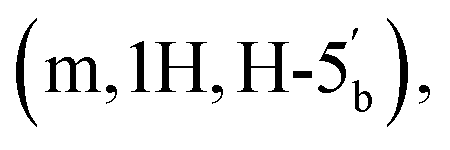 3.66–3.57
3.66–3.57 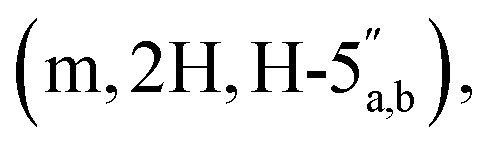 3.53 (q, 0.7H, J = 6.32 Hz, NCH2CH3), 3.19 (s, 3H, CH3O), 2.60–2.47 (m, 2H, CH2P), 1.53 and 1.33 (2 × s, 2 × 3H, C(CH3)2-Adn), 1.36 and 1.23 (2 × s, 2 × 3H, C(CH3)2-Rib). 31P NMR (161 MHz, d6-DMSO) δ 7.60 (s). 13C NMR (100 MHz, d6-DMSO) δ 169.60 (COO) HMBC, 156.04 (C-6), 152.76 (C-2), 149.13 (C-4), 140.15 (C-8), 118.75 (C-5), 113.23 (Adn C(CH3)2), 111.22 (Rib C(CH3)2), 108.45 (C-1′′), 88.75 (C-1′), 85.18 (C-4′′), 84.42 (C-3′′), 83.52 (C-4′), 83.30 (C-2′), 81.46 (C-2′′), 81.04 (C-3′), 64.00 (C-5′′), 63.50 (C-5′), 54.15 (CH3O), 36.32 (d, J = 112.0 Hz, CH2P a and b, HMBC), 27.03 and 25.21 (C(CH3)2-Adn), 26.22 and 24.61 (C(CH3)2-Rib). HRMS (ES−) calcd for C24H33N5O12P 614.1869, found [M − H]− 614.1885. Analytical HPLC, Rt = 8.62 min.:5 → 35/65 v/v). The title compound 1 was afforded as colourless glass, as a mixture of α and β anomers in the ratio α:β = 2:3, (3.1 mg, 36%, ⅓ Et3N salt). DOSY 1H NMR (water suppression, 500 MHz, D2O) δ 8.32 (s, 1H, H-8), 8.18 (s, 1H, H-2), 6.03 (d, 1H, J = 4.5 Hz, H-1′), 5.16 (d, 0.4H, J = 4.0 Hz, H-1′′α), 5.10 (d, 0.6H, J = 2.5 Hz, H-1′′β), 4.77 (t, 1H, obscured by D2O peak, J = 2.5 Hz, H-2′), 4.45 (t, 1H, J = 5.0 Hz, H-3′), 4.41–4.32 (m, 3H, H-4′, 5′), 4.12 (t, 0.6H, J = 5.0 Hz, H-3′′β), 4.07–4.04 (m, 0.4H, H-4′′α), 4.01–3.75
3.53 (q, 0.7H, J = 6.32 Hz, NCH2CH3), 3.19 (s, 3H, CH3O), 2.60–2.47 (m, 2H, CH2P), 1.53 and 1.33 (2 × s, 2 × 3H, C(CH3)2-Adn), 1.36 and 1.23 (2 × s, 2 × 3H, C(CH3)2-Rib). 31P NMR (161 MHz, d6-DMSO) δ 7.60 (s). 13C NMR (100 MHz, d6-DMSO) δ 169.60 (COO) HMBC, 156.04 (C-6), 152.76 (C-2), 149.13 (C-4), 140.15 (C-8), 118.75 (C-5), 113.23 (Adn C(CH3)2), 111.22 (Rib C(CH3)2), 108.45 (C-1′′), 88.75 (C-1′), 85.18 (C-4′′), 84.42 (C-3′′), 83.52 (C-4′), 83.30 (C-2′), 81.46 (C-2′′), 81.04 (C-3′), 64.00 (C-5′′), 63.50 (C-5′), 54.15 (CH3O), 36.32 (d, J = 112.0 Hz, CH2P a and b, HMBC), 27.03 and 25.21 (C(CH3)2-Adn), 26.22 and 24.61 (C(CH3)2-Rib). HRMS (ES−) calcd for C24H33N5O12P 614.1869, found [M − H]− 614.1885. Analytical HPLC, Rt = 8.62 min.:5 → 35/65 v/v). The title compound 1 was afforded as colourless glass, as a mixture of α and β anomers in the ratio α:β = 2:3, (3.1 mg, 36%, ⅓ Et3N salt). DOSY 1H NMR (water suppression, 500 MHz, D2O) δ 8.32 (s, 1H, H-8), 8.18 (s, 1H, H-2), 6.03 (d, 1H, J = 4.5 Hz, H-1′), 5.16 (d, 0.4H, J = 4.0 Hz, H-1′′α), 5.10 (d, 0.6H, J = 2.5 Hz, H-1′′β), 4.77 (t, 1H, obscured by D2O peak, J = 2.5 Hz, H-2′), 4.45 (t, 1H, J = 5.0 Hz, H-3′), 4.41–4.32 (m, 3H, H-4′, 5′), 4.12 (t, 0.6H, J = 5.0 Hz, H-3′′β), 4.07–4.04 (m, 0.4H, H-4′′α), 4.01–3.75 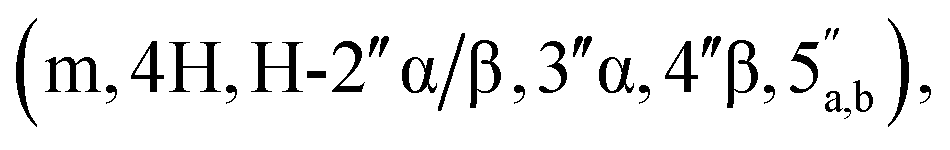 3.12 (q, 2H, J = 7.0 Hz, NCH2CH3), 2.81 and 2.79 (2 × dd, 2H, JH–P = 15.5 and 16 Hz, Jgem = 5.0 and 4.5 Hz, CH2P α, β), 1.20 (t, 3H, J = 7.0 Hz, NCH2CH3). 31P NMR (203 MHz, D2O) δ 14.40 (s), 14.30 (s). 13C NMR (126 MHz, D2O) δ 169.70 (COO) HMBC, 155.53 (C-6), 152.73 (C-2), 149.02 (C-4), 139.99 (C-8), 118.87 (C-5), 101.05 (C-1′′β), 96.28 (C-1′′α), 87.58 (C-1′), 81.85 and 81.16 (C-4′, 4′′α and β), 75.06 (C-2′′β), 73.44 (C-2′), 70.65, 70.44, 70.00, 69.85 (C-2′′α, 3′′α/β, 3′), 65.31 (d, J = 4.5 Hz, C-5′′), 64.00 (d, J = 5.5 Hz, C-5′), 46.65 (NCH2CH3), 35.03 and 34.34 (d, JC–P = 121.4 Hz, CH2P), 35.27 and 34.29 (d, JC–P = 122.1 Hz, CH2P), 8.19 (NCH2CH3). HRMS (ES−) calcd for C17H23N5O12P 520.1086, found [M − H]− 520.1097. Analytical HPLC: Rt = 4.14 min.:0 → 0:1 v/v) to afford the title compound 8 as a colourless glass (112 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 4.96 (s, 1H, H-1), 4.67 (d, 1H, J = 6.0 Hz, H-2), 4.59 (d, 1H, J = 6.0 Hz, H-3), 4.35 (t, 1H, J = 7.2 Hz, H-4), 4.21–4.13 (m, 6H, H-5, CH2CH3), 3.31 (s, 3H, OCH3), 2.98 (d, 2H, J = 21.6, CH2P), 1.46 (s, 3H, C(CH3)2), 1.34 (t, 6H, J = 7.2 Hz, CH2CH3), 1.30 (s, 3H, C(CH3)2). 31P NMR (161 MHz, CDCl3) δ 19.22. 13C NMR (100 MHz, CDCl3) δ 165.72 (COO), 112.90 (C-(CH3)2), 109.79 (C-1), 85.50 (C-3), 84.29 (C-4), 82.15 (C-2), 65.88 (C-5), 63.19 (d, JC–P = 6.06 Hz, CH2CH3), 55.34 (OCH3), 34.67 (d, JC–P = 133.79 Hz, CH2P), 26.73 (CH3), 25.29 (CH3), 16.70 (CH2CH3). HRMS (ES+) calcd for C15H28O9P 383.1465, found [M + H]+ 383.1474.:2:1 v/v/v + 0.1% Et3N) and the product was eluted using iPrOH–H2O–aq. ammonia (7:2:1 v/v/v + 0.1% Et3N) to afford phosphonic acid 9 (603 mg, 68%, mono-triethylammonium salt) which was used for the next reaction without any further purification. 1H NMR (400 MHz, d6-DMSO) δ 4.91 (s, 1H, H-1), 4.75 (d, 1H, J = 7.5 Hz, H-2), 4.57 (d, 1H, J = 7.5 Hz, H-3), 4.22 (t, 1H, J = 9.0 Hz, H-4), 3.98–3.86 (m, 2H, H-5), 3.22 (s, 3H, MeO), 2.78 (q, 2H, J = 9.0 Hz, CH2P), 1.37 (s, 3H, C(CH3)2), 1.25 (s, 3H, C(CH3)2). 31P NMR (161 MHz, d6-DMSO) δ 8.36 (s). 13C NMR (400 MHz, d6-DMSO) δ 168.51 (d, J = 6.5 Hz, COO), 111.44 (C(CH3)2), 108.73 (C-1), 84.44 (C-3), 83.37 (C-4), 81.23 (C-2), 64.03 (C-5), 54.23 (MeO), 44.82 (CH2P), 26.20 and 24.63 (2 × C(CH3)2). HRMS (ES−) calcd for C11H18O9P 325.0694, found [M − H]− 325.0702.:1 → 6:4 v/v + 0.1% Et3N) to afford the title compound 10 as a white solid (52 mg, 56%). 1H NMR (500 MHz, D2O) δ 8.41 (s, 1H, H-8), 8.24 (s, 1H, H-2), 6.27 (d, 1H, J = 3.0 Hz, H-1′), 5.43–5.41 (m, 1H, H-2′), 5.20 (dd, 1H, J = 5.0 Hz, J = 2.0 Hz, H-3′), 4.93 (s, 1H, H-1′′), 4.71–4.66 (m, 1H, H-4′), 4.58 (d, J = 6.0 Hz, H-2′′), 4.43 (d, 1H, J = 6.0 Hz, H-3′′), 4.18–4.12 (m, 3H, H-4′′, 5′), 4.00–32.96 (m, 1H, H-5′′a), 3.86–3.81 (m, 1H, H-5′′b), 3.23 (s, 3H, CH3O), 2.84 (dq, 2H, JH–P = 20.5 Hz, J = 13.5 Hz, Jgem = 5.0 Hz, CH2P), 1.68 (s, 3H, C(CH3)2-Adn), 1.40 (s, 3H, C(CH3)2-Adn), 1.46 (s, 3H, C(CH3)2-Rib), 1.27 (s, 3H, C(CH3)2-Rib). 31P NMR (161 MHz, D2O) δ 14.11 (s). 13C NMR (126 MHz, D2O) δ 169.17 (d, J = 6.8 Hz, COO), 155.56 (C-6), 152.79 (C-2), 148.78 (C-4), 139.86 (C-8), 118.55 (C-5), 114.76 (C(CH3)2-Adn), 112.96 (C(CH3)2-Rib), 108.69 (C-1′′), 90.16 (C-1′), 84.90 (d, J = 8.78 Hz, C-4′), 84.06 (C-3′′), 83.96 (C-2′), 83.40 (C-4′′), 81.46 (C-3′), 80.94 (C-2′′), 65.17 (C-5′′), 64.80 (d, J = 5.41 Hz, C-5′), 54.69 (CH3O), 34.89 and 33.93 (d, JC–P = 120.5 Hz, CH2P), 26.14 and 24.35 (2 × C(CH3)2-Adn), 25.06 and 23.41 (2 × C(CH3)2-Rib); HRMS (ES−) calcd for C24H33N5O12P 614.1869, found [M − H]− 614.1889.:5 → 35:65 v/v) to afford the title compound 2 as a colourless glass, as a mixture of α and β anomers α/β = 2/3 (7.6 mg, 60%, ⅓ Et3N salt). DOSY 1H NMR (water suppression, 500 MHz, D2O) δ 8.37 (s, 1H, H-8), 8.15 (s, 1H, H-2), 6.04 (d, 1H, J = 5.5 Hz, H-1′), 5.21–5.19 (m, 0.4H, H-1′′α), 5.11 (d, 0.6H, J = 1.5 Hz, H-1′′β), 4.67 (t, 1H, J = 5.0 Hz, H-2′), 4.44–4.40 (m, 1H, H-3′), 4.31–4.27 (m, 1H, H-4′), 4.24
3.12 (q, 2H, J = 7.0 Hz, NCH2CH3), 2.81 and 2.79 (2 × dd, 2H, JH–P = 15.5 and 16 Hz, Jgem = 5.0 and 4.5 Hz, CH2P α, β), 1.20 (t, 3H, J = 7.0 Hz, NCH2CH3). 31P NMR (203 MHz, D2O) δ 14.40 (s), 14.30 (s). 13C NMR (126 MHz, D2O) δ 169.70 (COO) HMBC, 155.53 (C-6), 152.73 (C-2), 149.02 (C-4), 139.99 (C-8), 118.87 (C-5), 101.05 (C-1′′β), 96.28 (C-1′′α), 87.58 (C-1′), 81.85 and 81.16 (C-4′, 4′′α and β), 75.06 (C-2′′β), 73.44 (C-2′), 70.65, 70.44, 70.00, 69.85 (C-2′′α, 3′′α/β, 3′), 65.31 (d, J = 4.5 Hz, C-5′′), 64.00 (d, J = 5.5 Hz, C-5′), 46.65 (NCH2CH3), 35.03 and 34.34 (d, JC–P = 121.4 Hz, CH2P), 35.27 and 34.29 (d, JC–P = 122.1 Hz, CH2P), 8.19 (NCH2CH3). HRMS (ES−) calcd for C17H23N5O12P 520.1086, found [M − H]− 520.1097. Analytical HPLC: Rt = 4.14 min.:0 → 0:1 v/v) to afford the title compound 8 as a colourless glass (112 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 4.96 (s, 1H, H-1), 4.67 (d, 1H, J = 6.0 Hz, H-2), 4.59 (d, 1H, J = 6.0 Hz, H-3), 4.35 (t, 1H, J = 7.2 Hz, H-4), 4.21–4.13 (m, 6H, H-5, CH2CH3), 3.31 (s, 3H, OCH3), 2.98 (d, 2H, J = 21.6, CH2P), 1.46 (s, 3H, C(CH3)2), 1.34 (t, 6H, J = 7.2 Hz, CH2CH3), 1.30 (s, 3H, C(CH3)2). 31P NMR (161 MHz, CDCl3) δ 19.22. 13C NMR (100 MHz, CDCl3) δ 165.72 (COO), 112.90 (C-(CH3)2), 109.79 (C-1), 85.50 (C-3), 84.29 (C-4), 82.15 (C-2), 65.88 (C-5), 63.19 (d, JC–P = 6.06 Hz, CH2CH3), 55.34 (OCH3), 34.67 (d, JC–P = 133.79 Hz, CH2P), 26.73 (CH3), 25.29 (CH3), 16.70 (CH2CH3). HRMS (ES+) calcd for C15H28O9P 383.1465, found [M + H]+ 383.1474.:2:1 v/v/v + 0.1% Et3N) and the product was eluted using iPrOH–H2O–aq. ammonia (7:2:1 v/v/v + 0.1% Et3N) to afford phosphonic acid 9 (603 mg, 68%, mono-triethylammonium salt) which was used for the next reaction without any further purification. 1H NMR (400 MHz, d6-DMSO) δ 4.91 (s, 1H, H-1), 4.75 (d, 1H, J = 7.5 Hz, H-2), 4.57 (d, 1H, J = 7.5 Hz, H-3), 4.22 (t, 1H, J = 9.0 Hz, H-4), 3.98–3.86 (m, 2H, H-5), 3.22 (s, 3H, MeO), 2.78 (q, 2H, J = 9.0 Hz, CH2P), 1.37 (s, 3H, C(CH3)2), 1.25 (s, 3H, C(CH3)2). 31P NMR (161 MHz, d6-DMSO) δ 8.36 (s). 13C NMR (400 MHz, d6-DMSO) δ 168.51 (d, J = 6.5 Hz, COO), 111.44 (C(CH3)2), 108.73 (C-1), 84.44 (C-3), 83.37 (C-4), 81.23 (C-2), 64.03 (C-5), 54.23 (MeO), 44.82 (CH2P), 26.20 and 24.63 (2 × C(CH3)2). HRMS (ES−) calcd for C11H18O9P 325.0694, found [M − H]− 325.0702.:1 → 6:4 v/v + 0.1% Et3N) to afford the title compound 10 as a white solid (52 mg, 56%). 1H NMR (500 MHz, D2O) δ 8.41 (s, 1H, H-8), 8.24 (s, 1H, H-2), 6.27 (d, 1H, J = 3.0 Hz, H-1′), 5.43–5.41 (m, 1H, H-2′), 5.20 (dd, 1H, J = 5.0 Hz, J = 2.0 Hz, H-3′), 4.93 (s, 1H, H-1′′), 4.71–4.66 (m, 1H, H-4′), 4.58 (d, J = 6.0 Hz, H-2′′), 4.43 (d, 1H, J = 6.0 Hz, H-3′′), 4.18–4.12 (m, 3H, H-4′′, 5′), 4.00–32.96 (m, 1H, H-5′′a), 3.86–3.81 (m, 1H, H-5′′b), 3.23 (s, 3H, CH3O), 2.84 (dq, 2H, JH–P = 20.5 Hz, J = 13.5 Hz, Jgem = 5.0 Hz, CH2P), 1.68 (s, 3H, C(CH3)2-Adn), 1.40 (s, 3H, C(CH3)2-Adn), 1.46 (s, 3H, C(CH3)2-Rib), 1.27 (s, 3H, C(CH3)2-Rib). 31P NMR (161 MHz, D2O) δ 14.11 (s). 13C NMR (126 MHz, D2O) δ 169.17 (d, J = 6.8 Hz, COO), 155.56 (C-6), 152.79 (C-2), 148.78 (C-4), 139.86 (C-8), 118.55 (C-5), 114.76 (C(CH3)2-Adn), 112.96 (C(CH3)2-Rib), 108.69 (C-1′′), 90.16 (C-1′), 84.90 (d, J = 8.78 Hz, C-4′), 84.06 (C-3′′), 83.96 (C-2′), 83.40 (C-4′′), 81.46 (C-3′), 80.94 (C-2′′), 65.17 (C-5′′), 64.80 (d, J = 5.41 Hz, C-5′), 54.69 (CH3O), 34.89 and 33.93 (d, JC–P = 120.5 Hz, CH2P), 26.14 and 24.35 (2 × C(CH3)2-Adn), 25.06 and 23.41 (2 × C(CH3)2-Rib); HRMS (ES−) calcd for C24H33N5O12P 614.1869, found [M − H]− 614.1889.:5 → 35:65 v/v) to afford the title compound 2 as a colourless glass, as a mixture of α and β anomers α/β = 2/3 (7.6 mg, 60%, ⅓ Et3N salt). DOSY 1H NMR (water suppression, 500 MHz, D2O) δ 8.37 (s, 1H, H-8), 8.15 (s, 1H, H-2), 6.04 (d, 1H, J = 5.5 Hz, H-1′), 5.21–5.19 (m, 0.4H, H-1′′α), 5.11 (d, 0.6H, J = 1.5 Hz, H-1′′β), 4.67 (t, 1H, J = 5.0 Hz, H-2′), 4.44–4.40 (m, 1H, H-3′), 4.31–4.27 (m, 1H, H-4′), 4.24  4.71–3.96
4.71–3.96  3.90–3.88 (m, 0.6H, H-2′′β), 3.11 (q, 2H, J = 7.0 Hz, NCH2CH3), 2.87–2.79 (m, 2H, CH2P), 1.19 (t, 3H, J = 7.5, NCH2CH3). 31P NMR (203 MHz, D2O) δ 14.61 (s), 14.50 (s). 13C NMR (126 MHz, D2O) δ 169.72 (COO), 155.52 (C-6), 152.77 (C-2), 149.1 (C-4), 139.74 (C-8), 118.69 (C-5), 101.18 (C-1′′β), 96.34 (C-1′′α), 87.02 (C-1′), 83.76 (C-4′), 79.83 and 79.44 (C-3′′α/β, 4′′α/β), 74.93 (C-2′′β), 74.20 (C-2′), 70.50 and 70.28 and 70.10 (C-3′′α/β, 2′′α, 3′), 65.30
3.90–3.88 (m, 0.6H, H-2′′β), 3.11 (q, 2H, J = 7.0 Hz, NCH2CH3), 2.87–2.79 (m, 2H, CH2P), 1.19 (t, 3H, J = 7.5, NCH2CH3). 31P NMR (203 MHz, D2O) δ 14.61 (s), 14.50 (s). 13C NMR (126 MHz, D2O) δ 169.72 (COO), 155.52 (C-6), 152.77 (C-2), 149.1 (C-4), 139.74 (C-8), 118.69 (C-5), 101.18 (C-1′′β), 96.34 (C-1′′α), 87.02 (C-1′), 83.76 (C-4′), 79.83 and 79.44 (C-3′′α/β, 4′′α/β), 74.93 (C-2′′β), 74.20 (C-2′), 70.50 and 70.28 and 70.10 (C-3′′α/β, 2′′α, 3′), 65.30  64.58 and 63.84 (C-5′′α/β), 46.63 (NCH2CH3), 35.14 and 34.18 (d, JC–P = 121.1 Hz, CH2P a and b), 8.18 (NCH2CH3). HRMS (ES−) calcd for C17H23N5O12P 520.1086, found [M − H]− 520.1086. Analytical HPLC: RT = 3.68 min.
64.58 and 63.84 (C-5′′α/β), 46.63 (NCH2CH3), 35.14 and 34.18 (d, JC–P = 121.1 Hz, CH2P a and b), 8.18 (NCH2CH3). HRMS (ES−) calcd for C17H23N5O12P 520.1086, found [M − H]− 520.1086. Analytical HPLC: RT = 3.68 min.Pharmacology
Author contributions
J. M. W. and B. V. L. P. designed the study. O. B. and J. M. W. equally contributed and O. B. synthesized the ADPR analogues supervised by J. M. W.; M. D. R. carried out patch-clamp experiments with R. F. and A. H. G.; O. B., J. M. W. and B. V. L. P. wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (Project number 335447717; SFB1328, project A01 to A. H. G, SFB1328, project A05 to R. F) and the Wellcome Trust. BVLP is a Wellcome Trust Senior Investigator (Grant 101010). Research in the Guse/Fliegert labs is also supported by the Joachim-Herz-Foundation, Infectophysics consortium, project 4; and EU project INTEGRATA - DLV-813284.References
- S. Partida-Sanchez, A. Gasser, R. Fliegert, C. C. Siebrands, W. Dammermann, G. Shi, B. J. Mousseau, A. Sumoza-Toledo, H. Bhagat, T. F. Walseth, A. H. Guse and F. E. Lund, Chemotaxis of mouse bone marrow neutrophils and dendritic cells is controlled by ADP-ribose, the major product generated by the CD38 enzyme reaction, J. Immunol., 2007, 179, 7827–7839 CrossRef CAS PubMed.
- B. Buelow, Y. Song and A. M. Scharenberg, The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes, J. Biol. Chem., 2008, 283, 24571–24583 CrossRef CAS PubMed.
- C. J. Lord and A. Ashworth, PARP inhibitors: synthetic lethality in the clinic, Science, 2017, 355, 1152–1158 CrossRef CAS PubMed.
- H. C. Lee, Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization, J. Biol. Chem., 2012, 287, 31633–31640 CrossRef CAS PubMed.
- J. M. Swarbrick, R. Graeff, C. Garnham, M. P. Thomas, A. Galione and B. V. L. Potter, Click cyclic ADP-ribose: a neutral second messenger mimic, Chem. Commun., 2014, 50, 2458–2461 RSC.
- G. I. Karras, G. Kustatscher, H. R. Buhecha, M. D. Allen, C. Pugieux, F. Sait, M. Bycroft and A. G. Ladurner, The macro domain is an ADP-ribose binding module, EMBO J., 2005, 24, 1911–1920 CrossRef CAS PubMed.
- A. L. Perraud, A. Fleig, C. A. Dunn, L. A. Bagley, P. Launay, C. Schmitz, A. J. Stokes, Q. Q. Zhu, M. J. Bessman, R. Penner, J. P. Kinet and A. M. Scharenberg, ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology, Nature, 2001, 411, 595–599 CrossRef CAS PubMed.
- J. C. Belrose and M. F. Jackson, TRPM2: a candidate therapeutic target for treating neurological diseases, Acta Pharmacol. Sin., 2018, 39, 722–732 CrossRef CAS PubMed.
- B. Tóth, I. Iordanov and L. Csanády, Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2′-phosphate, J. Gen. Physiol., 2015, 145, 419–430 CrossRef PubMed.
- T. Rosenbaum, Activators of TRPM2: getting it right, J. Gen. Physiol., 2015, 145, 485–487 CrossRef PubMed.
- R. Fliegert, A. Bauche, A. M. W. Perez, J. M. Watt, M. D. Rozewitz, R. Winzer, M. Janus, F. Gu, A. Rosche, A. Harneit, M. Flato, C. Moreau, T. Kirchberger, V. Wolters, B. V. L. Potter and A. H. Guse, 2′-Deoxyadenosine 5′-diphosphoribose is an endogenous TRPM2 superagonist, Nat. Chem. Biol., 2017, 13, 1036–1044 CrossRef CAS PubMed.
- F. J. P. Kühn, J. M. Watt, B. V. L. Potter and A. Lückhoff, Different substrate specificities of the two ADPR binding sites in TRPM2 channels of Nematostella vectensis and the role of IDPR, Sci. Rep., 2019, 9, 12 CrossRef PubMed.
- C. Moreau, T. Kirchberger, J. M. Swarbrick, S. J. Bartlett, R. Fliegert, T. Yorgan, A. Bauche, A. Harneit, A. H. Guse and B. V. L. Potter, Structure-Activity Relationship of Adenosine 5′-diphosphoribose at the Transient Receptor Potential Melastatin 2 (TRPM2) Channel: Rational Design of Antagonists, J. Med. Chem., 2013, 56, 10079–10102 CrossRef CAS PubMed.
- O. Baszczyňski, J. M. Watt, M. D. Rozewitz, A. H. Guse, R. Fliegert and B. V. L. Potter, Synthesis of Terminal Ribose Analogues of Adenosine 5′-Diphosphate Ribose as Probes for the Transient Receptor Potential Cation Channel TRPM2, J. Org. Chem., 2019, 84, 6143–6157 CrossRef PubMed.
- R. Fliegert, J. M. Watt, A. Schöbel, M. D. Rozewitz, C. Moreau, T. Kirchberger, M. P. Thomas, W. Sick, A. C. Araujo, A. Harneit, B. V. L. Potter and A. H. Guse, Ligand-induced activation of human TRPM2 requires the terminal ribose of ADPR and involves Arg1433 and Tyr1349, Biochem. J., 2017, 474, 2159–2175 CrossRef CAS PubMed.
- L. R. Comstock and J. M. Denu, Synthesis and biochemical evaluation of O-acetyl-ADP-ribose and N-acetyl analogs, Org. Biomol. Chem., 2007, 5, 3087–3091 RSC.
- O. P. Chevallier and M. E. Migaud, Synthesis of simple adenosine diphosphate ribose analogues, Nucleosides, Nucleotides Nucleic Acids, 2008, 27, 1127–1143 CrossRef CAS PubMed.
- T. Shimizu, R. M. Dietz, I. Cruz-Torres, F. Strnad, A. K. Garske, M. Moreno, V. R. Venna, N. Quillinan and P. S. Herson, Extended therapeutic window of a novel peptide inhibitor of TRPM2 channels following focal cerebral ischemia, Exp. Neurol., 2016, 283, 151–156 CrossRef CAS PubMed.
- K. Pahnke and C. Meier, Synthesis of a Bioreversibly Masked Lipophilic Adenosine Diphosphate Ribose Derivative, ChemBioChem, 2017, 18, 1616–1626 CrossRef CAS PubMed.
- T. S. Elliott, A. Slowey, Y. Ye and S. J. Conway, The use of phosphate bioisosteres in medicinal chemistry and chemical biology, RSC Med. Chem., 2012, 3, 735–751 RSC.
- For an up to date list search, http://www.rcsb.org/, for AMPCPR.
- B. Tóth, I. Iordanov and L. Csanády, Putative chanzyme activity of TRPM2 cation channel is unrelated to pore gating, Proc. Natl. Acad. Sci., 2014, 111, 16949–16954 CrossRef PubMed.
- I. Iordanov, C. Mihalyi, B. Toth and L. Csanady, The proposed channel-enzyme transient receptor potential melastatin 2 does not possess ADP ribose hydrolase activity, eLife, 2016, 5, e17600 CrossRef PubMed.
- X. Luo, M. Li, K. Zhan, W. Yang, L. Zhang, K. Wang, P. Yu and L. Zhang, Selective inhibition of TRPM2 channel by two novel synthesized ADPR analogues, Chem. Biol. Drug Des., 2018, 91, 552–566 CrossRef CAS PubMed.
- J. P. Quinn, A. N. Kulakova, N. A. Cooley and J. W. McGrath, New ways to break an old bond: the bacterial carbon-phosphorus hydrolases and their role in biogeochemical phosphorus cycling, Environ. Microbiol., 2007, 9, 2392–2400 CrossRef CAS PubMed.
- A. M. Riley, H. Wang, J. D. Weaver, S. B. Shears and B. V. L. Potter, First synthetic analogues of diphosphoinositol polyphosphates: interaction with PP-InsP5 kinase, Chem. Commun., 2012, 48, 11292–11294 RSC.
- D. V. Patel, R. J. Schmidt, S. A. Biller, E. M. Gordon, S. S. Robinson and V. Manne, Farnesyl Diphosphate-Based Inhibitors of Ras Farnesyl Protein Transferase, J. Med. Chem., 1995, 38, 2906–2921 CrossRef CAS PubMed.
- K. W. Pankiewicz, K. Lesiak and K. A. Watanabe, Efficient Synthesis of Methylenebis(phosphonate) Analogues of P1, P2-Disubstituted Pyrophosphates of Biological Interest. A Novel Plausible Mechanism, J. Am. Chem. Soc., 1997, 119, 3691–3695 CrossRef CAS.
- A. M. Riley, H. Wang, S. B. Shears and B. V. L. Potter, Synthesis of an α-phosphono-α,α-difluoroacetamide analogue of the diphosphoinositol pentakisphosphate 5-InsP7, RSC Med. Chem., 2019, 10, 1165–1172 RSC.
- Y. Huang, P. A. Winkler, W. Sun, W. Lü and J. Du, Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium, Nature, 2018, 562, 145–149 CrossRef CAS PubMed.
- Y. Huang, B. Roth, W. Lü and J. Du, Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel, eLife, 2019, 8, e50175 CrossRef PubMed.
- W. Li, Y. Niu, D.-C. Xiong, X. Cao and X.-S. Ye, Highly Substituted Cyclopentane–CMP Conjugates as Potent Sialyltransferase Inhibitors, J. Med. Chem., 2015, 58, 7972–7990 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H-NMR, 13C-NMR and 31P-NMR spectra for compounds 1, 2 and 5–10. See DOI: 10.1039/c9ra09284f |
| ‡ Present address: Department of Organic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, 128 43 Prague 2, Czech Republic. |
| This journal is © The Royal Society of Chemistry 2020 |