
Degradable pH and redox dual responsive nanoparticles for efficient covalent drug delivery†
Jinhong
Du
 a,
Bonnie
Choi
a,
Yuxuan
Liu
a,
Anchao
Feng
*a and
San H.
Thang
ab
a,
Bonnie
Choi
a,
Yuxuan
Liu
a,
Anchao
Feng
*a and
San H.
Thang
ab
aBeijing Advanced Innovation Center for Soft Matter Science and Engineering, College of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: fengac@mail.buct.edu.cn
bSchool of Chemistry, Monash University, Clayton Campus, VIC 3800, Australia
First published on 15th February 2019
Abstract
Examples of successful therapies using macromolecular prodrugs to which the drug is linked via covalent bonds have been reported; however, there are safety concerns which render them disfavourable for clinical applications. Therefore, it is essential to reassess this class of drugs by designing compounds that combine covalent linking with reducible linkers. Herein, the hydrophobic drug paclitaxel was modified into a polymerizable monomer and subsequently copolymerized with pH-sensitive monomers and redox-sensitive disulfide-based cyclic monomers. The resulting diblock copolymer can self-assemble into vesicles incorporating the hydrophobic drug PTX into the vesicle walls. On decreasing the pH value, the size of the vesicular drug container varied with the change in the hydrophobicity–hydrophilicity balance. Treatment with reducing agents mediated the disassembly of the aggregates facilitating the release of the drug moiety. Furthermore, in vitro cell experiments showed that the nanoparticles exhibited cytotoxicity, indicating that they have potential as a drug delivery system for anti-cancer treatments.
Introduction
One of the leading diseases causing the highest number of deaths in developed countries is cancer.1 One of the most effective chemotherapeutic agents is paclitaxel (PTX) utilized in the treatment of different types of cancer.2 PTX is a tricyclic diterpenoid and is almost insoluble in aqueous medium. Due to this hydrophobicity, the intravenous administration must be performed by dissolving PTX in a solution containing a formulation vehicle (Cremophor El) and ethanol to aid delivery; however, a severe drawback of this is that there are excipient-associated side effects and reduced therapeutic efficiency.3 There are two main strategies to improve the solubility of drugs: one strategy involves physically encapsulating the drug into the hydrophobic cavities of drug delivery systems.4 The other strategy involves conjugating drug molecules to other materials such as polymers by covalent bonds to improve the absorption, distribution, metabolization, and excretion and to overcome bioavailability and toxicity issues; these are generally termed prodrugs.5,6 These drug molecules are well protected by the conjugate and thus have improved stability. One of the first examples of a prodrug is aspirin, introduced by Bayer in 1899,7 and since then, tremendous efforts have been made over the last few decades in prodrug design and synthesis. Over the last 20 years, over thirty new prodrugs have been approved by the FDA.7 The prodrug concept utilizes the grafting of a molecule (promoiety) onto a biologically active drug molecule aiding it to reach the pharmacological target. One of the challenges is to ensure that the promoiety can be removed once it has reached the target to regenerate the active drug molecule. Stimuli responsiveness has been used to trigger the release of prodrugs, which include oxidative/reductive agents, pH value, temperature and enzyme responsiveness.1,8–13 Amongst these stimuli, pH responsiveness is one of the most widely utilized, exploiting the difference in pH values between the intracellular and extracellular environment and between pH values in different cells or tissues. For example, the pH of human blood (pH 7.0–7.4) is higher than that of the extracellular environment of tumor cells (pH 6.5–7.0)14–16 and the pH of endosomes and lysosomes have lower values (early endosome pH 5.5–6.3 and late endosome pH < 5.5).17 There are a number of pH-sensitive functional groups, such as acetals,18 poly(ketals),19–21 and vinyl ethers;22 however, many of them do not react in the desired pH range or produce toxic byproducts.23 Amino groups have been introduced into prodrugs as ionizable groups to impart pH sensitivity.24,25 One method described by Wang and co-workers, utilized dual pH-sensitive polymer–doxorubicin conjugate nanoparticles (NPs) as an efficient anti-cancer drug delivery system.26,27 In these NPs, all of the allyl groups have been transformed to amino groups under acidic conditions, which allows the NPs to change from negative charge to positive charge and can therefore enter the cells more easily by endocytosis. However, there was low grafting efficiency due to the steric hindrance of the conjugate. They also modified the drug into a monomer first, and then used it to copolymerize with other monomers.3 They found that this method increased the efficiency and the drug-loading rate was well controlled during the synthesis. Gao and co-workers have prepared tunable, ultra-pH responsive fluorescent nanoparticles, incorporating tertiary amines into the hydrophobic block of the polymers as ionizable groups to tune the morphology of the NPs. Tertiary amines with hydrophobic constituents are introduced as the ionizable hydrophobic block. For example, with different tertiary amino segments, their pH transition points are 5.2, 6.4, 6.9, and 7.2 for 2-(dibutylamino) ethyl methacrylate (DBA), 2-(diisopropyl amino)ethyl methacrylate (DPA), 2-(hexamethyleneimino) ethyl methacrylate and 2-(pentamethyleneimino) ethyl methacrylate (C6A), respectively.23,28Polymers have been extensively explored as drug conjugates as they can be biocompatible and have multifunctional uses.29,30 A drug conjugate must be easily eliminated from the body. To this end, different types of degradable mechanisms such as enzymatic degradation,31–34 photodegradation,35,36 thermal degradation,37,38 pH degradation21,39 and redox degradation have been intensely investigated.40–42 In terms of redox degradation, several functional groups have been utilized including silyl ethers,7 acetals and disulfide bonds.43,44 The degradation of disulfide bonds under reductive conditions has been widely used in anti-cancer prodrugs.45 What's more, the pH-mediated disassembly of the vesicles may improve the accessibility of the polymer reducible linkers, ultimately facilitating the reduction of these moieties. With this approach in the intracellular environment, the disulfide bonds can be broken to release the anticancer drug with the help of the tumor's intracellular reductive reducing substances like glutathione (GSH).3
To date, the only example of a paclitaxel-containing polymer prodrug to reach phase III clinical trials has been Opaxio (paclitaxel polyglumex). However, the marketing authorization application for Opaxio was withdrawn from the European Medicines Agency in 2009.52 Herein, we report a PTX-loaded hydrophilic–hydrophobic diblock copolymer incorporating reduction-sensitive linkages (disulphide bonds) synthesized via RAFT polymerization. It can self-assemble into vesicles incorporating the hydrophobic drug PTX into the vesicle walls. In an acidic environment, the balance of hydrophobicity and hydrophilicity is disturbed, leading to the destruction of the aggregated structures. Drug containing conjugates are released following treatment with reducing agents (Scheme 1). Furthermore, in vitro cell experiments proved this to be a potential drug delivery system for cancer treatment.
 | ||
| Scheme 1 Synthetic routes employed for the preparation of POEGMA8-b-P(DPAEMA73-co-PTXMA0.12-co-MTC0.11) diblock copolymers (a); schematic illustration of the fabrication of degradable pH/redox dual responsive drug conjugates via RAFT polymerization and their dual stimuli responsiveness (b). | ||
Experimental section
Materials
Oligo(ethylene glycol) methyl ether methacrylate (OEGMA) (Mn = 475 g mol−1, J&K), 2-(diisopropylamino) ethyl methacrylate (DPAEMA) (97%, Aldrich), paclitaxel (98%, Beijing OuHe Technology Co., Ltd), [(2-carboxyethyl)thiomethyl]acrylic acid (99%, J&K), oxalyl chloride (98%, J&K), N,N′-dicyclo-hexylcarbodimide (DCC, 99%, J&K), and 4-dimethylaminopyridine (DMAP, >99%, J&K) were used as received. 4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl] pentanoic acid (DTTCP) was prepared according to the previously reported literature.46 The solvents including dichloromethane (DCM), tetrahydrofuran (THF), N,N-dimethylformamide (DMF), chloroform, acetone, toluene, ethyl acetate, hexane, ethanol and methanol were purchased from Sinopharm Chemical Reagent Co., Ltd and purified before use. NIH3T3 cell lines were purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd.Analysis techniques
1H NMR spectra were recorded on a Bruker AVANCE III system (400 MHz). For 1H NMR spectra, chemical shifts (δ) are quoted in parts per million (ppm) downfield of tetramethylsilane, using the residual protonated solvent as the internal standard (CDCl3 at 7.27 ppm). The size of the self-assemblies was measured by dynamic light scattering (DLS) performed on a Malvern Zetasizer Nano series (Nano-ZS) instrument. The samples were illuminated with a 633 nm He–Ne laser, and the scattering light at 90° angle was recorded using an avalanche photodiode detector. Gel permeation chromatography (GPC) was performed on a system comprising a Shimadzu LC-20AD pump and a Shimadzu RID-20A refractive index detector and an SPD-20A ultraviolet detector. The eluent was N,N-dimethylacetamide (DMAC) containing LiCl (0.05 mol L−1) at 50 °C (flow rate: 1 mL min−1). Number average (Mn) and weight average (Mw) molecular weights were evaluated using Shimadzu software with low dispersity poly(methyl methacrylate) standards ranging from 3000 to 106 g mol−1. Transmission electron microscopy (TEM) was performed with a Hitachi HT7700 instrument at a voltage of 120 kV. The specimen was prepared by drop-casting the sample solution onto a carbon-coated copper grid and then staining with 1 wt% phosphotungstic acid for 1 min and air-drying before observation.Synthesis of monomers
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n-hexane, 2:1) to give PTXMA as a white powder (0.34 g, 36%).
:n-hexane, 1:3 and 1:20). The product was dissolved in DCM and precipitated in n-hexane (1:9). Finally, slow evaporation yielded a white crystalline solid (150 mg, 15%).47
:n-hexane, 2:1) to give PTXMA as a white powder (0.34 g, 36%).
:n-hexane, 1:3 and 1:20). The product was dissolved in DCM and precipitated in n-hexane (1:9). Finally, slow evaporation yielded a white crystalline solid (150 mg, 15%).47
Synthesis of polymers
| Group | Polymer composition | [OEGMA]/[RAFT] | [DPAMA]/[CTA] | Feed ratioa | Time [h] | Conversionb [%] |
M
nb (NMR) |
M
nc (GPC) |
Đ |
|---|---|---|---|---|---|---|---|---|---|
|
a Ratio of [DPAEMA]:[POEGMA8 Macro-CTA]:[PTXMA]:[MTC].
b Calculated from the 1H NMR spectra (see the ESI for more details).
c Obtained from DMAc GPC.
d Data cannot be obtained because of the high viscosity of the solution.
|
|||||||||
| 1 | POEGMA8 Macro-CTA | 9:1 |
8 | 87 | 4203 | 5700 | 1.28 | ||
| 2 | POEGMA8-b-PDPAEMA90 | 100:1 |
24 | 90 | 23373 |
10700 |
1.09 | ||
| POEGMA8-b-PDPAEMA178 | 200:1 |
24 | 89 | 42117 |
13900 |
1.13 | |||
| POEGMA8-b-PDPAEMA258 | 300:1 |
24 | 86 | 59157 |
—d | — | |||
| POEGMA8-b-PDPAEMA340 | 400:1 |
24 | 85 | 76623 |
— | — | |||
| 3 | POEGMA8-b-P(DPAEMA73-co-PTXMA0.12-co-MTC0.11) | 100:1.0:1.0:0.5 |
24 | 73 | 19897 |
13600 |
1.40 | ||
| POEGMA8-b-P(DPAEMA145-co-PTXMA0.21-co-MTC0.32) | 200:1.0:2.0:1.0 |
24 | 73 | 31178 |
14900 |
1.41 | |||
| POEGMA8-b-P(DPAEMA165-co-PTXMA0.63-co-MTC0.57) | 300:1.0:3.0:1.5 |
24 | 55 | 40104 |
20200 |
1.48 | |||
| POEGMA8-b-P(DPAEMA187-co-PTXMA0.75-co-MTC0.75) | 400:1.0:4.0:2.0 |
24 | 47 | 44956 |
26900 |
1.46 | |||
Results and discussion
Synthesis of monomers
The drug containing a methacrylate monomer (PTXMA) was synthesized in a one-step coupling reaction between PTX and methacrylic acid. The relative integration of the resonances corresponding to the protons of the drug and the carbon–carbon double bond gives a reaction efficiency of around 98% (Fig. 1). One excellent strategy to incorporate disulfide linkages into the backbone of polymers is by the ring opening polymerization (ROP) of cyclic monomers based on allylic sulfides.48,49 A cyclic allylic disulfide monomer MTC, containing a 15 membered ring and two ester units, was prepared according to the procedure developed by Hawker and co-workers.47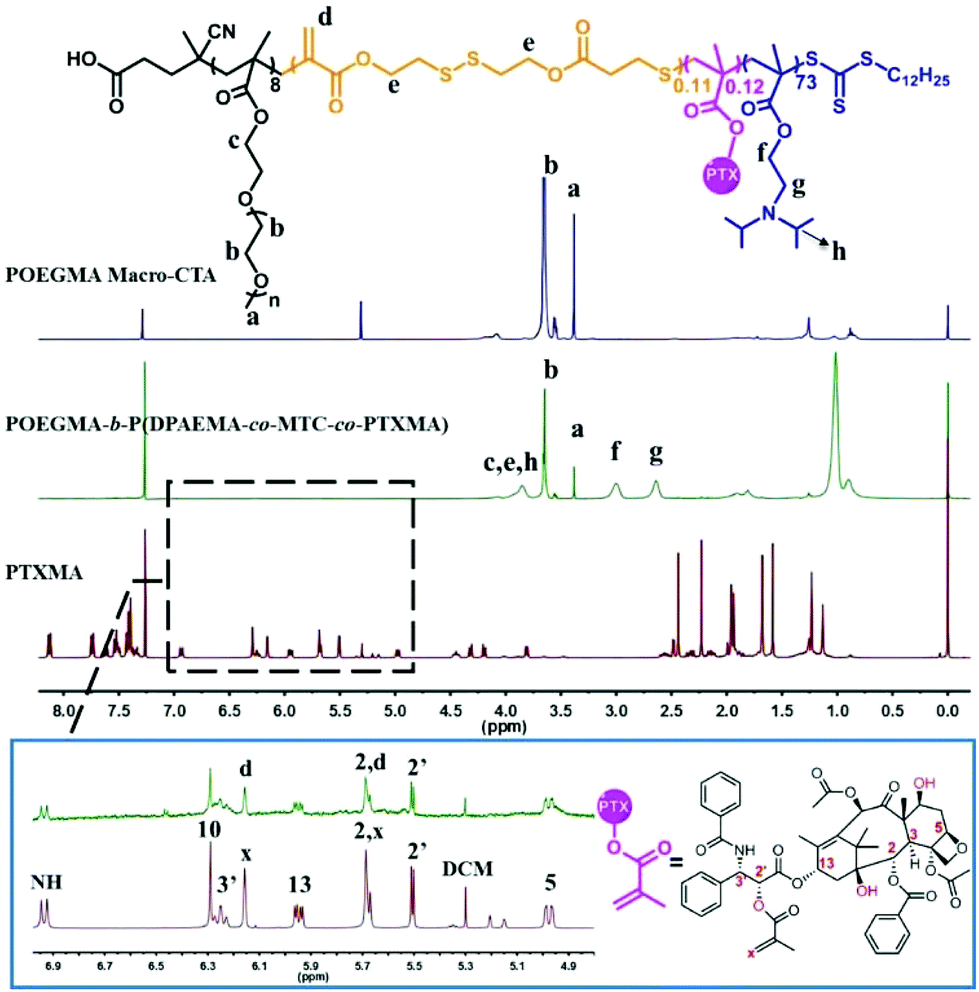 | ||
| Fig. 1 1H NMR spectra (400 MHz, CDCl3) of POEGMA8 Macro-CTA, POEGMA8-b-P(DPAEMA73-co-PTXMA0.12-co-MTC0.11) and the PTXMA monomer are shown respectively. The figure in the rectangle is the partially enlarged view of POEGMA8-b-P(DPAEMA73-co-PTXMA0.12-co-MTC0.11) and the PTXMA monomer. | ||
Construction of diblock copolymers
With the drug-loaded monomer and the cyclic disulfide monomer in hand, the diblock copolymer POEGMA8-b-P(DPAEMA73-co-PTXMA0.12-co-MTC0.11) (A) was obtained through a two-step polymerization. First, the hydrophilic polymer poly(oligo(ethylene glycol) methyl ether methacrylate (POEGMA) was prepared by RAFT polymerization using DTTCP as the chain transfer agent (CTA) and AIBN as the initiator. The RAFT polymerization proceeded in a controlled manner to obtain POEGMA with high conversion (87%) and narrow polydispersity (Đ = 1.28). 1H NMR spectroscopy confirmed the designed degree of polymerization to be 8 repeat units (Fig. 1). The polymer was purified by precipitation in diethyl ether and analyzed by GPC. The number average molecular weight (Mn) determined by GPC is in close agreement with the Mn calculated by 1H NMR spectroscopy (Table 1, Group 1).RAFT polymerization allows the chain extension of homopolymers to yield block copolymers. 2-(Diisopropylamino) ethyl methacrylate (DPAEMA) is a readily polymerizable acid sensitive monomer which can incorporate amino groups into a polymeric system. Its corresponding polymer PDPAEMA has a hydrophilic/hydrophobic transition pH point around 6.2, making it an attractive polymer for drug delivery applications.50 Thus it was selected as the monomer to impart pH sensitivity to our block copolymers. A series of POEGMA-b-PDPAEMA with different molecular weights were prepared as control block copolymers. The polymerization proceeded with good control of molecular weight, high conversion and narrow polydispersity (Table 1, Group 2).
For the diblock copolymer A, the hydrophobic block was prepared by the copolymerization of the drug-loaded monomer PTXMA, pH sensitive monomer DPAEMA and redox sensitive cyclic monomer MTC using POEGMA8 macro-CTA. The Mn and monomer conversion were calculated using 1H NMR spectroscopy. It can be seen from Table 1 (Group 3) that the monomer conversion decreased with the increase of the feed ratio. Meanwhile, on comparing the control group with the same feed ratio of DPAEMA, the monomer conversion was found to be relatively low. This is attributed to the introduction of the low activity monomer MTC. To achieve high monomer conversion and the desired length of the hydrophobic segments, the ratio [DPAEMA]:[POEGMA8 Macro-CTA]:[PTXMA]:[MTC] was fixed at 100:1.0:1.0:0.5. The 1H NMR spectrum of the resulting diblock copolymer A is shown in Fig. 1 with the relevant signals labeled. In comparison with the 1H NMR spectrum of POEGMA8 Macro-CTA, the appearance of signals at 3.33 ppm (a) and 3.72 ppm (b) are attributed to the protons from the OEGMA repeat unit, indicating the successful incorporation of the POEGMA block through the RAFT polymerization process. From the partially enlarged 1H NMR spectrum of the diblock copolymer, the characteristic peaks of PTX are observed. With a predetermined DP of POEGMA8, using the integrals of the characteristic peaks, the exact DP of the MTC monomer and the PTXMA monomer was calculated. The average DP of DPAEMA, MTC and PTXMA in each polymer chain was determined to be 73, 0.11 and 0.12, respectively. According to the calculated results, some polymer chains do not have PMTC or PPTXMA segments; others may have one or even two. Previously published results show that only a small quantity of the MTC cyclic monomer should be added, as high quantities affect the controllability of the molecular weight, distribution and conversion.51 In addition, excessive introduction may lead to an increase in the cytotoxicity of the materials.
Polymeric self-assemblies with dual stimuli responsiveness
According to our design, the diblock copolymer POEGMA8-b-P(DPAEMA73-co-PTXMA0.12-co-MTC0.11) is amphiphilic and can self-assemble into aggregates in aqueous solution. For the self-assembly, the diblock drug conjugate was first dissolved in THF (2 mL), and to the solution, distilled water (2 mL) was added dropwise over 30 min. The solution was dialyzed in distilled water for 24 h (water was changed every 6 h) to remove the organic solvent, and finally an opaque colloidal solution was formed. The formation of the aggregated particles was indicated by the strong Tyndall effect. To visualize the morphology and size, the self-assembled samples were deposited on a copper grid (stained with 1% phosphotungstic acid) and analyzed using TEM. As shown in Fig. 2A, spherical vesicles with diameters around 100 nm were observed. What's more, due to the drying steps in the sample preparation process for TEM, the collapse of the vesicle wall can also be observed. It also proved from another side that the assembly is a hollow vesicle structure, while the solid core–shell structure does not have such a collapse phenomenon. The drug conjugate is designed as a diblock copolymer with 8 aqueous soluble repeat units and 73 hydrophobic repeat units. The hydrophobic domain is much longer than the aqueous soluble domain, which resulted in vesicular structures. From the DLS data, the volume average hydrodynamic diameter was 103 nm, which is consistent with the TEM result. As shown in Fig. 2B, spheres and vesicles with diameters less than 40 nm were observed after the redox responsiveness. The disulfide bonds protected in the hydrophobic domain have been cleaved which will decrease the length of the hydrophobic domain. Thus, the morphology of the NPs changed from vesicles to spheres.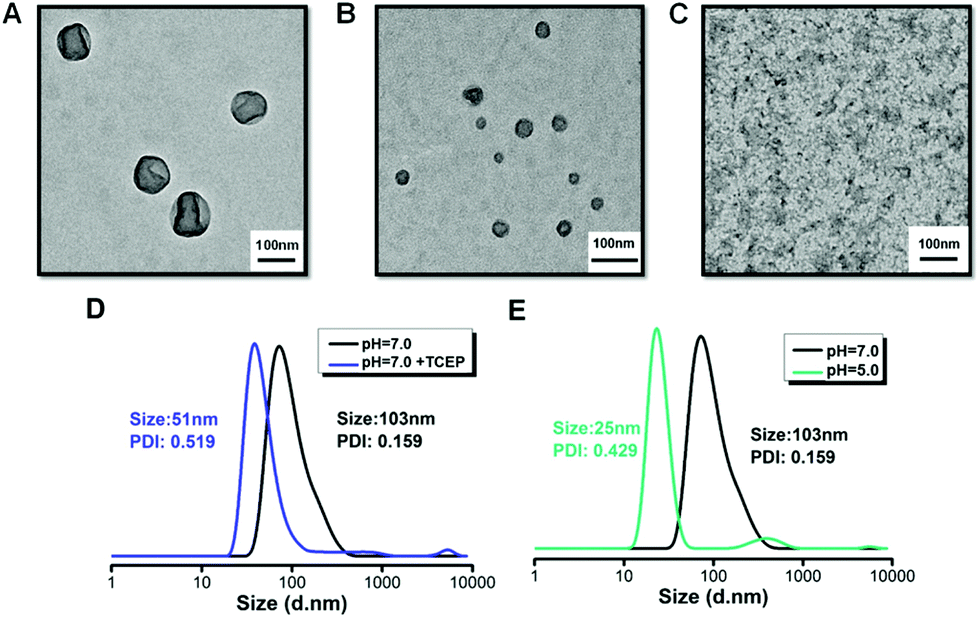 | ||
| Fig. 2 (A) TEM images of POEGMA8-b-P(DPAEMA73-co-PTXMA0.12-co-MTC0.11) self-assemblies in aqueous solution (pH = 7.0) after 24 h, (B) aqueous solution (pH = 7.0) with TCEP after 24 h and (C) aqueous solution (pH = 5.0) after 24 h. (D, E) The evolution volume particle size distributions characterized by DLS. | ||
The MTC cyclic monomer has a disulfide bond which is redox-sensitive, under the presence of a mild reducing agent such as glutathione (GSH), tris(2-carboxyethyl)phosphine hydrochloride (TCEP) or dithiothreitol (DTT). It can cleave the hydrophobic backbone into several short chains.3 In this study, after the addition of TCEP (TCEP/MTC molar ratio = 5.0) over 24 h, the results from TEM and DLS were recorded, as shown in Fig. 2B and D, respectively. From the TEM image, we observed a reduction in the diameter of the vesicles compared with the original state. The DLS result confirmed this trend, as the diameter of the aggregates decreased from 103 nm to 51 nm. Upon cleavage of the disulfide bonds, the hydrophobic block was partially cleaved, releasing short polymer segments from the vesicles and the diameter of the aggregates decreased. To verify the redox-responsive cleavage of the disulfide bond in this study, POEGMA8-b-PDPAEMA90, a diblock copolymer with a similar molecular weight, was employed under the same reaction conditions. Although self-assemblies with similar size and morphology were formed, no morphology change was observed under the redox conditions.
To elucidate whether the morphology is affected by different pH conditions, we used TEM to monitor the results. The results from a sample in a phosphate buffered solution of pH 5.0 are shown in Fig. 2C. The solubility of the macromolecular prodrug changes under acidic conditions as the DPAEMA segment becomes positively charged, changing from hydrophobic to hydrophilic, which can be clearly observed as the sample changes from turbid to clear (Fig. S14†). From the TEM image (Fig. 2C), we did not observe NPs with vesicle morphology as in Fig. 2A, which suggests that all the NPs have disassembled. The pH-mediated disassembly of the vesicles was confirmed by the DLS result (Fig. 2E), showing that the diameter of the NPs has decreased from 103 nm to 25 nm. Thus we have successfully proved the pH/redox dual stimuli responsiveness of POEGMA8-b-P(DPAEMA73-co-PTXMA0.12-co-MTC0.11), which is summarized in the illustration (Scheme 1). In tumour cells, the intracellular conditions are more acidic and reductive than those of non-cancerous cells. Thus, we postulate that under these conditions, the dual pH/redox responsive polymer–PTX conjugate NPs can enter the cell by endocytosis releasing the PTX drug conjugate after the pH-mediated disassembly and reductive cleavage of the disulfide bonds.
In vitro anticancer efficacy study
Certainly, the anticancer efficacy of our prodrug should be assured and studied. NIH3T3 cells, comprising a widely used cell line, were applied for the in vitro cytotoxicity study. All the cells were cultured in DMEM solution. Different concentrations of self-assemblies of POEGMA8-b-P(DPAEMA73-co-PTXMA0.12-co-MTC0.11) were cultured in the cell solution for 48 h. At the same time, the self-assemblies of POEGMA8-b-PDPAEMA90 and the drug containing monomer PTXMA served as the control groups. As shown in Fig. 3, the absorbance of both drug conjugates POEGMA8-b-P(DPAEMA73-co-PTXMA0.12-co-MTC0.11) (A) and the PTXMA monomer (B) decreased gradually with the increase in concentration, indicating that their cytotoxicity increased. On the other hand, we observed that POEGMA8-b-PDPAEMA90 showed negligible cytotoxicity (Fig. 3C). These results show that the cytotoxicity of the drug conjugate is stronger than that of the PTXMA monomer. The cytotoxicity of the drug conjugate exceeds that of the small molecule PTXMA which can be explained as follows: under the acidic and reductive intracellular conditions of the NIH3T3 cell lines, the NPs will disassemble, as they are now soluble in aqueous media, exposing the disulfide bonds previously protected by the hydrophobic domains. Thus, it is more facile to cleave these breakable sites releasing the drug conjugate from the macromolecular prodrug. The free PTX and PTXMA are both hydrophobic molecules. Under acidic conditions, the fragmented macromolecular prodrug has a short hydrophilic polymer which improves its solubility. As the macromolecular prodrug is more soluble in aqueous media than the free PTX and PTXMA, the efficacy is higher.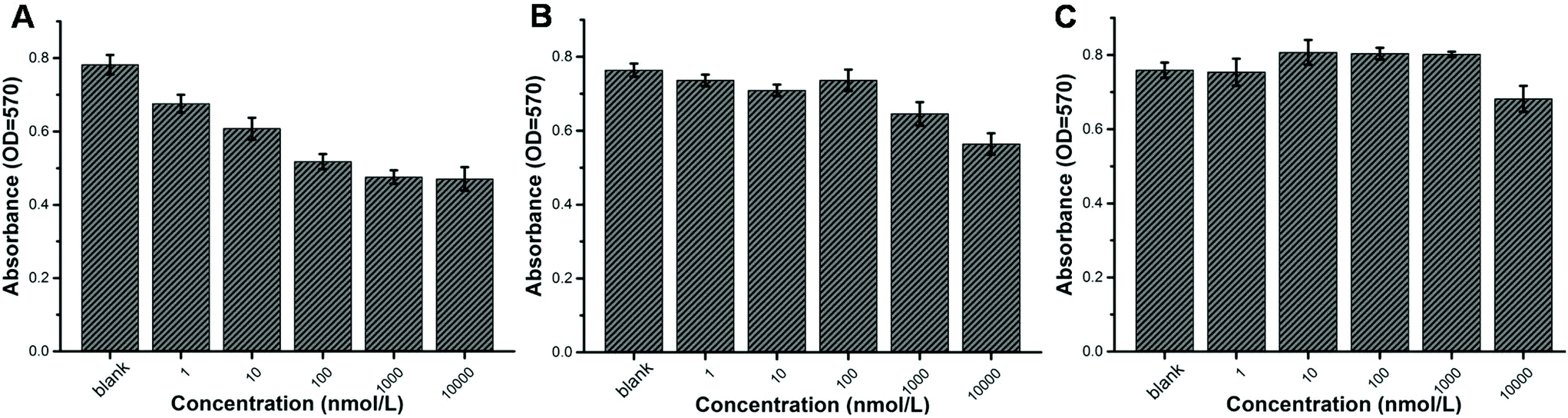 | ||
| Fig. 3 Cell viability characterized using an enzyme-linked immunoassay apparatus after adding samples. NIH3T3 cells were incubated with POEGMA8-b-P(DPAEMA73-co-PTXMA0.12-co-MTC0.11) conjugate NPs (A), PTXMA monomers (B) and POEGMA8-b-PDPAEMA90 (C) and subjected to an MTT assay. Samples of different concentrations from 0 to 10000 nmol L−1 were cultured in the cell solution for 48 h. | ||
Conclusions
In summary, this work has demonstrated an efficient covalent drug delivery system using degradable pH and redox dual responsive NPs. The prepared drug conjugate can self-assemble into vesicles of about 100 nm which could be endocytosed into cells. On decreasing the pH value, the size of the vesicular drug container changes as the hydrophobicity–hydrophilicity balance is disturbed. The aggregates can be further disintegrated by treatment with reducing agents, triggering the release of the drug-conjugated moiety. As cancer cells have lower pH and a higher concentration of reductive substances in the intracellular environment, our proof-of-concept design by combining the tumor microenvironment and pH/redox dual responsiveness provides a novel and versatile approach for efficient anticancer treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21704001), the Fundamental Research Funds for the Central Universities (buctrc201724) and the Beijing Advanced Innovation Center for Soft Matter Science and Engineering.Notes and references
- L. Bildstein, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2011, 63, 3–23 CrossRef CAS PubMed.
- A. M. L. Seca and D. Pinto, Int. J. Mol. Sci., 2018, 19, 263 CrossRef PubMed.
- B. Sun, C. Luo, H. Yu, X. Zhang, Q. Chen, W. Yang, M. Wang, Q. Kan, H. Zhang, Y. Wang, Z. He and J. Sun, Nano Lett., 2018, 18, 3643–3650 CrossRef CAS PubMed.
- W. Lu, X. Wang, R. Cheng, C. Deng, F. Meng and Z. Zhong, Polym. Chem., 2015, 6, 6001–6010 RSC.
- S. Mura, F. Zouhiri, S. Lerondel, A. Maksimenko, J. Mougin, C. Gueutin, D. Brambilla, J. Caron, E. Sliwinski, A. Lepape, D. Desmaele and P. Couvreur, Bioconjugate Chem., 2013, 24, 1840–1849 CrossRef CAS PubMed.
- F. Dosio, L. H. Reddy, A. Ferrero, B. Stella, L. Cattel and P. Couvreur, Bioconjugate Chem., 2010, 21, 1349–1361 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- A. Wadouachi and J. Kovensky, Molecules, 2011, 16, 3933–3968 CrossRef CAS.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- Y. Wang, S. Li, P. Zhang, H. Bai, L. Feng, F. Lv, L. Liu and S. Wang, Adv. Mater., 2018, 30, 1705418 CrossRef PubMed.
- W. Yuan, Z. Zhao, J. Yuan, S. Gu, F. Zhang, X. Xie and J. Ren, Polym. Int., 2011, 60, 194–201 CrossRef CAS.
- G. G. Hedir, M. C. Arno, M. Langlais, J. T. Husband, R. K. O'Reilly and A. P. Dove, Angew. Chem., Int. Ed., 2017, 56, 9178–9182 CrossRef CAS PubMed.
- L. Hao, C. Yegin, I. C. Chen, J. K. Oh, S. Liu, E. Scholar, L. Zhang, M. Akbulut and B. Jiang, Ind. Eng. Chem. Res., 2018, 57, 9231–9239 CrossRef CAS.
- E. S. Lee, K. Na and Y. H. Bae, Nano Lett., 2005, 5, 325–329 CrossRef CAS PubMed.
- Y. Tian, F. Su, W. Weber, V. Nandakumar, B. R. Shumway, Y. Jin, X. Zhou, M. R. Holl, R. H. Johnson and D. R. Meldrum, Biomaterials, 2010, 31, 7411–7422 CrossRef CAS PubMed.
- E. S. Lee, Z. Gao, D. Kim, K. Park, I. C. Kwon and Y. H. Bae, J. Controlled Release, 2008, 129, 228–236 CrossRef CAS PubMed.
- M. A. Quadir, S. W. Morton, Z. J. Deng, K. E. Shopsowitz, R. P. Murphy, T. H. Epps 3rd and P. T. Hammond, Mol. Pharmaceutics, 2014, 11, 2420–2430 CrossRef CAS PubMed.
- E. R. Gillies and J. M. Frechet, Bioconjugate Chem., 2005, 16, 361–368 CrossRef CAS PubMed.
- M. J. Heffernan and N. Murthy, Bioconjugate Chem., 2005, 16, 1340–1342 CrossRef CAS PubMed.
- J. Sankaranarayanan, E. A. Mahmoud, G. Kim, J. M. Morachis and A. Almutairi, ACS Nano, 2010, 4, 5930–5936 CrossRef CAS PubMed.
- A. M. Jazani and J. K. Oh, Macromolecules, 2017, 50, 9427–9436 CrossRef CAS.
- P. S. Junhwa Shin and D. H. Thompson, J. Controlled Release, 2003, 91, 187–200 CrossRef.
- K. Zhou, H. Liu, S. Zhang, X. Huang, Y. Wang, G. Huang, B. D. Sumer and J. Gao, J. Am. Chem. Soc., 2012, 134, 7803–7811 CrossRef CAS PubMed.
- V. Bütün, S. P. Armes and N. C. Billingham, Polymer, 2001, 42, 5993–6008 CrossRef.
- M. S. Kim, S. J. Hwang, J. K. Han, E. K. Choi, H. J. Park, J. S. Kim and D. S. Lee, Macromol. Rapid Commun., 2006, 27, 447–451 CrossRef CAS.
- J. Z. Du, X. J. Du, C. Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560–17563 CrossRef CAS PubMed.
- J. Z. Du, T. M. Sun, W. J. Song, J. Wu and J. Wang, Angew. Chem., 2010, 122, 3703–3708 CrossRef.
- K. Zhou, Y. Wang, X. Huang, K. Luby-Phelps, B. D. Sumer and J. Gao, Angew. Chem., Int. Ed., 2011, 50, 6109–6114 CrossRef CAS PubMed.
- W. C. Chan, Science, 1998, 281, 2016–2018 CrossRef CAS PubMed.
- A. Bernardos, E. Aznar, M. D. Marcos, R. Martinez-Manez, F. Sancenon, J. Soto, J. M. Barat and P. Amoros, Angew. Chem., Int. Ed., 2009, 48, 5884–5887 CrossRef CAS PubMed.
- M. A. Gauthier and H.-A. Klok, Polym. Chem., 2010, 1, 1352–1373 RSC.
- C. Wang, Q. Chen, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 8612–8615 CrossRef CAS PubMed.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O'Reilly, ACS Cent. Sci., 2018, 4, 718–723 CrossRef CAS PubMed.
- I. Insua, M. Petit, L. D. Blackman, R. Keogh, A. Pitto-Barry, R. K. O'Reilly, A. F. A. Peacock, A. M. Krachler and F. Fernandez-Trillo, ChemNanoMat, 2018, 4, 807–814 CrossRef CAS PubMed.
- S. Febvay, D. M. Marini, A. M. Belcher and D. E. Clapham, Nano Lett., 2010, 10, 2211–2219 CrossRef CAS PubMed.
- D. V. Volodkin, A. G. Skirtach and H. Möhwald, Angew. Chem., 2009, 121, 1839–1841 CrossRef.
- B. Jeong, Y. H. Bae and S. W. Kim, J. Controlled Release, 2000, 63, 155–163 CrossRef CAS PubMed.
- S. W. Choi, Y. Zhang and Y. Xia, Angew. Chem., Int. Ed., 2010, 49, 7904–7908 CrossRef CAS PubMed.
- L. Liu, C. Kong, M. Huo, C. Liu, L. Peng, T. Zhao, Y. Wei, F. Qian and J. Yuan, Chem. Commun., 2018, 54, 9190–9193 RSC.
- Y. L. Li, L. Zhu, Z. Liu, R. Cheng, F. Meng, J. H. Cui, S. J. Ji and Z. Zhong, Angew. Chem., Int. Ed., 2009, 48, 9914–9918 CrossRef CAS PubMed.
- G. Saito, J. A. Swanson and K.-D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS PubMed.
- J. Wu, J. Yuan, B. Ye, Y. Wu, Z. Xu, J. Chen and J. Chen, Front. Pharmacol., 2018, 9, 663 CrossRef PubMed.
- J. L. Cohen, A. Almutairi, J. A. Cohen, M. Bernstein, S. L. Brody, D. P. Schuster and J. M. Frechet, Bioconjugate Chem., 2008, 19, 876–881 CrossRef CAS PubMed.
- J. K. Oh, D. J. Siegwart, H. I. Lee, G. Sherwood, L. Peteanu, J. O. Hollinger, K. Kataoka and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 5939–5945 CrossRef CAS PubMed.
- N. J. Warren, J. Rosselgong, J. Madsen and S. P. Armes, Biomacromolecules, 2015, 16, 2514–2521 CrossRef CAS PubMed.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- J. M. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805–9812 CrossRef CAS PubMed.
- R. A. Evans, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1994, 27, 7935–7937 CrossRef CAS.
- R. A. Evans and E. Rizzardo, Macromolecules, 1996, 29, 6983–6989 CrossRef CAS.
- Y. Ma, Y. Tang, N. C. Billingham, S. P. Armes, A. L. Lewis, A. W. Lloyd and J. P. Salvage, Macromolecules, 2003, 36, 3475–3484 CrossRef CAS.
- L. P. Ratcliffe, C. Couchon, S. P. Armes and J. M. Paulusse, Biomacromolecules, 2016, 17, 2277–2283 CrossRef CAS PubMed.
- C. J. Langer, K. J. O'Byrne, M. A. Socinski, S. M. Mikhailov, K. Lesniewski-Kmak, M. Smakal, T. E. Ciuleanu, S. V. Orlov, M. Dediu, D. Heigener, A. J. Eisenfeld, L. Sandalic, F. B. Oldham, J. W. Singer and H. J. Ross, J. Thorac. Oncol., 2008, 3, 623–630 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01583j |
| This journal is © The Royal Society of Chemistry 2019 |