
Effects of synthesis methodology on microporous organic hyper-cross-linked polymers with respect to structural porosity, gas uptake performance and fluorescence properties†
Shuangshuang
Hou
,
Shumaila
Razzaque
and
Bien
Tan
 *
*
Key Laboratory of Material Chemistry for Energy Conversion and Storage, Ministry of Education, Hubei Key Laboratory of Material Chemistry and Service Failure, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan, 430074, China. E-mail: bien.tan@mail.hust.edu.cn; Fax: +86 27 87543632; Tel: +86 27 87558172
First published on 5th February 2019
Abstract
The structural characteristics of hyper-cross-linked polymers (HCPs) make them interesting for a wide variety of applications. To form knitted HCPs various synthesis approaches including solvent knitting, a knitting method using formaldehyde dimethyl acetal (FDA) as an external cross-linker and the Scholl coupling reaction have been used. However, a systematic study of these strategies has not been explored. Here HCPs have been made using facile synthesis methodologies and characterized to reveal resulting Brunauer–Emmett–Teller (BET) surface areas up to 2278 m2 g−1 and 2064 m2 g−1 with CO2 adsorption capacities of 20.89 wt% and 18.95 wt% (at 273.15 K/1.00 bar), respectively. Detailed information is also given with respect to distinctions in textural properties, gas uptake performances and optical properties. More importantly, our comparative studies have revealed the significance and efficiency of solvent knitting when used to fabricate HCP materials with good porosities and gas uptake abilities. Among these approaches, the knitting method with FDA as an external cross-linker is the most time-efficient technique, whereas the Scholl coupling reaction has advantages for the production of fluorescent polymers. Based on the proposed mechanisms, changes in structural features can also be rationally explained.
Introduction
Porous hyper-cross-linked polymers (HCPs) that can be prepared in a facile manner using the Friedel–Crafts reaction under mild conditions are attracting increasing interest.1 Due to extensive cross-linking, the resultant porous polymer's polymer chains will not collapse into a dense and nonporous state, while additionally their highly cross-linked nature confers an unusually high thermal stability that is not commonly found in organic polymers.2 With the advantages of a large surface area, low cost, relatively low heats of adsorption, excellent chemical robustness (e.g. resistance to strong acids and bases),3 they have found increasingly wide applications in gas capture and separation,4–7 energy storage,7–9 volatile organic compound adsorption,10,11 uptake of chemical warfare agents,12 catalysis,13–15 and drug release.16 These properties have sparked in recent years an interest in their rational design and synthesis.17The development of HCPs dates back to the introduction of hyper-cross-linked polystyrene in the early 1970s.2 Since then, much effort has been devoted to its development. For example, Cooper and co-workers showed the carbonization of HCPs with KOH activation can be used to generate highly porous structures with a BET surface area of 4334 m2 g−1 adsorbing 3.5 mmol g−1 CO2 at 298 K/1.00 bar and 3.6 wt% H2 at 77.3 K/1.00 bar.18 Jiang et al., presented a novel and facile strategy for making high surface area porous carbon nanotubes (PCNTs) with outstanding porosities and an excellent electrochemical performance by direct carbonization of 1D hyper-cross-linked polymer nanotubes.19 Our group has previously developed a cobalt coordinated polymer with a BET surface area of 1360 m2 g−1, which efficiently catalyzed the coupling of CO2 with various substituted epoxides at room temperature and atmospheric pressure.20 Currently many interesting strategies are being pursued to obtain high surface area polymers with tailored porosities. Building blocks, such as aromatic hydrocarbons,21–23 polycyclic aromatic hydrocarbons,19,21,24 and heterocyclic compounds,25,26 are used to synthesize HCPs. Among these materials a microporous organic polymer derived from N4,N4′-di-1-naphthalenyl-N4,N4′-di-2-naphthalenyl-[1,1′-biphenyl]-4,4′-diamine has a BET surface area of 1717 m2 g−1 and a CO2 adsorption of 18.85 wt% at 273.15 K/1.00 bar.27 Further attempts to optimize structural properties, solvents,21–23,28,29 external cross-linkers,22,28 and catalysts,21,23,24,28,29 have been made, e.g. a fluoranthene-based polymer synthesized from anhydrous AlCl3 in dichloromethane showed a CO2 adsorption of 24.79 wt% at 273.15 K/1.00 bar under optimized capture conditions.24
To make HCPs with high surface areas and controlled porosities, many synthesis strategies such as the ‘knitting method’ with formaldehyde dimethyl acetal (FDA) as an external cross-linker,21 the Scholl coupling reaction22 and the solvent knitting method23 have been used. In 2011, our group put forward the knitting method, using FDA as an external cross-linker and a Lewis acid (anhydrous FeCl3) catalyst in 1,2-dichloroethane.21 The resulting HCP materials had well formed microporous structures with various enclosed functional groups. This strategy allows for the use of a variety of ‘building blocks’ under facile reaction conditions and inexpensive reagents.21 Consequently, this strategy has been used to synthesize diverse polymer networks. In 2014, our group incorporated functional groups in microporous organic polymers (MOPs), in a protocol based on the Scholl coupling reaction catalyzed by anhydrous AlCl3 in the presence of chloroform.22 Recently in 2017, our group proposed the solvent knitting method to make 2D layered HCPs by ultrasonic-assisted solvent exfoliation.23 These HCPs are usually made using Friedel–Crafts alkylation with the use of dichloroalkane as an economical solvent, stable electrophilic reagent, and external cross-linker. Such materials’ porous structures and surface areas can be controlled by altering factors such as the molar ratio of the catalyst (AlCl3), the sizes of the building blocks, and the solvent's chain length. Because this method is simple, one-step and cost-effective, it offers opportunities for knitting polymers with high surface areas and gas adsorption capacities. This knitting strategy is considered a low-cost, scalable and efficient polymerization technique for creating HCP materials for real applications.23
These distinct approaches possess the following distinguishing features. The knitting strategy with FDA as an external cross-linker is conducted in atmospheric air. When catalyzed by FeCl3 and traces of atmospheric water a greater driving force for the reaction is created. Based on the Friedel–Crafts reaction, this method with FDA cross-linking and building block molecules usually gives a moderate reaction rate.21 The Scholl coupling reaction and the solvent knitting method are performed under a nitrogen atmosphere. In the absence of water AlCl3 will not be hydrolysed, which will favor the reactions. Scholl coupling, with the elimination of a C–C bond, has the fastest reaction rate without any cross-linker.22 In contrast, the solvent knitting method with an improved Friedel–Crafts alkylation and a low activity cross-linker (CH2Cl2), has the slowest reaction rate, which is helpful to enhance the free-packing of building blocks and knitting polymers with large surface areas and abundant micropores.23
The existing methodologies result in HCPs with abundant micropores and high surface areas. More significantly, great interest has been aroused to explore these strategies to gain insights into the design and development of HCP materials. This work focuses on the generation of microporous networks using the previously mentioned strategies that employ aromatic hydrocarbons such as 1,4-diphenylbenzene and 1,3-diphenylbenzene to fabricate novel microporous HCPs. It is worth noting that the prepared polymers have BET surface areas up to 2278 m2 g−1 and 2064 m2 g−1 and CO2 adsorption capacities of 20.89 wt% and 18.95 wt% at 273.15 K/1.00 bar, which have rarely been reported. Furthermore, with respect to porous structures, gas uptakes and the fluorescence of the resultant polymers, this work has demonstrated that: (I) the solvent knitting method has a higher efficiency for knitting polymers giving better porous structures and gas uptake performances; (II) the knitting method with FDA as an external cross-linker is the most time efficient; (III) the Scholl coupling reaction can be used to prepare fluorescent polymers. We finally offer a detailed insight into the reaction mechanisms that lead to these distinctions.
Experimental section
Materials
1,4-Diphenylbenzene (1,4-DPB), 1,3-diphenylbenzene (1,3-DPB) and formaldehyde dimethyl acetal (FDA) were purchased from Aladdin Chemical Reagent Corp. (Shanghai, China) and used as received. Dichloromethane (CH2Cl2), 1,2-dichloroethane (DCE), chloroform (CHCl3), hydrochloride (HCl), methanol, ethyl acetate, dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF), N,N-diethylformamide (DEF), ethanol, anhydrous ferric chloride (FeCl3) and anhydrous aluminum chloride (AlCl3) were acquired from the Sinopharm Chemical Reagent Ltd Co. (Shanghai, China) and used as received. Unless stated otherwise, all the solvents and chemicals were of analytical grade and obtained from local suppliers and used without further purification.Preparation of polymers
Synthesis of polymer 1 by the solvent knitting method. Under a nitrogen atmosphere, 1,4-DPB (2 mmol, 0.460 g) was dispersed in CH2Cl2 (8 mL) for 30 minutes, anhydrous AlCl3 (24 mmol, 3.204 g) was immediately added to the solution which was then allowed to react at 20 °C for 4 h, 30 °C for 8 h, 40 °C for 12 h, 60 °C for 12 h, and 80 °C for 24 h under vigorous stirring. After cooling to room temperature, the solid product was quenched using 20 mL of HCl–H2O (v/v = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), washed several times with deionized water and ethanol, further purified by Soxhlet extraction with ethanol for 48 h, and finally dried in a vacuum oven at 70 °C for 24 h. The polymer was obtained as a black solid. Yield: 140%.
:1), washed several times with deionized water and ethanol, further purified by Soxhlet extraction with ethanol for 48 h, and finally dried in a vacuum oven at 70 °C for 24 h. The polymer was obtained as a black solid. Yield: 140%.
Polymer 2 was prepared in a manner similar to polymer 1 by treating 1,3-DPB (2 mmol, 0.460 g) with anhydrous AlCl3 (24 mmol, 3.204 g) in CH2Cl2 (8 mL). The polymer material was again obtained as a black solid. Yield: 135%.
Polymer 3 was synthesized by the knitting method with FDA as an external cross-linker. Under an air atmosphere, anhydrous FeCl3 (18 mmol, 2.925 g) was added to a solution of 1,4-DPB (2 mmol, 0.460 g) and FDA (18 mmol, 1.370 g) in DCE (8 mL). The mixture was heated at 45 °C for 5 h and then 80 °C for 19 h with vigorous stirring. After cooling to room temperature, the solid product was washed with methanol several times, further purified by Soxhlet extraction with methanol for 48 h, and finally dried in a vacuum oven at 70 °C for 24 h. The polymer material was obtained as a brown solid. Yield: 137%.
Polymer 4 was also synthesized by the knitting method as described above for polymer 3, using 1,3-DPB (2 mmol, 0.460 g), anhydrous FeCl3 (12 mmol, 1.950 g) and FDA (12 mmol, 0.913 g) in DCE (8 mL). The polymer was again obtained as a brown solid. Yield: 140%.
Polymer 5 was synthesized by the Scholl coupling reaction method. Under a nitrogen atmosphere, anhydrous AlCl3 (24 mmol, 3.204 g) was added to a solution of 1,4-DPB (2 mmol, 0.460 g) in CHCl3 (8 mL) with vigorous stirring at 58 °C, the reaction proceeded for 48 h. The solid product was washed once with ethanol, twice with HCl–H2O (v/v = 2:1), and then three times with ethanol; followed by ethanolic Soxhlet extraction with ethanol for 48 h, prior to drying in a vacuum oven at 70 °C for 24 h. The polymer was obtained as a black solid. Yield: 97%.
Polymer 6 was likewise prepared by the Scholl coupling reaction as in the above procedure for polymer 5 using 1,3-DPB (2 mmol, 0.460 g) and anhydrous AlCl3 (24 mmol, 3.204 g) in CHCl3 (8 mL). The polymer was obtained as a brown solid. Yield: 98%.
Characterization
Fourier transform infrared (FT-IR) spectra were recorded using a Bruker VERTEX 70 FT-IR spectrometer (wavenumber range 4000–500 cm−1) at room temperature and atmospheric pressure. Solid state 13C cross-polarization/magic-angle spinning nuclear magnetic resonance (13C CP/MAS NMR) spectra were recorded on a WB 400 MHz Bruker Avance II spectrometer and collected with a spinning rate of 20 kHz by using a 2.5 mm double-resonance MAS probe. The field-emission scanning electron microscopy (FE-SEM) images were recorded on a FEI Sirion 200 field-emission scanning electron microscope operating at 10 kV. All the samples were dried in a vacuum oven at 70 °C for 24 h and then sputter-coated with platinum before measurement. The transmission electron microscopy (TEM) images were recorded on a Tecnai G2 F30 microscope (FEI Corp. Holland) operating at 200 kV. Thermogravimetric analysis (TGA) was performed from room temperature to 850 °C, using a PerkinElmer Instrument Pyris1 TGA with a heating rate of 10 °C min−1 under a nitrogen atmosphere. The solid state fluorescence spectra were recorded using an FP-6500 fluorescence spectrometer (Jasco, JPN). The absolute solid state fluorescence quantum yield of polymers was tested by using an Absolute PL Quantum Yield Spectrometer F4600 (Hitachi, JPN) at room temperature and atmospheric pressure. All the samples were degassed at 120 °C for a minimum of 8 h under vacuum of 10−5 bar prior to being measured. N2 isotherms were collected on a Micromeritics ASAP 2020 M analyzer at 77.3 K. Gas (CO2, N2 and CH4) adsorption and desorption isotherms were measured on a Micromeritics ASAP 2020 M analyzer at 273.15 and 298.15 K. Pore size distribution was calculated from N2 adsorption isotherms using a Tarazona nonlocal density functional theory (NLDFT) model assuming a slit pore geometry. Total pore volumes were calculated from nitrogen sorption isotherms at relative pressure P/P0 = 0.995.Results and discussion
In this work, HCP materials were prepared by three synthesis methods to gain better insights into the distinctions in structural porosities, gas uptakes and fluorescence properties of microporous organic HCPs. Of note, the Scholl coupling polymerization reaction proceeds22 without the addition of an external cross-linker. Therefore, the monomers react directly with a fast reaction rate. While the knitting method, based on the Friedel–Crafts reaction, has FDA as an external cross-linker,21 and in contrast with the Scholl coupling reaction, it shows a slower reaction rate. The solvent knitting method employs an improved Friedel–Crafts alkylation,23 in which dichloromethane plays the role of both an external cross-linker and solvent. Due to the lower reaction activity of dichloromethane compared to FDA, it has the slowest reaction rate, which is advantageous to form networks with layered structures. Since the reaction mechanism is closely associated with the materials’ structure, the mechanisms associated with these methodologies vary, which may explain the diversity of the resultant polymer networks.Scheme 1 shows the ‘building blocks’ and proposed pathways leading to the formation of HCPs by the solvent knitting method, the knitting method with FDA as an external cross-linker and the Scholl coupling reaction. The building blocks are soluble in organic solvents such as ethyl acetate, dimethyl sulfoxide, N,N-dimethylformamide and N,N-diethylformamide, while the obtained polymers are insoluble in these solvents. It has been demonstrated that the polymerization normally leads to a decrease in polymer solubility.
 | ||
| Scheme 1 Proposed synthesis pathways to microporous organic HCP networks, together with the building blocks’ chemical structures. | ||
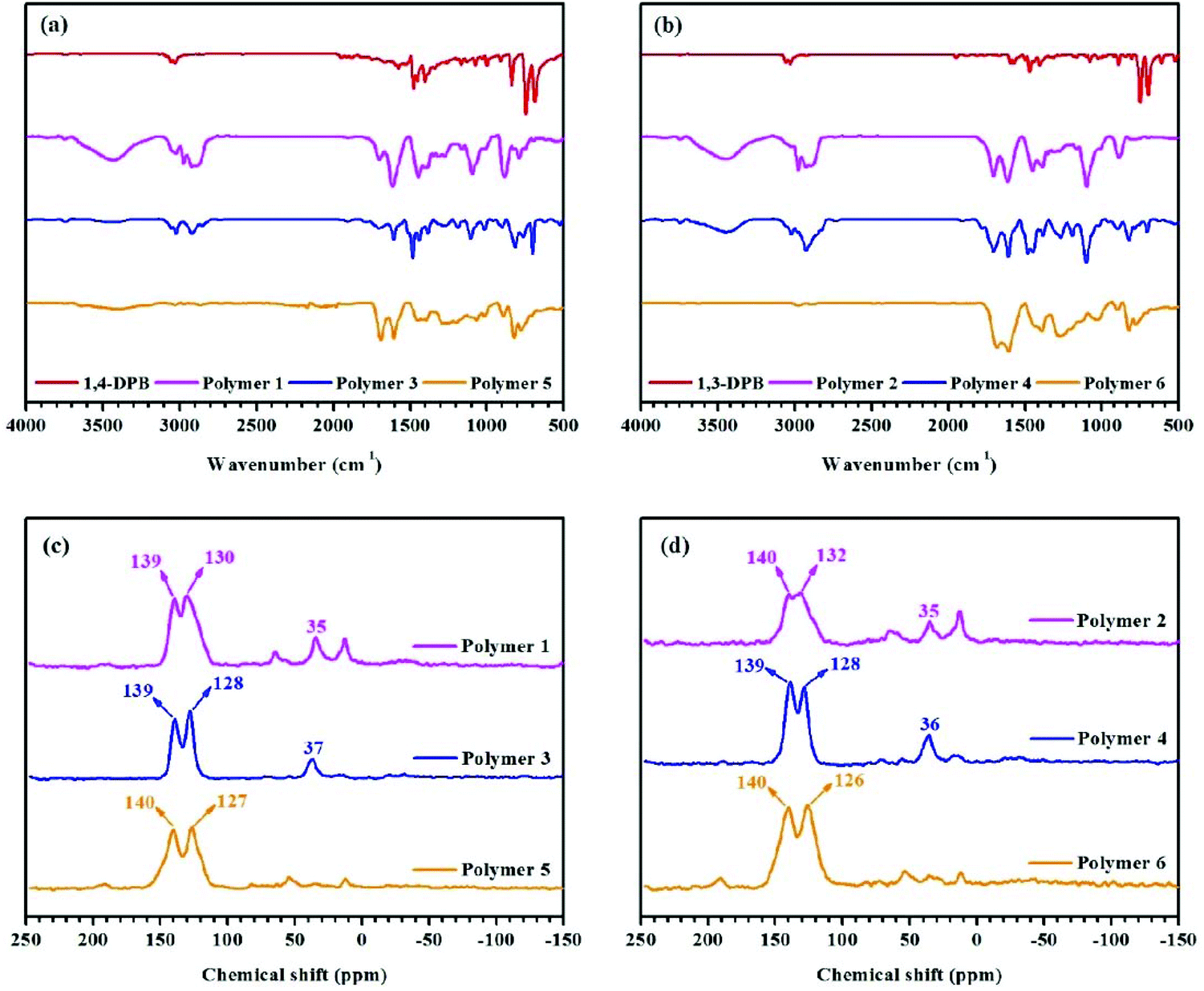
FT-IR spectra were recorded to confirm the starting materials and polymers’ structural features. Taking 1,3-DPB-based polymers as an example. The broad bands at 3600–3100 cm−1 in polymers 2 and 4 are largely attributed to adsorbed water in the networks created. The bands at 3100–3000 cm−1 are the C–H stretching vibrations of the benzene ring, while the bands at ∼1508 cm−1, 1477 cm−1 and 1437 cm−1 are assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of the benzene ring, which is found in both polymeric networks 2, 4, 6 and their ‘building blocks’ (Fig. 1b). The absorption bands at 2910 cm−1 and 2829 cm−1 are the C–H stretching vibrations of the methylene bridges, seen in polymers 2 and 4 (Fig. 1b), and the FT-IR spectra of 1,4-DPB and 1,4-DPB-based polymer materials are shown in Fig. 1a.
C stretching vibrations of the benzene ring, which is found in both polymeric networks 2, 4, 6 and their ‘building blocks’ (Fig. 1b). The absorption bands at 2910 cm−1 and 2829 cm−1 are the C–H stretching vibrations of the methylene bridges, seen in polymers 2 and 4 (Fig. 1b), and the FT-IR spectra of 1,4-DPB and 1,4-DPB-based polymer materials are shown in Fig. 1a.
 | ||
| Fig. 1 FT-IR spectrum of (a) 1,4-DPB and 1,4-DPB-based polymer materials, and (b) 1,3-DPB and 1,3-DPB-based polymer materials; solid state cross-polarization (CP) 13C MAS natural abundance NMR spectrum of (c) 1,4-DPB-based polymer materials, and (d) 1,3-DPB-based polymer materials. | ||
Following the FT-IR study, solid state 13C CP/MAS NMR spectroscopy was used to investigate the structural characteristics of the polymers. Well-resolved signals are seen in Fig. 1d. For example, the singlets at ∼137 ppm belong to the substituted aromatic carbon and the singlets near to 130 ppm are attributed to the unsubstituted aromatic carbon,15 while the resonance at ∼35 ppm in polymer 2 is ascribed to the methylene linkers – derived from CH2Cl2 (solvent and an external cross-linker).23,27 Meanwhile, there are also doublets in the range 70–10 ppm in polymer 2, attributable to methylene bridges (resulting from hyper-cross-linking) and residual dichloromethane. As previously stated, dichloromethane acts as an external cross-linker and solvent in the solvent knitting strategy. Similarly, in polymer 4 the signal at ∼36 ppm is assigned to the methylene linkers, derived from CH3OCH2OCH3 (an external cross-linker).25 Moreover, the resonances at ∼126 and 140 ppm in polymer 6 are respectively due to the non-coupled and coupled aromatic carbons.22 A detailed graphical explanation of the polymers derived from 1,4-DPB is given in Fig. 1c.
Taking together the FT-IR and solid state 13C CP/MAS NMR spectra we have shown the successful generation of HCPs by precursor cross-linking using three different techniques.
The surface morphologies and porous nature of the polymers were studied by FE-SEM and TEM. Fig. S2–S4 (ESI†) show FE-SEM images of these networks, which have the irregular blocks aggregated together. Fig. S1 (ESI†) shows the morphologies of their parent monomers, which are amorphous in nature. By comparing the morphologies of polymers and their building blocks we can deduce that these synthesis methodologies did not distinctly influence the morphology of polymer materials. Fig. S5–S7 (ESI†) show TEM images of the polymers showing the amorphous and porous structures of the HCPs obtained by using different strategies.
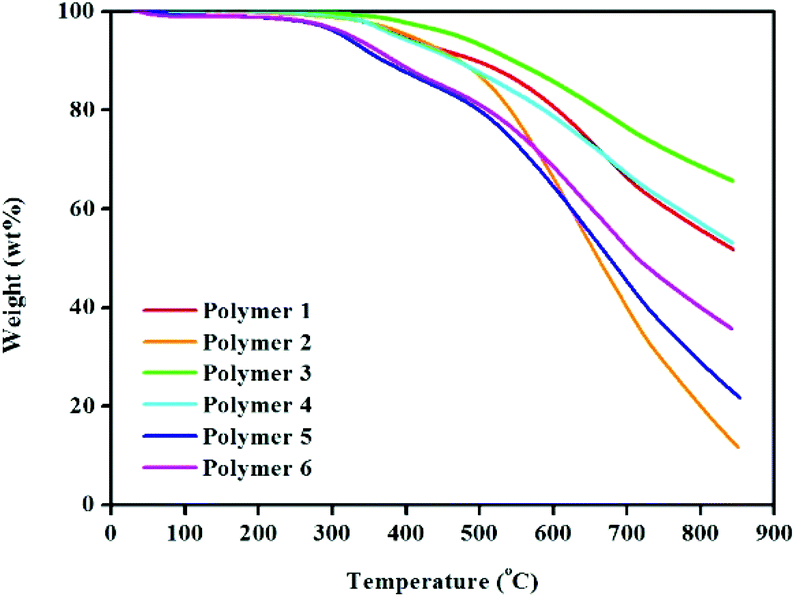
TGA analysis (Fig. 2) was used to record the thermal stability of all the HCPs. As can be seen, polymer materials from polymer 1 to polymer 4 are stable up to approximately 400 °C under a nitrogen atmosphere, which is a little higher than those of polymers 5 and 6. Thus it is evident that, polymer networks from 1 to 4 prepared using the knitting strategy tend to be more stable than polymers 5 and 6 synthesized by the Scholl coupling reaction. This is primarily attributed to the different degrees of polymer cross-linking resulting from the use of differing synthesis methodologies. Furthermore, their obvious thermal stability (with weight loss only occurring with temperatures above 400 °C) is ascribed to polymeric network degradation.
 | ||
| Fig. 2 TGA analysis of materials from polymer 1 to polymer 6. | ||
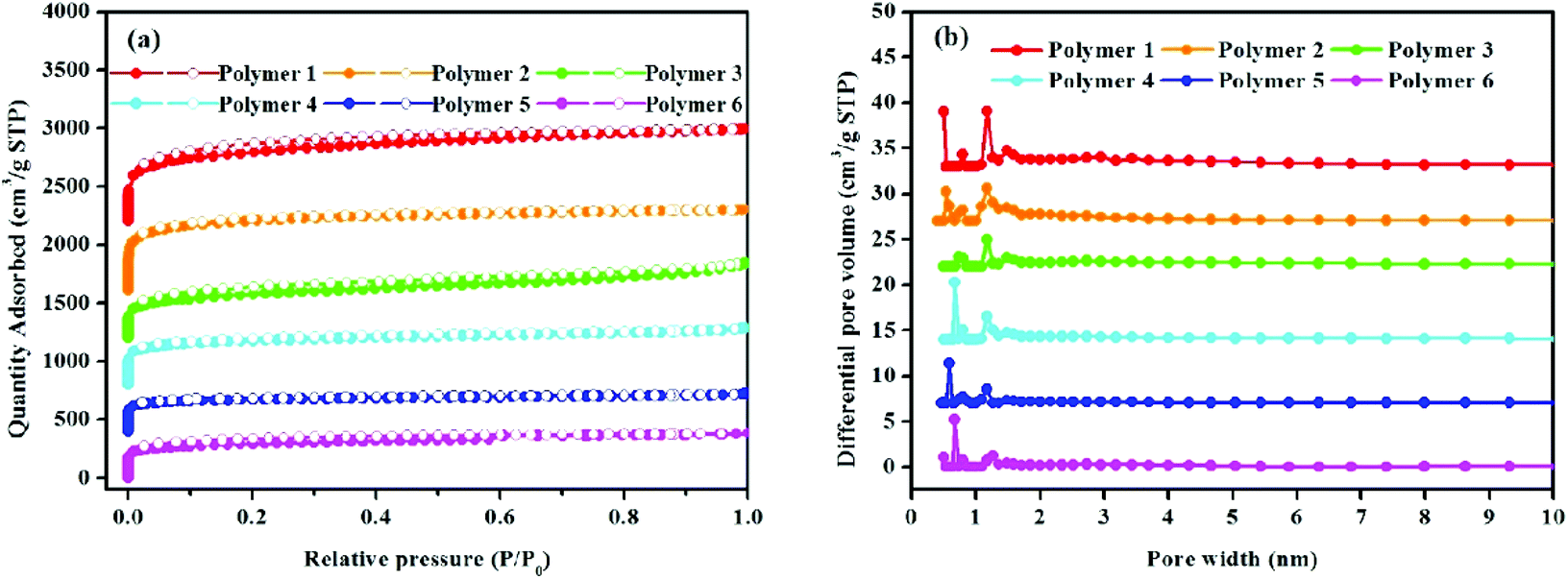
The porous nature of these hyper-cross-linked polymers was shown by nitrogen adsorption and desorption analysis at 77.3 K and 1.00 bar. Fig. 3a shows the high nitrogen gas uptake at a low relative pressure (P/P0 < 0.001) thereby highlighting the microporous structure of the resultant polymers. The observed hysteresis loops in the middle of all isotherms imply the mesopores. No obvious rise in the nitrogen adsorption isotherms at high relative pressures (P/P0 > 0.9) can be seen, which suggests that macropores are nearly absent in these polymers. The pore size distribution of polymers was estimated using N2 adsorption isotherms employing the NLDFT model. As seen from Fig. 3b, these polymers with different textures have hierarchical pore size distributions. Besides the peaks (2–10 nm) in the mesoporous region, the pore size is mainly located within 2 nm in the microporous section, including the ultramicroporous region, whose diameter is no more than 0.7 nm. Table 1 lists the porosity parameters of all HCP samples. For clarity, 1,3-DPB-based polymers are discussed in detail below. The BET surface areas of the polymeric networks varied from 1092 m2 g−1 to 2278 m2 g−1, the Langmuir surface areas ranged from 1335 m2 g−1 to 2797 m2 g−1, the micropore areas were from 692 m2 g−1 to 1443 m2 g−1, the total pore volumes ranged between 0.58 cm3 g−1 and 1.08 cm3 g−1, with micropore volumes between 0.28 cm3 g−1 to 0.57 cm3 g−1. It was found that when compared to polymers produced by the knitting method, with FDA as an external cross-linker and the Scholl coupling reaction, polymers prepared by the solvent knitting method have the highest BET surface areas, Langmuir surface areas, micropore areas, pore volumes and micropore volumes. Similarly, 1,4-DPB-based polymer networks including polymers 1, 3, and 5 also follow the same trend. This is probably due to the polymerization strategies. As already mentioned, the solvent knitting method proceeds without the addition of an external cross-linker. In this strategy, dichloromethane plays the role of both the solvent and cross-linking agent. The activity of dichloromethane is very low, thus it has the slowest reaction rate, thereby enhancing the cross-linking degree, while improving the free-packing of building blocks with the resulting polymers tending to produce abundant micropores and a much higher surface area (Table 1).
 | ||
| Fig. 3 (a) Nitrogen adsorption and desorption isotherms at 77.3 K and 1.00 bar, and (b) pore distribution curves of the pore size calculated using nonlocal density functional theory (slit pore models, differential pore volumes, pore width). Filled symbols represent adsorption and open symbols represent desorption. | ||
| Samples | S BET (m2 g−1) | S L (m2 g−1) | MAc (m2 g−1) | PVd (cm3 g−1) | MPVe (cm3 g−1) | CO2 uptakef (wt%) | CO2 uptakeg (wt%) | Q st (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|
| a Apparent surface areas calculated from nitrogen adsorption isotherms at 77.3 K using the BET equation. b Surface areas calculated from nitrogen adsorption isotherms at 77.3 K using the Langmuir equation. c t-Plot micropore areas. d Pore volumes calculated from the nitrogen isotherm at P/P0 = 0.995 and 77.3 K. e t-Plot micropore volumes calculated from the nitrogen isotherm at P/P0 = 0.050. f CO2 uptake determined volumetrically using a Micromeritics ASAP 2020 M analyzer at 1.00 bar and 273.15 K. g CO2 uptake determined volumetrically using a Micromeritics ASAP 2020 M analyzer at 1.00 bar and 298.15 K. h Isosteric heat of adsorption of polymer materials determined volumetrically using a Micromeritics ASAP 2020 M analyzer at 273.15 and 298.15 K. | ||||||||
| Polymer 1 | 2064 | 2780 | 921 | 1.23 | 0.41 | 18.95 | 9.54 | 26.30–26.04 |
| Polymer 2 | 2278 | 2797 | 1443 | 1.08 | 0.57 | 20.89 | 11.83 | 21.97–21.60 |
| Polymer 3 | 1315 | 1771 | 533 | 0.99 | 0.24 | 13.70 | 7.87 | 27.22–24.90 |
| Polymer 4 | 1421 | 1726 | 935 | 0.75 | 0.37 | 16.47 | 9.26 | 26.20–24.95 |
| Polymer 5 | 1040 | 1227 | 778 | 0.49 | 0.30 | 15.04 | 8.82 | 27.34–25.97 |
| Polymer 6 | 1092 | 1335 | 692 | 0.58 | 0.28 | 15.11 | 9.00 | 28.01–26.68 |
Porous materials with ultramicroporous structures are often used for gas sorption and storage e.g. in CO2 sorption. Microporous organic HCPs were examined in this study for their potential for CO2 capture at 273.15 and 298.15 K up to a pressure of 1.13 bar (Fig. 4 and Table 1). For each sample, it can be seen, the CO2 adsorption capacity continuously increased with the feed pressure, however saturation is not seen in experimental pressures ranging up to 1.13 bar, clearly indicating that a larger CO2 uptake can be expected with additional elevations in CO2 pressure. As a result, it was demonstrated that polymer 2 with a BET surface area of 2278 m2 g−1 and a Langmuir surface area of 2797 m2 g−1 had a CO2 adsorption of 20.89 wt% at 273.15 K/1.00 bar. This is comparable to many reported materials such as the carbazolic porous organic framework Cz-POF-3 (4.77 mmol g−1, SBET = 1927 m2 g−1 at 273.15 K/1.00 bar),30 CP-CMP5 (20.11 wt%, SBET = 2241 m2 g−1 at 273.15 K/1.13 bar),31 CP-CMP6 (19.14 wt%, SBET = 1764 m2 g−1 at 273.15 K/1.13 bar),31 CPOP-1 (21.2 wt%, SBET = 2220 m2 g−1 at 273.15 K/1.00 bar),32 and PPN-CH2DETA (15.8 wt%, SBET = 555 m2 g−1 at 298.15 K/1.00 bar).33 In addition, the CO2 uptake is much higher than some types of porous materials with ultrahigh surface areas or high total pore volumes under similar conditions, e.g. COF-102 (6.8 wt%, SBET = 3622 m2 g−1),34 PAF-1 (9.1 wt%, SBET = 4077 m2 g−1),35 CMPN-3 (2.36 cm3 g−1, 3.82 wt%),36 and COP-10 (about 9.1 wt%, SBET = 3337 m2 g−1 at 298.15 K/1.00 bar).37 The CO2 adsorption capacity of polymer 2 is much higher than many HCP materials such as 1,3,5-triphenylbenzene-based HCP (15.9 wt%, SBET = 1059 m2 g−1),21 tetraphenylmethane-based HCP (1.66 mmol g−1, SBET = 1679 m2 g−1)38 and binaphthol-based HCP (17.4 wt%, SBET = 1015 m2 g−1).19 Moreover, the CO2 uptake ability of polymer 2 also outperforms commercially available BPL carbon (2.09 mmol g−1, at 273 K/1 bar),26 POSS-based HPPs (0.62–1.04 mmol g−1),39,40 and organic–inorganic hybrid porous Fc-HPP (4.87 wt%, SBET = 1015 m2 g−1, at 273 K/101 kPa),41 thus highlighting a comparative CO2 uptake capacity. From the gas sorption analysis, it is evident that, the solvent knitting method enhances CO2 capture to a greater extent than does either the knitting method with FDA (an external cross-linker) or the Scholl coupling reaction. Also of note is that the CO2 uptake of polymer 3 is the lowest among all the samples, even though its surface area is higher than that of either polymer 4 or 5. This is primarily due to the polymer's pore size distribution. Because a previous study has shown that, in addition to the surface area, the pore size and pore size distribution are also closely related to the CO2 adsorption capacity of microporous organic polymers.42 From Fig. 3b, it can be seen, polymer 3 with a pore size >0.7 nm lacks ultramicropores, while polymers with a pore size <0.7 nm possess ultramicropores. Since a pore size <1 nm preferentially adsorbs CO2 molecules,43 especially the ultramicropore, whose diameter is comparable to the kinetic diameter of CO2 and is therefore prone to increasing the interactions between the CO2 molecules and the pore walls,44 it is not hard to understand, that although the surface area of polymer 3 is moderate its CO2 uptake performance is the lowest. To better understand the binding affinity of polymers towards CO2, the isosteric heat (Qst) of polymers was calculated by fitting CO2 adsorption isotherms obtained at 273.15 and 298.15 K using the Clausius–Clapeyron equation,45 which was found to be in the ranges of 26.30–26.04, 21.97–21.60, 27.22–24.90, 26.20–24.95, 27.34–25.97, and 28.01–26.68 kJ mol−1 for polymer 1 to polymer 6 (Fig. S8,†Table 1). These results make it clear that there is a normal physical adsorption interaction between the CO2 molecules and polymer networks. While in comparison with polymers having a higher isosteric heat of adsorption such as Py-1 (36 kJ mol−1),26 PI-1 (34 kJ mol−1),46 and PECONF-4 (34 kJ mol−1),47 the moderate Qst values of the prepared polymers are probably due to the lack of heteroatoms e.g. nitrogen atoms, which are known to improve the binding affinity towards CO2 molecules by promoting dipole–quadrupole interactions.26
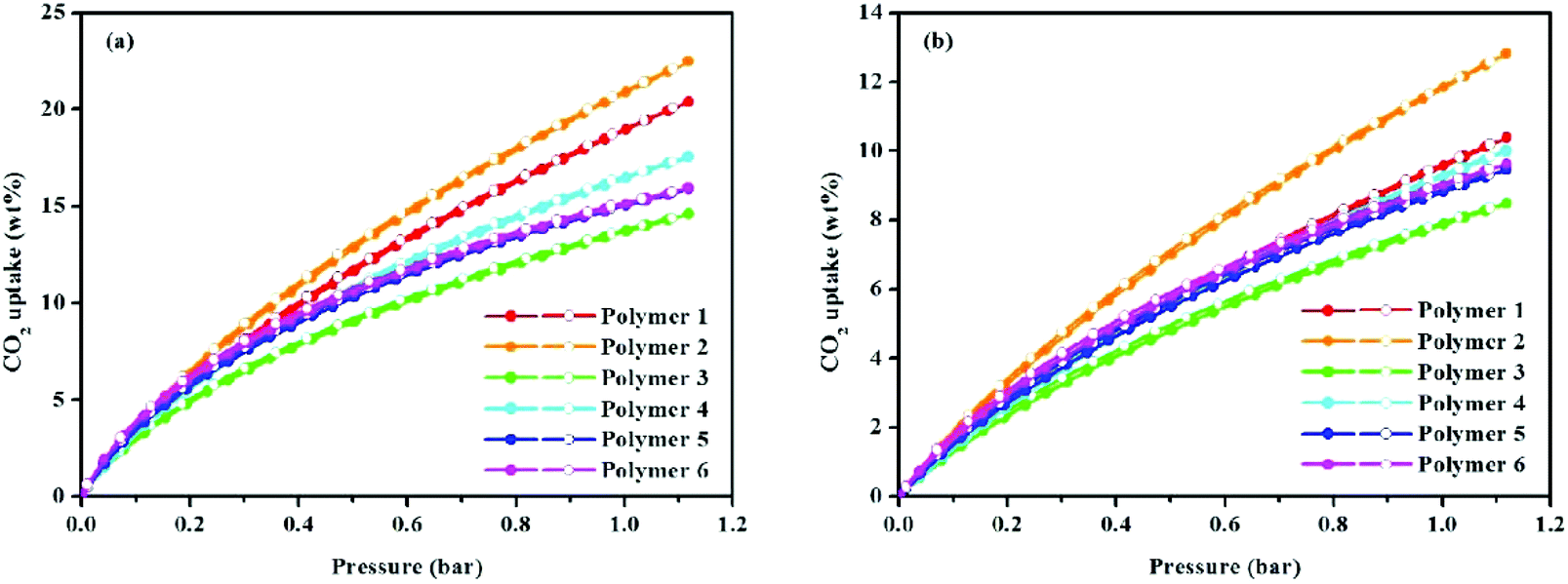 | ||
| Fig. 4 (a) Volumetric CO2 adsorption isotherms and desorption isotherms up to 1.13 bar at 273.15 K, and (b) volumetric CO2 adsorption isotherms and desorption isotherms up to 1.13 bar at 298.15 K. | ||
In addition to the studies on CO2 uptake capacity and isosteric heat of adsorption, gas adsorption selectivity including CO2/N2 and CO2/CH4 is another significant parameter relevant to polymer performance. Single-component physisorption isotherms for CO2, N2 and CH4 were measured by volumetric methods at 273.15 and 298.15 K, see Fig. 5. Gas adsorption values are listed in Table 2. The gas adsorption selectivity of the polymers towards CO2 over N2 and CH4 was calculated using the initial slope ratios from the Henry's law constants of the pure gas adsorption isotherms at the same temperature and a low pressure coverage of less than 0.3 bar. From Fig. S9–S14 (ESI†) and Table 2, the CO2/N2 selectivities at 273.15 K are ∼16.7, 16.9, 26.1, 17.2, 21.6, and 19.9 for polymers 1 to 6, respectively. The experimental results also prove that polymer 3 has the highest CO2/N2 selectivity i.e. 26.1 at 273.15 K. As stated, the incorporation of heteroatoms into the polymer's framework is an effective way to enhance the gas sorption selectivity of the polymer networks. The fabricated polymers mainly comprise carbon, hydrogen and a lack of heteroatoms. Though much lower than KFUPM-1 (141),48 PVAm/PEI-g-ZIF-8/mPSf (79.9),49 TrzPOP-1 (61),50 bare Pebax membranes (57),51 and PMOP (47.1),52 the CO2/N2 value of polymer 3 at 273.15 K is comparable to those of heteroatom-containing scaffolds such as CPOP-1 (25),32 pTOC (27),53 Network-2 (26),29 Network-4 (27),29 NOP-3 (27.1),54 PI-ADNT (25),55 APOP-1-OH (26.0),56 and NPTN-3 (25)57 at 273 K, 1,1-bi-2-naphthol (26),58 Glc-3 (27),59 and Ara-1 (26)59 at 298 K, and better than the porous polymer frameworks (PPFs) (14.5–20.4),60 NPTN-2 (22),57 JUC-141 (21.62),61 and CTFs (16.1–24.1)62 at 273 K, hydroxyl group functionalized polymers (16),58 Gal-1 (21),59 Glc-2 (18),59 Glc-1 (18),59 and CTF-20-400 (19)63 at 298 K. As shown in Fig. S9–S14 (ESI†) and Table 2, the CO2/CH4 selectivities at 273.15 K are ∼4.2, 4.1, 6.3, 4.0, 4.8, and 4.5 for materials from polymers 1 to 6 respectively. It is apparent that polymer 3 has the best CO2/CH4 selectivity at 273.15 K. Although lower than those of materials such as IITKGP-8 (17.7),64 MaSOFPd50 (15),65 6FDA-durene (12.45 ± 0.29),66 and [Co2(5,4-PMIA)2(TPOM)0.5]·xsolvent (9),67 the CO2/CH4 selectivity value of polymer 3 at 273.15 K is comparable to APOP-1-F (6.4),56 ALP-1 (6.0),68 CP-CMP-7 (6.0),31 and APOP-1-ONa (6.7)56 at 273 K, APOP-1-ONa (6.6),56 APOP-1-F (6.3),56 and ALP-3 (6.0)68 at 298 K, and exceeds TBMIDM (4.5),69 Azo-CMP2 (5.3),70 P-2 (3.0),71 FCDTPA-K-900 (3.4),28 TBMIDE (4.3),69 BO-CMP-2 (4.9),72 and OBO-CMP-1 (4.7)72 at 273 K, P-1 (4.0),71 and APOP-4 (4.7),56 P-2 (3.0),71 and TPI-3@IC (2.0)73 at 298 K. To understand the relationship between the structure and performance, following the gas sorption selectivity of polymers at 273.15 K, the CO2/N2 and CO2/CH4 selectivities of all polymers at 298.15 K were assessed. As seen in Fig. S15–S20 (ESI†) and Table 2, the CO2/N2 and CO2/CH4 selectivity values are in the ranges of 12.0–22.1 and 3.2–5.6 from polymers 1 to 6. It is apparent that polymer 3 has a CO2/N2 selectivity of 22.1 and a CO2/CH4 selectivity of 5.6, values that are comparable to other MOPs.
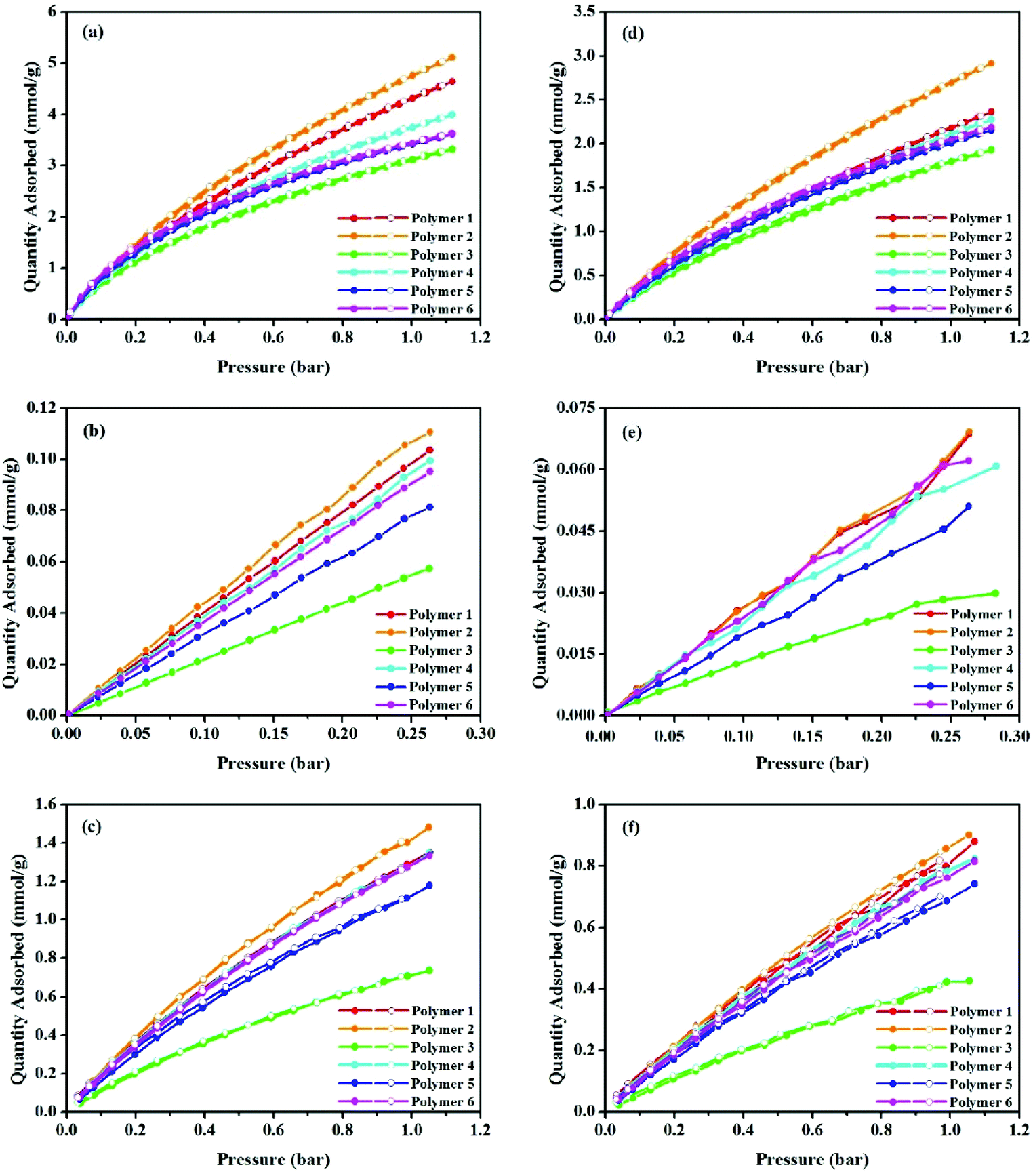 | ||
| Fig. 5 Adsorption isotherms of CO2 (a), N2 (b), CH4 (c) gases at 273.15 K, and CO2 (d), N2 (e), CH4 (f) gases at 298.15 K for polymer 1 to polymer 6. Filled symbols represent adsorption and open symbols represent desorption. | ||
| Samples | N2 uptakea (wt%) | N2 uptakeb (wt%) | CH4 uptakec (wt%) | CH4 uptaked (wt%) | CO2/N2e | CO2/N2f | CO2/CH4g | CO2/CH4h |
|---|---|---|---|---|---|---|---|---|
| a N2 uptake determined volumetrically using a Micromeritics ASAP 2020 M analyzer at 273.15 K and 0.25 bar. b N2 uptake determined volumetrically using a Micromeritics ASAP 2020 M analyzer at 298.15 K and 0.25 bar. c CH4 uptake determined volumetrically using a Micromeritics ASAP 2020 M analyzer at 273.15 K and 1.00 bar. d CH4 uptake determined volumetrically using a Micromeritics ASAP 2020 M analyzer at 298.15 K and 1.00 bar. e Adsorption selectivity of CO2 over N2 calculated by employing the initial slope ratio from the Henry's law constants at 273.15 K and a low pressure coverage of less than 0.3 bar. f Adsorption selectivity of CO2 over N2 calculated by employing the initial slope ratio from the Henry's law constants at 298.15 K and a low pressure coverage of less than 0.3 bar. g Adsorption selectivity of CO2 over CH4 calculated by employing the initial slope ratio from the Henry's law constants at 273.15 K and a low pressure coverage of less than 0.3 bar. h Adsorption selectivity of CO2 over CH4 calculated by employing the initial slope ratio from the Henry's law constants at 298.15 K and a low pressure coverage of less than 0.3 bar. | ||||||||
| Polymer 1 | 0.28 | 0.18 | 2.08 | 1.30 | 16.7 | 12.0 | 4.2 | 3.2 |
| Polymer 2 | 0.30 | 0.18 | 2.27 | 1.38 | 16.9 | 15.2 | 4.1 | 3.7 |
| Polymer 3 | 0.15 | 0.08 | 1.14 | 0.68 | 26.1 | 22.1 | 6.3 | 5.6 |
| Polymer 4 | 0.27 | 0.16 | 2.06 | 1.26 | 17.2 | 12.8 | 4.0 | 3.4 |
| Polymer 5 | 0.22 | 0.13 | 1.80 | 1.11 | 21.6 | 16.7 | 4.8 | 3.9 |
| Polymer 6 | 0.25 | 0.17 | 2.05 | 1.23 | 19.9 | 13.7 | 4.5 | 3.9 |
From the above gas sorption polymer data, the selectivity of CO2/N2 is much higher than that of CO2/CH4 at both 273.15 and 298.15 K. There are many influencing factors such as the pore size and physicochemcial nature of polymers, the kinetic diameter of the gas molecules, and differences in thermodynamic behaviour of the pairs of gases.74 To have a convincing explanation and better understanding the selective adsorption behavior and the critical temperature (Tc), which is a thermodynamic factor, should be taken into consideration. Specifically, the critical temperatures of common gas molecules like CO2, N2 and CH4 are 304.2, 190.6 and 126.2 K, which can be ranked in the order Tc (CO2) > Tc (CH4) > Tc (N2).75 It has been documented that the gas solubility coefficient in a polymer is positively correlated with its critical temperature.76 Due to having the lowest Tc value, the N2 molecule has the smallest uptake performance. Due to having the highest Tc value, which makes it easy to condense CO2 within the small pores, the CO2 molecule has the highest adsorption capacity,77 making it largely responsible for the higher selectivity of CO2/N2 when compared to its corresponding CO2/CH4 gas pair.
Studies have shown that light-emitting MOPs are promising candidates for applications in optoelectronics,78,79 chemosensing,80,81 light-harvesting antennas,82 molecular sensing devices,83 and ion conductivity.84 Due to their extensive conjugation we decided to explore the polymers’ fluorescence properties. After studying the solid state fluorescence emission spectra (Fig. 6) it was found that, polymers 5 and 6 were fluorescent with λem = 280 nm and λex = 466 nm, while almost no fluorescence was observed for polymers 1 to 4. Meanwhile, the absolute solid state fluorescence quantum yield of polymers 5 and 6 was found to be 0.23 and 0.18 respectively, which demonstrates that polymers knitted by the Scholl coupling reaction may also have potential in sensing, electric conduction, and other applications.
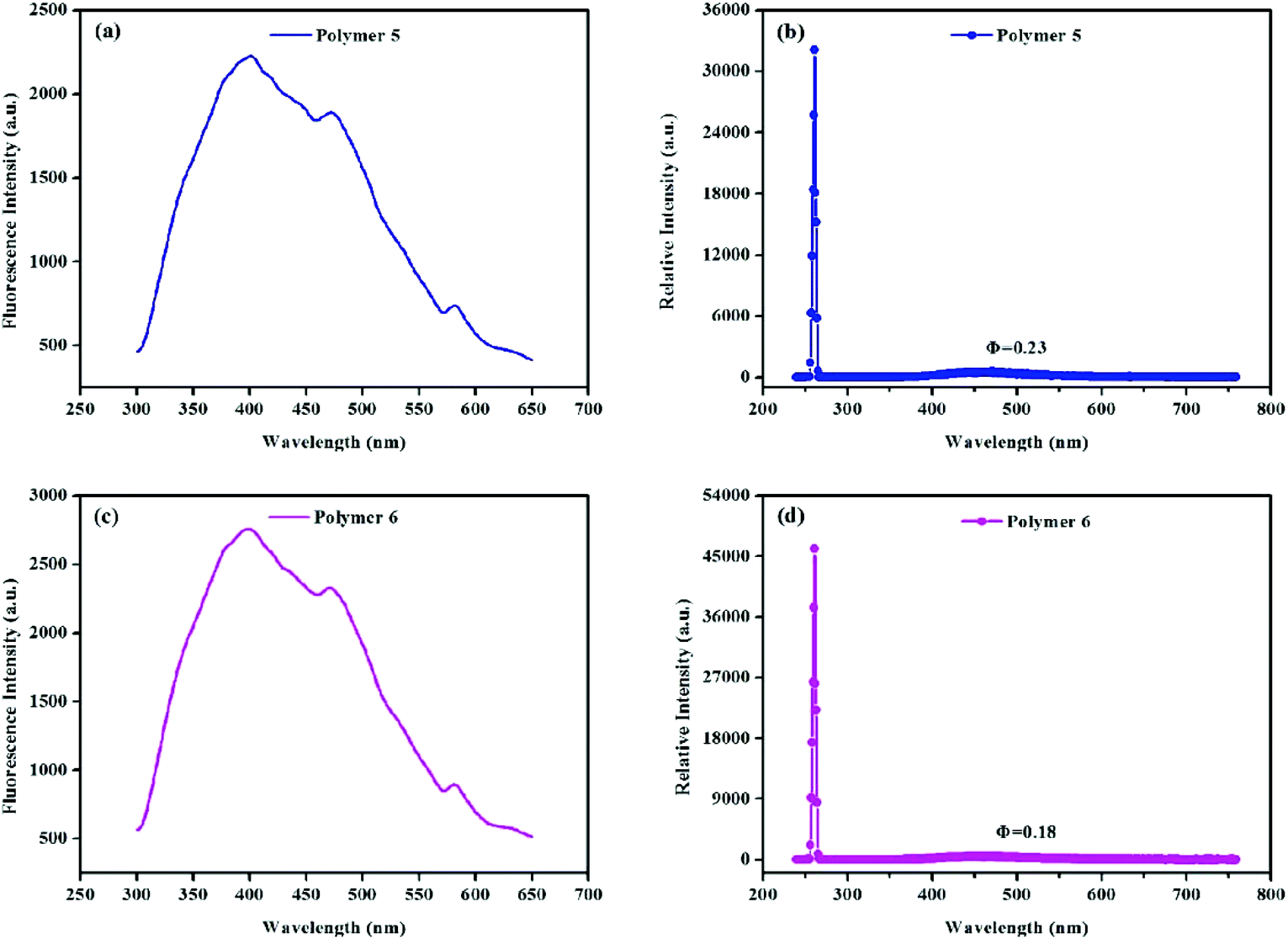 | ||
| Fig. 6 Solid state fluorescence excitation spectrum of polymer 5 (a, λem = 280 nm, λex = 466 nm), the absolute solid state fluorescence quantum yield of polymer 5 (b, Φ = 0.23), the solid state fluorescence excitation spectrum of polymer 6 (c, λem = 280 nm, λex = 466 nm), and the absolute solid state fluorescence quantum yield of polymer 6 (d, Φ = 0.18). | ||
Based on our findings, these HCPs prepared by using different strategies have distinctly different structural features and gas uptake abilities that result from their building blocks’ chemical and aggregation structures42 and their consequent influence on the commonly employed Friedel–Crafts alkylation reaction. During the solvent knitting reaction method, CH2Cl2, which acts as both the solvent and an external cross-linker, firstly reacts with AlCl3 to produce complex CH2Cl2·AlCl3. Since the interaction force between the chlorine atom and the methylene has been weakened, the CH2Cl2·AlCl3 will form CH22+ and AlCl4−, and then the CH22+ will react with the building block (R). By substituting hydrogen atoms from R, the methylene groups will cross-link R and form polymer networks, with hydrogen chloride as the only byproduct. Due to the methylene groups, the π-conjugated systems and consequently the fluorescence of the resultant polymers will be destroyed. For the knitting method with FDA as an external cross-linker, it is a simple one-step (two stage 45 °C for 5 h and then 80 °C for 19 h) Friedel–Crafts reaction catalyzed by anhydrous FeCl3 in 1,2-dichloroethane. In this reaction, the external cross-linker CH3OCH2OCH3 activated by FeCl3 will turn into CH3O+CH2+OCH3, by reacting with R, CH3O+ will change into the only byproduct CH3OH by hydrolysis in the presence of trace water, leaving the building blocks bridged by methylene groups. Because of the methylene groups, the π-conjugated systems in polymer materials are interrupted. The Scholl coupling is typically performed at 58 °C for 48 h in which R will react with AlCl3 in AlCl3 [H+] and chloroform, normally resulting R+ and AlCl3H. R+ then engages in a nucleophile substitution reaction with another R, resulting in the elimination of two aryl-bound hydrogen atoms to form a new aryl–aryl bond, leading to novel polymer networks. Since the building blocks are cross-linked by a carbon–carbon single bond, the polymers can have extended π-conjugated systems and three-dimensional networks of π–π stacked aryl rings, which are conducive to maintaining fluorescence. It is worth mentioning that, due to the bridging by methylene groups, polymers obtained by the solvent knitting method and the knitting method with FDA as an external cross-linker tend to have similar pore size distributions (Fig. 3b). Due to the connection type (carbon–carbon single bonds), polymers based on the Scholl coupling reaction tend to have narrow pore size distributions (Fig. 3b). Additionally, the solvent knitting method requires 60 h, the knitting method with FDA as an external cross-linker needs 24 h and the Scholl coupling reaction requires 48 h. Therefore, the solvent knitting method is the most time-consuming while the knitting method with FDA as an external cross-linker is the most time-efficient.
Conclusions
A series of hyper-cross-linked polymers based on two aromatic hydrocarbon building blocks and three versatile methodologies i.e. the solvent knitting method, the knitting method with FDA as an external cross-linker and the Scholl coupling reaction were made. For the resulting polymers the BET surface area can be as high as 2064 m2 g−1 and 2278 m2 g−1, the CO2 uptake capacity can be up to 18.95 wt% and 20.89 wt% at 273.15 K/1.00 bar. The systematic and comprehensive study which focused on the porous texture, gas adsorption capacity and fluorescence properties of the prepared materials shows that the solvent knitting method generally produces polymers with better porosities and gas uptake performances. The knitting method with FDA as an external cross-linker is the fastest to generate polymer networks. The Scholl coupling reaction facilitates the formation of knitting fluorescent polymers. It is important to note that these distinctions among the generated microporous HCPs may be largely due to the underlying organic reaction mechanisms. Our study is an attempt to provide original insights into the rational design and synthesis of microporous organic HCPs and offer a route to novel future polymers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Sincere thanks are given to the Analysis and Testing Center, Huazhong University of Science and Technology for their assistance in materials characterization. This work was financially supported by the Program for National Natural Science Foundation of China (no. 21474033), the International S&T Cooperation Program of China (2016YFE 0124400), and the Program for HUST Interdisciplinary Innovation Team (2016JCTD104).Notes and references
- L. X. Tan and B. E. Tan, Hypercrosslinked Porous Polymer Materials: Design, Synthesis, and Applications, Chem. Soc. Rev., 2017, 46(11), 3322–3356 RSC.
- S. J. Xu, Y. L. Luo and B. E. Tan, Recent Development of Hypercrosslinked Microporous Organic Polymers, Macromol. Rapid Commun., 2013, 34(6), 471–484 CrossRef CAS PubMed.
- J. Y. Wang, L. Huang, R. Y. Yang, Z. Zhang, J. W. Wu, Y. S. Gao, Q. Wang, D. O'Hare and Z. Y. Zhang, Recent Advances in Solid Sorbents for CO2 Capture and New Development Trends, Energy Environ. Sci., 2014, 7(11), 3478–3518 RSC.
- W. J. Wang, M. Zhou and D. Q. Yuan, Carbon Dioxide Capture in Amorphous Porous Organic Polymers, J. Mater. Chem. A, 2017, 5(4), 1334–1347 RSC.
- S. Mane, Z. Y. Gao, Y. X. Li, D. M. Xue, X. Q. Liu and L. B. Sun, Fabrication of Microporous Polymers for Selective CO2 Capture: The Significant Role of Crosslinking and Crosslinker Length, J. Mater. Chem. A, 2017, 5(44), 23310–23318 RSC.
- L. Ding, H. Gao, F. F. Xie, W. Q. Li, H. Bai and L. Li, Porosity-Enhanced Polymers from Hyper-Cross-Linked Polymer Precursors, Macromolecules, 2017, 50(3), 956–962 CrossRef CAS.
- S. Bracco, D. Piga, I. Bassanetti, J. Perego, A. Comotti and P. Sozzani, Porous 3D Polymers for High Pressure Methane Storage and Carbon Dioxide Capture, J. Mater. Chem. A, 2017, 5(21), 10328–10337 RSC.
- W. Q. Li, A. J. Zhang, H. Gao, M. J. Chen, A. Liu, H. Bai and L. Li, Massive Preparation of Pitch-Based Organic Microporous Polymers for Gas Storage, Chem. Commun., 2016, 52(13), 2780–2783 RSC.
- Y. W. Yang, B. E. Tan and C. D. Wood, Solution-Processable Hypercrosslinked Polymers by Low Cost Strategies: A Promising Platform for Gas Storage and Separation, J. Mater. Chem. A, 2016, 4(39), 15072–15080 RSC.
- H. Gao, L. Ding, W. Q. Li, G. F. Ma, H. Bai and L. Li, Hyper-Cross-Linked Organic Microporous Polymers Based on Alternating Copolymerization of Bismaleimide, ACS Macro Lett., 2016, 5(3), 377–381 CrossRef CAS.
- H. Gao, L. Ding, H. Bai, A. H. Liu, S. Z. Li and L. Li, Pitch-Based Hyper-Cross-Linked Polymers with High Performance for Gas Adsorption, J. Mater. Chem. A, 2016, 4(42), 16490–16498 RSC.
- C. Wilson, M. J. Main, N. J. Cooper, M. E. Briggs, A. I. Cooper and D. J. Adams, Swellable Functional Hypercrosslinked Polymer Networks for the Uptake of Chemical Warfare Agents, Polym. Chem., 2017, 8(12), 1914–1922 RSC.
- Z. F. Jia, K. W. Wang, B. E. Tan and Y. L. Gu, Hollow Hyper-Cross-Linked Nanospheres with Acid and Base Sites as Efficient and Water-Stable Catalysts for One-Pot Tandem Reactions, ACS Catal., 2017, 7(5), 3693–3702 CrossRef CAS.
- H. Y. Li, B. Meng, S. H. Chai, H. L. Liu and S. Dai, Hyper-Crosslinked Beta-Cyclodextrin Porous Polymer: An Adsorption-Facilitated Molecular Catalyst Support for Transformation of Water-Soluble Aromatic Molecules, Chem. Sci., 2016, 7(2), 905–909 RSC.
- B. Y. Li, Z. H. Guan, W. Wang, X. J. Yang, J. L. Hu, B. E. Tan and T. Li, Highly Dispersed Pd Catalyst Locked in Knitting Aryl Network Polymers for Suzuki-Miyaura Coupling Reactions of Aryl Chlorides in Aqueous Media, Adv. Mater., 2012, 24(25), 3390–3395 CrossRef CAS PubMed.
- Y. W. Yang, L. Y. Feng, J. Ren, Y. F. Liu, S. B. Jin, L. Su, C. D. Wood and B. E. Tan, Soluble Hyperbranched Porous Organic Polymers, Macromol. Rapid Commun., 2018, 39(21), 1800441 CrossRef PubMed.
- A. G. Slater and A. I. Cooper, Function-Led Design of New Porous Materials, Science, 2015, 348(6238), aaa8075 CrossRef PubMed.
- J. S. M. Lee, M. E. Briggs, T. Hasell and A. I. Cooper, Hyperporous Carbons from Hypercrosslinked Polymers, Adv. Mater., 2016, 28(44), 9804–9810 CrossRef CAS PubMed.
- X. Y. Wang, P. Mu, C. Zhang, Y. Chen, J. H. Zeng, F. Wang and J. X. Jiang, Control Synthesis of Tubular Hyper-Cross-Linked Polymers for Highly Porous Carbon Nanotubes, ACS Appl. Mater. Interfaces, 2017, 9(24), 20779–20786 CrossRef CAS PubMed.
- S. L. Wang, K. P. Song, C. X. Chen, Y. Shu, T. Li and B. E. Tan, A Novel Metalporphyrin-Based Microporous Organic Polymer with High CO2 Uptake and Efficient Chemical Conversion of CO2 under Ambient Conditions, J. Mater. Chem. A, 2017, 5(4), 1509–1515 RSC.
- B. Y. Li, R. N. Gong, W. Wang, X. Huang, W. Zhang, H. M. Li, C. X. Hu and B. E. Tan, A New Strategy to Microporous Polymers: Knitting Rigid Aromatic Building Blocks by External Cross-Linker, Macromolecules, 2011, 44(8), 2410–2414 CrossRef CAS.
- B. Y. Li, Z. H. Guan, X. J. Yang, W. D. Wang, W. Wang, I. Hussain, K. P. Song, B. E. Tan and T. Li, Multifunctional Microporous Organic Polymers, J. Mater. Chem. A, 2014, 2(30), 11930–11939 RSC.
- S. L. Wang, C. X. Zhang, Y. Shu, S. L. Jiang, Q. Xia, L. J. Chen, S. B. Jin, I. Hussain, A. I. Cooper and B. E. Tan, Layered Microporous Polymers by Solvent Knitting Method, Sci. Adv., 2017, 3(3), e1602610 CrossRef PubMed.
- S. S. Hou, S. L. Wang, X. J. Long and B. E. Tan, Knitting Polycyclic Aromatic Hydrocarbon-Based Microporous Organic Polymers for Efficient CO2 Capture, RSC Adv., 2018, 8(19), 10347–10354 RSC.
- Y. L. Luo, S. C. Zhang, Y. X. Ma, W. Wang and B. E. Tan, Microporous Organic Polymers Synthesized by Self-Condensation of Aromatic Hydroxymethyl Monomers, Polym. Chem., 2013, 4(4), 1126–1131 RSC.
- Y. L. Luo, B. Y. Li, W. Wang, K. B. Wu and B. E. Tan, Hypercrosslinked Aromatic Heterocyclic Microporous Polymers: A New Class of Highly Selective CO2 Capturing Materials, Adv. Mater., 2012, 24(42), 5703–5707 CrossRef CAS PubMed.
- S. S. Hou and B. E. Tan, Naphthyl Substitution-Induced Fine Tuning of Porosity and Gas Uptake Capacity in Microporous Hyper-Cross-Linked Amine Polymers, Macromolecules, 2018, 51(8), 2923–2931 CrossRef CAS.
- X. Yang, M. Yu, Y. Zhao, C. Zhang, X. Y. Yang and J. X. Jiang, Remarkable Gas Adsorption by Carbonized Nitrogen-Rich Hypercrosslinked Porous Organic Polymers, J. Mater. Chem. A, 2014, 2(36), 15139–15145 RSC.
- S. W. Yao, X. Yang, M. Yu, Y. H. Zhang and J. X. Jiang, High Surface Area Hypercrosslinked Microporous Organic Polymer Networks Based on Tetraphenylethylene for CO2 Capture, J. Mater. Chem. A, 2014, 2(21), 8054–8059 RSC.
- X. Zhang, J. Z. Lu and J. Zhang, Porosity Enhancement of Carbazolic Porous Organic Frameworks Using Dendritic Building Blocks for Gas Storage and Separation, Chem. Mater., 2014, 26(13), 4023–4029 CrossRef CAS.
- M. Yu, X. Y. Wang, X. Yang, Y. Zhao and J. X. Jiang, Conjugated Microporous Copolymer Networks with Enhanced Gas Adsorption, Polym. Chem., 2015, 6(17), 3217–3223 RSC.
- Q. Chen, M. Luo, P. Hammershoj, D. Zhou, Y. Han, B. W. Laursen, C. G. Yan and B. H. Han, Microporous Polycarbazole with High Specific Surface Area for Gas Storage and Separation, J. Am. Chem. Soc., 2012, 134(14), 6084–6087 CrossRef CAS PubMed.
- W. G. Lu, J. P. Sculley, D. Q. Yuan, R. Krishna, Z. W. Wei and H. C. Zhou, Polyamine-Tethered Porous Polymer Networks for Carbon Dioxide Capture from Flue Gas, Angew. Chem., Int. Ed., 2012, 51(30), 7480–7484 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, Storage of Hydrogen, Methane, and Carbon Dioxide in Highly Porous Covalent Organic Frameworks for Clean Energy Applications, J. Am. Chem. Soc., 2009, 131(25), 8875–8883 CrossRef CAS PubMed.
- T. Bei, C. Y. Pei, D. L. Zhang, J. Xu, F. Deng, X. F. Jing and S. L. Qiu, Gas Storage in Porous Aromatic Frameworks (PAFs), Energy Environ. Sci., 2011, 4(10), 3991–3999 RSC.
- Y. F. Chen, H. X. Sun, R. X. Yang, T. T. Wang, C. J. Pei, Z. T. Xiang, Z. Q. Zhu, W. D. Liang, A. Li and W. Q. Deng, Synthesis of Conjugated Microporous Polymer Nanotubes with Large Surface Areas as Absorbents for Iodine and CO2 Uptake, J. Mater. Chem. A, 2015, 3(1), 87–91 RSC.
- Z. H. Xiang, R. Mercado, J. M. Huck, H. Wang, Z. H. Guo, W. C. Wang, D. P. Cao, M. Haranczyk and B. Smit, Systematic Tuning and Multifunctionalization of Covalent Organic Polymers for Enhanced Carbon Capture, J. Am. Chem. Soc., 2015, 137(41), 13301–13307 CrossRef CAS PubMed.
- X. F. Jing, D. L. Zou, P. Cui, H. Ren and G. S. Zhu, Facile Synthesis of Cost-Effective Porous Aromatic Materials with Enhanced Carbon Dioxide Uptake, J. Mater. Chem. A, 2013, 1(44), 13926–13931 RSC.
- F. Alves, P. Scholder and I. Nischang, Conceptual Design of Large Surface Area Porous Polymeric Hybrid Media Based on Polyhedral Oligomeric Silsesquioxane Precursors: Preparation Tailoring of Porous Properties, and Internal Surface Functionalization, ACS Appl. Mater. Interfaces, 2013, 5(7), 2517–2526 CrossRef CAS PubMed.
- D. X. Wang, W. Y. Yang, L. G. Li, X. Zhao, S. Y. Feng and H. Z. Liu, Hybrid Networks Constructed from Tetrahedral Silicon-Centered Precursors and Cubic POSS-Based Building Blocks via Heck Reaction: Porosity, Gas Sorption, and Luminescence, J. Mater. Chem. A, 2013, 1(43), 13549–13558 RSC.
- X. R. Yang and H. Z. Liu, Ferrocene-Functionalized Silsesquioxane-Based Porous Polymer for Efficient Removal of Dyes and Heavy Metal Ions, Chem. – Eur. J., 2018, 24(51), 13504–13511 CrossRef CAS PubMed.
- G. Y. Li, B. Zhang, J. Yan and Z. G. Wang, Microporous Polyimides with Functional Groups for the Adsorption of Carbon Dioxide and Organic Vapors, J. Mater. Chem. A, 2016, 4(29), 11453–11461 RSC.
- T. Ben, H. Ren, S. Q. Ma, D. P. Cao, J. H. Lan, X. F. Jing, W. C. Wang, J. Xu, F. Deng, J. M. Simmons, S. L. Qiu and G. S. Zhu, Targeted Synthesis of a Porous Aromatic Framework with High Stability and Exceptionally High Surface Area, Angew. Chem., Int. Ed., 2009, 48(50), 9457–9460 CrossRef CAS PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Chemical Functionalization Strategies for Carbon Dioxide Capture in Microporous Organic Polymers, Polym. Int., 2013, 62(3), 345–352 CrossRef CAS.
- V. Krungleviciute, L. Herous, A. D. Migone, C. T. Kingston and B. Simard, Isosteric Heat of Argon Adsorbed on Single-Walled Carbon Nanotubes Prepared by Laser Ablation, J. Phys. Chem. B, 2005, 109(19), 9317–9320 CrossRef CAS PubMed.
- A. Laybourn, R. Dawson, R. Clowes, J. A. Iggo, A. I. Cooper, Y. Z. Khimyak and D. J. Adams, Branching Out with Aminals: Microporous Organic Polymers from Difunctional Monomers, Polym. Chem., 2012, 3(2), 533–537 RSC.
- P. Mohanty, L. D. Kull and K. Landskron, Porous Covalent Electron-Rich Organonitridic Frameworks as Highly Selective Sorbents for Methane and Carbon Dioxide, Nat. Commun., 2011, 2(1), 401 CrossRef PubMed.
- M. M. Abdelnaby, A. M. Alloush, N. A. A. Qasem, B. A. Al-Maythalony, R. B. Mansour, K. E. Cordova and O. C. S. Al Hamouz, Carbon Dioxide Capture in the Presence of Water by an Amine-Based Crosslinked Porous Polymer, J. Mater. Chem. A, 2018, 6(15), 6455–6462 RSC.
- Y. Q. Gao, Z. H. Qiao, S. Zhao, Z. Wang and J. X. Wang, In Situ Synthesis of Polymer Grafted ZIFs and Application in Mixed Matrix Membrane for CO2 Separation, J. Mater. Chem. A, 2018, 6(7), 3151–3161 RSC.
- S. K. Das, P. Bhanja, S. K. Kundu, S. Mondal and A. Bhaumik, Role of Surface Phenolic-OH Groups in N-Rich Porous Organic Polymers for Enhancing the CO2 Uptake and CO2/N2 Selectivity: Experimental and Computational Studies, ACS Appl. Mater. Interfaces, 2018, 10(28), 23813–23824 CrossRef CAS PubMed.
- A. Sabetghadam, X. L. Liu, M. Benzaqui, E. Gkaniatsou, A. Orsi, M. M. Lozinska, C. Sicard, T. Johnson, N. Steunou, P. A. Wright, C. Serre, J. Gascon and F. Kapteijn, Influence of Filler Pore Structure and Polymer on the Performance of MOF-Based Mixed-Matrix Membranes for CO2 Capture, Chem. – Eur. J., 2018, 24(31), 7949–7956 CrossRef CAS PubMed.
- S. J. Luo, Q. N. Zhang, Y. Z. Zhang, K. P. Weaver, W. A. Phillip and R. L. Guo, Facile Synthesis of a Pentiptycene-Based Highly Microporous Organic Polymers for Gas Storage and Water Treatment, ACS Appl. Mater. Interfaces, 2018, 10(17), 15174–15182 CrossRef CAS PubMed.
- Z. Wang, H. Ma, T. L. Zhai, G. Cheng, Q. Xu, J. M. Liu, J. K. Yang, Q. M. Zhang, Q. P. Zhang, Y. S. Zheng, B. E. Tan and C. Zhang, Networked Cages for Enhanced CO2 Capture and Sensing, Adv. Sci., 2018, 5(7), 1800141 CrossRef PubMed.
- S. H. Xiong, X. Fu, L. Xiang, G. Yu, J. P. Guan, Z. G. Wang, Y. Du, X. Xiong and C. Y. Pan, Liquid Acid-Catalysed Fabrication of Nanoporous 1,3,5-Triazine Frameworks with Efficient and Selective CO2 Uptake, Polym. Chem., 2014, 5(10), 3424–3431 RSC.
- C. J. Shen and Z. G. Wang, Tetraphenyladamantane-Based Microporous Polyimide and Its Nitro-Functionalization for Highly Efficient CO2 Capture, J. Phys. Chem. C, 2014, 118(31), 17585–17593 CrossRef CAS.
- W. C. Song, X. K. Xu, Q. Chen, Z. Z. Zhuang and X. H. Bu, Nitrogen-Rich Diaminotriazine-Based Porous Organic Polymers for Small Gas Storage and Selective Uptake, Polym. Chem., 2014, 4(17), 4690–4696 RSC.
- S. F. Wu, Y. Liu, G. P. Yu, J. G. Guan, C. Y. Pan, Y. Du, X. Xiong and Z. G. Wang, Facile Preparation of Dibenzoheterocycle-Functional Nanoporous Polymeric Networks with High Gas Uptake Capacities, Macromolecules, 2014, 47(9), 2875–2882 CrossRef CAS.
- R. Dawson, L. A. Stevens, T. C. Drage, C. E. Snape, M. W. Smith, D. J. Adams and A. I. Cooper, Impact of Water Coadsorption for Carbon Dioxide Capture in Microporous Polymer Sorbents, J. Am. Chem. Soc., 2012, 134(26), 10741–10744 CrossRef CAS PubMed.
- H. Y. Li, B. Meng, S. M. Mahurin, S. H. Chai, K. M. Nelson, D. C. Baker, H. L. Liu and S. Dai, Carbohydrate Based Hyper-Crosslinked Organic Polymers with -OH Functional Groups for CO2 Separation, J. Mater. Chem. A, 2015, 3(42), 20913–20918 RSC.
- Y. L. Zhu, H. Long and W. Zhang, Imine-Linked Porous Polymer Frameworks with High Small Gas (H2, CO2, CH4, C2H2) Uptake and CO2/N2 Selectivity, Chem. Mater., 2013, 25(9), 1630–1635 CrossRef CAS.
- N. A. Zhao, F. X. Sun, P. Li, X. Mu and G. S. Zhu, An Amino-Coordinated Metal-Organic Framework for Selective Gas Adsorption, Inorg. Chem., 2017, 56(12), 6938–6942 CrossRef CAS PubMed.
- S. J. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Porous, Fluorescent, Covalent Triazine-Based Frameworks Via Room-Temperature and Microwave-Assisted Synthesis, Adv. Mater., 2012, 24(17), 2357–2361 CrossRef CAS PubMed.
- G. B. Wang, K. Leus, S. N. Zhao and P. Van Der Voort, Newly Designed Covalent Triazine Framework Based on Novel N-Heteroaromatic Building Blocks for Efficient CO2 and H2 Capture and Storage, ACS Appl. Mater. Interfaces, 2018, 10(1), 1244–1249 CrossRef CAS PubMed.
- S. Chand, A. Pal and M. C. Das, A Moisture-Stable 3D Microporous Co-II-Metal-Organic Framework with Potential for Highly Selective CO2 Separation under Ambient Conditions, Chem. – Eur. J., 2018, 24(22), 5982–6986 CrossRef CAS PubMed.
- D. Reinhard, W. S. Zhang, Y. Vaynzof, F. Rominger, R. R. Schroder and M. Mastalerz, Triptycene-Based Porous Metal-Assisted Salphen Organic Frameworks: Influence of the Metal Ions on Formation and Gas Sorption, Chem. Mater., 2018, 30(8), 2781–2790 CrossRef CAS.
- A. A. Shamsabadi, F. Seidi, M. Nozari and M. Soroush, A New Pentiptycene-Based Dianhydride and Its High-Free-Volume Polymer for Carbon Dioxide Removal, ChemSusChem, 2018, 11(2), 472–482 CrossRef CAS PubMed.
- J. G. Duan, R. Yan, L. L. Qin, Y. Wang, L. L. Wen, S. X. Cheng, H. Xu and P. Y. Feng, Highly Selective Gaseous and Liquid-Phase Separation over a Novel Cobalt(II) Metal-Organic Framework, ACS Appl. Mater. Interfaces, 2018, 10(27), 23009–23017 CrossRef CAS PubMed.
- P. Arab, M. G. Sekizkardes, T. Islamoglu and H. M. El-Kaderi, Copper(I)-Catalyzed Synthesis of Nanoporous Azo-Linked Polymers: Impact of Textural Properties on Gas Storage and Selective Carbon Dioxide Capture, Chem. Mater., 2014, 26(3), 1385–1392 CrossRef CAS.
- Y. Zhao, X. Y. Wang, C. Zhang, F. Y. Xie, R. Kong and J. X. Jiang, Isoindigo-Based Microporous Organic Polymers for Carbon Dioxide Capture, RSC Adv., 2015, 5(121), 100322–100329 RSC.
- X. Y. Wang, Y. Zhao, L. L. Wei, C. Zhang and J. X. Jiang, Nitrogen-Rich Conjugated Microporous Polymers: Impact of Building Blocks on Porosity and Gas Adsorption, J. Mater. Chem. A, 2015, 3(42), 21185–21193 RSC.
- S. L. Qiao, Z. K. Du and R. Q. Yang, Design and Synthesis of Novel Carbazole-Spacer-Carbazole Type Conjugated Microporous Networks for Gas Storage and Separation, J. Mater. Chem. A, 2014, 2(6), 1877–1885 RSC.
- S. J. Yang, X. S. Ding and B. H. Han, Conjugated Microporous Polymers with Extended π–Structures for Organic Vapor Adsorption, Macromolecules, 2018, 51(3), 947–953 CrossRef CAS.
- S. F. Wu, S. Gu, A. Q. Zhang, G. P. Yu, Z. G. Wang, J. G. Jian and C. Y. Pan, Rational Construction of Microporous Imide-Bridged Covalent-Organic Polytriazines for High-Enthalpy Small Gas Absorption, J. Mater. Chem. A, 2015, 3(2), 878–885 RSC.
- G. Y. Li, B. Zhang, J. Yan and Z. G. Wang, Tetraphenyladamantane-Based Polyaminals for Highly Efficient Captures of CO2 and Organic Vapors, Macromolecules, 2014, 47(19), 6664–6670 CrossRef CAS.
- J. Jeans, An Introduction to the Kinetic Theory of Gases, Cambridge University Press, London, 1982 Search PubMed.
- K. Y. Yuan, C. Liu, J. H. Han, G. P. Yu, J. Y. Wang, H. M. Duan, Z. G. Wang and X. G. Jian, Phthalazinone Structure-Based Covalent Triazine Frameworks and Their Gas Adsorption and Separation Properties, RSC Adv., 2016, 6(15), 12009–12020 RSC.
- A. Vishnyakov, P. Ravikovitch and A. V. Neimark, Molecular Level Models for CO2 Sorption in Nanopores, Langmuir, 1999, 15(25), 8736–8742 CrossRef CAS.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. L. Jiang, A Photoconductive Covalent Organic Framework: Self-Condensed Arene Cubes Composed of Eclipsed 2D Polypyrene Sheets for Photocurrent Generation, Angew. Chem., Int. Ed., 2009, 48(30), 5439–5442 CrossRef CAS PubMed.
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. P. Dong and D. L. Jiang, Photoelectric Covalent Organic Frameworks: Converting Open Lattices into Ordered Donor-Acceptor Heterojunctions, J. Am. Chem. Soc., 2014, 136(28), 9806–9809 CrossRef CAS PubMed.
- S. Dalapati, E. Q. Jin, M. Addicoat, T. Heine and D. L. Jiang, Highly Emissive Covalent Organic Frameworks, J. Am. Chem. Soc., 2016, 138(18), 5797–5800 CrossRef CAS PubMed.
- S. Dalapati, S. B. Jin, J. Gao, Y. H. Xu, A. Nagai and D. L. Jiang, An Azine-Linked Covalent Organic Frameworks, J. Am. Chem. Soc., 2013, 135(46), 17310–17313 CrossRef CAS PubMed.
- L. Chen, Y. Honsho, S. Seki and D. L. Jiang, Light-Harvesting Conjugated Microporous Polymers: Rapid and Highly Efficient Flow of Light Energy with a Porous Polyphenylene Framework as Antenna, J. Am. Chem. Soc., 2010, 132(19), 6742–6748 CrossRef CAS PubMed.
- X. M. Liu, Y. H. Xu and D. L. Jiang, Conjugated Microporous Polymers as Molecular Sensing Devices: Microporous Architecture Enables Rapid Response and Enhances Sensitivity in Fluorescence-On and Fluorescence-Off Sensing, J. Am. Chem. Soc., 2012, 134(21), 8738–8741 CrossRef CAS PubMed.
- Q. Xu, S. S. Tao, Q. H. Jiang and D. L. Jiang, Ion Conduction in Polyelectrolyte Covalent Organic Frameworks, J. Am. Chem. Soc., 2018, 140(24), 7429–7432 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01730a |
| This journal is © The Royal Society of Chemistry 2019 |