
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic single process additive manufacturing of hydro-responsive Ag nanoparticle composites by digital visible light processing 3D printing†
Sokhna I. Y.
Diouf
a,
Darrick J.
Williams
a,
Alejandra
Londoño-Calderon
 a,
Michael T.
Pettes
a,
Sönke
Seifert
b and
Millicent A.
Firestone
*ac
a,
Michael T.
Pettes
a,
Sönke
Seifert
b and
Millicent A.
Firestone
*ac
aLos Alamos National Laboratory, Los Alamos, NM 87545, USA
bArgonne National Laboratory, Lemont, IL 60439, USA
cLawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. E-mail: mafirestone@lbl.gov
First published on 4th September 2020
Abstract
Single unit process fabrication of a responsive Ag nanoparticle (NP) chemical hydrogel composite is achieved through digital visible light processing (DLP) 3D printing of a self-assembled nanostructured (multilamellar) resin doped with Ag(I). The In situ production of plasmonic Ag NPs synergistically enhances structural fidelity (resolution) of the 3D builds, acting as optical absorbers to control depth of light penetration. The storage modulus (stiffness) of the Ag NP hydrogel increases up to a factor of 3 compared to the hydrogel alone. The multiscale architected composite contains nanoscale features that augment the DLP generated geometry and microscale features and displays reversible changes in the collective Ag NP plasmon resonance. This work lays the foundation for the realization of multi-scale, multi-constituent, multi-functional materials through additive manufacturing.
Polymer additive manufacturing (AM) of self-assembled resins paired with in situ synthesis of metal NPs offers a means to carry out single process fabrication of functional hierarchically-structured composites. Of particular interest is the fabrication of metal NP hydrogels, a versatile class of materials having demonstrated utility in a wide range of applications spanning biomedical (e.g., tissue regeneration) to optical and/or electronic materials.1–4 The ability to manufacture nanoscale materials, structures, devices, and ultimately systems to advance commercialization of nanotechnology depends on our ability to reliably, and cost-effectively carry out high-throughput production at scale.5 Achievement of these goals requires moving beyond traditional iterative/sequence unit operations towards single process strategies. Herein, we demonstrate an integrated nano-fabrication approach for the single process production of solvent-responsive Ag NP chemical hydrogel composites through the amalgamation of bottom-up self-assembly with DLP 3D printing to achieve the in situ synthesis of plasmonic metal (Ag) NPs. The successful implementation of visible light vat photopolymerization using a custom synthesized resin doped with Ag+ paired with a visible light photo-initiating system succeeds in capturing the self-assembled nanostructure of the resin mixture, and in situ generates spatially localized plasmonic Ag NPs within the 3D printed build. Most importantly, the in situ synthesis of plasmonic Ag NPs synergistically act to regulate of the depth of light penetration during DLP 3D printing, leading to macroscopically observable improvements in build structural fidelity. In addition, the Ag NPs enhance the hydrogel mechanical characteristics without detriment to solvent responsivity, manifesting as reversible changes in collective plasmonic properties of the composite.
Examples of additive nano-manufacturing for the production of functional 3D printed NP polymer composites have been reported.6,7 The majority of these works has focused on the 3D printing of photoactive resins, extrudable inks, or thermoplastics doped with ex situ synthesized nanofillers. For example, ex situ synthesized Au NPs blended with polyvinyl alcohol (PVA) have been printed by fused deposition modelling (FDM) to yield dichroic plastics.8 Carbon nanotubes doped into chitosan have been extrusion printed to generate conductive composites.9,10 Preparative approaches using ex situ synthesized NPs often suffer from uncontrolled filler aggregation, phase separation, and limited NP loading levels.1 Alternatively, the in situ synthesis of NPs offers a means to improve filler loading levels, reduce unwanted NP aggregation, eliminate phase separation, and control NP spatial organization and geometric arrangement in the matrix.1 The in situ generation of Ag NPs during vat photo-polymerization (3D printing using DLP), employing commercially-available acrylate resins doped with silver ions has been carried out. Fantino et al. demonstrated the DLP 3D printing of a mixture of polyethylene diacrylate (PEGDA) doped with AgNO3 produces geometrically complex builds that only upon post-processing with UV light promoted the reduction of Ag+ to Ag NPs randomly dispersed throughout the builds.11 The duration of UV illumination was used to control the extent of Ag NP formation. Extended irradiation times produced metallic (silver mirror-like) structures determined to be electronically conductive, suggesting attainment of Ag NP percolation paths. Taormina et al. extended this work achieving silver reduction during 3D printing by UV-light stereolithography (SLA), thereby eliminating the post-processing step.12 Here too, the in situ synthesized Ag NPs were randomly distributed (unorganized) within the 3D printed materials. Devising a resin system that can succeed in controlling the spatial localization of the in situ synthesized NPs, and/or exploit the production of the NPs during printing to synergistically enhance properties (i.e., mechanical or print fidelity) of the composite has not been reported.
3D printing of internally structured polymers has recently been demonstrated.13 A custom synthesized resin formulation composed of a mixture of self-assembled ionic liquid (IL) monomer (vinyldecylimidazolium chloride), a crosslinker, PEGDA, dispersed in water generated a 2D hexagonally structured lyotropic mesophase.13 3D printing by visible light DLP photo-initiated free radical polymerization of the self-assembled liquid-crystalline lyotropic mesophase captured geometrically complex 3D builds possessing a hexagonal perforated lamellar (HPL) nanostructure.13 This work confirmed the utility of combining bottom-up self-assembly with top-down 3D printing as a low-cost strategy for introducing high-resolution nanoscale features (2–5 nm) into otherwise low resolution (microscale) features in a 3D printed build.13 3D printed polymers containing internal nanostructure offers a mean to promote organization of other nanosized functional components. In the present work, we seek to build upon our prior efforts using visible light DLP printing of self-assembled custom synthesized resins to demonstrate single process 3D printing of an internally structured (multi-lamellar) chemical hydrogel14 concomitant with in situ generation of spatially confined Ag NPs. There is increasing recognition that achieving greater control over the spatial location and/or geometric arrangement of NPs within a polymer matrix offers opportunities for nanocomposite fabrication exhibiting emergent or multi-functionality.1 Further elaborated is how production of the Ag NPs during 3D printing acts to improve mechanical characteristics of the hydrogel and synergistically enhances structural fidelity of the prints. All of these benefits are achieved without detriment to the hydro-responsivity of the 3D printed Ag NP composite, engendering collective plasmonic modulation.
A self-assembled,14,15 visible-light reactive vat photo-polymerizable resin mixture is prepared in several steps as outlined in Fig. 1. Specifically, a slightly viscous aqueous (74.6% (w/w) H2O) mixture containing a saturated phospholipid (1,2-dimyristoyl-sn-glycero-3-phosphocholine), DMPC (1) (6.7% (w/w)), freshly synthesized diacrylated poly(ethylene oxide)75-b-poly(propylene oxide)30-b-poly(ethylene oxide)75, PEO75–PPO30–PEO75, F68(Acr)2 triblock copolymer (2) (14.7% (w/w)), a zwitterionic co-surfactant, lauryldimethylamine-N-oxide, LDAO (3) (3.7% (w/w)), and a co-macromonomer, poly(ethylene glycol)diacrylate (PEGDA Mn 700) (4) (0.60% (w/w)) is prepared to which a visible light photo-initiator system composed of 1.54 mM eosin Y ethanolic solution (5) (1.2% (w/w)), and triethanolamine, TEOA (6) (0.44% (w/w)) is added. The multicomponent mixture is pink (from the visible photo-initiator, λmax = 510 nm) and macroscopically homogenous (Fig. 1A). Immediately prior to vat photo-polymerization AgNO3 is added to achieve a concentration ranging between 1–100 mM. Builds of varying complexity can be 3D printed, such as a honeycomb (Fig. 1B and C), and a flower-of-life (geometric shape composed of multiple evenly spaced, overlapping circles arranged with six-fold symmetry, Fig. 1D and E). The printed objects are orange-brown in colour and have sufficient mechanical strength to be self-supporting, yet compliant (storage modulus of 228 MPa at 27 °C, Fig. S1A, ESI†). The builds exhibit good structural integrity with accuracy achieved in all dimensions (Fig. S2, ESI†). 3D prints prepared without AgNO3 are colourless (Fig. 1F and G) and considerably less mechanically strong (storage modulus of 69 MPa at 27 °C, Fig. S1B, ESI†) and display a loss in structural fidelity (Fig. 1F, G and Fig. S3, ESI†). The three-fold increase in storage modulus on 3D prints prepared with 9.3 mM Ag+ signals the in situ produced filler (Ag NPs) acts to increase the stiffness of the hydrogel, imparting a good reinforcing effect. The effect of increasing the Ag+ content, continues to improve the storage modulus, compared to the hydrogel alone. At very high concentrations of Ag+ (i.e., ∼50–100 mM), a reduction in the storage modulus at 27 °C is noted (187–162 MPa) but remains strengthened over the prints lacking the Ag (Fig. S1C, ESI†). A reduction in storage modulus at high filler content has been noted previously for a variety of nanocomponent fillers and attributed to competition between detrimental filler–polymer interactions and reinforcing percolated fractal (jammed) network of the fillers.16
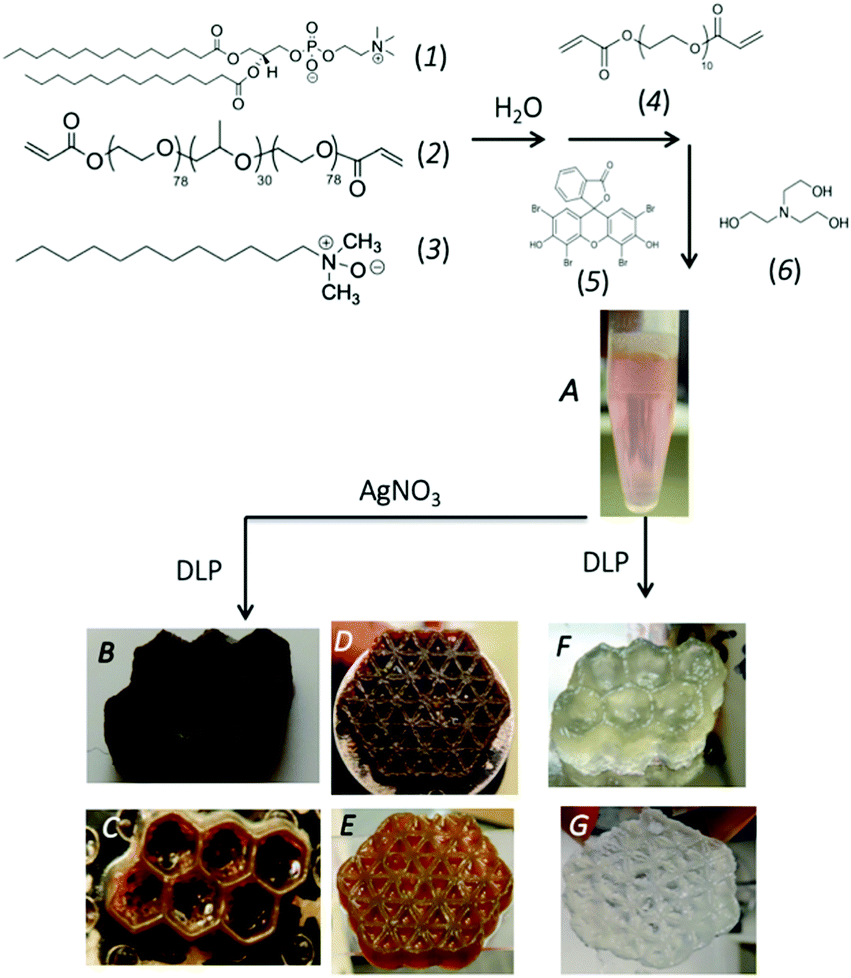 | ||
| Fig. 1 Synthetic scheme for the 3D visible light DLP printing of Ag NP chemical hydrogel. The self-assembled mesophase is prepared by combining (1) DMPC, (2) F68(Acr)2, and (3) LDAO in water. To the mixture, a water soluble crosslinker, PEGDA (4) is added. Finally, the visible light photo-initiator, a mixture of (5) eosin Y and (6) TEOA is added to the mesophase (A). The mesophase is vat photopolymerized to yield a chemically crosslinked hydrogel (F and G). Doping the mesophase (A) with AgNO3 prior to printing yields the Ag NP hydrogel composite (B–E). | ||
Successful crosslinking of the F68(Acr)2 and PEGDA to form a chemical (polymer network) gel during printing, eliminating a post-processing step (i.e., UV curing) was verified by ATR/FT-IR spectroscopy. Signature modes include the appearance of a strong vibrational mode at 1738 cm−1, confirming production of unconjugated acryloyl carbonyls (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), and a weak mode at 1763 cm−1 associated with saturated ester groups (Fig. S4, ESI†).14,17 The orange-brown colour implies successful in situ photoreduction of Ag(I) to Ag(0) and production Ag NPs. The application and mechanistic details of photo-sensitizer (here, eosin Y) mediated photochemical reduction of silver is presented elsewhere.13,18 The formation of Ag NPs is confirmed by TEM collected on particles extracted from a printed composite and by optical spectroscopy (Fig. 2). The particles are highly poly-dispersed, quasi-spherical Ag(0), ranging from 2–50 nm in diameter with a mean value of 16.6 ± 6.7 nm (N = 200 particles), Fig. 2A, inset. The Ag NPs are either single crystalline or polycrystalline containing structural defects such as twining or stacking faults. Completeness of Ag(I) reduction to Ag(0) and NP stability (i.e., loss from the matrix) was evaluated by optical spectroscopy of water extracts collected on swollen hydrogels.19,20 Given the lack of expected optical signatures (data not shown), no appreciable loss of Ag(I) or Ag NPs were found over the concentration ranges examined herein.
O stretching), and a weak mode at 1763 cm−1 associated with saturated ester groups (Fig. S4, ESI†).14,17 The orange-brown colour implies successful in situ photoreduction of Ag(I) to Ag(0) and production Ag NPs. The application and mechanistic details of photo-sensitizer (here, eosin Y) mediated photochemical reduction of silver is presented elsewhere.13,18 The formation of Ag NPs is confirmed by TEM collected on particles extracted from a printed composite and by optical spectroscopy (Fig. 2). The particles are highly poly-dispersed, quasi-spherical Ag(0), ranging from 2–50 nm in diameter with a mean value of 16.6 ± 6.7 nm (N = 200 particles), Fig. 2A, inset. The Ag NPs are either single crystalline or polycrystalline containing structural defects such as twining or stacking faults. Completeness of Ag(I) reduction to Ag(0) and NP stability (i.e., loss from the matrix) was evaluated by optical spectroscopy of water extracts collected on swollen hydrogels.19,20 Given the lack of expected optical signatures (data not shown), no appreciable loss of Ag(I) or Ag NPs were found over the concentration ranges examined herein.
 | ||
| Fig. 2 (A) Representative TEM image collected on grid cast particles released from Ag NP hydrogel composite using a 17 (v/v)% aqueous hydrazine solution. Histogram (N = 200) of nanoparticle size distribution (inset). (B) Normalized extinction spectrum collected on Ag NP hydrogel composite (prepared using 9.35 mM AgNO3) fully hydrated (swollen in water, blue curve), after 5 h of ambient air dehydration (brown curve), and after 48 h dehydrated (orange circles). | ||
The internal structure of the builds was interrogated by X-ray scattering, a facile, reliable means to interrogate 3D volumetric structural hierarchy in composites as rigorously confirmed through complementary direct space imaging techniques.19,21 A representative circularly averaged SWAXS profile (Fig. 3A) collected on a de-hydrated (24 h) printed honeycomb structure displays a number of diffraction peaks. The Bragg reflections are positioned at q = 0.098 Å−1 (001), 0.20 Å−1 (002), 0.30 Å−1 (003), 0.40 Å−1 (004), 0.49 Å−1 (005), indexing to a multi-lamellar structure with a d-spacing of 64.1 Å. The nanostructure compares well to the SAXS determined structure previously reported for the mesophase (physical gel).22 Thus, paring self-assembly with 3D printing offers a low cost strategy for introducing sub-printer resolution features, here nanometre channels (i.e., water layers of ∼1.8 nm and amphiphile layers of ∼4.6 nm),22 into a build. The dimensions of these features are considerably smaller than achievable by stereolithography which produces features on the range of 0.6–50 μm.23,24 In the low q region, below the multi-lamellar diffraction signatures, a broad (weak) correlation peak is found at q ∼ 0.046 Å−1 (∼136 Å). This feature signals crystallized EO chains confined within the inter-lamellar water channels.20 In the WAXS region, additional diffraction peaks are resolved. Bragg peaks located at 1.37 Å−1 (4.59 Å) and 1.67 Å−1 (3.76 Å) arise from the crystallized F68(Acr)2 PEO blocks.17 The peak at q = 1.52 Å−1 (4.13 Å) signals ordered amphiphile hydrocarbon chains comprising the bilayer (i.e., the phospholipid and co-surfactant). A chain–chain correlation distance of 4.13 Å indicates the amphiphile bilayer is in an un-tilted Lβ phase.17 The peak at 2.68 Å−1 is assigned to the (111) reflection of fcc metallic Ag, confirming the presence of crystalline Ag NPs.25 For comparison, a representative SAXS profile collected on a 3D build lacking Ag NPs is given in Fig. S5 (ESI†). The SAXS pattern contains integrally ordered diffraction peaks located at q = 0.11 Å−1 (001), 0.22 Å−1 (002), 0.32 Å−1 (003), 0.43 Å−1 (004) with a d-spacing of 58.1 Å. A strong, broad correlation peak at q = 0.062 Å−1 (101.2 Å) consistent with crystallized EO chains confined in the inter-lamellar water channels.
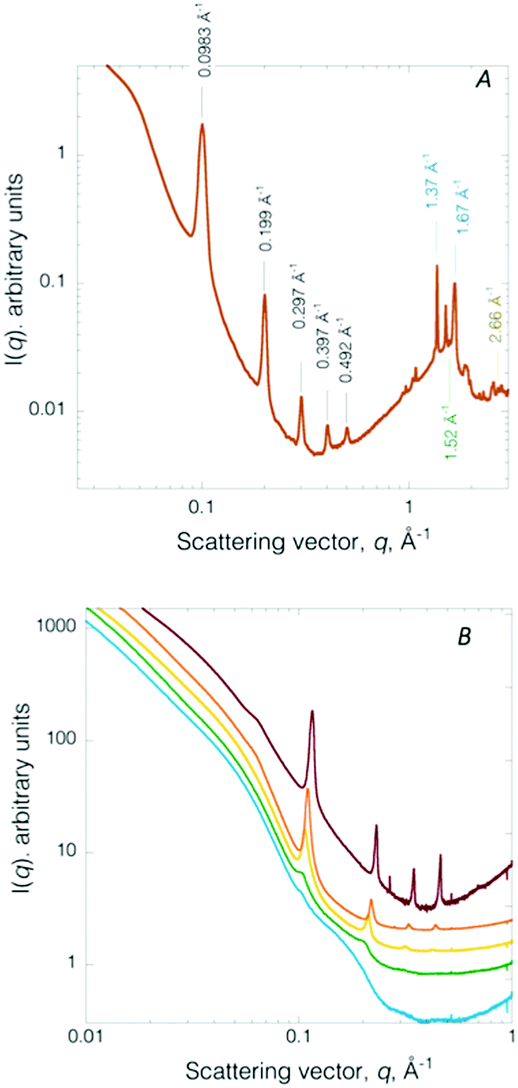 | ||
| Fig. 3 (A) Representative SWAXS profile collected on a de-hydrated (24 h) printed Ag NP hydrogel composite honeycomb structure. (B) Temporal evolution of SAXS pattern recorded during dehydration under ambient atmosphere of a fully water swollen composite. Fully water swollen (blue). After 1 h 40 min (green), 2 h 30 min (yellow), 3 h 18 min (orange), 4 h 10 min (red) deswelling. | ||
As previously noted, the gels are hydro-responsive, reversibly swelling in water. Immediately post-printing, the 3D printed Ag NP hydrogel retains ∼64 wt% water, as determined by TGA (Fig. S6A, ESI†). After 48 h of ambient air storage the builds becomes dark brown due to loss of water (to a final water content of ∼4 wt%, Fig. S6B, ESI†) and shrinkage of the builds (Fig. S2, ESI†). Nanoscale structure evolution during ambient dehydration is monitored by in situ SAXS (Fig. 3B). The hydro-responsive nanostructure evolution is consistent with that previously reported for monolithic crosslinked non-ionic triblock copolymer-based chemical gels14 and Au NP hydrogel composites (i.e., not 3D printed).20 Briefly, the fully water swollen Ag NP 3D printed chemical hydrogel SAXS pattern contains a broad feature spanning the scattering vector range 0.098 Å−1 to 0.26 Å−1. The SAXS profile corresponds to form factor scattering from multilayer amphiphilic bilayers where the water layer thickness is so expanded large vesicles of irregular size are produced (Fig. 3B, blue).14 After 1 h 40 min of de-swelling, two low intensity peaks emerge at q = 0.105, 0.20 Å−1 (Fig. 3B, green), signalling re-formation of a multi-lamellar structure. The two diffraction peaks continue to sharpen and increase intensity and a third Bragg peak emerges at q = 0.32 Å−1, at 2 h 30 min of de-swelling (Fig. 3B, yellow). After 3 h 18 min of de-swelling, the diffraction peaks continue to narrow, signifying increased spatial coherence (Fig. 3B, orange). Finally, at 4 h 10 min, four sharp diffraction peaks are resolved (Fig. 3B, red). The peaks are shifted to slightly higher scattering vector (due to dehydration) and the PEO crystallization peak is now observable (Fig. 3B, red curve). Thus, the well-ordered multi-lamellar structure is fully recovered upon de-swelling. Structure modulation is fully reversible over many swelling–deswelling cycles.
The influence of hydration state on the collective optical properties of the encapsulated Ag NPs was evaluated by Vis-NIR spectroscopy (Fig. 2B). A water swollen 3D print possesses a surface plasmon resonance (SPR) peak centred at 412 nm. The expected SPR for non-aggregated, spherical Ag NPs of diameter 16.6 ± 6.7 nm (as determined by TEM) is ∼400 nm.26 Here, however, given the NP size dispersity, which ranges from 2–50 nm, the SPR is expected to span from 390 to 419 nm.26 Unlike Au NPs, other factors contribute to changes in the SPR for Ag NPs. The SPR for Ag NPs is known, for example, to be particularly sensitive to particle-adsorbate interactions (e.g., changes in the dielectric properties of the metal NPs induced by chemi- or physi-sorption) and changes in the immediate chemical environment of the particles. Thus, the red shifted SPR observed for the water swollen composite most likely signals the Ag NPs are localized within the PEO chains, a finding which has previously been established for in situ synthesized Au NPs.20 Furthermore, prior work evaluating SPR changes on Ag NPs of ∼15 nm in diameter coated with PEG showed a UV-Vis spectrum containing a single band with a peak maximum at 414 nm.27 Thus, the SPR observed here, located at 412 nm, represents particles located within the PEO/PEG chains of the nanostructured hydrogel. After de-swelling for 5 h the optical spectrum (Fig. 2B, brown) retains the primary SPR at 412 nm but the peak is considerably broader with a long tail spanning over 500–800 nm. The broadened SPR suggests dehydration induced aggregation of the PEO/PEG confined Ag NPs.28 Previously we have observed time-dependent SPR changes in Au NPs synthesized within F98 PEO–PPO–PEO triblock copolymers during dehydration.20 Specifically, pronounced red-shifting, peak broadening, and the appearance of a second maximum in the plasmon band arise from polymer mediated interparticle coupling.28 Finally, the optical spectrum obtained for a fully de-swollen composite (Fig. 2B, orange curve) contains a triplet of peaks with the highest peak intensity at 525 nm flanked by features a 470 nm and 630 nm. The pronounced changes in the optical properties arise from compaction of the PEO/PEG layers within the multilamellar structure that confine the Ag NPs, promoting NP agglomeration. The aggregated Ag NPs can be fully reversed (de-aggregated) over many cycles by swelling in water with no significant changes in the collective plasmon resonance. Thus, the nanostructured resin permits deterministic placement (localization within the PEG/PEO layer) during in situ synthesis of the metal plasmonic NPs. Silver ion concentration at ∼10 mM yields a composite with an appropriate balance of properties; enhanced storage modulus yet sufficient Ag NP number density for solvent driven aggregation-de-aggregation.19,20
Further noted, the in situ production of Ag NPs, possessing a SPR in the visible region, function as embedded optical absorbers controlling the penetration depth of the incident 3D printer light (λ > 395 nm). Enhancement of 3D print-resolution through the introduction of ex situ synthesized optical absorbing (molecular) species to resin formulations has been reported in the literature.29 The light absorbing molecules were used to control optical penetration during photo-printing (hence polymerization depth) within individual layers.29 Specifically, with increasing absorber concentration the depth of light penetration was correspondingly limited leading to significantly reduced microchannel size (z-direction) and an increase in the x–y plane resolution of the projected image.29 Thus, we hypothesize the observed increase in structural integrity of the 3D builds containing the Ag NPs (vs. the hydrogel alone) is attributed to the Ag NPs acting as optical absorbing species thereby controlling the incident light penetration within the layer. Given that the depth of light penetration, DL, is inversely related to the molar absorptivity times the concentration of the absorber the current system offers considerable adjustability over DL since the Ag NPs size can be varied so as to regulated the εA and as demonstrated here, concentration can be varied over a wide range.30 Further studies validating an estimated DL (e.g., DL ∼ 0.25 μm is estimated for builds prepared employing 10 mM [Ag+] yielding 20 nm Ag NP, (ESI,† experimental)) are ongoing and will be reported elsewhere.
In summary, through the careful design of resin formulation, lower value feedstocks, individually lacking advanced functionality are transformed into a higher value free-form nano- to micro-architected product using a single unit process fabrication strategy. Specifically, the approach succeeds in introducing nanoscale features unattainable by current DLP technologies through resin self-assembly. The DLP 3D printer promotes the photo-initiated in situ synthesis of Ag NPs spatially confined within the PEO/PEG domains during printing of the nanostructured hydrogel. This is distinct from the majority of NP composites that contain randomly distributed nanoscopic components. Spatial localization of plasmonic metal NPs within the structured hydrogel allows for solvent-driven modulation of the collective plasmonic characteristics and also serves to enhance the hydrogel mechanical properties (stiffness). Most interestingly, in situ production of visible plasmonic NPs act as embedded optical absorbers regulating light penetration during 3D printing, thereby synergistically improving print fidelity (resolution). This work illustrates how careful design of resin chemistry for additive manufacturing can lead to synergistic materials fabrication, potentially providing access to a richer array of responsive plasmonic nanocomposites that may prove useful in optical data storage, chemical sensing, and in vivo medical imaging,31 all while being amenable for nanomanufacturing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was preformed, in part, at the Center for Integrated Nanotechnologies (CINT), a DOE Office of Science User Facility. This research used resources of the Advanced Photon Source (12ID), a DOE Office of Science User Facility operated by Argonne National Laboratory. LANL is operated by Triad, LLC for the National Nuclear Security Administration of the U. S. Department of Energy (DOE) under contract DE-AC52-06NA25396.Notes and references
- M. A. Firestone, S. C. Hayden and D. L. Huber, MRS Bull., 2015, 40, 760–767 CrossRef CAS.
- P. Thoniyot, M. J. Tan, A. A. Karim, D. J. Young and X. J. Loh, Adv. Sci., 2015, 2, 1400010–1400023 CrossRef.
- H.-L. Tan, S.-Y. Teow and J. Pushpamalar, Bioengineering, 2019, 6, 17 CrossRef CAS.
- J. K. Chang, J. K. He, M. Mao, W. X. Zhou, Q. Lei, X. Li, D. C. Li, C. K. Chua and X. Zhao, Materials, 2018, 11, 170455–170465 Search PubMed.
- J. A. Liddle and G. M. Gallatin, ACS Nano, 2016, 10, 2995–3014 CrossRef CAS.
- S. Almohammed, M. Alruwaili, E. G. Reynaud, G. Redmond, J. H. Rice and B. J. Rodriguez, ACS Appl. Nano Mater., 2019, 2, 5029–5034 CrossRef CAS.
- A. Vyatskikh, S. Delalande, A. Kudo, X. Zhang, C. M. Portela and J. R. Greer, Nat. Commun., 2018, 9, 442–447 CrossRef.
- L. Kool, A. Bunschoten, A. H. Velders and V. Saggiomo, Beilstein J. Nanotechnol., 2019, 10, 442–447 CrossRef CAS.
- Q. H. Wu, S. B. Zou, F. P. Gosselin, D. Therriault and M. C. Heuzey, J. Mater. Chem. C, 2018, 6, 12180–12186 RSC.
- Q. H. Wu, D. Therriault and M. C. Heuzey, ACS Biomater. Sci. Eng., 2018, 4, 2643–2652 CrossRef CAS.
- E. Fantino, A. Chiappone, I. Roppolo, D. Manfredi, R. Bongiovanni, C. F. Pirri and F. Calignano, Adv. Mater., 2016, 28, 3712–3717 CrossRef CAS.
- G. Taormina, C. Sciancalepore, F. Bondioli and M. Messori, Polym, 2018, 10, 212–226 Search PubMed.
- B. G. Ndefru, B. S. Ringstrand, S. I. Y. Diouf, S. Seifert, J. H. Leal, T. A. Semelsberger, T. A. Dreier and M. A. Firestone, Mol. Syst. Des. Eng., 2019, 4, 580–585 RSC.
- S. Grubjesic, B. Lee, S. Seifert and M. A. Firestone, Soft. Matter, 2011, 7, 9695–9705 RSC.
- M. A. Firestone, A. C. Wolf and S. Seifert, Biomacromolecules, 2003, 4, 1539–1549 CrossRef CAS.
- C. G. Robertson, C. J. Lin, M. Rackaitis and C. M. Roland, Macromolecules, 2008, 41, 2727–2731 CrossRef CAS.
- S. I. Y. Diouf, D. J. Williams, S. Seifert, A. Londoño-Calderon, M. T. Pettes, C. J. Sheehan and M. A. Firestone, Polym. Chem., 2019, 10, 6456–6472 RSC.
- P. K. Sudeep and P. V. Kamat, Chem. Mater., 2005, 17, 5404–5410 CrossRef CAS.
- S. Lee, M. D. Cummins, G. A. Willing and M. A. Firestone, J. Mater. Chem., 2009, 19(43), 8092–8101 RSC.
- S. Grubjesic, B. S. Ringstrand, K. L. Jungjohann, S. M. Brombosz, S. Seifert and M. A. Firestone, Nanoscale, 2016, 8, 2601–2612 RSC.
- P. M. Welch, T. A. Dreier, H. D. Magurudeniya, M. G. Firth, J. Ilavsky, S. Seifert, A. J. Singh, B. S. Ringstrand, C. J. Hanson, J. A. Hollingsworth and M. A. Firestone, Macromolecules, 2020, 53(8), 2822–2833 CrossRef CAS.
- B. Lee and M. A. Firestone, Biomacromolecules, 2008, 9, 1541–1550 CrossRef.
- C. Sun, N. Fang, D. M. Wu and X. Zhang, Sens. Actuators, A, 2005, 121, 113–120 CrossRef CAS.
- R. M. Hensleigh, H. C. Cui, J. S. Oakdale, J. C. C. Ye, P. G. Campbell, E. B. Duoss, C. M. Spadaccini, X. Y. Zheng and M. A. Worsley, Mater. Horiz., 2018, 5, 1035–1041 RSC.
- S. Pal, R. Nisi, M. Stoppa and A. Licciulli, ACS Omega, 2017, 2, 3632–3639 CrossRef CAS.
- D. Paramelle, A. Sadovoy, S. Gorelik, P. Free, J. Hobley and D. G. Fernig, Analyst, 2014, 139, 4855–4861 RSC.
- M. D. Xu, J. Liu, X. K. Xu, S. H. Liu, F. Peterka, Y. R. Ren and X. F. Zhu, Materials, 2018, 11, 1990–2001 CrossRef.
- D. G. Angelescu, M. Vasilescu, M. Anastasescu, R. Baratoiu, D. Donescu and V. S. Teodorescu, Colloids Surf., A, 2012, 394, 57–66 CrossRef CAS.
- H. Gong, M. Beauchamp, S. Perry, A. T. Woolley and G. P. Nordin, RSC Adv., 2015, 5, 106621–106632 RSC.
- S. C. Ligon, R. Liske, J. Stampfl, M. Gurr and R. Mulhopf, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS.
- I. Pastoriza-Senton, C. Kinnear, J. Perez-Juste, P. Mulvaney and L. M. Liz-Marzan, Nat. Rev., 2018, 375–391 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DMA and TGA profiles, build dimension, ATR/FT-IR spectrum, SAXS. See DOI: 10.1039/d0ma00549e |
| This journal is © The Royal Society of Chemistry 2020 |