
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuppressing vanadium crossover using sulfonated aromatic ion exchange membranes for high performance flow batteries†
Tongshuai
Wang
a,
Junyoung
Han
b,
Kihyun
Kim
b,
Andreas
Münchinger
c,
Yuechen
Gao
a,
Alain
Farchi
d,
Yoong-Kee
Choe
e,
Klaus-Dieter
Kreuer
*c,
Chulsung
Bae
*b and
Sangil
Kim
 *a
*a
aDepartment of Chemical Engineering, University of Illinois at Chicago, Chicago, IL 60607, USA. E-mail: sikim@uic.edu
bDepartment of Chemistry & Chemical Biology, Department of Chemical & Biological Engineering, Rensselaer Polytechnic Institute, Troy, NY 12180, USA. E-mail: baec@rpi.edu
cMax-Planck-Institute for Solid State Research, Heisenbergstrasse 1, 70569 Stuttgart, Germany. E-mail: kreuer@fkf.mpg.de
dUniversité Grenoble Alpes, CEA, IRIG-DEPHY-MEM, 38000 Grenoble, France
eNational Institute of Advanced Industrial Science and Technology, Tsukuba, Japan
First published on 1st September 2020
Abstract
Liquid flow batteries have potential to achieve high energy efficiency as a large-scale energy storage system. However, the ion exchange membranes (IEMs) currently used in flow batteries do not have high ion selectivity and conductance at the same time, owing to the trade-off between ionic membrane resistance and ion selectivity. Here, we report a rationally designed sulfonated aromatic polymer membrane which can greatly mitigate the trade-off limitation and achieve high performance vanadium RFB. Small-angle X-ray scattering studies and density functional theory calculations indicated that the narrowly distributed aqueous ionic domain of just the right width (<7 Å) and the strong hydrogen bond interaction of vanadium species with a unique polymer side chain structure play a key role in improving the ion selectivity. Our systematic studies of the polymer structures, morphologies, and transport properties provide valuable insight that can aid in elucidating the structure–property relationship of IEMs and in establishing design criteria for the development of high-performance membranes.
Introduction
Membrane-based electrochemical devices have shown great potential for use in many next-generation energy conversion and storage systems, such as fuel cells,1 redox flow batteries,2,3 and reverse electrodialysis,4 owing to their high energy efficiency and large-scale adaptability. For a broader application of these technologies, ion exchange membranes (IEMs) with high ion conductivity and selectivity properties are critical for enhancing the electrochemical performance of electrolyte cells. Conventional polymeric IEMs, however, suffer from the trade-off between ion conductivity (membrane resistance) and selectivity of co/counter-ions and fuels, which greatly reduces the efficiency of energy conversion and storage systems.5,6Among energy storage systems, vanadium redox flow batteries (VRFBs) have been recognized as one of the most promising technologies for grid-scale energy storage, owing to excellent design flexibility and safety, benefited from their decoupled energy storage and power generation.7–11 By using vanadium ions as the active species in both catholyte and anolyte solutions, VRFBs eliminate the risk of detrimental cross-contamination effects, thus greatly improving their service life.12,13 As a key component in VRFBs, IEMs should have low vanadium permeability for the separation of the vanadium species in the catholyte and anolyte solutions while allowing selective transport of charge-carrier ions with high ionic conductivity.14 Unfortunately, most of the cation exchange membranes (CEMs) do not meet these criteria, owing to their intrinsic trade-off between proton conductance and the selectivity of protons over vanadium ions (H+/Vn+), thus hampering the widespread implementation of VRFBs. For instance, although Nafion®, a perfluorosulfonated polymer CEM, is commonly used in VRFBs because of its high proton conductivity, chemical stability, and commercial availability, the high vanadium permeability of the Nafion® membrane causes undesired coulombic efficiency (CE) drop and fast capacity decay.14–16 To minimize the crossover of vanadium ions, a thicker Nafion membrane (e.g., Nafion 117) has to be employed at the expense of higher membrane resistance, raising the VRFB stack cost. Currently, Nafion membranes account for 40% of the total cost of a VRFB cell stack.17 To overcome the limitations of Nafion membranes, extensive efforts have been made to improve ion selectivity by introducing ion selective surface coatings18 or inorganic fillers, such as SiO2,19 TiO2,20 and ZrO2.21,22 Although the modified Nafion membranes show a suppressed vanadium permeability, the additives can cause adverse effect of higher cell resistance, which does not help to overcome the trade-off limitation.14 Alternatively, hydrocarbon-based anion exchange membranes (AEMs)23 have been investigated for VRFBs because of their cation (e.g. vanadyl cation) exclusion effect (Donnan exclusion). However, in highly concentrated electrolytes of VRFBs (typically ≥3 M), the Donnan exclusion effect is weak and cannot compensate for the lower ion conductivity of typical AEMs compared to Nafion. As a consequence, the decrease in voltage efficiency (VE) and energy efficiency (EE) makes AEMs less appealing for VRFB applications at high current densities.24 Recently, a polyamide thin-film composite membrane exhibited an EE of VRFB greater than 80% at 260 mA cm−2 by optimizing the thickness of the polyamide selective layer.25 However, the polyamide membrane still exhibits the trade-off limitation. Furthermore, the amide bonds in polyamide are known to be susceptible to hydrolysis which could result in stability issues under strong acidic conditions.26 Therefore, for deployment of durable, high-performance VRFB systems, the design and fabrication of advanced IEMs, which can overcome the intrinsic trade-off limitations, via the optimization of various parameters at molecular levels (e.g. ionic group, ion exchange capacity, and aqueous ionic domain width) is highly critical.
In this paper, we report a novel class of sulfonated aromatic polymer CEMs and demonstrate their superior battery performance in achieving both high VE and high CE and overcoming the trade-off limitation barrier of IEMs. To establish the relationship between the molecular structures and the selective ion transport properties, we rationally designed a series of polymers composed of a biphenyl backbone with different side chains (Fig. 1a). Among the polymers investigated, the biphenyl polymer with the longest side chain containing an additional aromatic ring and a perfluorinated segment (BP-ArF4 of Fig. 1a) exhibited good proton conductivity while also maintaining extraordinary high proton/vanadium ion selectivity. The unique feature of this ionic polymer includes a hydrophobic/hydrophilic separation on a small length scale typical of hydrocarbons with a high degree of order and the super-acidity characteristic typical of perfluorosulfonate ionomers. The unique combination of these characteristics affords BP-ArF4 a remarkable battery performance (CE ∼ 99%, EE up to 93.9%) exceeding that of all other IEMs in VRFB applications. Small-angle X-ray scattering (SAXS) and density functional theory (DFT) calculations provide a semi-quantitative understanding of the observed performance.
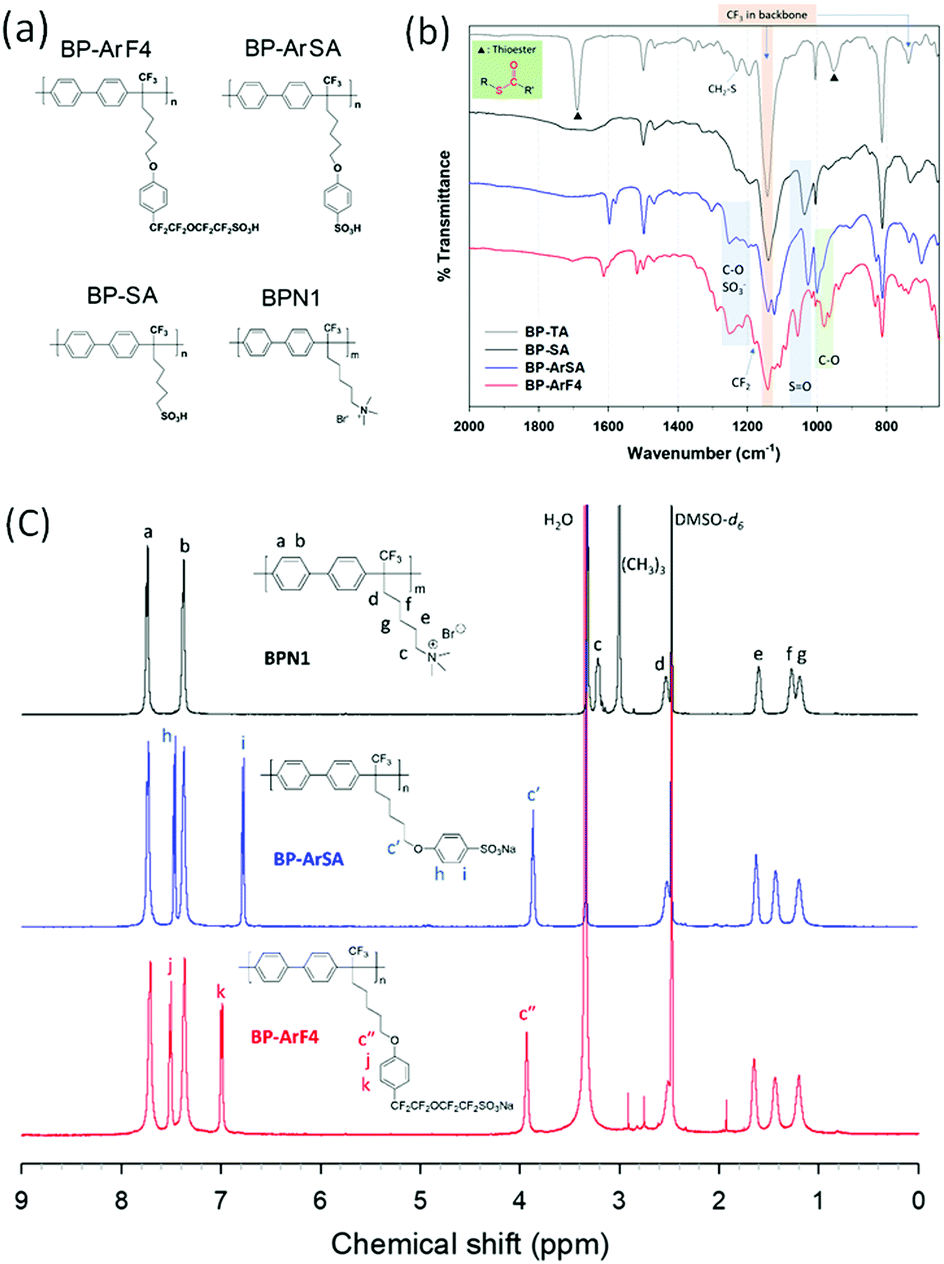 | ||
| Fig. 1 Chemical structures and FT-IR spectra of the synthesized polymers. (a) Chemical structures of the polymers. (b) FT-IR spectra of the synthesized BP-TA (gray) and cation exchange polymers: BP-SA (black), BP-ArSA (blue), and BP-ArF4 (red). (c) 1H NMR spectra (DMSO-d6) of the synthesized polymers: BPN1 (black), BP-ArSA (Na salt form, blue), and BP-ArF4 (red; the peaks at 1.96, 2.78, and 2.94 ppm are from residual DMAc solvent from solvent casting). | ||
Results and discussion
Polymer synthesis and characterization
As shown in Fig. S1 (ESI†), three examples of cation exchange polymers (BP-SA, BP-ArSA, and BP-ArF4) were prepared from the biphenyl polymer precursor BPBr-100, which has a bromoalkyl group on the side chain. For BP-SA, the brominated side chain of the precursor polymer was replaced with –SO3H, and it has the shortest side chain, with a flexible –(CH2)5– spacer between the backbone and sulfonate group, among the three investigated CEMs. BP-ArSA has an additional aromatic ring in the sulfonate side chain, while BP-ArF4 contains a perfluoroalkylsulfonate group in addition to an aromatic ring in the side chain. As all three sulfonated polymers share the same biphenyl backbone structure and were prepared from the precursor polymer BPBr-100, they should have no difference in molecular weight. The difference in properties of these polymers should be derived only from the side chain structures. Through the minor differences in the side chain structures, we studied how they could impact ion conductivity and vanadium ion permeability, which can be translated to battery efficiencies. We also prepared BPN1, an AEM counterpart of BP-SA, from BPBr-100 to study the effect of sulfonate vs. quaternary ammonium side chains on membrane properties and battery performance.Because BP-SA has poor solubility in organic solvents, BP-SA membranes were prepared by the oxidation of BP-TA film (Fig. S1, ESI†). BP-TA was prepared via a nucleophilic substitution reaction of BPBr-100 and potassium thioacetate; whereafter, it was cast into a film and immersed in a hydrogen peroxide solution for the oxidation of –SC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CH3 of BP-TA to –SO3H of BP-SA.27 Successful conversion to a sulfonic acid group in BP-SA was confirmed using FT-IR spectroscopy and titrated ion exchange capacity (IEC) (Fig. 1b), in which the thioester S–(CO)–C stretching bands of BP-TA at 1690 and 952 cm−1 disappeared, and the SO stretching bands of –SO3H in BP-SA were clearly visible at 1200 and 1037 cm−1. BP-ArSA was synthesized using a one-step nucleophilic substitution of BP-Br with sodium 4-hydroxybenzenesulfonate, as shown in Fig. S1 (ESI†). Because BP-ArSA (in –SO3Na form) is soluble in DMSO, the chemical structure of BP-ArSA was confirmed using 1H NMR and FT-IR spectroscopies (Fig. 1c). In addition to SO stretching bands at 1220 and 1026 cm−1, C–O stretching bands were observed at 1252 and 1000 cm−1 in the FT-IR spectrum. To synthesize BP-ArF4, we first synthesized BP-Ar-Ivia the nucleophilic substitution of BPBr-100 with 4-indophenol and then subjected the aryl iodide in the polymer pendant chain to Ullmann coupling reaction with compound 1 (Fig. S1, ESI†). Structural characterization of BP-ArF4 was confirmed using 1H and 19F NMR (in –SO3Na form) and FT-IR (in –SO3H form) spectroscopies (Fig. S1 and S3, ESI†). The FT-IR spectrum showed the presence of –SO3H (SO stretching bands at 1216 and 1056 cm−1), aryl ether bonds (C–O stretching bands at 1251 and 1090 cm−1), and a perfluoroalkyl ether moiety (CF2–O stretching at 980–960 cm−1). BPN1 was prepared by following the procedure mentioned in our previous report.28
O)CH3 of BP-TA to –SO3H of BP-SA.27 Successful conversion to a sulfonic acid group in BP-SA was confirmed using FT-IR spectroscopy and titrated ion exchange capacity (IEC) (Fig. 1b), in which the thioester S–(CO)–C stretching bands of BP-TA at 1690 and 952 cm−1 disappeared, and the SO stretching bands of –SO3H in BP-SA were clearly visible at 1200 and 1037 cm−1. BP-ArSA was synthesized using a one-step nucleophilic substitution of BP-Br with sodium 4-hydroxybenzenesulfonate, as shown in Fig. S1 (ESI†). Because BP-ArSA (in –SO3Na form) is soluble in DMSO, the chemical structure of BP-ArSA was confirmed using 1H NMR and FT-IR spectroscopies (Fig. 1c). In addition to SO stretching bands at 1220 and 1026 cm−1, C–O stretching bands were observed at 1252 and 1000 cm−1 in the FT-IR spectrum. To synthesize BP-ArF4, we first synthesized BP-Ar-Ivia the nucleophilic substitution of BPBr-100 with 4-indophenol and then subjected the aryl iodide in the polymer pendant chain to Ullmann coupling reaction with compound 1 (Fig. S1, ESI†). Structural characterization of BP-ArF4 was confirmed using 1H and 19F NMR (in –SO3Na form) and FT-IR (in –SO3H form) spectroscopies (Fig. S1 and S3, ESI†). The FT-IR spectrum showed the presence of –SO3H (SO stretching bands at 1216 and 1056 cm−1), aryl ether bonds (C–O stretching bands at 1251 and 1090 cm−1), and a perfluoroalkyl ether moiety (CF2–O stretching at 980–960 cm−1). BPN1 was prepared by following the procedure mentioned in our previous report.28
Ion transport properties
To characterize the ion transport properties of the membranes, we evaluated the IEC, vanadium ion permeability, proton conductivity, and area resistance of the prepared membranes (Table 1). Commercial Nafion 117 and Nafion 212 membranes, which are widely used in VRFBs, were used as reference samples. Low vanadium ion crossover is a critical property of IEMs to achieve high CE and low capacity loss for VRFB applications. Among all evaluated CEMs, BP-SA showed the highest vanadium permeability of 110.1 × 10−7 cm2 min−1 possibly due to the highest IEC and swelling ratio. In comparison, BP-ArSA exhibited a markedly lower vanadium permeability of 13.5 × 10−7 cm2 min−1, thus indicating that the incorporation of an aromatic group and an ether linkage in the side chain can greatly improve the vanadium ion rejection property of the membrane. BP-ArF4 with an additional perfluoroalkyl pendant chain further suppressed the vanadium ion permeability to 10.2 × 10−7 cm2 min−1, a value 4.0 times and 3.2 times lower than those for Nafion 212 and Nafion 117, respectively. Hydrated vanadium ion and proton transports in the sulfonated aromatic polymers have been found to be heavily affected by the dimension of the separated hydrophobic and hydrophilic domains in the microstructures.29 In general, swelling ratio values are closely related to water uptake and, in Table 1, we use the swelling ratio to correlate the dimensional stabilities of membranes with ion selectivity. The swelling ratios for BP-ArSA (12.9%) and BP-ArF4 (10.2%) were significantly lower than that of BP-SA (18.6%), suggesting a smaller aqueous domain within the polymer matrix in spite of the same backbone structure of all three biphenyl CEMs. We also tested BPN1, an AEM with the identical molecular structure of BP-SA, to investigate the structure–membrane property relationship of sulfonate vs. quaternary ammonium side chains in aromatic polymers. Although BPN1 and BP-SA have comparable linear swelling ratios, BPN1 exhibited a significantly lower vanadium permeability (23.8 × 10−7 cm2 min−1). This is most likely because the highly concentrated positive quaternary ammonium functional groups of BPN1 AEM impede the transport of the multi-valent vanadium cations as a result of the Donnan exclusion.30,31| Samples | Thickness (μm) | IEC (meq. g−1) | VO2+ permeability (×10−7 cm2 min−1) | Ionic conductivity (mS cm−1) | α (×104 min S cm−3) | Area resistance (Ω cm2) | Linear swelling ratio (%) |
|---|---|---|---|---|---|---|---|
| a Refers to proton conductivity. b Refers to anion conductivity. | |||||||
| BP-ArF4 | 88 ± 1 | 1.25 ± 0.05 | 10.2 ± 0.8 | 55.3 ± 1.2a | 5.42 | 0.28 ± 0.02 | 10.2 |
| BP-ArSA | 90 ± 1 | 2.01 ± 0.06 | 13.5 ± 0.5 | 67.6 ± 0.9a | 5.08 | 0.26 ± 0.02 | 12.9 |
| BP-SA | 60 ± 2 | 2.29 ± 0.19 | 110.1 ± 2.5 | 94.4 ± 2.1a | 0.75 | 0.23 ± 0.01 | 18.6 |
| BPN1 | 97 ± 2 | 2.60 ± 0.05 | 23.8 ± 3.4 | 14.5 ± 1.4b | 0.61 | 0.67 ± 0.07 | 17.4 |
| Nafion 212 | 55 ± 1 | 0.90 ± 0.02 | 41.2 ± 2.0 | 73.5 ± 3.1a | 1.78 | 0.23 ± 0.05 | 14.3 |
| Nafion 117 | 195 ± 1 | 0.89 ± 0.02 | 32.1 ± 1.7 | 59.2 ± 2.2a | 1.84 | 0.89 ± 0.09 | 15.1 |
As the length of the side chain increases in the biphenyl polymer series, their IECs decrease due to lower ion exchange groups (–SO3H) in unit mass (volume): BP-SA (2.29 meq. g−1), BP-ArSA (2.01 meq. g−1), and BP-ArF4 (1.25 meq. g−1). As expected, the proton conductivity of the CEMs follows the order of IEC: BP-ArF4 < BP-ArSA < BP-SA (Table 1). The proton conductivities of BP-ArSA and BP-ArF4 were 67.6 and 55.3 mS cm−1, respectively, which are comparable to that of Nafion 117 (59.2 mS cm−1) and Nafion 212 (73.5 mS cm−1). BP-SA showed the highest proton conductivity (94.4 mS cm−1) among all studied CEMs as shown in Table 1 due to the highest IEC.
We defined the ion selectivity (α) of the membranes as the ratio of proton conductivity and vanadium permeability, and the values are listed in Table 1. Although BP-ArF4 and BP-ArSA have a lower IEC and lower proton conductivity than BP-SA, their ion selectivity values were almost seven times higher than that of BP-SA. This result suggests that the positive contributions from lower swelling (hydration number) and vanadium permeability due to the incorporation of an aromatic ring and additional perfluoroalkyl moiety in the flexible side chain of the polymers far outweigh the decreases in IEC and ion conductivity. In spite of lower proton conductivity, the ion selectivity of BP-ArF4 is more than three times higher than those of Nafion 212 and Nafion 117. Areal specific resistance (ASR) of the membranes was measured using electrochemical impedance spectroscopy (Table 1). The ASR of BPN1 (0.67 Ω cm2) was measured for SO42− anion transport of the AEM. Owing to the intrinsically low mobility of SO42− anions (8.27 × 10−8 m2 s−1 V−1) in aqueous solution, which is approximately one-fourth the proton mobility,32 the ASR of BPN1 was significantly higher than those of biphenyl CEMs with a similar thickness. Although the thickness of BP-ArF4 and BP-ArSA membranes (approximately 90 μm) was higher than that of Nafion 212 (55 μm), they all showed comparable ASR values in the range of 0.23–0.28 Ω cm2. These ASR values were significantly lower than that of Nafion 117 (0.89 Ω cm2), the standard CEM in VRFB with a thickness of 195 μm to minimize vanadium ion crossover in the membrane.
VRFB performance
Fig. 2a–c illustrate VRFB single cell tests with various membrane separators. Among all membranes evaluated, BP-ArF4 showed the best overall performance far exceeding Nafion 117, the most commonly used benchmark separator membrane. Because the transport of vanadium species may occur through diffusional processes even in the absence of current, coulombic losses are expected to dominate at low current density, whereas VE is expected to decrease with increasing charging/discharging current, owing to the charge transfer overpotential and transport resistance within the anolyte, catholyte, and membrane (Fig. 2a and b). As listed in Table 1, Nafion 212, BP-SA, BP-ArSA, and BP-ArF4 have similar ranges of ASR (0.23–0.28 Ω cm2), and thus they all exhibited higher VE than the thicker Nafion 117 (195 μm thick, 0.89 Ω cm2 ASR). It was found that, although BP-SA has a smaller ASR than BPN1, the VE of BP-SA is lower than BPN1. The unexpected low VE of BP-SA is presumably due to the voltage loss that resulted from the fast crossover of active species.33 The effect of enhanced ion selectivity of BP-ArF4 on VRFB performance was clearly visible in CE. Among all tested membranes, the CE of VRFBs with the BP-ArF4 membrane approached 100% at a current density >60 mA cm−2. Even at a lower current density of 40 mA cm−2, the BP-ArF4 membrane still exhibited a very high CE of 98.6%, as compared with 95.0% exhibited by Nafion 117 and 85.9% by Nafion 212. Notably, the CE of BP-ArF4 was even higher than that of BPN1 (96.2%) at 40 mA cm−2. The high CE of BP-ArF4 can also be reflected from the charge–discharge curves (Fig. S4, ESI†). The discharge capacity of BP-ArF4 is almost identical to the charge capacity, indicating that the loss of the discharge capacity has been minimized.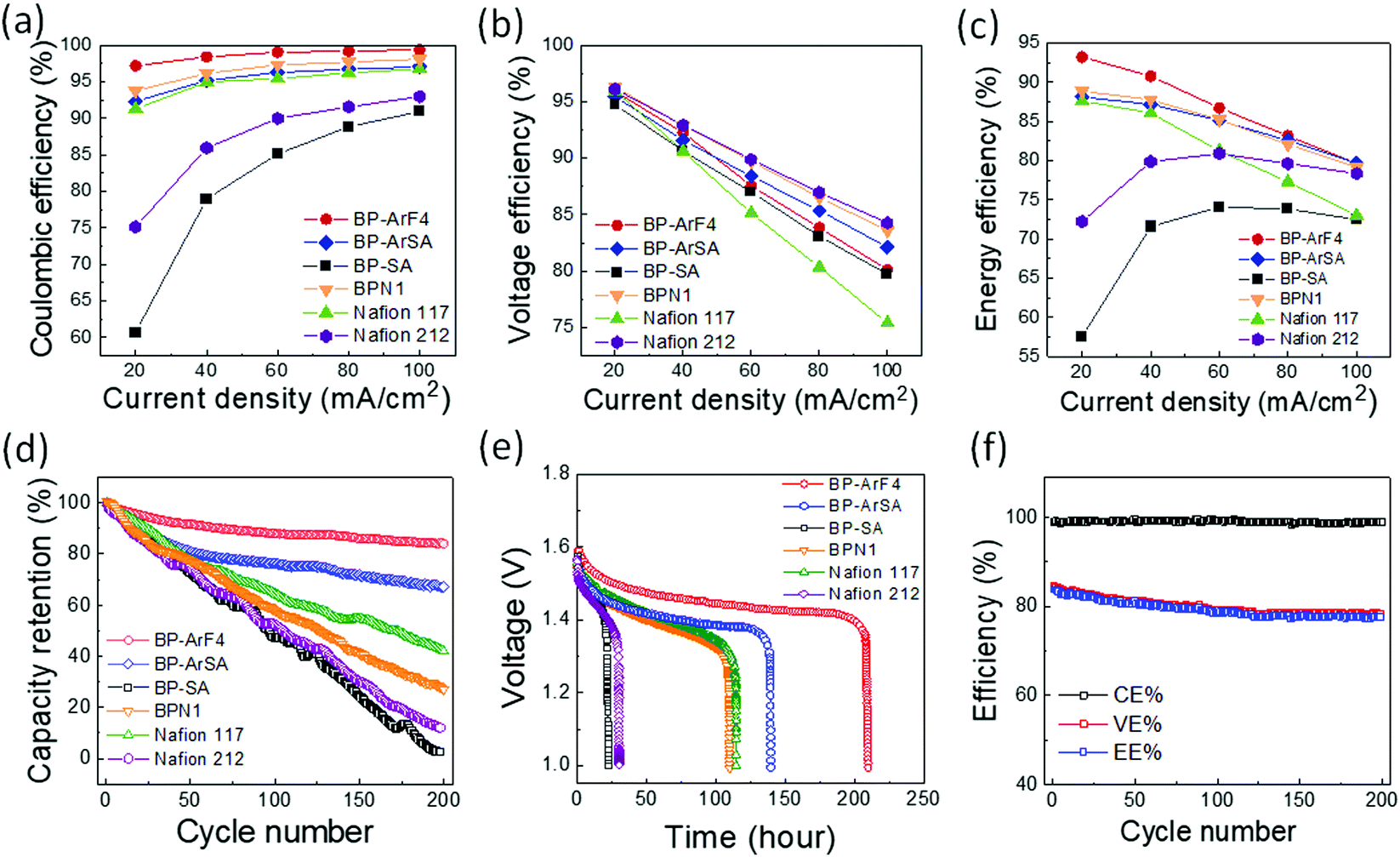 | ||
| Fig. 2 VRFB single cell performance. (a) CE for PEMs at a current density from 20 to 100 mA cm−2, (b) VE for PEMs at a current density from 20 to 100 mA cm−2, (c) EE for PEMs at a current density from 20 to 100 mA cm−2, (d) capacity retention for IEMs at 100 mA cm−2, (e) self-discharge curves of PEMs, and (f) cycling stability of VRFB with the BP-ArF4 membrane at 100 mA cm−2. | ||
Since the CE and EE values of the BP-ArF4, BP-ArSA, BPN1, and Nafion 212 are similar at 100 mA cm−2 (Fig. 2a and c), we monitored the capacity retention of the IEMs to further evaluate their performance, particularly at a high current density. VRFB single cells loaded with each IEM were charged and discharged at 100 mA cm−2 for 200 cycles. As shown in Fig. 2d, BP-ArF4 has the lowest capacity decay rate (0.08% per cycle) followed by BP-ArSA (0.16%), Nafion 117 (0.29%), BPN1 (0.36%), Nafion 212 (0.44%), and BP-SA (0.49%). The capacity decay of the VRFB cell is typically a result of the charge imbalance due to the crossover of vanadium ions as well as water transfer.34 Benefiting from remarkably lower vanadium permeability and swelling ratio, BP-ArF4 and BP-ArSA exhibited a slow capacity decay rate and maintained high capacity retention after 200 cycles, 84% and 67%, respectively. In contrast, BP-SA showed a faster capacity decay and lower capacity retention, presumably due to the more rapid vanadium and water crossover, which can deteriorate the charge imbalance between positive and negative half cells. Nafion 117 was able to achieve a higher capacity retention than Nafion 212 (40% vs. 10% after 200 charge–discharge cycles) by lowering vanadium ion crossover with increased thickness. Additionally, we evaluated the self-discharge rate of the IEMs which is closely related to the loss of energy capacity of VRFB.35,36Fig. 2e shows the self-discharge curves of VRFB with IEMs starting from ∼90% of the state of charge (SOC). Since the cells were held statically (zero current) during the self-discharge test, the difference of overpotential between each membrane is minimized. Therefore, the initial open circuit voltage (OCV) for all membranes approaches 1.6 V. However, because of the different crossover rates of active vanadium species through membrane separators, the OCV of VRFB cells decreased over time at varying speed. Toward the end of the test, OCV sharply decreased below 1.0 V. When OCV tests were compared with 1.0 V as a cut-off voltage, BP-ArF4 exhibited the longest self-discharge time (209.5 h) among all tested IEMs: BP-ArSA (139.5 h), Nafion 117 (114.5 h), BPN1 (109.9 h), Nafion 212 (30.5 h), and BP-SA (22.2 h). Interestingly, the 90 μm-thick BP-ArF4 membrane showed almost twice longer self-discharging time than the 195 μm-thick Nafion 117, owing to its superior ability to mitigate vanadium crossover. Overall, the results of capacity retention and self-discharge tests agree well with the trend of vanadium permeability results provided in Table 1; the lower the vanadium permeability of the membrane, the higher the capacity retention and the longer the self-discharge time in a VRFB single cell.
We also evaluated the cycling stability and chemical stability of BP-ArF4 in oxidative/acidic V5+/H2SO4 solution and compared with Nafion membranes. The high CE of BP-ArF4 remained stable (nearly constant close to 100%) during the 200 charge–discharge cycles at 100 mA cm−2, which indicates negligible crossover of vanadium ions through the membrane during the operation and good chemical stability in the strong oxidizing acidic environment (Fig. 2f). The observed decrease of VE and EE during the first 100 cycles is most likely due to the electrochemical degradation of carbon electrodes, as encountered in other works.37,38 While EE of BP-ArF4 remained almost stable at 80% after 110 cycles and slightly decreased to 79% at 200 cycles, the EEs of VRFB cycling stability of the Nafion 212 and 117 membranes continuously decreased to 73% and 68% after 200 cycles, respectively (Fig. S5, ESI†). When the chemical stability of the IEMs was evaluated in 0.1 M V5+ in 4 M H2SO4, all our biphenyl CEMs showed significantly lower V4+ generation than other hydrocarbon PEMs such as BPSH6039 (Fig. S6, ESI†). We believe that the enhanced chemical stability stems from the absence of chemically labile aryl ether bonds in the polymer backbone. As all biphenyl CEMs reported here have polymer backbones made only of carbon–carbon bonds, they should be chemically more resistant to strongly acidic oxidizing environments than aryl ether-containing polymer CEMs such as BPSH60. Among biphenyl CEMs, BP-SA which does not have any heteroatom other than CF3– and –SO3H moieties in the chemical structure exhibited the greatest chemical resistance against oxidation. Notably, the chemical stability of BP-SA is almost comparable to that of perfluorinated polymer Nafion, which is unprecedented for a hydrocarbon-based CEM. Overall, both the battery cycling test and off-cell chemical stability test clearly suggest that our biphenyl CEMs are quite stable in VRFBs during long-term operation.
SAXS and DFT studies: selective ion transport mechanism
On the basis of our recent work on selective ion transport,40,41 we suggest that the exceptionally good battery performance of BP-ArF4 is due to its unique morphology created by the polymer structure of rigid biphenyl backbone and long tethered perfluoroalkylsulfonate side chain and optimum ion exchange capacity. When equilibrated with a 1.6 M VOSO4/4 M H2SO4 electrolyte, the hydration number (swelling) of the CEMs was low (Table 2) and a substantial amount of co-ions (sulfate/bisulfate anions) entered the membrane with a corresponding amount of counter-ions (mostly H3O+ and vanadyl species). Interestingly, there was a strong indication that VO2+ counter-ions were well hydrated and dissociated (they did not bind to the –SO3− fixed ionic group), whereas excess VO2+ was strongly associated with SO42− co-ions by forming neutral VOSO4 species within the aqueous domains of the CEMs. Then, vanadium crossover mainly occurred through VOSO4 diffusion, with a minor contribution from charged vanadium species, whereas the major contribution to the ionic current through the membrane was from protons, which were present as counter-ions and part of the excess H2SO4 entering the membrane as a consequence of the breakdown of Donnan exclusion at high acid concentrations (i.e., 4 M H2SO4). We also demonstrated that the transport of vanadium-containing species was largely decreased when the extension of aqueous domains fell below the hydrated sizes of these species (sieving-effect), which was also observed in the subnano-sieving PIM-1 membrane.42 This effect began to occur at a domain width of approx. 0.7 nm, which remained wide enough for rapid local H3O+ diffusion.| Samples | Density | H+ form in water | In 4 M H2SO4/1.6 M VOSO4 | |||||
|---|---|---|---|---|---|---|---|---|
| ρ polymer (g cm−3) | q (Å−1) | w H2O (%) | λ | d aq (nm) | q (Å−1) | w electrolyte (%) | d aq (nm) | |
| Nafion 117 | 2.1 | 0.124 | 25 | 20.4 | 2.10 | 0.149 | 21 | 1.20 |
| BP-ArF4 | 1.4 | 0.156 | 28 | 17.3 | 1.41 | 0.190 | 19 | 0.63 |
| BP-ArSA | 1.2 | 0.162 | 32 | 13.0 | 1.41 | 0.210 | 28 | 0.75 |
| BP-SA | 1.2 | 0.184 | 50 | 24.3 | 1.85 | 0.224 | 37 | 0.94 |
With their pronounced hydrophobic/hydrophilic separation, as indicated in Table 2, perfluorosulfonic acid membranes, Nafion 117 and Nafion 212, have much wider aqueous domains, despite the lower swelling in highly concentrated electrolytes (i.e., 1.6 M VOSO4/4 M H2SO4), thus explaining their low CE values. In contrast, the flexible and long side chains of Nafion efficiently decouple hydrophobic aggregation within the polymeric domain from ionic ordering and water coalescence within the aqueous ionic domain, thus leading to a relatively smooth interface between the two domains and good connectivity of the aqueous ionic domain. Consequently, even with low IEC, Nafion membranes show less of a conductivity decrease than other IEMs as hydration decreases.43 With respect to low hydration conditions, such as those in VRFBs, relatively high proton conductivity can still be achieved, thus leading to high VE for a given current density (Fig. 2b). Therefore, a key design criterion for obtaining both high CE and VE is the morphology of the IEMs, which should have narrow aqueous ionic domains that efficiently block the transport of vanadium species (e.g., VOSO4, VOaq2+, VOaq2+, Vaq3+, Vaq2+) but are still wide enough and well-connected for high proton conductivity.
To investigate how the average width of the aqueous ionic domain daq and the degree of order/disorder affect ion selectivity and VRFB performance, we recorded SAXS patterns for CEMs immersed in pure water and in 1.6 M VOSO4/4 M H2SO4 (Fig. 3). From the structural correlation length d obtained from the position of the ionomer peak in q-space, daq is estimated by using eqn (1).
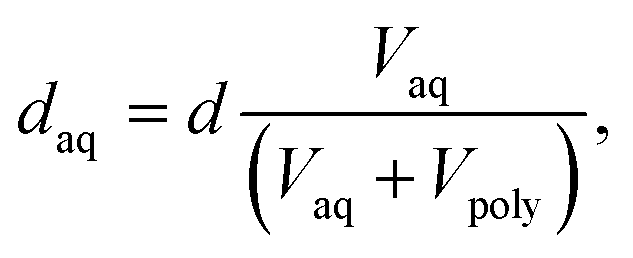 | (1) |
 | ||
| Fig. 3 SAXS patterns of: (a) Nafion 117 in water and 4 M H2SO4/1.6 M VOSO4, (b) BP-ArF4, BP-ArSA, and BP-SA membranes in the acid-form immersed in water, and (c) equilibrated in 1.6 M VO2SO4/4 M H2SO4 electrolyte. (d) Total room temperature conductivity of Nafion 117 and BP-ArF4 membranes equilibrated in solutions of VOSO4 with different molarities. (e) Illustration of the selective ion transport mechanism of BP-ArF4 membrane. The narrow aqueous ionic channel (0.63 nm) and functionalities of BP-ArF4 can efficiently block VOSO4 transport, while keeping high proton conductivity. | ||
This estimation is based on the assumption that the structural correlation length d of the present ionomers scales in a linear fashion with the volume fraction of the aqueous ionic domain Vaq as observed for other CEMs with sulfonic-acid as fixed ionic groups.44 This corresponds to the local anisotropic swelling, which had been claimed to be characteristic for the locally flat morphologies with the dominant swelling component parallel to the normal.44
The relevant data are shown in Table 2. BP-ArF4 has the unique molecular structure of a hydrocarbon backbone and flexible pendant side chains with a perfluorinated terminating part (Fig. 1a). Although the BP-ArF4 membrane does not show the smallest structural correlation length d, its daq is the smallest as compared with other membranes, owing to its lower water volume fraction, essentially reflecting the low IEC. Notably, the ionomer peak of BP-ArF4 is relatively narrow, and the second order peak is clearly visible (Fig. 3b and c), thus indicating a well-ordered morphology with a narrow distribution of the aqueous ionic domain width. This finding may be a reason for its superior blocking properties for vanadium species, thus leading to the highest CE over the entire current densities (20–100 mA cm−2). Despite low IEC, BP-ArF4 maintains good ion conductivity because of the super-acidic perfluorosulfonate group which leads to its reasonably high VE. These characteristics of CE and VE lead to a better compromise than for other types of membranes. Therefore, the results of SAXS studies indicate that the unique molecular structure of BP-ArF4 with narrow aqueous ionic domains can afford the combination of properly sized aqueous ionic transport pathways and the high degree of order. The morphological “sieving” effect can be characterized by a relatively distinct threshold with respect to the width of the aqueous ionic domain.43 It is the very nature of this effect that only a narrow distribution of this width allows for a high blocking effect close to the percolation threshold. Otherwise, one has to further reduce the average width of the transport pathways so as to avoid transport through the fraction of wider parallel pathways. This would further reduce proton conductivity which is also the highest for a narrow domain width distribution because of the non-linear dependence of the water diffusion coefficient on the water volume fraction (domain width).43 As shown in Fig. 3b and c, the morphology of the BP-ArF4 membrane could provide both properly sized aqueous ionic domains and a high degree of order. The molecular structure of this ionomer represents a rare case of a hydrocarbon backbone with flexible pendant side chains with a perfluorosulfonate terminating group (see chemical structure of BP-ArF4 in Fig. 1). This allows for the formation of a well-developed ordered hydrophobic/hydrophilic separation, while hydrocarbon structures with sulfonic groups tethered to the main chain via a short chain, such as BP-SA, are by far less ordered on the nano-meter scale as indicated by broad ionomer peaks of the SAXs patterns (Fig. 3b and c). It should also be mentioned that the super-acidity of the terminating group of BP-ArF4 guarantees quasi complete dissociation at reduced water activity characteristics for VRFB electrolytes.
We further investigated the transport phenomena of vanadium species in two extreme cases, Nafion 117 (the largest daq) and BP-ArF4 (the smallest daq), by estimating VOSO4 transport rates. Because both membranes enabled complete exchange into VO2+ form in an excess of 0.1 M VOSO4 with limited uptake of excess VOSO4 (<10% of the fixed ion concentration), the total conductivity mainly represented the mobility of VOaq2+ in both structures (the volumetric density of ionic groups in both membranes was very close, i.e., approximately 1.9 meq. mL−1). As shown in Fig. 3d, the total conductivity of BP-ArF4 (2.8 mS cm−1) is lower than that of Nafion 117 (6 mS cm−1) by approximately a factor of two. A more interesting observation was that the increase in the VOSO4 concentration from 0.1 to 1.6 M left the total conductivity of Nafion 117 unchanged while the value of BP-ArF4 largely decreased to 1.3 mS cm−1. In the highly concentrated electrolytes, we observed a substantial amount of co-ion (SO42−) uptake, mainly from neutral VOSO4 and diminished water uptake (approximately 10%) as a response to the decreased water activity in the electrolyte solution. Thus, in more highly concentrated electrolytes with a further decrease in the membrane's hydration, the conductivity contribution of vanadium species (e.g., VO2+) can be further decreased in the BP-ArF4 after its de-swelling. The lower swelling can decrease the width of the aqueous ionic domain to an extent (e.g., 0.63 nm in Table 2), which substantially impedes vanadium ions, whereas protons still can be transported through the channels. This result is consistent with the recent molecular dynamics simulation study which suggests that zeolite-based IEMs with pore size <0.7 nm are crucial for the selective hydronium ion transport over vanadium ions.45
DFT calculation results provided additional insights into vanadium ion transport in terms of the chemical functionalities of polymer chains. A previous study has reported that the vanadium(IV) ion in sulfuric acid solution forms a [VO(SO4)(H2O)4]H2O complex.46 Our further optimized vanadium ion structures indicated that the energetics of the vanadium ion was the lowest when its spin state was a doublet (Fig. S7, ESI†), and its structural parameters, such as the VO bond length and V–O bond length, were in good agreement with previously reported values.47 Fig. S8–S11 (ESI†) illustrate the optimized structures of the vanadium ion complexes with different moieties of the sulfonated aromatic IEMs including Nafion and Table S1 (ESI†) lists the interaction energies (detailed calculation methods and optimized structures of IEMs can be found in the ESI†). The interaction energies between BP-ArF4 and vanadium ion were in the range of 22.6–33.8 kcal mol−1, which are significantly higher than those of other IEMs (14.2–21.4 kcal mol−1). The higher interaction energy is due to the strong binding energy through π⋯H and π⋯F hydrogen bonding in BP-ArF4 and vanadium ion complexes (Fig. S7, ESI†), which hinders the diffusion of vanadium ions through the polymer membrane. Among several complexes, comp3-BP-ArF4, the optimized structure of which is depicted in Fig. S8c (ESI†), showed a remarkably large binding energy (33.8 kcal mol−1). In this complex, three hydrogen atoms of the vanadium ion interact with the fluorine atoms of BP-ArF4, which should contribute to the interaction to a large extent. In addition, the fourth hydrogen atom (HD) interacts with the neighboring aromatic moiety. The formation of three π⋯F hydrogen bonding between vanadium ions and BP-ArF4 is possible because of the uniquely designed side chain structure of BP-ArF4 in which the flexible –CF2CF2OCF2CF2SO3H side chain is attached to the neighboring aromatic ring. In contrast, the interaction of Nafion with vanadium complexes occurs around its fluorine atoms, owing to the absence of an aromatic ring in the backbone or side chains (Fig. S11, ESI†). All complexes of Nafion showed that two hydrogen atoms of the vanadium complex interact with the –CF3 group of Nafion. Thus, the computed binding energies indicated that the interaction between Nafion and the vanadium complex is weaker (13.7 and 21.0 kcal mol−1 for complexes with comp1 and comp2, respectively) than that of BP-ArF4 (Table S1, ESI†). Although the fluorine atoms in the –CF3 group typically have a sizeable negative charge, their hydrogen bonding ability with hydrogen donor molecules is rather weak.48
As graphically illustrated in Fig. 3e, the extraordinary high ion selectivity property and VRFB performance of BP-ArF4 are believed to be derived from the synergistic effects of its unique morphology and functionalities in the polymer chain which greatly hinder the vanadium ion transport, while keeping high proton conductivity. We also investigated the differences of VOSO4 transport in BP-ArF4 and Nafion 117 by comparing the permeation activation energies. The activation energies of VOSO4 transport for BP-ArF4 and Nafion 117 membranes were calculated using Arrhenius plots obtained by measuring the VOSO4 permeability at different temperatures. As shown in Table S2 (ESI†), BP-ArF4 has almost two times higher activation energy (9.14 kcal mol−1, 0.40 eV) than Nafion 117 (4.5 kcal mol−1, 0.20 eV) and Nafion 212 (5.4 kcal mol−1, 0.23 eV), which agrees with the conclusion from our simulation data provided in Table S1 (ESI†) and the conductivity measurement from VOSO4 equilibrated solutions shown in Fig. 3c.
Breaking the trade-off limitations in IEMs
Finally, to evaluate the battery performance with the ion transport properties of BP-ArF4, we compared its trade-off of ion selectivity and membrane resistivity with other high-performance CEMs and AEMs reported to date (Fig. 4). To make a fair comparison, the performance of IEMs that were enhanced with additional components or by thickness optimization (e.g., mixed matrix membranes, blended membranes, and nanocomposite membranes) are not included in the figure. As all data of BP-ArF4 in this report were obtained from membranes in the form of a free-standing film directly related to the polymer's unique properties, they should be compared with the performance of membranes made of a single polymer. For example, low areal resistance (3.9 × 10−2 Ω cm2) and high ion conductivity (0.27 S cm−1) values of a recently reported polyamide thin-film composite membrane were obtained by normalizing the resistance and conductance with the entire thickness of the composite membrane (support layer + polyamide selective layer) rather than using a thin selective polyamide layer.49 Considering that the intrinsic resistivity and ion conductivity of the polyamide layer are ∼706 Ω cm2 and 1.4 × 10−3 S cm−1, respectively, a trade-off comparison between BP-ArF4 and thin-film composite membranes does not provide insightful information about the properties of the materials.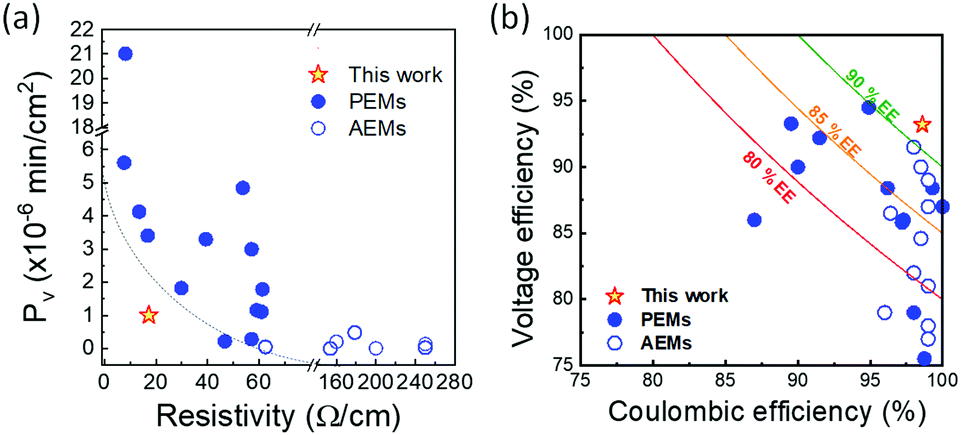 | ||
| Fig. 4 (a) Trade-off between vanadium diffusivity and resistivity. (b) VRFB performance relationship among CE, VE, and EE at 40–60 mA cm−2. Detailed information on the trade-off curves is available in Tables S3 and S4 (ESI†). | ||
As shown in Fig. 4a, BP-ArF4 has the best combination of low membrane resistivity and low vanadium permeability outperforming all CEM and AEM materials reported so far. Generally, CEMs show low resistivity and high vanadium permeability, while AEMs show high resistivity and low vanadium permeability. Notably, the performance of BP-ArF4 overcomes the conventional trade-off limitation of ion selectivity and resistivity of IEMs. The high proton selectivity over vanadium ions by BP-ArF4 demonstrates its great potential for application in VRFBs. Indeed, Fig. 4b shows a trade-off relationship between CE and VE, which is similar to the ion selectivity/conductivity trade-off. With a few exceptions, IEMs with high VE tend to show low CE due to high crossover of vanadium species and vice versa. However, BP-ArF4 illustrates a remarkable combination of high CE with high VE, resulting in one of the highest EEs reported in the literature. It outperforms most of the CEMs and AEMs for VRFB systems reported to date and mitigates the trade-off limitation on battery performance due to the highly selective proton/vanadium ion transport.
Conclusions
In summary, we developed a novel class of sulfonated aromatic polymers that have a good combination of high proton conductivity and ion selectivity, and can greatly mitigate the trade-off limitations of conventional IEMs. The effects of the pendant group structure of the polymer on the IEM's ion selectivity and battery performance were investigated. The incorporation of an aromatic ring and a short perfluoroalkyl chain on the pendant group of BP-ArF4 greatly improved the proton/vanadium ion selectivity, which is three times higher than that of Nafion®, the standard CEM in VRFB. With excellent vanadium ion blocking capacity and good proton conductivity, BP-ArF4 achieved high CE and VE values in a VRFB single cell. SAXS and DFT calculation studies revealed that the narrowly distributed aqueous ionic domain width of BP-ArF4 and the strong interaction between the side chain of the polymer and vanadium ion complexes through π⋯H or π⋯F hydrogen bonding synergistically improve proton/vanadium ion selectivity. This study suggests an effective path toward the design of high-performance IEMs that can overcome the trade-off limit of ion selectivity/conductivity in other energy conversion and storage systems. Enhanced evaluations on the IEM performance in VRFB (e.g. longer cyclic stability test, volumetric imbalance measurement) will further validate their practical-level cycling stability and efficiency. Additional optimization, such as nanocomposite membranes, using the unique material platform of BP-ArF4 as a base material can lead to more advanced IEMs with further improvement in efficiencies of energy storage systems.Experimental section
Reagents
The biphenyl precursor polymer BPBr-100 (Mn = 60 kg mol−1, Mw/Mn = 2.0) was synthesized using an acid-catalyzed polycondensation method as mentioned in our previous reports.28,50 Potassium thioacetate (Sigma-Aldrich, 98%), formic acid (VWR, 88%), hydrogen peroxide aqueous solution (ACROS, 35 wt%), sodium 4-hydroxybenzenesulfonate (TCI, >98%), 4-iodophenol (Sigma-Aldrich, 99%), tetrafluoro-2-(tetrafluoro-2-iodoethoxy)ethanesulfonyl fluoride (Sigma-Aldrich, 97%), and copper powder (Sigma-Aldrich, 99%, 200 mesh) were used as received without further purification. Sodium hydroxide, potassium hydroxide and potassium carbonate (anhydrous) were purchased from Fisher Scientific. Ethanol, methanol, tetrahydrofuran (THF), dimethyl sulfoxide (DMSO) and N,N-dimethylacetamide (DMAc) were of reagent grade and used without further purification. Commercial Nafion® 117 and 212 membranes were purchased from Ion Power Inc. (DE, USA). Vanadyl sulfate (VOSO4, 99.9%) and magnesium sulfate (MgSO4, 99.5%) were purchased from Alfa Aesar (MA, USA). Sulfuric acid (H2SO4, 98.0%) was purchased from Fisher Scientific (PA, USA).Polymer synthesis and characterization
Synthesis of BP-TA. BPBr-100 (5.00 g, 13.0 mmol), potassium thioacetate (5.19 g, 45.4 mmol) and THF (150 mL) were added to a 250 mL round-bottom flask. After the precursor polymer was fully dissolved, the reaction flask was connected to a reflux condenser and was heated to 50 °C for 12 h. After cooling to room temperature, methanol (450 mL) was added to the solution to precipitate the polymer, which was then collected by vacuum filtration and dried. The crude polymer product was dissolved in THF (95 mL) and filtered through a cotton plug to remove insoluble solids. The filtrate was poured into methanol to precipitate BP-TA which was collected by filtration and dried under vacuum at 80 °C for 15 h (yield: 4.86 g). 1H NMR (CDCl3): δ (ppm) = 7.57 (d, 4H), 7.36 (d, 4H), 2.8 (t, 2H), 2.43 (m, 2H), 1.52 (m, 2H), 1.37 (m, 2H), 1.24 (m, 2H).
Fabrication of BP-SA membrane. Once BP-TA was converted to the sulfonic acid form in BP-SA, it exhibited poor solubility in all organic solvents. Thus, the oxidation of the thioacetate group of BP-TA to the –SO3H of BP-SA was conducted in a membrane form after casting BP-TA film from a 5 wt% THF solution onto a glass plate. After the membrane was dried, the oxidation was performed with a mixture of 6 M formic acid and hydrogen peroxide aqueous solution (35 wt%) in a glass container at 50 °C for 3 h. The membrane was then removed from the container and washed extensively with water.
Synthesis of BP-ArSA. BPBr-100 (3.00 g, 7.80 mmol), sodium 4-hydroxybenzenesulfonate (5.00 g, 25.5 mmol), and potassium hydroxide (1.80 g, 32.0 mmol) were added to a 250 mL round-bottom flask. Anhydrous DMSO (75 mL) and THF (15 mL) were added into the flask; then the reaction mixture was stirred at 40 °C for 24 h after which THF was removed under reduced pressure. The concentrated polymer in DMSO solution was stirred for an additional 12 h. After cooling to room temperature, the solution was poured into acetone (450 mL) to precipitate the polymer, which was washed with methanol to remove excess sodium 4-hydroxybenzenesulfonate. The polymer product was filtered and dried under vacuum at 50 °C for 12 h before fabricating into membrane (yield: 3.8 g). 1H NMR (DMSO-d6): δ (ppm) = 7.75 (d, 4H), 7.48 (d, 2H), 7.37 (d, 4H), 6.79 (d, 2H), 3.87 (t, 2H), 2.53 (m, 2H), 1.63 (m, 2H), 1.44 (m, 2H), 1.20 (m, 2H).
Synthesis of BP-Ar-I. BPBr-100 (10.0 g, 26.2 mmol), 4-iodophenol (17.75 g, 80.6 mmol), and potassium carbonate (11.15 g, 80.6 mmol) were added to a 500 mL round-bottom flask which was connected with a reflux condenser under a positive flow of nitrogen. Anhydrous DMAc (200 mL) was added to the flask and the reaction mixture was stirred at room temperature until all reagents were completely dissolved and then heated to 90 °C for 3 h. After cooling to room temperature, the solution was poured into methanol containing HCl (12 mL) to precipitate the polymer. The collected polymer was re-dissolved in THF (220 mL), filtered through a cotton plug, and precipitated by dropping into methanol. Precipitated polymer was filtered and dried in a vacuum oven at 50 °C overnight to afford BP-ArI (yield: 13.5 g). 1H NMR (DMSO-d6): δ (ppm) = 7.57 (d, 4H), 7.49 (d, 2H), 7.39 (d, 4H), 6.60 (d, 2H), 3.82 (t, 2H), 2.47 (m, 2H), 1.71 (m, 2H), 1.47 (m, 2H), 1.31 (m, 2H).
Synthesis of sodium tetrafluoro-2-(tetrafluoro-2-iodoethoxy)ethanesulfonate (1). Tetrafluoro-2-(tetrafluoro-2-iodoethoxy)ethanesulfonyl fluoride (5.00 g, 11.7 mmol) and NaOH solution (0.95 g in 6.5 mL H2O; 12.8 wt%) were added to a 25 mL round-bottom flask connected with a reflux condenser, and then the reaction mixture was heated to 95 °C for 16 h in an oil bath. After the reaction completed, water was removed under vacuum at 60 °C. Ethanol (20 mL) was added to the concentrated reaction mixture to precipitate sodium fluoride, which was removed by filtration through a cotton plug to give a clear solution. Removal of ethanol using a rotary evaporator gave compound 1 in sodium sulfonate form as an off-white powder which was dried in a vacuum oven at 50 °C overnight (yield: 5.20 g, 99.3%). 19F NMR (D2O): δ (ppm) = −68.38 (t, 2F), −83.17 (t, 2F), −86.31 (t, 2F), −118.46 (t, 2F).
Synthesis of BP-ArF4. BP-Ar-I (3.05 g, 5.75 mmol), Cu (3.65 g, 57.0 mmol), and DMAc (18 mL) were added to a 100 mL 2-neck round-bottom flask under a positive flow of nitrogen. After BP-Ar-I was dissolved, the reaction mixture was stirred at 95 °C for 1 h. DMAc (8.0 mL) solution containing compound 1 (5.13 g, 11.5 mmol) was then added dropwise slowly via a syringe at 95 °C. The reaction mixture was stirred at 95 °C for 2 h, then at 120 °C for 48 h and then at 160 °C for 24 h.51 The reaction mixture was cooled to room temperature and additional DMAc (20 mL) was added to dilute the polymer solution which was centrifuged at 9000 rpm for 30 min to separate copper powder. The centrifuged polymer solution was filtered through a short silica plug to remove any fine copper residue and poured into Petri-dishes evenly to cast polymer films. The BP-ArF4 membranes (in –SO3Na form) were dried in a vacuum oven at 80 °C, washed with warm water, and finally dried under vacuum (yield: 4.07 g). 1H NMR (DMSO-d6): δ (ppm) = 7.71 (d, 4H), 7.52 (d, 2H), 7.37 (d, 4H), 7.01 (d, 2H), 3.94 (t, 2H), 2.52 (m, 2H), 1.66 (m, 2H), 1.45 (m, 2H), 1.21 (m, 2H). 19F NMR (DMSO-d6): δ (ppm) –64.57 (s, 3F, –CF3), –82.25 (t, 2F), –87.08 (t, 2F), –111.82 (t, 2F), –117.92 (t, 2F).
Synthesis of BPN1. BPN1 was prepared using a modified procedure mentioned in our previous report.50 BPBr-100 (5.00 g, 13.0 mmol) was dissolved with DMAc (50 mL) in a 250 mL round-bottom flask. TMA in ethanol solution (33 wt%, 20 mL) was added to the flask and the reaction mixture was stirred at room temperature for 24 h. The polymer was precipitated by adding a mixture of hexane/THF solution (7
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 in v/v), which was collected by filtration and dried under vacuum at 40 °C overnight. 1H NMR (DMSO-d6): δ (ppm) = 7.56 (d, 4H), 7.39 (d, 4H), 3.22 (t, 2H), 3.01 (s, –CH3, 9H) 2.54 (m, 2H), 1.61 (m, 2H), 1.28 (m, 2H), 1.20 (m, 2H).
:3 in v/v), which was collected by filtration and dried under vacuum at 40 °C overnight. 1H NMR (DMSO-d6): δ (ppm) = 7.56 (d, 4H), 7.39 (d, 4H), 3.22 (t, 2H), 3.01 (s, –CH3, 9H) 2.54 (m, 2H), 1.61 (m, 2H), 1.28 (m, 2H), 1.20 (m, 2H).
Polymer molecular structure characterization. 1H NMR spectra were obtained using a Varian Unity 500 MHz spectrometer, and chemical shifts were referenced to the NMR solvent peaks; CDCl3 (at 7.26 ppm) and DMSO-d6 (at 2.48 ppm). FT-IR spectra were obtained for the investigation of the functional groups of the synthesized polymer using a Thermo Nicolet Nexus 4700 FT-IR Spectrometer with ATR tool. The average molecular weight of the precursor polymer (BPBr-100) was determined using gel permeation chromatography (GPC, Viscotek T60A instrument), with THF as the eluent and a polystyrene standard curve.
Membrane fabrication and characterization
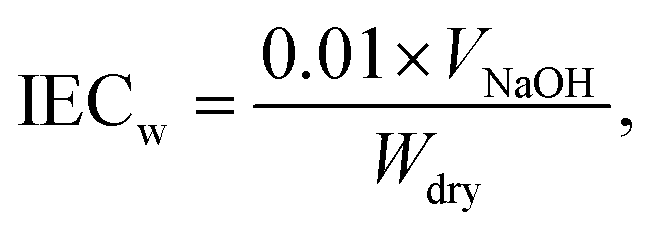 | (2) |
Linear swelling (%) was obtained by measuring the x–y and diagonal length of dry and wet membrane samples. Water uptake (WH2O in wt%) was measured by comparing the weights of wet vs. dry membranes at room temperature according to:
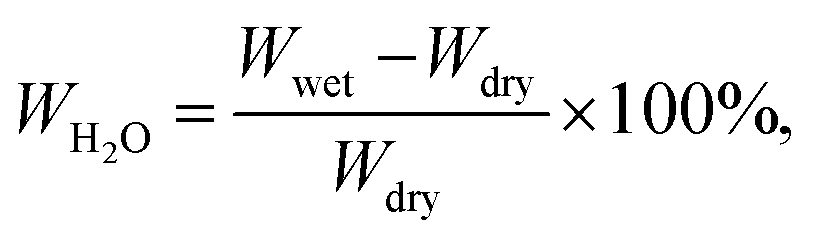 | (3) |
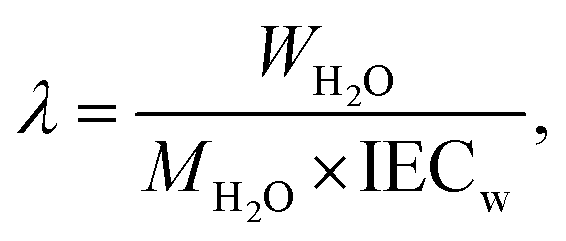 | (4) |
Ion transport properties and VRFB performance
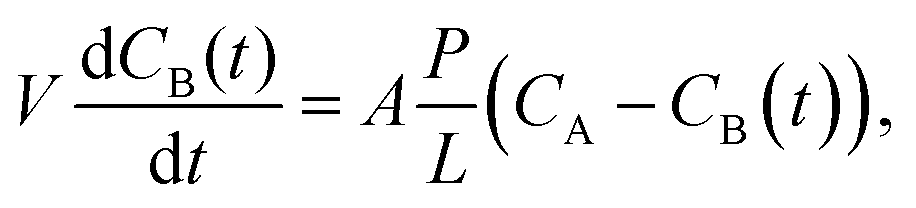 | (5) |
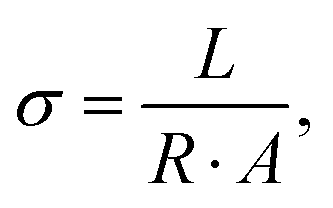 | (6) |
It is worth mentioning that a membrane has different swelling ratio at different temperatures. Hence, a change in membrane dimension was considered during calculating membrane permeability at each temperature. The activation energies of Nafion 117, Nafion 212, and BP-ArF4 are listed in Table S2 (ESI†). The temperature dependence of the vanadium permeability of Nafion 117, Nafion 212, and BP-ArF4 is displayed in Fig. S9 (ESI†).
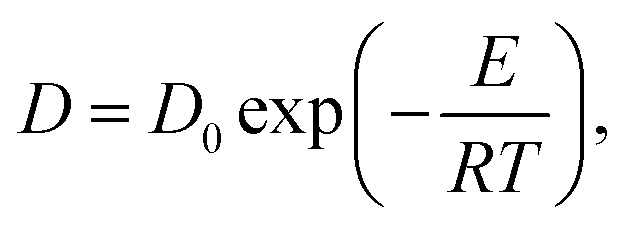 | (7) |
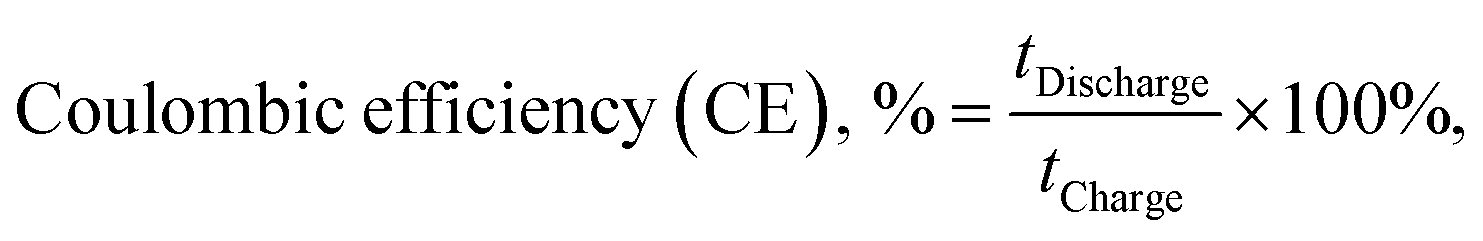 | (8) |
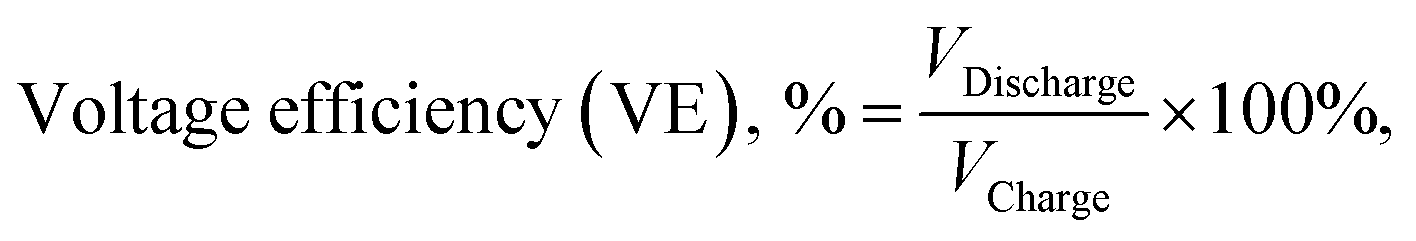 | (9) |
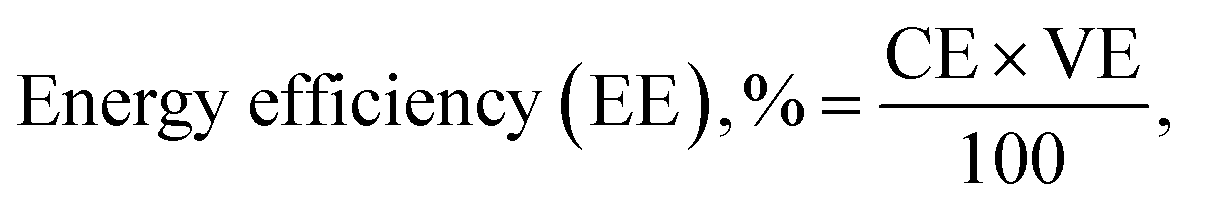 | (10) |
Small angle X-ray scattering. Small angle X-ray scattering (SAXS) was carried out using a home-built camera using a rotating anode (Bruker FR591-Nonius) operating at 3 kW with a source of Cu-Kα radiation (λ = 1.5418 Å). The X-ray optics consists of a set of two horizontal and vertical 16 cm long Ni-filtered total reflection mirrors (Kirkpatrick-Baez mirrors) positioned at 180 cm from the X-ray source. The size of the source (0.2 × 0.2 mm2) and its distance from the X-ray optics (180 cm) provide a low divergence of the X-ray beam (∼0.05 mrad), which is a key parameter (with the high-brightness) to optimize the SAXS and GISAXS applications.
The beam line is equipped with a set of 4 beam defining slits (close to mirror optics) and a set of 4 anti-scattering slits in front of the sample position (Xenocs motorized non-scattering slits). The distance between the two sets of slits is 180 cm, thus providing very low-background intensity even at low scattering angle. Scattering patterns were recorded using a two-dimensional 14 × 14 cm2 area (Bruker VANTEC-2000) mikroGap® detector. The mikroGap® technology gives a very fine Point Spread Function which allows the measurement of even weak scattering intensities close to strong diffraction peaks with pixel size resolution of either 70 μm, 140 μm or 280 μm. The flux on the sample is ∼3 × 107 ph s−1 with a beam size (FWHM) ∼ 600 × 600 μm2. The sample detector distance was placed at 1 m and 3.5 m to explore q values from about q = 0.01 up to 0.4 Å−1, where q is the momentum transfer q = (4πsinθ)/λ and 2θ is the total scattering angle. The sample-detector distance was calibrated using silver behenate as the standard. The normalization of the scattering data was applied by first measuring the transmission with a semi-transparent beam stop, followed by the thickness of each sample, and finally using a Lupolen sample as the calibrating sample. After equilibration in pure water or in a 1.6 M VOSO4/4 M H2SO4 aqueous solution, two or three membrane samples with 4 mm diameter (total thickness around 300 μm) were placed in a brass cell between 5 μm thick Mylar sheets serving as windows. The mass difference of the membranes in the dry proton and the soaked state was attributed to electrolyte uptake. For calculating the volume fraction of vanadium electrolyte in the membrane, its density was taken to be identical to that of the 1.6 M VOSO4/4 M H2SO4 aqueous solution (1.4 g cm−3).
Density functional theory (DFT) calculations. For density functional theory (DFT) calculations, we employed the ωB97XD functional59 and 6-31G(d,p) basis sets for S, N, C, H, and O atoms.60,61 For vanadium, core electrons were represented using the LANL08 effective core potential (ECP) and for valence electrons of vanadium, we used LANL08 basis sets augmented by f function.62–64 Geometry optimizations and additional frequency calculations to confirm that the optimized structure is a minimum were conducted using the Gaussian 16 program.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by grants from the National Science Foundation (grant No. CBET-1706910 with Dr Christina Payne as program manager), the U.S. Department of Energy, Office of Efficiency and Renewable Energy (EERE), Fuel Cell Technology Office (FCTO), Award No. DE-EE0007647 (Program manager: Neha Rustagi), ARPA-E (IONICS DE-AR0000769 and REFUEL DE-AR0000805), and New York State Energy Research and Development Authority (NYSERDA; grant No. 127734). T. Wang acknowledges support from the UIC Provost's Graduate Research Award program. S. Kim and C. Bae thank StorEn for discussions.Notes and references
- C. H. Park, C. H. Lee, M. D. Guiver and Y. M. Lee, Prog. Polym. Sci., 2011, 36, 1443–1498 CrossRef.
- R. M. Darling, K. G. Gallagher, J. A. Kowalski, S. Ha and F. R. Brushett, Energy Environ. Sci., 2014, 7, 3459–3477 RSC.
- M. Skyllas-Kazacos, M. H. Chakrabarti, S. A. Hajimolana, F. S. Mjalli and M. Saleem, J. Electrochem. Soc., 2011, 158, R55–R79 CrossRef.
- B. E. Logan and M. Elimelech, Nature, 2012, 488, 313–319 CrossRef.
- G. M. Geise, M. A. Hickner and B. E. Logan, ACS Appl. Mater. Interfaces, 2013, 5, 10294–10301 CrossRef.
- P. Długołęcki, K. Nymeijer, S. Metz and M. Wessling, J. Membr. Sci., 2008, 319, 214–222 CrossRef.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef.
- W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Adv. Funct. Mater., 2013, 23, 970–986 CrossRef.
- K. Lin, R. Gómez-Bombarelli, E. S. Beh, L. Tong, Q. Chen, A. Valle, A. Aspuru-Guzik, M. J. Aziz and R. G. Gordon, Nat. Energy, 2016, 1, 16102 CrossRef.
- W. Wang and V. Sprenkle, Nat. Chem., 2016, 8, 204 CrossRef.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195 CrossRef.
- N. Wang, J. Yu, Z. Zhou, D. Fang, S. Liu and Y. Liu, J. Membr. Sci., 2013, 437, 114–121 CrossRef.
- S. Roe, C. Menictas and M. Skyllas-Kazacos, J. Electrochem. Soc., 2016, 163, A5023–A5028 CrossRef.
- X. Li, H. Zhang, Z. Mai, H. Zhang and I. Vankelecom, Energy Environ. Sci., 2011, 4, 1147–1160 RSC.
- X. Wei, Z. Nie, Q. Luo, B. Li, B. Chen, K. Simmons, V. Sprenkle and W. Wang, Adv. Energy Mater., 2013, 3, 1215–1220 CrossRef.
- M. Ulaganathan, V. Aravindan, Q. Yan, S. Madhavi, M. Skyllas-Kazacos and T. M. Lim, Adv. Mater. Interfaces, 2016, 3, 1500309 CrossRef.
- S.-H. Cha, J. Nanomater., 2015, 2015, 207525 Search PubMed.
- Q. Luo, H. Zhang, J. Chen, P. Qian and Y. Zhai, J. Membr. Sci., 2008, 311, 98–103 CrossRef.
- J. Xi, Z. Wu, X. Qiu and L. Chen, J. Power Sources, 2007, 166, 531–536 CrossRef.
- X. Teng, Y. Zhao, J. Xi, Z. Wu, X. Qiu and L. Chen, J. Membr. Sci., 2009, 341, 149–154 CrossRef.
- M. A. Aziz and S. Shanmugam, J. Power Sources, 2017, 337, 36–44 CrossRef.
- S.-H. Cha, J. Nanomater., 2015, 2015, 1 CrossRef.
- D. Chen, M. A. Hickner, E. Agar and E. C. Kumbur, Electrochem. Commun., 2013, 26, 37–40 CrossRef.
- Z. Li, W. Dai, L. Yu, J. Xi, X. Qiu and L. Chen, J. Power Sources, 2014, 257, 221–229 CrossRef.
- Q. Dai, Z. Liu, L. Huang, C. Wang, Y. Zhao, Q. Fu, A. Zheng, H. Zhang and X. Li, Nat. Commun., 2020, 11, 1–9 Search PubMed.
- Y. Zeng, L. Wang, L. Zhang and J. Q. Yu, J. Membr. Sci., 2018, 546, 225–233 CrossRef.
- A. Buonerba, V. Speranza and A. Grassi, Macromolecules, 2013, 46, 778–784 CrossRef.
- W.-H. Lee, E. J. Park, J. Han, D. W. Shin, Y. S. Kim and C. Bae, ACS Macro Lett., 2017, 6, 566–570 CrossRef.
- X. Ling, C. Jia, J. Liu and C. Yan, J. Membr. Sci., 2012, 415–416, 306–312 CrossRef.
- M. S. Cha, H. Y. Jeong, H. Y. Shin, S. H. Hong, T.-H. Kim, S.-G. Oh, J. Y. Lee and Y. T. Hong, J. Power Sources, 2017, 363, 78–86 CrossRef.
- B. Zhang, S. Zhang, D. Xing, R. Han, C. Yin and X. Jian, J. Power Sources, 2012, 217, 296–302 CrossRef.
- J. Si, Y. Lv, S. Lu and Y. Xiang, J. Power Sources, 2019, 428, 88–92 CrossRef.
- D. Chen, M. A. Hickner, E. Agar and E. C. Kumbur, J. Membr. Sci., 2013, 437, 108–113 CrossRef.
- B. Jiang, L. Wu, L. Yu, X. Qiu and J. Xi, J. Membr. Sci., 2016, 510, 18–26 CrossRef.
- C. Li, A. L. Ward, S. E. Doris, T. A. Pascal, D. Prendergast and B. A. Helms, Nano Lett., 2015, 15, 5724–5729 CrossRef CAS.
- D. You, H. Zhang, C. Sun and X. Ma, J. Power Sources, 2011, 196, 1578–1585 CrossRef CAS.
- O. Nibel, S. M. Taylor, A. Pătru, E. Fabbri, L. Gubler and T. J. Schmidt, J. Chem. Soc., Faraday Trans., 2017, 164, A1608 CAS.
- I. Derr, M. Bruns, J. Langner, A. Fetyan, J. Melke and C. Roth, J. Power Sources, 2016, 325, 351–359 CrossRef CAS.
- C. H. Park, S. Y. Lee, D. S. Hwang, D. W. Shin, D. H. Cho, K. H. Lee, T.-W. Kim, T.-W. Kim, M. Lee and D.-S. Kim, Nature, 2016, 532, 480–483 CrossRef.
- A. Münchinger and K.-D. Kreuer, J. Membr. Sci., 2019, 592, 117372 CrossRef.
- A. Münchinger, T. Wang, S. Kim and K.-D. Kreuer, 2019, in preparation.
- I. S. Chae, T. Luo, G. H. Moon, W. Ogieglo, Y. S. Kang and M. Wessling, Adv. Energy Mater., 2016, 6, 1600517 CrossRef.
- K.-D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104, 4637–4678 CrossRef CAS.
- K. D. Kreuer and G. Portale, Adv. Funct. Mater., 2013, 23, 5390–5397 CrossRef CAS.
- K. R. Hinkle, C. J. Jameson and S. Murad, J. Phys. Chem. C, 2014, 118, 23803–23810 CrossRef CAS.
- X. Wu, J. Wang, S. Liu, X. Wu and S. Li, Electrochim. Acta, 2011, 56, 10197–10203 CrossRef CAS.
- C. Choi, S. Kim, R. Kim, Y. Choi, S. Kim, H.-y. Jung, J. H. Yang and H.-T. Kim, Renewable Sustainable Energy Rev., 2017, 69, 263–274 CrossRef CAS.
- H.-J. Schneider, Chem. Sci., 2012, 3, 1381–1394 RSC.
- Q. Dai, Z. Liu, L. Huang, C. Wang, Y. Zhao, Q. Fu, A. Zheng, H. Zhang and X. Li, Nat. Commun., 2020, 11, 13 CrossRef CAS.
- W.-H. Lee, Y. S. Kim and C. Bae, ACS Macro Lett., 2015, 4, 814–818 CrossRef CAS.
- L. Assumma, C. Iojoiu, R. Mercier, S. Lyonnard, H. D. Nguyen and E. Planes, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1941–1956 CrossRef CAS.
- G.-J. Hwang and H. Ohya, J. Membr. Sci., 1996, 120, 55–67 CrossRef CAS.
- J. Tolliday, E. Woods and E. Hartung, Trans. Faraday Soc., 1949, 45, 148–155 RSC.
- T. Wang, S. J. Moon, D.-S. Hwang, H. Park, J. Lee, S. Kim, Y. M. Lee and S. Kim, J. Membr. Sci., 2019, 583, 16–22 CrossRef CAS.
- M. Rychcik and M. Skyllas-Kazacos, J. Power Sources, 1988, 22, 59–67 CrossRef CAS.
- M. S. Cha, S. W. Jo, S. H. Han, S. H. Hong, S. So, T.-H. Kim, S.-G. Oh, Y. T. Hong and J. Y. Lee, J. Power Sources, 2019, 413, 158–166 CrossRef CAS.
- S. Kim, T. B. Tighe, B. Schwenzer, J. Yan, J. Zhang, J. Liu, Z. Yang and M. A. Hickner, J. Appl. Electrochem., 2011, 41, 1201–1213 CrossRef CAS.
- T. Mohammadi and M. S. Kazacos, J. Appl. Electrochem., 1997, 27, 153–160 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- A. McLean and G. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS.
- A. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00508h |
| This journal is © The Royal Society of Chemistry 2020 |