
Poly(trimethylene carbonate)-based polymers engineered for biodegradable functional biomaterials
K.
Fukushima
Department of Polymer Science and Engineering, Graduate School of Science and Engineering, Yamagata University, Yonezawa, Yamagata 992-8510, Japan. E-mail: fukushima@yz.yamagata-u.ac.jp
First published on 18th August 2015
Abstract
Aliphatic polycarbonates have drawn attention as biodegradable polymers that can be applied to a broad range of resorbable medical devices. In particular, poly(trimethylene carbonate) (PTMC), its copolymers, and its derivatives are currently studied due to their unique degradation characteristics that are different from those of aliphatic polyesters. Furthermore, their flexible and hydrophobic nature has driven the application of PTMC-based polymers to soft tissue regeneration and drug delivery. This review presents the diverse applications and functionalization strategies of PTMC-based materials in relation to recent advances in medical technologies and their subsequent needs in clinical settings.
 K. Fukushima | Kazuki Fukushima received his Bachelor's and Master's degrees in Polymer Science and Engineering in 2002 and 2004 and his Doctorate in Applied Science for Functionality in 2007 from Kyoto Institute of Technology, Japan under the supervision of Prof. Yoshiharu Kimura. He subsequently conducted postdoctoral research at IBM Almaden Research Center with Dr James L. Hedrick in 2007–2011. In 2011, he was appointed as an Assistant Professor of Yamagata University. He currently focuses on the synthesis and applications of functionalized biodegradable polymers. He has received the Award for Encouragement of Research in Polymer Science from the Society of Polymer Science, Japan in 2015. |
1. Introduction
Synthetic biodegradable polymers are widely used and studied for their biomedical applications, ranging from drug delivery to tissue engineering.1 Aliphatic polyesters such as polylactides (PLAs) and poly(ε-caprolactone) (PCL) have been used most widely for a long time due to their easy accessibility and decent mechanical properties.2 Recently, aliphatic polycarbonates have drawn more attention as another class of degradable and resorbable materials. They are biodegradable and some are derived from carbon dioxide and epoxides or anhydrides.3,4 Thus, like PLA, aliphatic polycarbonates are also recognized to be bio-based biodegradable polymers.Poly(trimethylene carbonate) (PTMC), its copolymers, and its derivatives are extensively studied for their biomedical applications. PTMC can be prepared by a ring-opening polymerization (ROP) of trimethylene carbonate (TMC) using both conventional organometallic catalysts and emerging organocatalysts.5,6 Currently, TMC is also regarded as a bio-based monomer, since the starting material 1,3-propanediol can be derived from carbohydrates and some of the carbonylation reagents used to close the ring are potentially produced from carbon dioxide.7–9 PTMC is a hydrophobic non-crystalline polymer with a glass transition temperature (Tg) of around −20 °C.10,11 Therefore, PTMC is usually used as a soft material in a scaffold application for soft tissue regeneration and as a hydrophobic segment of amphiphilic block copolymers for drug delivery. The unique degradation behavior of PTMC such as resistance to non-enzymatic hydrolysis, generation of non-acidic degradation products, and enzymatic degradation with a surface erosion mechanism12 opens a new window for applications in biomedical devices that could never be achieved with aliphatic polyesters. For instance, slow degradation prolongs its lifetime, non-acidic degradation products reduce the risk of adverse reactions in in vivo applications, and the mechanical properties of the polymer tend to be maintained with surface erosion.12
However, progress in current medical technologies has led to the requirement for more complex and higher level functional materials. Thus, the integration of multiple functions has been explored for synthetic biodegradable polymers by various approaches, including polymer blends, composites, copolymerization, and functional pendant groups, in order to respond to a broad range of applications. Since most synthetic biodegradable polymers are prepared by ROP of the corresponding cyclic monomers, several efforts have been made to develop heterocyclic monomers with functional pendant groups such as analogues of lactide (LA),13,14 glycolide (GA),15–17 and ε-caprolactone (CL).18,19 TMC analogues can also be obtained from substituted 1,3-propanediols so as to yield PTMC derivatives with functional pendant groups via the subsequent ROP.20 Generally, the TMC analogues are relatively stable under ambient conditions as compared to the substituted lactide and glycolide, which allows for easy handling and storage. Furthermore, recent advances in controlled polymerization techniques enable the construction of complex macromolecular architectures.21 Hence, the number of studies on PTMC-based polymers as biomaterials increases every year. This review presents recent studies on biomedical applications of PTMC, its copolymers, and its derivatives, focusing particularly on the progress in the last few years.
2. Aims of modifying TMC-based polymers
2.1. Alteration of mechanical properties
As mentioned above, PTMC is flexible and soft. Sometimes, the mechanical properties of PTMC do not meet a required range for the target materials, particularly in bulk applications. The simplest way of tuning the mechanical properties of PTMC is to change its molecular weight. A significant difference in PTMC bulk mechanical properties can be observed between PTMC materials with a molecular weight of a few kilo-Daltons (kDa) and those with a molecular weight of several hundreds of kDa.10,11 This difference is attributed to the molecular weight dependence of the glass transition. Nevertheless, there are few examples of the use of unmodified PTMC in biomaterial applications.Cross-linking is commonly employed to provide elasticity to the PTMC-based polymers in most cases. The mechanical properties are controllable by the cross-linking density that can be regulated by the concentration of the cross-linker, geometry of the reactive points for cross-linking in the prepolymer structure, and the molecular weight between the cross-linking points.22 An interesting study showed that PTMC cross-linking affects the differentiation of adipose stem cells (ASCs) into smooth muscle cells (SMCs).23 ASCs seeded on UV-cross-linked PTMC of low molecular weight (20 kDa) differentiated into SMCs more than those seeded on high molecular weight PTMC (250 kDa) cross-linked by gamma irradiation. Surface flexibility seems to influence cell attachment and differentiation (Young's moduli were 4.3 MPa and 6.6 MPa, respectively).24,25 Although the difference in the mechanical properties is not significant, the molecular mobility at the surface upon hydration may substantially differ from that in the dry state. Therefore, the mechanical properties should be assessed at 37 °C in an aqueous environment to determine whether the materials can be utilized under in vivo conditions.
Copolymerization has also been adopted to improve the mechanical strength. In this case, random copolymers or segmented multi-block copolymers are included. Since PTMC is prepared through ROP of TMC, heterocyclic monomers such as LA, GA, and lactones are often used as comonomers. Similarly, the formation of segmented polyurethanes results in a soft segment elastomer, which is another option to improve the mechanical properties of PTMC. The target range of the mechanical properties varies depending on the targeted tissues. The copolymerization and cross-linking should be used in combination in order to provide the ideal properties to the materials.26,27
2.2. Control of biodegradation
PTMC biodegradation only occurs from the surface and in the presence of enzymes, by hydrolysis into 1,3-propanediol and carbon dioxide.28 Therefore, the PTMC degradation process is slower than that of poly(L-lactide) (PLLA) and other aliphatic polyesters that are degraded through bulk erosion by both enzymatic and non-enzymatic hydrolysis. Additionally, autocatalysis by carboxyl end groups is involved in the degradation of the polyesters. The copolymerization of TMC and the other cyclic esters can regulate the biodegradation with the comonomer ratio. Regarding aliphatic polyesters, the copolymerization with TMC is beneficial in decreasing acidic degradation products and in tuning the degradation period.High molecular weight PTMC is known to degrade faster than low molecular weight PTMC in vivo12 mainly via macrophage-mediated enzymatic and oxidative degradation.29,30 Vyner et al. recently proved that this difference is associated with the conformation of adsorbed enzymes on the surface rather than with macrophage behavior.31 The enzymes appear to recognize the surface stiffness that is relevant to the polymer chain mobility upon hydration. The surface hydration is known to control protein adsorption and conformation changes.32 Therefore, more flexible low molecular weight PTMC, possessing more hydroxyl termini, is liable to absorb water and, thereby, is less subjected to enzymatic degradation. This surface hydration dependence of the enzymes could extend to PTMC derivatives with hydrophilic side chains, resulting in slow biodegradation. In fact, the degradation needs to be estimated from the composition of carbonate bonds in the main chain and the extent of surface hydration and wettability.
2.3. Extension of chemical/biological properties
Typically, PTMC is recognized as a cell friendly material. High levels of cell viability and cell adhesion are observed on the PTMC surface. Thus, PTMC is considered to be biocompatible. Therefore, PTMC-coating is used to improve the biocompatibility of some inorganic biomaterials, including biometals and bioactive ceramics that have a good mechanical strength and high bioactivity, but a brittle surface in some cases.33 Compared to coatings of polyesters, the PTMC-coating presents a better efficacy in terms of protecting the inorganic biomaterials against corrosion that usually occurs faster than desired under biological conditions. This property is attributed to the absence of acidic degradation products from PTMC.28As aforementioned, the biological response is affected by surface properties, including stiffness, topology, hydration, and charge as well as the primary chemical structure. Indeed, some modifications may unexpectedly weaken the other properties. Therefore, multiple modifications are required for practical application. PTMC is hydrophobic and thereby adsorbs proteins and cells non-specifically.34 Conjugation of hydrophilic polymers such as poly(ethylene glycol) (PEG) increases hydration that suppresses the adhesion of proteins and cells at the interface.35,36
Sometimes, PTMC-based polymers with amphiphilicity form micelles in water, which are mostly applied in drug delivery. Functionalization of end groups can significantly alter the biochemical properties and solubility of the PTMC-based polymers when the molecular weight is low because the effect of the functional groups depends on the density of the functional groups. Alternatively, the introduction of functional pendant groups into the PTMC backbone expands not only the solubility, but also the extent of interaction with (bio)molecules and cells at the interface more than the end functionalization. Although synthesis of TMC analogues with the functional pendant group is usually necessary, a broader range of applications can be potentially targeted. Regardless of the approaches, the composition and density of the functional groups relative to the PTMC framework roughly dictate the physicochemical properties of the PTMC-based polymers.
3. Strategies for the functionalization of PTMC-based polymers and for polymer architecture design
Functionalization approaches of the PTMC-based polymers are classified into two types: peripheral decoration and vertical decoration. The former includes end functionalization, cross-linking, and copolymerization that are mostly carried out for PTMC. The latter involves preparation of the PTMC derivatives with functional side chains and the post-modifications of the side chains, which will create a new class of biodegradable polycarbonate materials with a broad range of physicochemical properties.3.1. Cross-linking
Cross-linking of PTMC is performed through UV irradiation and gamma irradiation (Fig. 1A and B). UV irradiation needs a double bond moiety in the polymer structure, while strong gamma radiation generates free radicals for cross-linking by itself. Cross-linking density, which dictates the mechanical strength, is regulated only by irradiation time and beam strength for the gamma irradiation. UV cross-linking is more controllable, in which the architecture and molecular weight of prepolymers regulate the cross-linking density. Both irradiations are useful for simultaneous sterilization of the polymers, which is one of the advantages of this type of cross-linking.37 | ||
| Fig. 1 Typical examples of PTMC-based polymers applied to various biomedical applications in this review. The TMC repeating units are shown in red. | ||
The ROP of TMC initiated by an alcohol initiator brings a hydroxyl end group. UV reactive groups are then easily introduced by esterification with (meth)acryloyl chloride (or anhydride) as the most common approach.23,38,39 Cinnamoyl chloride is also employed as an alternative UV reactive moiety.40–42 The radical addition reaction of the double bonds can be performed by UV irradiation, and photosensitizers such as benzophenone are sometimes added to prompt the reaction. Multi-arm alcohols serve as initiators to provide PTMC with multiple UV reactive terminals. The initiation terminal also plays a role of a cross-linking point when the number of hydroxyl groups in the initiator is more than three. There is another way of cross-linking where the reactive side chains are bridged with each other41 or through the multi-functional cross-linkers by UV irradiation and/or click reactions described later (see Fig. 4B).43–46 In combination with advancing 3D printing technology, the UV irradiation assisted cross-linking is applied for stereolithography where resin deposition is concomitantly conducted, producing tailor-made 3D scaffolds with a tunable mechanical strength.47–50
3.2. Copolymerization
Since the ROP of TMC is performed through transesterification, copolymerization with other cyclic esters yields a random sequence (Fig. 1C–F), particularly when the ROP is performed using tin catalysts upon heating.51,52 The comonomers often used include LA, GA, and CL. Emerging organocatalytic ROP sometimes induces gradient or blocky sequences, resulting from different monomer reactivities to the catalysts.21Amphiphilic block copolymers are mostly studied among PTMC-containing block copolymers for biomaterial applications (Fig. 1G–I). PEG has been the first choice as a hydrophilic block for a long time because PEG is non-ionic and compatible with the blood.53 Due to difficulties in synthesis and hazard control, the preparation of heterofunctional monohydroxy PEG by ROP of ethylene oxide, leading to end-functionalized PEG-PTMC block copolymers (Fig. 1G), is rarely performed at the laboratory level. Instead, various heterofunctional PEGs are commercially available, although some are expensive. Currently, poly(L-glutamic acid) (PGlu), which has a carboxyl residue at the side chain, is also used as the hydrophilic segment. Recent progress in controlled polymerization of amino acid N-carboxyanhydride (NCA)54 may facilitate the use of PGlu and allows for easy access to the end functionalization of PGlu-PTMC block copolymers (Fig. 1H).
For graft copolymers with degradable polymer branches, the “grafting-from” approach is the most popular, using polymers with hydroxyl side chains as an initiator of ROP. Poly(vinyl alcohol) (PVA) and poly(2-hydroxyethyl methacrylate) (PHEMA) are famous as hydroxy side-functionalized polymers for applications in biocompatible medical devices. Cellulose has also drawn attention as a macroinitiator55 for the “grafting-from” ROP of TMC (Fig. 1J)56 since homogeneous ROP using cellulose in ionic liquids was established.57,58
3.3. End functionalization
Attaching functionality to the chain ends is a straightforward approach to modifying or adding chemical functions to PTMC. The typical end functionalities attached contain UV reactive terminal groups for cross-linking, multi-arm initiators for divergent architecture (Fig. 1B, F and L), multiple tertiary amine terminals for dissolution in water and binding DNA (Fig. 1K), and isocyanate terminals (Fig. 1L) for covalent conjugation with proteins (–NH2). There are two ways for the end functionalization, the use of alcohols with a functional or reactive moiety and esterifying the terminal hydroxyl groups with acid halides presenting a functional or reactive moiety. In addition, further functionalization can sometimes be conducted via the reactive groups introduced at the chain end. Since the carbonate bonds are relatively more stable than ester bonds, several end modification approaches are available for PTMC.3.4. Incorporation of functional side chains
Side chain functionalization of PTMC is usually performed through ROP of TMC analogues with a functional residue. The TMC analogues are derived from substituted 1,3-propanediols through ring-closing reactions (Fig. 2-(I)) with carbonylation agents59 such as phosgene derivatives,60–62 bis-carbonates,9,63 and N,N′-carbonyldiimidazole.64 The synthesis and ROPs (Fig. 2-(II)) of the TMC analogues have previously been reviewed in detail elsewhere.20,65,66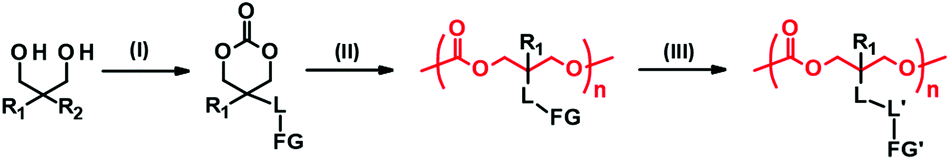 | ||
| Fig. 2 A typical synthetic pathway to PTMC derivatives bearing a functional side chain. (I) Cyclization, (II) ring-opening polymerization, and (III) post-modification. R1, R2: substituents. L, L′: linkers. FG, FG′: functional groups. | ||
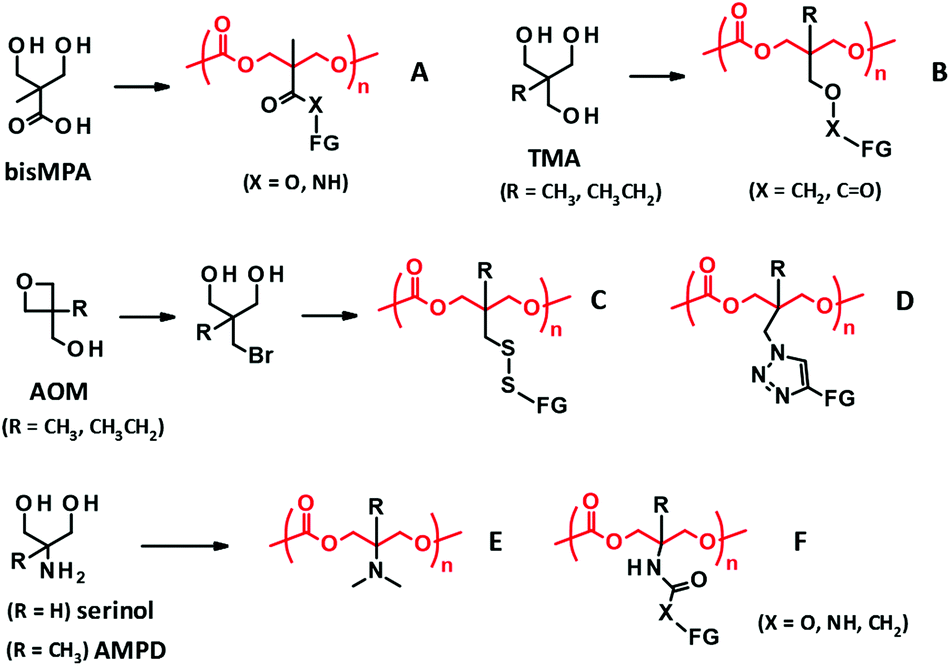 | ||
| Fig. 3 PTMC derivatives bearing a functional side chain derived from bisMPA (A), TMA (B), AOM (C, D), and serinol and AMPD (E, F). FG denotes functional group. | ||
2,2-Bis(hydroxymethyl)propionate (bisMPA) has been used as a building block of biocompatible dendrimers,67 which could support the potential safety of bisMPA as a constituent of biodegradable biomaterials. From the synthetic aspect, the carboxyl group of bisMPA is favorable for introducing functional groups.61 As with (meth)acrylate analogues, a wide variety of functional groups are reasonably installed through ester and amide linkers using commercially available alcohols and amines. For these reasons, the bisMPA-derived PTMC derivatives (Fig. 3A) have been predominantly studied as typical functionalized PTMC derivatives for applications in biomaterials. Recent trends appear to shift to post-modification strategies to attach drugs and generate the desired functions indirectly, but with high efficiency (Fig. 2-(III)).
Trimethylol alkanes (TMA) provide PTMC derivatives, tagging functional groups through an ether linker46 or a reverse ester (oxycarbonyl) linker42,68 (Fig. 3B). As compared to the bisMPA-derived TMC analogues, synthesis of the TMA-derived TMC analogues is somewhat tedious, because of a troublesome ether formation and equal reactivity of three methylol (hydroxymethyl) groups. Furthermore, a limited number of functional groups can be introduced. Hence, not many monomers and PTMC derivatives have been reported. The ether linker presents a low risk of being involved in transesterification during the ROP to result in scrambling chains and cross-linking, which could be an advantage of the TMA-derived TMC analogues. However, these side reactions would not occur by steric hindrance of the α-methyl group for the bisMPA-derived TMC analogues. Indeed, there are no reports on the side reactions, at least when using organocatalysts.5
Recently, new synthetic approaches to TMC analogues have been developed using 3-alkyl-3-oxetanemethanol (AOM), which is converted to 2-bromomethyl-2-alkyl-1,3-propanediol. The bromo group is then altered to thiol and azide groups to yield PTMC derivatives possessing disulfide69 and triazole linkers70 with functional groups (Fig. 3C and D).
Serinol (2-amino-1,3-propanediol) and 2-amino-2-methyl-1,3-propanediol (AMPD) have also proved useful as other starting materials towards functionalized PTMC derivatives. The primary amino group can be transformed to a tertiary amine and polymerized the cyclized monomer by an enzymatic ROP. The resultant polymer (Fig. 3E) is water soluble, with a molecular weight of around 5000 Da and minimal cytotoxicity.71 Furthermore, the amino groups of serinol and AMPD are effective in selective reactions with esters and bis-carbonates to form amides and carbamates without the protection of the hydroxyl groups.72 The synthesis of the corresponding TMC analogues is relatively straightforward compared to that of others, and thus exploring the applications of the PTMC derivatives (Fig. 3F) is very much expected. In addition to the reagents cited above, there are further commercially available substituted 1,3-diols. New platforms for the development of PTMC derivatives with functional side chains will emerge if the facile synthetic route is established and the versatility of the substituent is discovered.
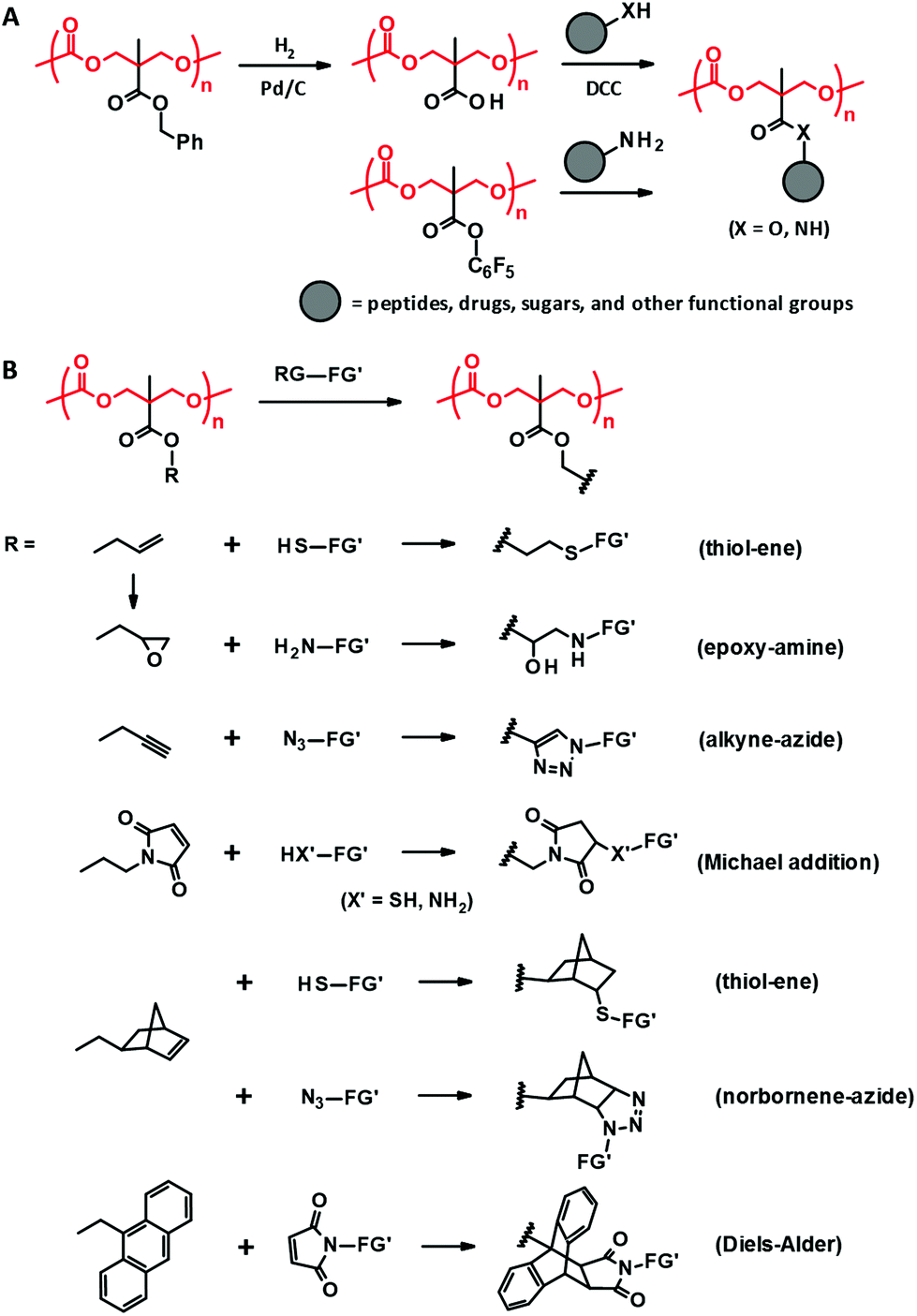 | ||
| Fig. 4 Post-functionalization of PTMC derivatives derived from bisMPA through DCC coupling and amidation (A) and “click” reactions (B). RG: reactive group, FG′: functional group. | ||
The aforementioned coupling reactions based on esterification and amidation are less efficient in terms of completing the reaction than “click” reactions, including thiol–ene,45,79 alkyne–azide cycloaddition,43,79,80 Michael addition,81,82 Diels–Alder,79,83 and epoxide–amine reactions (Fig. 4B).44 Indeed, these “click” post-modifications are completed in one step with high conversion for the bisMPA-derived PTMC derivatives bearing reactive side groups, e.g., allyl,44,45 propargyl,43,80 norbornene,79 maleimide,81,82 anthracene,83 and epoxy.44 Since the polycarbonate backbone is subjected to decomposition under acidic or basic conditions, the post-modifications should be performed under ambient and neutral conditions with no harsh catalysts. Thus, the “click” reactions have actually been preferred to load peptides,81 ferrocene,82 and spiropyran.80 Furthermore, due to the reaction characteristics, these reactions are also exploited for in situ cross-linking to form hydrogels and 3D scaffolds using multi-functional reagents as cross-linkers such as dithiols and diamines.43
TMA-derived PTMC derivatives can extend the range of functionality introduced at the side chain by post-modification of the acryloyl pendant through Michael reaction with thiols and amines.84 The acryloyl group is added to a pendant hydroxyl group with acryloyl chloride. The acryloyl side chain is also applicable to photo cross-linking.68 Combination with an allyl functionalized bisMPA-derived PTMC derivative enables multiple and selective post-functionalization by both thiol–ene and Michael reactions (Fig. 5A).85
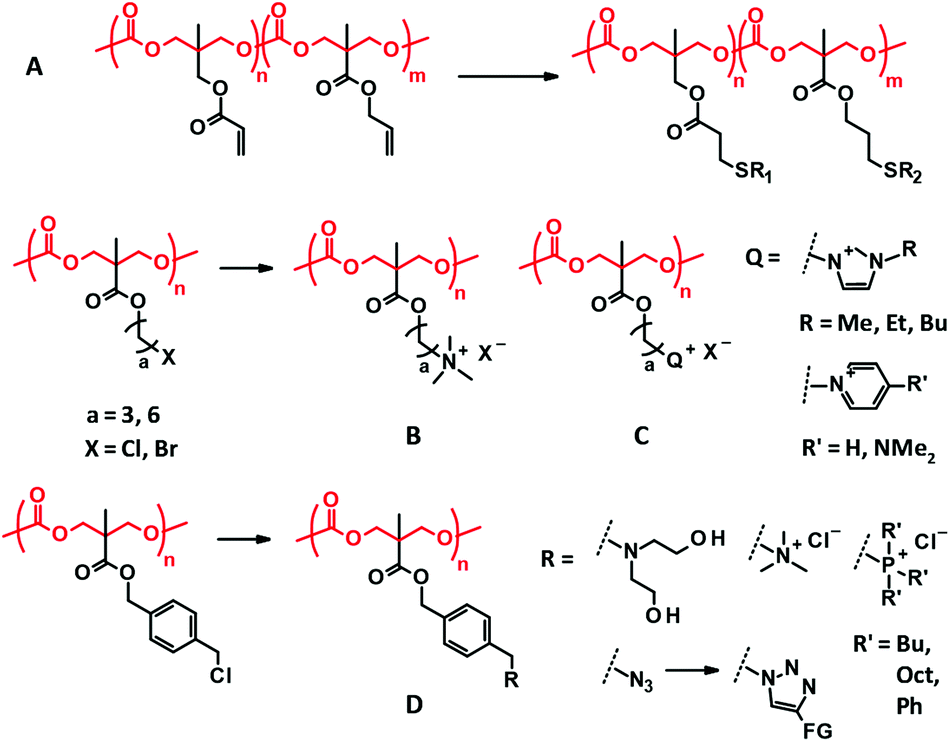 | ||
| Fig. 5 Other selected examples of post-modifications of the PMTC derivatives. (A) Multi-functionalization of acrylate and allyl pendants of TMA and bisMPA-derived PTMC derivatives. (B, C) Quaternization of bisMPA-derived PTMC derivatives for antimicrobial applications. (D) Facile transformation of the benzyl chloride side chain. | ||
Quaternization reaction between tertiary amine and alkyl halides is known to be a relatively highly efficient reaction. Less bulky amines and alkyl bromides are favored for the high conversion of the reaction. In most cases, the quaternized side chains are used for antimicrobial purposes due to their cationic nature (Fig. 5B and C). The introduced additional pendant groups have not been considered important so far. Benzyl chloride side chains have recently been presented as a new versatile precursor for the facile and efficient post-modification of the bisMPA-derived PTMC derivatives.86 Due to its higher reactivity compared to that of alkyl chloride, benzyl chloride can be readily converted to tertiary amines, quaternary ammonium and phosphonium, and azide followed by triazole under mild conditions in which the PTMC backbone remains intact (Fig. 5D).
The thiol–disulfide exchange reaction is also famous for a highly efficient reaction and is employed for post-modifications. The AOM-derived PTMC derivative with a pyridyl disulfide side chain has been designed for post-modification with a thiol-functionalized PEG to introduce PEG branches.69 In contrast, the thiol functionalized side chains of the bisMPA-derived PTMC derivative have also been effective in the exchange reaction with functionalized disulfides.87 However, the direction of the reversible reactions, including thiol–disulfide exchange and Diels–Alder reaction, is determined by the equilibrium based on concentration and temperature, which may affect the stability of the products after the reaction. Some post-modifications form a bulky junction that may influence the side chain properties, including the added functionality. Hence, it would be better to consider the properties of the forming junction when the type of post-modification is selected.
4. Applications of the PTMC-based polymers for biomedical devices
4.1. Scaffold for tissue regeneration
Along with the advance in stem cell research, tissue regeneration using biodegradable scaffolds and hydrogels is now being boosted.88 For tissue regeneration, the backing materials usually need to retain a certain level of mechanical properties for several months. PTMC could be suitable as scaffold materials because of its slow degradation profile and surface erosion mechanism that usually has only a little effect on the mechanical properties of the polymer during the healing. However, the mechanical strength of PTMC is mostly considered insufficient89 even as a scaffold for soft tissue regeneration. Hence, cross-linking and copolymerization with LA and GA are often employed to meet the required mechanical strength.10,88,90–92Copolymerization also plays a significant role in regulating the degradation of PLLA and poly(glycolide) (PGA) that is rapidly degraded by non-enzymatic hydrolysis through bulk erosion, inducing a drastic decline in mechanical strength.93 Furthermore, the surge of local acid concentration in the body caused by the degradation is not negligible, provoking various adverse reactions.91,94 Therefore, studies on PTMC-containing scaffold materials are increasingly expanding.
A composite of the high molecular weight PTMC with 50 wt% (30 vol%) of biphasic calcium phosphate (BCP) and its laminate with poly(D,L-lactide) (PDLLA) were then tested for reconstruction of orbital floor defects with 2.5–3.0 cm2 size in a sheep model (Fig. 6A and B).100 BCP, which is a constituent of the human bone and consists of β-tricalcium phosphate and hydroxyapatite, enables not only toughness and rigidity with the increased flexural modulus of 6–17 MPa,101 but also provides osteoinductive properties to the PTMC membranes. As a consequence of the excellent bone formation and the retained mechanical stability 9 months after implantation, the PTMC–BCP composite and its laminate with PDLLA were proven to be appropriate as osteoinductive materials for the orbital floor reconstruction.
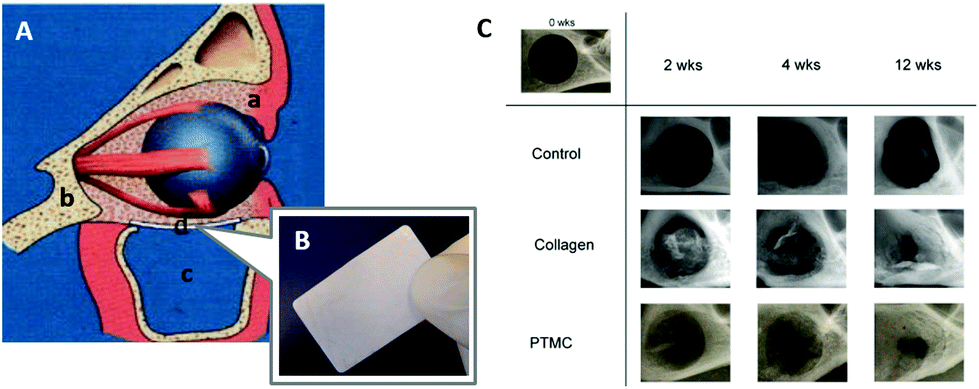 | ||
| Fig. 6 Schematic representation of the reconstruction of the human orbital floor (A): soft tissue (a), bone tissue (b), the maxillary sinus (c), and an implant (d). An image of the fabricated PTMC/calcium phosphate composite (B). Microradiographic X-ray images of the mandibular defect of rats treated by membranes (C). (A, B) Reprinted with permission from ref. 101. Copyright (2012) Wiley Periodicals, Inc. (C) Reprinted with permission from ref. 107. Copyright (2011) Elsevier Ltd. | ||
Incorporating TMC units allows the formation of a PLA-based scaffold more compatible with osteoblast cells. A random copolymer composed of L-lactide (LLA), D,L-lactide (DLLA), and TMC (PLDLT, Fig. 1D) with a weight-average molecular weight (Mw) of more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da has been examined as a scaffold for culturing human osteoblast-like SaOS-2 cells in vitro.51 TMC units (30 mol%) were incorporated, which imparted sufficient softness to the scaffold. The viability of the osteoblast-like cells grown on the PLDLT scaffold was greater than that of cells grown on poly(LLA-co-DLLA) (PLDLA). Moreover, the alkaline phosphatase (ALP) activity level, which is known as a differentiation marker for bone tissues,102 was also greater for the cells grown on the PLDLT scaffold than for cells grown on the PLDLA. These results may be explained by both the increased softness of the scaffold and the decrease in acidic degradation products as a result of copolymerization with TMC.
000 Da has been examined as a scaffold for culturing human osteoblast-like SaOS-2 cells in vitro.51 TMC units (30 mol%) were incorporated, which imparted sufficient softness to the scaffold. The viability of the osteoblast-like cells grown on the PLDLT scaffold was greater than that of cells grown on poly(LLA-co-DLLA) (PLDLA). Moreover, the alkaline phosphatase (ALP) activity level, which is known as a differentiation marker for bone tissues,102 was also greater for the cells grown on the PLDLT scaffold than for cells grown on the PLDLA. These results may be explained by both the increased softness of the scaffold and the decrease in acidic degradation products as a result of copolymerization with TMC.
Polymeric materials have been applied to guided bone regeneration (GBR), where bone regeneration is promoted in a space retained by a barrier membrane to prevent ingrowth of fibroblasts.103,104 For the GBR, the ideal barrier membrane entails resorbability, space retentivity for bone formation, prevention of ingrowth of fibroblasts, and retentive mechanical strength.105,106 Van Leeuwen et al. used PTMC with high molecular weight over 400000 Da in Mw as a barrier membrane for the GBR in vivo.107,108 The PTMC was compression molded and subjected to gamma irradiation for sterilization and simultaneous cross-linking (Fig. 1A).10 By using the PTMC barrier membrane, a created mandibular defect (5 mm diameter) in rats was covered by regenerated tissues after 12 weeks. This result is comparable to that obtained with a collagen membrane as the control (Fig. 6C). Furthermore, the PTMC membrane demonstrated almost two times greater space-maintaining properties than the collagen membrane at 2 and 4 weeks after implantation, resulting from the slow and surface erosive degradation of PTMC.
A porous scaffold (350 μm diameter, 54% porosity) was fabricated by stereolithography of a three-armed methacrylated PTMC with a number-average molecular weight (Mn) of 3000 Da (Fig. 1B) and tested for the culture of bovine chondrocytes in vitro.39 Favorable cell adhesion was observed after 3 weeks of incubation and the superficial pores of the scaffold were filled with the cells after 6 weeks. Moreover, sulfated glycosaminoglycans, which are major components of cartilage-specific proteoglycans,110,120,121 and fibrillar collagens, were formed on the scaffold after 6 weeks, suggesting that the cells recognize the scaffold as an appropriate growing environment.122 However, the compression modulus of the scaffold was not as high as that of natural cartilage tissue (450–800 kPa).110 The modulus was around 120 kPa at 21 °C in the dry state, and it was depressed to 47 kPa after immersion in water at 37 °C (47 kPa). Even after the chondrocytes adhered and proliferated, the modulus remained (68 kPa). This is mainly attributed to the plasticization of PTMC by water, which is often found in low molecular weight PTMC.32 Since 70–85% of the cartilage tissue is water,109,110 the mechanical properties of the scaffold should be targeted assuming its use in aqueous environment.
The PTMC porous scaffold has also been investigated for intervertebral disc repair, especially annulus fibrosus (AF) composed of fibrocartilage.123 The compression modulus of the scaffold fabricated by the stereolithography of a three-armed PTMC of Mn of about 5000 Da was adjusted to 0.21–0.31 MPa, which is comparable to that of human AF.124,125 The scaffold demonstrated good cell adhesion and proliferation during 14 days of culturing of human AF cells. Furthermore, Pirvu et al. recently tested the implantation of the PTMC scaffold by seeding mesenchymal stromal cells126 covered with a polyurethane (PU) membrane into a bovine annulotomy model. The hybrid device successfully restored the AF rupture with prevention of herniation of the nucleus pulposus (Fig. 7A and B).127 This system is proposed for the repair of AF rupture after herniotomy and partial discectomy.
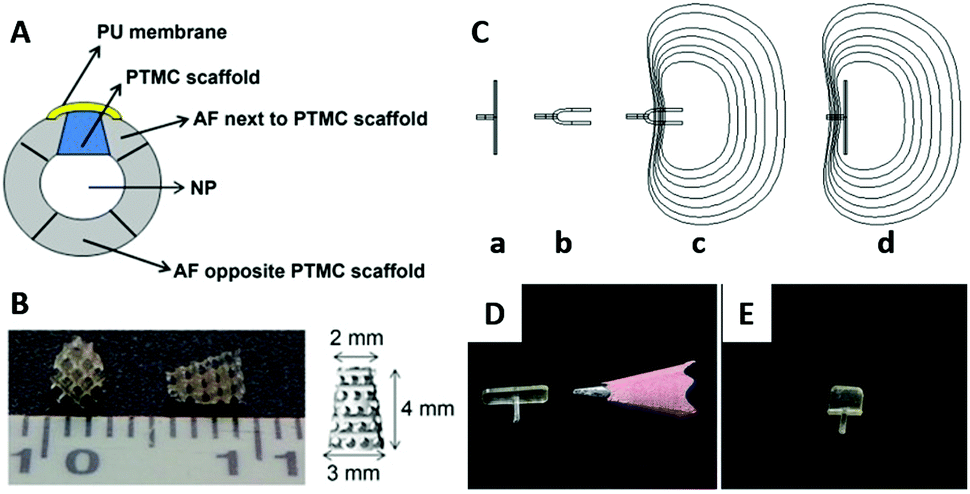 | ||
| Fig. 7 A schematic view of the intervertebral disc implanted with the PTMC scaffold and covered by a PU membrane (A); PU: polyurethane, NP: nucleus pulposus. Photographical images of the PTMC scaffold (B). Working concept of a shape memory AF closure device (C): the permanent shape above the Tg (a), the folded temporary shape fixed below Tg (b), minimal invasive implantation of the device with the temporary shape (c), and self-deployment of the device at the body temperature (d). Images of the AF closure devices of permanent shape (D) and temporary shape at 0 °C (E). Reprinted with permission from ref. 127 and 130. Copyright (2014) Elsevier Ltd. | ||
Photo cross-linked copolymers of DLLA and TMC (Fig. 1F) exhibit shape memory properties with switching temperatures at 0 and 40 °C,128 in addition to a tunable degradation speed.129 Using the poly(DLLA-co-TMC), the AF closure device consisting of a defect-covering membrane and holding grip has been fabricated and applied to the AF defect in a canine spine model. The device showed successful temperature-responsive sealing of the AF defect in vivo, supporting a minimal invasive treatment of the AF tear by the shape memory polymer (Fig. 7C–E).130
Rocha et al. determined that a copolymer of TMC and CL (11:89 mol%) (P(TMC-CL), Fig. 1C) significantly stimulates higher neuronal polarization and axonal elongation on the surface than PCL and PTMC alone.136 This polymer-specific cell response is explained by nanomechanical properties of the copolymer surface rather than surface topology that is generally known to play a key role in controlling axonal growth for other polymers.137,138 For instance, stiffness and hardness values for P(TMC-CL) measured by nanoindentation were 312 ± 56.4 N m−1 and 3.32 ± 0.373 × 106 N m−2, respectively, while those for PCL were 435 ± 40.4 N m−1 and 6.60 ± 2.11 × 106 N m−2, respectively. Another study139 revealed that microglia,140 immune cells in the central nervous system (CNS), seeded on the P(TMC-CL) is guided toward a pro-regenerative profile and engaged in phagocytosis of myelin that forms glial scar to inhibit CNS regeneration.139 It turns out that the nanomechanical properties of P(TMC-CL) also influence the microglia function. Consequently, the surface properties of P(TMC-CL) are involved in CNS regeneration both directly and indirectly.
000 Da (Fig. 1F) exhibited moderate Young's moduli that are similar to those of human arteries (0.4–0.8 MPa),142 and the favorable adhesion and proliferation of human mesenchymal stem cells were confirmed by using in vitro tests.38 This porous scaffold could be a breakthrough in developing synthetic degradable vascular grafts. However, how blood compatibility, particularly the antithrombotic property, should be achieved will be the next hurdle to be overcome for the application of small-diameter vascular grafts of less than 3 mm that have been longed for in the coronary bypass surgery and treatment in child patients.143,144
Biodegradable polymers have often been used in drug-eluting stents (DES) where the polymer is coated on metal stents incorporating drugs to prevent coagulation and abnormal proliferation of smooth muscle cells.145 The surface-eroding and slow-degrading properties of PTMC should be ideal for DES to manage slow and prolonged drug release. Moreover, since no acidic degradation products are generated, the corrosion of the base metal is less affected. Magnesium-based stents are known to be biodegradable, mechanically strong, and non-toxic.146 However, the corrosion-based degradation is fast in vivo, which incites rapid hydrogen formation, hemolysis due to a high pH, and loss of the required mechanical strength at the early stage of the detainment.147 The PTMC-coating has effectively suppressed the corrosion of the Mg-based stents, exhibiting significantly better performance than the coating by bulk-eroding PLA, PCL, and PGA that generate acidic degradation products.148
Fully polymer-based resorbable cardiovascular stents have been recently studied for a low risk vascular therapy where no materials remain after healing.149 The required mechanical properties become more complex than those for artificial vascular grafts. The stents need to expand and maintain the shape of the occluded part of the blood vessel. For this reason, shape-memory polymers have drawn attention as promising candidates for polymeric stents. Linear terpolymers of LLA, TMC, and GA (Fig. 1E) were then designed to demonstrate the shape-memory property around 40 °C with the high LLA composition.150 The shape-memory property was not affected by incorporation of drugs. This fully polymeric shape memory stent has been proven to be useful as DES.52 Due to its decent processability, industrial fabrication of the terpolymer-based stent appears feasible (Fig. 8).151
 | ||
| Fig. 8 A prototype stent fabricated from a high molecular weight poly(LLA-co-GA-co-TMC) (LLA/GA/TMC = 95/5/5). Reprinted with permission from ref. 151. Copyright (2013) Society of Plastics Engineers. | ||
4.2. Drug delivery and theranostics
Drug delivery and therapeutics are two of the largest areas in which biodegradable polymers are studied due to their biomedical applications at the experimental level. Recent efforts entail active drug release upon stimuli,76,80,158 integration of imaging agents,159 and targeting properties.160,161 The last two are relevant to theranostics that allows for therapy and diagnostics at the same time using one delivery vehicle.162,163 PTMC is used as a hydrophobic part of amphiphilic block copolymers to form spherical carriers, including micelles and vesicles, as well as other hydrophobic biodegradable polymers such as PLLA and PCL. In most cases, PEG and PGlu are used in the hydrophilic segment.164 PEGylated block copolymers forming micelles show extremely low cytotoxicity both in vitro and in vivo.165–167 Moreover, due to their stealth property, PEGylated drug carriers tend to circulate longer in the bloodstream, avoiding phagocytosis, although recent reports revealed that multiple doses induce anti-PEG IgM production, resulting in an accelerated blood clearance (ABC) phenomenon of the PEGylated carriers.168,169 PGlu-b-PTMC (Fig. 1H), which generally forms vesicles, exhibits no cytotoxicity, but provokes oxidative stress against hepatocellular carcinoma cells (HepG2) and human lymphoblast cells (TK6) and slightly high cytokine production in lung epithelial cells (A549).170 These findings indicate that the negative charge could be somewhat immunogenic and triggers an inflammatory response.170,171 However, the negative charge of the carboxylate is useful to enhance drug loading of doxorubicin (Dox) and pH-responsive release of drugs upon protonation.172Since the functions of targeting and cell recognition are applied to the surface of the carriers, the chemistry is not directly associated with PTMC that resides inside the carrier. Decoration of the terminal of PEG or PGlu by conjugating peptides173–175 and sugars176,177 (Fig. 1G) has been recently reported as new approaches for surface functionalization of PTMC-based drug carriers (Fig. 9). When the molecular weight of PTMC is quite small, hydrophilic terminal modification almost equates surface functionalization of micelle-like aggregates of PTMC to disperse in the aqueous system. Short PTMC (Mn < 3000 Da) with positively charged end groups (Fig. 1K) has been proposed as a new candidate for non-viral gene vectors178 in which entirely charged cationic polymers are often used.179–181 The hydrophobic PTMC segment functions as a part of the DNA-binding moiety in combination with the peripheral charged end groups. Consequently, the low charge density likely contributes to the high transfection efficacy.
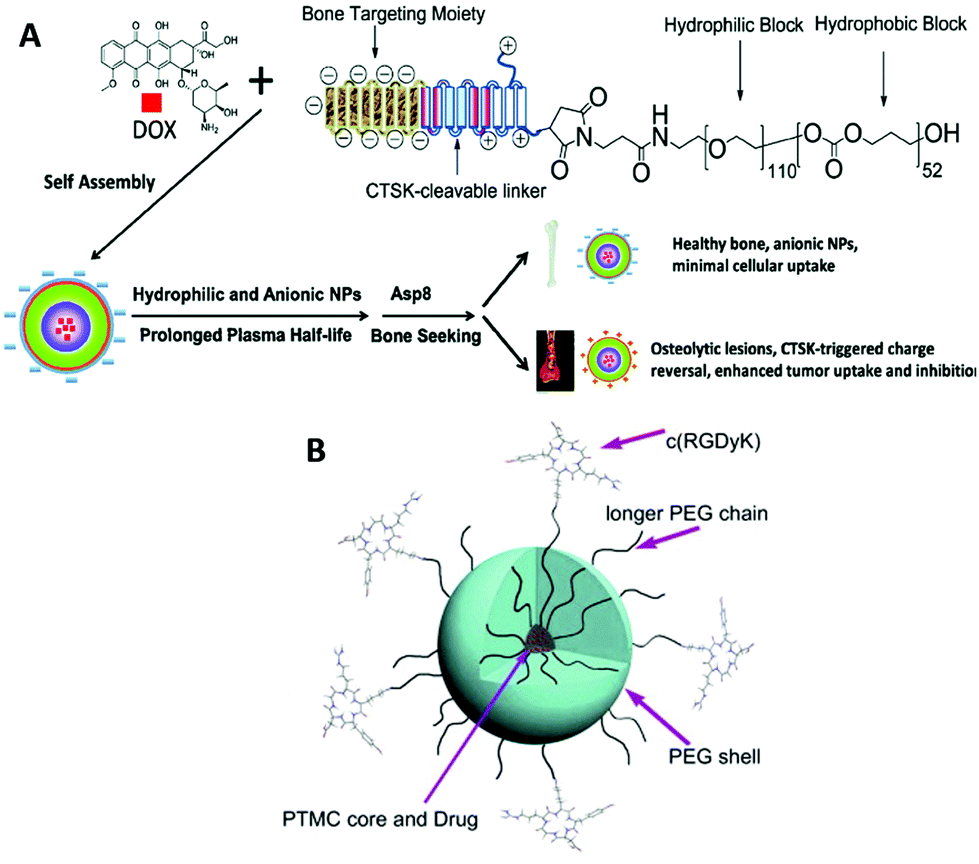 | ||
| Fig. 9 Illustration of peptide-decorated PEG-b-PTMC for bone metastasis chemotherapy (A). Paclitaxel delivery to target integrin-rich malignant glioma cells (B). Reprinted with permission from ref. 173. Copyright (2013) Elsevier Ltd. | ||
Due to the availability and a wide range of variation, PEG is predominantly used as the hydrophilic domain of the carriers and PEGylated block copolymers form micelles. The loading of the hydrophobic drug in the micelle core depends on the hydrophobic interaction between the drug and the hydrophobic segment when covalent conjugation of drugs is not exerted,182–184 while the drug release is susceptible to degradation of the hydrophobic core composed of biodegradable polymers. The advantage of PTMC-based drug carriers is the slow and moderate degradation of the hydrophobic core,185,186 promising constant and prolonged release of the drugs loaded. In contrast, PLA-based drug carriers often show an “initial burst” type release profile.
Conjugation of fluorophores in the micelle core helps to monitor the stability of the micelle. A quinine-functionalized PTMC derivative in the form of the amphiphilic random copolymer indicated minimal fluorescence in water because of self-quenching of the quinine pendants in the micelle core, whereas higher fluorescence emission was observed for the much shorter oligomer (dimer) and free quinine.159 This result suggests that the collapse of the micelle based on the polymer degradation can be traced in real time by the fluorescence emission level of the quinine pendants.
The disulfide bridge is often employed as a reduction responsive cleavable linkage.87 An AOM-derived PTMC derivative (Fig. 3C) with cleavable PEG side chains in the form of a block copolymer with PCL has demonstrated drug release by 1,4-dithiothreitol (DTT), a model reductive agent, resulting from the detachment of PEG and the subsequent dissociation of the micelle.69 In another case, a bisMPA-derived PTMC derivative with attached PLLA grafts through the disulfide bridge showed reductive removal of the PLLA grafts by DTT.87
Recently, thermoresponsive biodegradable hydrogels have drawn more attention as an alternative to non-degradable poly(N-isopropylacrylamide) (PNIPAAM)200 for its application as an injectable hydrogel-type drug carrier. Typically, they are handled as solution at room temperature outside the body and form hydrogels upon injection in the body, responding to the body temperature. A PTMC-containing triblock copolymer with a clinically used thermoresponsive hydrophilic polymer Pluronic F127® (PTMC-F127-PTMC, Fig. 1I), showing a sol–gel transition at 37 °C, locally accumulated at the injected position, presenting a stable drug release profile over 25 days and no adverse reactions during the in vivo test using a rabbit glaucoma model.201 Kim et al. first reported the thermoresponsive hydrogel system based on the PEG-tagged PTMC derivatives derived from bisMPA, displaying a lower critical solution temperature (LCST) around the body temperature, which is tuned by copolymerization of the TMC analogues with a long alkyl pendant.202 Ajiro et al. also reported that a TMA-derived PTMC derivative (Fig. 3B) bearing a methoxy terminated triethylene glycol unit at the side chain showed an LCST around the body temperature at a particular molecular weight range.203,204 This is the first example of the LCST-type polymer obtained by using PTMC derivatives comprising a single monomer unit.
4.3. Antimicrobial
One of the growing areas in biomaterials is polymeric antimicrobials, corresponding to the urgent demand in medical settings where emerging multi-drug resistant bacteria threaten patients worldwide.205 Quaternary ammonium tagged PTMC derivatives (Fig. 5B) have now been extensively studied as degradable polymeric antimicrobials since the first report in 2011.206 The first polymer demonstrated minimal hemolytic property and high antimicrobial activities against Gram-positive bacteria, including drug-resistant species such as methicillin-resistant Staphylococcus aureus (MRSA). As with other cationic polymers with methacrylate207–209 and vinyl ether210 backbones, recent challenges converge on improving the antimicrobial activities and broadening the antimicrobial spectrum without increasing the hemolytic properties. According to recent comprehensive reviews,211,212 amphiphilicity, which is defined as a hydrophilic–hydrophobic balance of the polymer, plays a crucial role in the selectivity of membranes between bacteria and mammalian cells. The amphiphilicity is tuned by copolymerization with hydrophobic monomers with an alkyl pendant (segregated monomer approach)209,210,213 and variation of the length of an alkyl spacer between the cationic charge center and the backbone208,214–216 or of peripheral alkyl arms extending from the charge center217 (the same center approach, Fig. 5B and C). Although no report has been published yet, some of the serinol-derived PTMC derivatives (Fig. 3E and F) can be potentially used as polymeric antimicrobials through quaternization and acidolysis of tert-butoxycarbonyl (Boc)-protected side chain affording ammonium salts.72Besides the modulation of the primary architecture of the cationic moiety, the introduction of biocompatible biomolecules,216,218 supramolecular approach,219 and hydrogelation220 has also been surveyed to increase and broaden the antimicrobial properties with a low level of hemolytic properties (Fig. 10). In particular, the supramolecular approach raised the importance of the shape of antimicrobials on the efficacy against some fungi.219,221 According to a recent study by Stupp et al., the cytotoxicity of cationic supramolecular aggregates depends on the strength of the interaction in the internal domain.222 The strong “internal binding affinity” maintains the stable cationic supramolecular structure that could rip out the cell membrane, while the weak affinity augments the dynamic nature of the cationic aggregates to release unimers by stronger interaction with cell membranes. When the cells are of human origin, the former is regarded as cytotoxic and the latter leads to no damage to the cells. Thus, this study suggests that various structural factors from the backbone structure to higher-order structures should be considered to comprehensively elucidate the hemocompatibility and biocidal activity of the polymeric antimicrobials. Otherwise, relatively long-life aliphatic polycarbonates may be regarded as unsuitable platforms as alternatives and/or supplements to conventional antibiotics. One of the preliminary results indicated a distinguished hydration behavior only for the carbonate linkage in the backbone, different from ester linkages, which may contribute to the high biocompatibility of PTMC.32,223
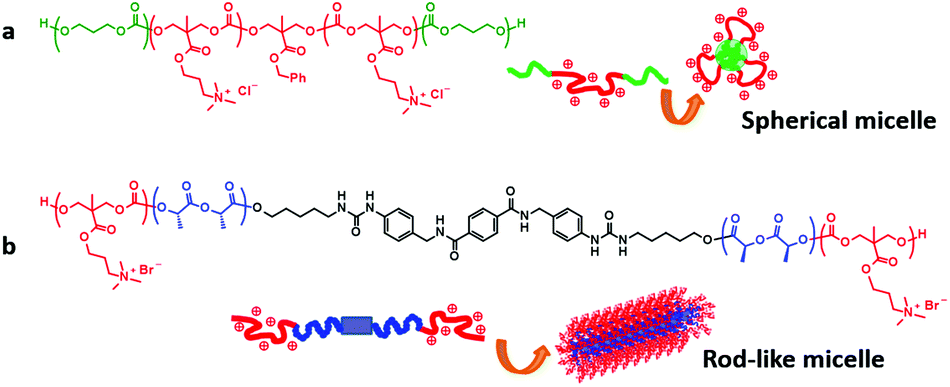 | ||
| Fig. 10 PTMC derivatives for antimicrobials in different active forms. (a) Spherical micelles and (b) rod-like micelles. Reprinted with permission from ref. 223. Copyright (2015) The Society of Polymer Science, Japan. | ||
5. Conclusions and outlook
This review focuses on PTMC and its derivatives with peripheral and/or vertical decorations developed in response to the diversifying demand for degradable biomedical devices. The modifications of the PTMC-based polymers affect the primary architecture, which alters the mechanical strength, degradation behavior, chemical properties, and affinity for cells and tissues. The peripheral decorations include block and random copolymerization, cross-linking, and terminal functionalization that often maintain the inherent characteristics of PTMC. Therefore, the peripheral decorations are valuable for the improvement and amendment of PTMC and for the addition of supplemental functions. Additionally, the modified PTMC materials are still easy to use for in vivo evaluation and the subsequent clinical testing. In contrast, the vertical decoration involves the introduction of side chains with some functionality onto the backbone of PTMC, providing polymers with a different physicochemical nature from that of PTMC. This means that any advantages of PTMC, including softness, compatibility with cells/tissues, and even biodegradability, are not technically guaranteed for the PTMC derivatives, although no generation of the acidic degradation products could be promised. As aforementioned, this is because the biological response highly depends on the surface chemistry of the materials, significantly involving side chain functionality. Thus, each PTMC derivative should be thoroughly examined in vitro to estimate the in vivo behavior. Nevertheless, PTMC derivatives have great potential to solve issues and technical limitations of the peripheral decorations. The next-generation biodegradable biomaterials can be created by integrating the knowledge obtained from the in vivo experiments designed to test the peripheral decorations and the synthetic solutions in the vertical decorations.In addition, when we consider the application of PTMC or design the PTMC derivatives, we should understand and take full advantage of the specific characteristics of the main chain such as flexibility, resistance to hydrolysis, hydration properties, biocompatibility, and non-generation of acidic degradation products. Otherwise, the aforementioned PTMC derivatives would become temporary once another platform is proven to be more valuable. It is also important to determine new characteristics observed only in the PTMC derivatives in order for PTMC derivatives to remain attractive as a biodegradable functional biomaterial compared to other polymer platforms with the same side chain architecture.
Acknowledgements
This study was supported by a JSPS Grant-in-Aid for Young Scientists (B) #25870078, the Takeda Science Foundation, and the Ito Science Foundation. The author would like to thank Editage (http://www.editage.jp) for English language editing of the manuscript.Notes and references
- H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Prog. Polym. Sci., 2012, 37, 237–280 CrossRef CAS.
- A. L. Sisson, M. Schroeter and A. Lendlein, in Handbook of Biodegradable Polymers, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 1–21 Search PubMed.
- D. J. Darensbourg and F.-T. Tsai, Macromolecules, 2014, 47, 3806–3813 CrossRef CAS.
- Y. Liu, K. Deng, S. Wang, M. Xiao, D. Han and Y. Meng, Polym. Chem., 2015, 6, 2076–2083 RSC.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- F. Nederberg, B. G. G. Lohmeijer, F. Leibfarth, R. C. Pratt, J. Choi, A. P. Dove, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2007, 8, 153–160 CrossRef CAS PubMed.
- D. J. Darensbourg, A. Horn Jr. and A. I. Moncada, Green Chem., 2010, 12, 1376–1379 RSC.
- C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed.
- K. Koga, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4351–4355 CrossRef CAS.
- A. P. Pêgo, D. W. Grijpma and J. Feijen, Polymer, 2003, 44, 6495–6504 CrossRef.
- K. J. Zhu, R. W. Hendren, K. Jensen and C. G. Pitt, Macromolecules, 1991, 24, 1736–1740 CrossRef CAS.
- Z. Zhang, R. Kuijer, S. K. Bulstra, D. W. Grijpma and J. Feijen, Biomaterials, 2006, 27, 1741–1748 CrossRef CAS PubMed.
- J. Zou, C. C. Hew, E. Themistou, Y. Li, C.-K. Chen, P. Alexandridis and C. Cheng, Adv. Mater., 2011, 23, 4274–4277 CrossRef CAS PubMed.
- R. J. Pounder and A. P. Dove, Biomacromolecules, 2010, 11, 1930–1939 CrossRef CAS PubMed.
- Y. Yu, J. Zou and C. Cheng, Polym. Chem., 2014, 5, 5854–5872 RSC.
- O. T. du Boullay, N. Saffon, J.-P. Diehl, B. Martin-Vaca and D. Bourissou, Biomacromolecules, 2010, 11, 1921–1929 CrossRef PubMed.
- F. Coumes, V. Darcos, D. Domurado, S. Li and J. Coudane, Polym. Chem., 2013, 4, 3705–3713 RSC.
- J. Hao, Y. Cheng, R. J. K. U. Ranatunga, S. Senevirathne, M. C. Biewer, S. O. Nielsen, Q. Wang and M. C. Stefan, Macromolecules, 2013, 46, 4829–4838 CrossRef CAS.
- F. Ercole, A. E. Rodda, L. Meagher, J. S. Forsythe and A. P. Dove, Polym. Chem., 2014, 5, 2809–2815 RSC.
- S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- S. Schüller-Ravoo, J. Feijen and D. W. Grijpma, Acta Biomater., 2012, 8, 3576–3585 CrossRef PubMed.
- S. J. German, M. Behbahani, S. Miettinen, D. W. Grijpma and S. P. Haimi, Macromol. Symp., 2013, 334, 133–142 CrossRef CAS.
- N. D. Evans, C. Minelli, E. Gentleman, V. LaPointe, S. N. Patankar, M. Kallivretaki, X. Chen, C. J. Roberts and M. M. Stevens, Eur. Cells Mater., 2009, 18, 1–13 CAS.
- X. Li, Y. Huang, L. Zheng, H. Liu, X. Niu, J. Huang, F. Zhao and Y. Fan, J. Biomed. Mater. Res., Part A, 2014, 102, 1092–1101 CrossRef PubMed.
- F. Wang, Z. Li, J. L. Lannutti, W. R. Wagner and J. Guan, Acta Biomater., 2009, 5, 2901–2912 CrossRef CAS PubMed.
- X. J. Loh, W. Guerin and S. M. Guillaume, J. Mater. Chem., 2012, 22, 21249–21256 RSC.
- J. Yang, W. Tian, Q. Li, Y. Li and A. Cao, Biomacromolecules, 2004, 5, 2258–2268 CrossRef CAS PubMed.
- R. Chapanian, M. Y. Tse, S. C. Pang and B. G. Amsden, Biomaterials, 2009, 30, 295–306 CrossRef CAS PubMed.
- E. Bat, T. G. van Kooten, J. Feijen and D. W. Grijpma, Biomaterials, 2009, 30, 3652–3661 CrossRef CAS PubMed.
- M. C. Vyner, A. Li and B. G. Amsden, Biomaterials, 2014, 35, 9041–9048 CrossRef CAS PubMed.
- K. Fukushima, M.-Y. Tsai, T. Ota, Y. Haga, K. Matsuzaki, Y. Inoue and M. Tanaka, Polym. J., 2015, 47, 469–473 CrossRef CAS.
- E. Pirhonen and P. Törmälä, J. Mater. Sci., 2006, 41, 2031–2036 CrossRef CAS.
- F. Nederberg, J. Watanabe, K. Ishihara, J. Hilborn and T. Bowden, J. Biomater. Sci., Polym. Ed., 2006, 17, 605–614 CrossRef CAS PubMed.
- R. Michel, S. Pasche, M. Textor and D. G. Castner, Langmuir, 2005, 21, 12327–12332 CrossRef CAS PubMed.
- P. Charles, V. Stubbs, C. Soto, B. Martin, B. White and C. Taitt, Sensors, 2009, 9, 645 CrossRef CAS PubMed.
- N. Zeng, A. van Leeuwen, R. R. M. Bos, D. W. Grijpma and R. Kuijer, Macromol. Symp., 2013, 334, 68–74 CrossRef CAS.
- B. L. Dargaville, C. Vaquette, F. Rasoul, J. J. Cooper-White, J. H. Campbell and A. K. Whittaker, Acta Biomater., 2013, 9, 6885–6897 CrossRef CAS PubMed.
- S. Schüller-Ravoo, S. M. Teixeira, J. Feijen, D. W. Grijpma and A. A. Poot, Macromol. Biosci., 2013, 13, 1711–1719 CrossRef PubMed.
- D. Tunc, C. Le Coz, M. Alexandre, P. Desbois, P. Lecomte and S. Carlotti, Macromolecules, 2014, 47, 8247–8254 CrossRef CAS.
- A. Garle, S. Kong, U. Ojha and B. M. Budhlall, ACS Appl. Mater. Interfaces, 2012, 4, 645–657 CAS.
- X. Hu, X. Chen, H. Cheng and X. Jing, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 161–169 CrossRef CAS.
- V. Truong, I. Blakey and A. K. Whittaker, Biomacromolecules, 2012, 13, 4012–4021 CrossRef CAS PubMed.
- D. M. Stevens, S. Tempelaar, A. P. Dove and E. Harth, ACS Macro Lett., 2012, 1, 915–918 CrossRef CAS PubMed.
- D. M. Stevens, A. Rahalkar, B. Spears, K. Gilmore, E. Douglas, M. Muthukumar and E. Harth, Polym. Chem., 2015, 6, 1096–1102 RSC.
- P. Olsén, K. Odelius and A.-C. Albertsson, Macromolecules, 2014, 47, 6189–6195 CrossRef.
- S. Skoog, P. Goering and R. Narayan, J. Mater. Sci.: Mater. Med., 2014, 25, 845–856 CrossRef CAS PubMed.
- J. L. Ifkovits and J. A. Burdick, Tissue Eng., 2007, 13, 2369–2385 CrossRef CAS PubMed.
- E. Sachlos and J. T. Czernuszka, Eur. Cells Mater., 2003, 5, 29–39 CAS ; discussion 39–40.
- S. M. Peltola, F. P. W. Melchels, D. W. Grijpma and M. Kellomäki, Ann. Med., 2008, 40, 268–280 CrossRef CAS PubMed.
- A. D. Messias, K. F. Martins, A. C. Motta and E. A. d. R. Duek, Int. J. Biomater., 2014, 2014, 7 CrossRef PubMed.
- J. Jaworska, K. Jelonek, M. Sobota, J. Kasperczyk, P. Dobrzynski, M. Musial-Kulik, A. Smola-Dmochowska, H. Janeczek and B. Jarzabek, J. Appl. Polym. Sci., 2015, 132 CrossRef CAS , 41902.
- C.-J. Pan, Y.-H. Hou, B.-B. Zhang, Y.-X. Dong and H.-Y. Ding, J. Mater. Chem. B, 2014, 2, 892–902 RSC.
- H. Lu and J. Cheng, J. Am. Chem. Soc., 2008, 130, 12562–12563 CrossRef CAS PubMed.
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- S. A. Pendergraph, G. Klein, M. K. G. Johansson and A. Carlmark, RSC Adv., 2014, 4, 20737–20743 RSC.
- Y. Luan, J. Wu, M. Zhan, J. Zhang, J. Zhang and J. He, Cellulose, 2013, 20, 327–337 CrossRef CAS.
- C. Yan, J. Zhang, Y. Lv, J. Yu, J. Wu, J. Zhang and J. He, Biomacromolecules, 2009, 10, 2013–2018 CrossRef CAS PubMed.
- W. Guerin, M. Helou, M. Slawinski, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2014, 5, 1229–1240 RSC.
- L. Pasquato, G. Modena, L. Cotarca, P. Delogu and S. Mantovani, J. Org. Chem., 2000, 65, 8224–8228 CrossRef CAS PubMed.
- R. C. Pratt, F. Nederberg, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2008, 114–116 RSC.
- S. Venkataraman, J. L. Hedrick and Y. Y. Yang, Polym. Chem., 2014, 5, 2035–2040 RSC.
- D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef CAS PubMed.
- J. V. Olsson, D. Hult, Y. Cai, S. Garcia-Gallego and M. Malkoch, Polym. Chem., 2014, 5, 6651–6655 RSC.
- J. Xu, E. Feng and J. Song, J. Appl. Polym. Sci., 2014, 131, 39822 Search PubMed.
- W. Chen, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2014, 190, 398–414 CrossRef CAS PubMed.
- H. Ihre, A. Hult and E. Söderlind, J. Am. Chem. Soc., 1996, 118, 6388–6395 CrossRef CAS.
- F. Chen, J. W. S. Hayami and B. G. Amsden, Biomacromolecules, 2014, 15, 1593–1601 CrossRef CAS PubMed.
- W. Chen, Y. Zou, J. Jia, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Macromolecules, 2013, 46, 699–707 CrossRef CAS.
- J. Mindemark and T. Bowden, Polym. Chem., 2012, 3, 1399–1401 RSC.
- X. Zhang, M. Cai, Z. Zhong and R. Zhuo, Macromol. Rapid Commun., 2012, 33, 693–697 CrossRef CAS PubMed.
- S. Venkataraman, N. Veronica, Z. X. Voo, J. L. Hedrick and Y. Y. Yang, Polym. Chem., 2013, 4, 2945–2948 RSC.
- L. Shen, Z. Li, F. Gong, F. Zhang, Q. Qin, S. Cheng and J. Ge, Polym. Degrad. Stab., 2013, 98, 1015–1021 CrossRef CAS.
- D. Chitkara, A. Mittal, S. W. Behrman, N. Kumar and R. I. Mahato, Bioconjugate Chem., 2013, 24, 1161–1173 CrossRef CAS PubMed.
- H. Xiao, H. Song, Q. Yang, H. Cai, R. Qi, L. Yan, S. Liu, Y. Zheng, Y. Huang, T. Liu and X. Jing, Biomaterials, 2012, 33, 6507–6519 CrossRef CAS PubMed.
- M. Wang, J. Sun, Y. Zhai, H. Lian, C. Luo, L. Li, Y. Du, D. Zhang, W. Ding, S. Qiu, Y. Liu, L. Kou, X. Han, R. Xiang, Y. Wang and Z. He, Biomacromolecules, 2015, 16, 1179–1190 CrossRef CAS PubMed.
- A. C. Engler, X. Ke, S. Gao, J. M. W. Chan, D. J. Coady, R. J. Ono, R. Lubbers, A. Nelson, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2015, 48, 1673–1678 CrossRef CAS.
- A. C. Engler, J. M. W. Chan, D. J. Coady, J. M. O'Brien, H. Sardon, A. Nelson, D. P. Sanders, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2013, 46, 1283–1290 CrossRef CAS.
- R. J. Williams, I. A. Barker, R. K. O'Reilly and A. P. Dove, ACS Macro Lett., 2012, 1, 1285–1290 CrossRef CAS.
- D. Hu, H. Peng, Y. Niu, Y. Li, Y. Xia, L. Li, J. He, X. Liu, X. Xia, Y. Lu and W. Xu, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 750–760 CrossRef CAS.
- D. Xing, L. Ma and C. Gao, Macromol. Biosci., 2014, 14, 1429–1436 CrossRef CAS PubMed.
- S. Onbulak, S. Tempelaar, R. J. Pounder, O. Gok, R. Sanyal, A. P. Dove and A. Sanyal, Macromolecules, 2012, 45, 1715–1722 CrossRef CAS.
- A. Dag, M. Aydin, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4476–4483 CrossRef CAS.
- S. Li, F. Meng, Z. Wang, Y. Zhong, M. Zheng, H. Liu and Z. Zhong, Eur. J. Pharm. Biopharm., 2012, 82, 103–111 CrossRef CAS PubMed.
- B. B. Uysal, U. S. Gunay, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1581–1587 CrossRef CAS.
- R. J. Ono, S. Q. Liu, S. Venkataraman, W. Chin, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2014, 47, 7725–7731 CrossRef CAS.
- A. C. Engler, J. M. W. Chan, K. Fukushima, D. J. Coady, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2013, 2, 332–336 CrossRef CAS.
- R. Jin, L. S. Moreira Teixeira, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, Biomaterials, 2010, 31, 3103–3113 CrossRef CAS PubMed.
- L.-Q. Yang, B. He, S. Meng, J.-Z. Zhang, M. Li, J. Guo, Y.-M. Guan, J.-X. Li and Z.-W. Gu, Polymer, 2013, 54, 2668–2675 CrossRef CAS.
- E. H. Busaina Dhariwala and T. Boland, Tissue Eng., 2004, 10, 1316–1322 CrossRef PubMed.
- E. Bat, B. H. M. Kothman, G. A. Higuera, C. A. van Blitterswijk, J. Feijen and D. W. Grijpma, Biomaterials, 2010, 31, 8696–8705 CrossRef CAS PubMed.
- Q. Hou, D. W. Grijpma and J. Feijen, Acta Biomater., 2009, 5, 1543–1551 CrossRef CAS PubMed.
- A. P. Pêgo, M. J. A. Van Luyn, L. A. Brouwer, P. B. van Wachem, A. A. Poot, D. W. Grijpma and J. Feijen, J. Biomed. Mater. Res., Part A, 2003, 67A, 1044–1054 CrossRef PubMed.
- X.-H. Qu, Q. Wu, K.-Y. Zhang and G. Q. Chen, Biomaterials, 2006, 27, 3540–3548 CAS.
- S. Talebian, M. Mehrali, S. Mohan, H. r. Balaji Raghavendran, M. Mehrali, H. M. Khanlou, T. Kamarul, A. M. Afifi and A. A. Abass, RSC Adv., 2014, 4, 49144–49152 RSC.
- T. Pyhältö, M. Lapinsuo, H. Pätiälä, M. Pelto, P. Törmälä and P. Rokkanen, Biomaterials, 2005, 26, 645–654 CrossRef PubMed.
- L. Wu and J. Ding, Biomaterials, 2004, 25, 5821–5830 CrossRef CAS PubMed.
- J. E. Bergsma, W. C. de Bruijn, F. R. Rozema, R. R. M. Bos and G. Boering, Biomaterials, 1995, 16, 25–31 CrossRef CAS PubMed.
- M. S. Taylor, A. U. Daniels, K. P. Andriano and J. Heller, J. Appl. Biomater., 1994, 5, 151–157 CrossRef CAS PubMed.
- A. C. van Leeuwen, H. Yuan, G. Passanisi, J. W. van der Meer, J. D. de Bruijn, T. G. van Kooten, D. W. Grijpma and R. R. Bos, Eur. Cells Mater., 2014, 27, 81–96 CAS ; discussion 96–87.
- A. C. van Leeuwen, R. R. M. Bos and D. W. Grijpma, J. Biomed. Mater. Res., Part B, 2012, 100B, 1610–1620 CrossRef CAS PubMed.
- A. Polini, D. Pisignano, M. Parodi, R. Quarto and S. Scaglione, PLoS One, 2011, 6, e26211 CAS.
- S. A. Kay, L. Wisner-Lynch, M. Marxer and S. E. Lynch, Practical Periodontics and Aesthetic Dentistry: PPAD, 1997, 9, 185–194 CAS ; quiz 196.
- T. von Arx, D. L. Cochran, R. K. Schenk and D. Buser, Int. J. Oral Maxillofac. Surg., 2002, 31, 190–199 CrossRef CAS PubMed.
- H.-L. Wang and L. Boyapati, Implant Dent., 2006, 15, 8–17 CrossRef PubMed.
- P. Gentile, V. Chiono, C. Tonda-Turo, A. M. Ferreira and G. Ciardelli, Biotechnol. J., 2011, 6, 1187–1197 CrossRef CAS PubMed.
- A. C. van Leeuwen, J. J. R. Huddleston Slater, P. F. M. Gielkens, J. R. de Jong, D. W. Grijpma and R. R. M. Bos, Acta Biomater., 2012, 8, 1422–1429 CrossRef CAS PubMed.
- A. C. Van Leeuwen, T. G. Van Kooten, D. W. Grijpma and R. R. M. Bos, J. Mater. Sci.: Mater. Med., 2012, 23, 1951–1959 CrossRef CAS PubMed.
- S. Park, C. T. Hung and G. A. Ateshian, Osteoarthritis Cartilage, 2004, 12, 65–73 CrossRef CAS PubMed.
- J. M. Mansour, Biomechanics of Cartilage, Lippincott Williams & Wilkins, Philadelphia, PA, 2003 Search PubMed.
- J. A. Stella, A. D'Amore, W. R. Wagner and M. S. Sacks, Acta Biomater., 2010, 6, 2365–2381 CrossRef PubMed.
- L. Kock, C. van Donkelaar and K. Ito, Cell Tissue Res., 2012, 347, 613–627 CrossRef CAS PubMed.
- S. P. Zustiak, Y. Wei and J. B. Leach, Tissue Eng., Part B, 2013, 19, 160–171 CrossRef CAS PubMed.
- L. Calderon, E. Collin, D. Velasco-Bayon, M. Murphy, D. O'Halloran and A. Pandit, Eur. Cells Mater., 2010, 20, 134–148 CAS.
- G. D. Nicodemus and S. J. Bryant, Tissue Eng., Part B, 2008, 14, 149–165 CrossRef CAS PubMed.
- C. Scotti, L. Mangiavini, F. Boschetti, F. Vitari, C. Domeneghini, G. Fraschini and G. Peretti, Knee Surgery, Sports Traumatology, Arthroscopy, 2010, 18, 1400–1406 CrossRef PubMed.
- B. G. Amsden, A. Sukarto, D. K. Knight and S. N. Shapka, Biomacromolecules, 2007, 8, 3758–3766 CrossRef CAS PubMed.
- S. J. Hollister, Nat. Mater., 2005, 4, 518–524 CrossRef CAS PubMed.
- Y.-J. Seol, T.-Y. Kang and D.-W. Cho, Soft Matter, 2012, 8, 1730–1735 RSC.
- C. D. Hoemann, J. Sun, V. Chrzanowski and M. D. Buschmann, Anal. Biochem., 2002, 300, 1–10 CrossRef CAS PubMed.
- P. Roughley, C. Hoemann, E. DesRosiers, F. Mwale, J. Antoniou and M. Alini, Biomaterials, 2006, 27, 388–396 CrossRef CAS PubMed.
- R. Jin, L. S. Moreira Teixeira, A. Krouwels, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, Acta Biomater., 2010, 6, 1968–1977 CrossRef CAS PubMed.
- H. J. Wilke, P. Neef, M. Caimi, T. Hoogland and L. E. Claes, Spine, 1999, 24, 755–762 CrossRef CAS PubMed.
- S. B. G. Blanquer, S. P. Haimi, A. A. Poot and D. W. Grijpma, Macromol. Symp., 2013, 334, 75–81 CrossRef CAS.
- D. Périé, D. Korda and J. C. Iatridis, J. Biomech., 2005, 38, 2164–2171 CrossRef PubMed.
- M. E. Bernardo and W. E. Fibbe, Cell Stem Cell, 2013, 13, 392–402 CrossRef CAS PubMed.
- T. Pirvu, S. B. G. Blanquer, L. M. Benneker, D. W. Grijpma, R. G. Richards, M. Alini, D. Eglin, S. Grad and Z. Li, Biomaterials, 2015, 42, 11–19 CrossRef CAS PubMed.
- M. Bao, X. Lou, Q. Zhou, W. Dong, H. Yuan and Y. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 2611–2621 CAS.
- A. P. Pêgo, A. A. Poot, D. W. Grijpma and J. Feijen, J. Controlled Release, 2003, 87, 69–79 CrossRef.
- S. Sharifi, T. G. van Kooten, H.-J. C. Kranenburg, B. P. Meij, M. Behl, A. Lendlein and D. W. Grijpma, Biomaterials, 2013, 34, 8105–8113 CrossRef CAS PubMed.
- R. Deumens, A. Bozkurt, M. F. Meek, M. A. E. Marcus, E. A. J. Joosten, J. Weis and G. A. Brook, Prog. Neurobiol., 2010, 92, 245–276 CrossRef PubMed.
- R. Li, Z. Liu, Y. Pan, L. Chen, Z. Zhang and L. Lu, Cell Biochem. Biophys., 2014, 68, 449–454 CrossRef CAS PubMed.
- R. A. Wach, A. Adamus, A. K. Olejnik, J. Dzierzawska and J. M. Rosiak, J. Appl. Polym. Sci., 2013, 127, 2259–2268 CrossRef CAS.
- P. Prang, R. Müller, A. Eljaouhari, K. Heckmann, W. Kunz, T. Weber, C. Faber, M. Vroemen, U. Bogdahn and N. Weidner, Biomaterials, 2006, 27, 3560–3569 CAS.
- J. Silver and J. H. Miller, Nat. Rev. Neurosci., 2004, 5, 146–156 CrossRef CAS PubMed.
- D. N. Rocha, P. Brites, C. Fonseca and A. P. Pêgo, PLoS ONE, 2014, 9, e88593 Search PubMed.
- E. Martínez, E. Engel, J. A. Planell and J. Samitier, Ann. Anat. – Anat. Anz., 2009, 191, 126–135 CrossRef PubMed.
- M. S. Lord, M. Foss and F. Besenbacher, Nano Today, 2010, 5, 66–78 CrossRef CAS.
- L. R. Pires, D. N. Rocha, L. Ambrosio and A. P. Pêgo, J. R. Soc., Interface, 2014, 12 Search PubMed.
- A. Aguzzi, B. A. Barres and M. L. Bennett, Science, 2013, 339, 156–161 CrossRef CAS PubMed.
- T. Courtney, M. S. Sacks, J. Stankus, J. Guan and W. R. Wagner, Biomaterials, 2006, 27, 3631–3638 CAS.
- W. A. Riley, R. W. Barnes, G. W. Evans and G. L. Burke, Stroke, 1992, 23, 952–956 CrossRef CAS PubMed.
- D. Aytemiz, W. Sakiyama, Y. Suzuki, N. Nakaizumi, R. Tanaka, Y. Ogawa, Y. Takagi, Y. Nakazawa and T. Asakura, Adv. Healthcare Mater., 2013, 2, 361–368 CrossRef CAS PubMed.
- N. Kasoju and U. Bora, Adv. Healthcare Mater., 2012, 1, 393–412 CrossRef CAS PubMed.
- W. H. Maisel and W. K. Laskey, Circulation, 2007, 115, e426–e427 CrossRef PubMed.
- M. P. Staiger, A. M. Pietak, J. Huadmai and G. Dias, Biomaterials, 2006, 27, 1728–1734 CrossRef CAS PubMed.
- A. Atrens, M. Liu and N. I. Zainal Abidin, J. Mater. Sci. Eng. B, 2011, 176, 1609–1636 CrossRef CAS.
- J. Wang, Y. He, M. F. Maitz, B. Collins, K. Xiong, L. Guo, Y. Yun, G. Wan and N. Huang, Acta Biomater., 2013, 9, 8678–8689 CrossRef CAS PubMed.
- S. Nishio, K. Kosuga, K. Igaki, M. Okada, E. Kyo, T. Tsuji, E. Takeuchi, Y. Inuzuka, S. Takeda, T. Hata, Y. Takeuchi, Y. Kawada, T. Harita, J. Seki, S. Akamatsu, S. Hasegawa, N. Bruining, S. Brugaletta, S. de Winter, T. Muramatsu, Y. Onuma, P. W. Serruys and S. Ikeguchi, Circulation, 2012, 125, 2343–2353 CrossRef CAS PubMed.
- E. Zini, M. Scandola, P. Dobrzynski, J. Kasperczyk and M. Bero, Biomacromolecules, 2007, 8, 3661–3667 CrossRef CAS PubMed.
- J. Dong, L. Liao, L. Shi, Z. Tan, Z. Fan, S. Li and Z. Lu, Polym. Eng. Sci., 2014, 54, 1418–1426 CAS.
- A. Lauto, D. Mawad and L. J. R. Foster, J. Chem. Technol. Biotechnol., 2008, 83, 464–472 CrossRef CAS.
- W. D. Spotnitz, Am. J. Surg., 2001, 182, S8–S14 CrossRef.
- D. M. Toriumi, W. F. Raslan, M. Friedman and M. Tardy, Arch. Otolaryngol., Head Neck Surg., 1990, 116, 546–550 CrossRef CAS.
- T. B. Bruns and J. M. Worthington, Am. Fam. Physician, 2000, 61, 1383–1388 CAS.
- A. I. Bochyńska, S. Sharifi, T. G. van Tienen, P. Buma and D. W. Grijpma, Macromol. Symp., 2013, 334, 40–48 CrossRef.
- S. B. Blanquer, S. Sharifi and D. W. Grijpma, J. Appl. Biomater. Funct. Mater., 2012, 10, 177–184 Search PubMed.
- Y. E. Aguirre-Chagala, J. L. Santos, B. A. Aguilar-Castillo and M. Herrera-Alonso, ACS Macro Lett., 2014, 3, 353–358 CrossRef CAS.
- J. A. Edward, M. K. Kiesewetter, H. Kim, J. C. A. Flanagan, J. L. Hedrick and R. M. Waymouth, Biomacromolecules, 2012, 13, 2483–2489 CrossRef CAS PubMed.
- W. Cheng, C. Yang, J. L. Hedrick, D. F. Williams, Y. Y. Yang and P. G. Ashton-Rickardt, Biomaterials, 2013, 34, 3697–3705 CrossRef CAS PubMed.
- Z. Y. Ong, C. Yang, S. J. Gao, X.-Y. Ke, J. L. Hedrick and Y. Yan Yang, Macromol. Rapid Commun., 2013, 34, 1714–1720 CrossRef CAS PubMed.
- I. Peták, R. Schwab, L. Őrfi, L. Kopper and G. Kéri, Nat. Rev. Drug Discovery, 2010, 9, 523–535 CrossRef PubMed.
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS PubMed.
- C. Sanson, C. Schatz, J.-F. Le Meins, A. Brûlet, A. Soum and S. Lecommandoux, Langmuir, 2010, 26, 2751–2760 CrossRef CAS PubMed.
- R. Langer and N. A. Peppas, AIChE J., 2003, 49, 2990–3006 CrossRef CAS.
- C. Gong, S. Shi, P. Dong, B. Kan, M. Gou, X. Wang, X. Li, F. Luo, X. Zhao, Y. Wei and Z. Qian, Int. J. Pharm., 2009, 365, 89–99 CrossRef CAS PubMed.
- X. Yang, D. Cao, N. Wang, L. Sun, L. Li, S. Nie, Q. Wu, X. Liu, C. Yi and C. Gong, J. Pharm. Sci., 2014, 103, 305–313 CrossRef CAS PubMed.
- T. Ishida and H. Kiwada, Int. J. Pharm., 2008, 354, 56–62 CrossRef CAS PubMed.
- E. Hara, A. Makino, K. Kurihara, F. Yamamoto, E. Ozeki and S. Kimura, Int. Immunopharmacol., 2012, 14, 261–266 CrossRef CAS PubMed.
- F. Goñi-de-Cerio, V. Mariani, D. Cohen, L. Madi, J. Thevenot, H. Oliveira, C. Uboldi, G. Giudetti, R. Coradeghini, E. Garanger, F. Rossi, M. Portugal-Cohen, M. Oron, R. Korenstein, S. Lecommandoux, J. Ponti, B. Suárez-Merino and P. Heredia, J. Nanopart. Res., 2013, 15, 1–17 CrossRef.
- H. Oliveira, J. Thevenot, E. Garanger, E. Ibarboure, P. Calvo, P. Aviles, M. Guillen and S. Lecommandoux, Pharm. Res., 2014, 31, 983–991 CrossRef CAS PubMed.
- D. Bacinello, E. Garanger, D. Taton, K. C. Tam and S. Lecommandoux, Eur. Polym. J., 2015, 62, 363–373 CrossRef CAS.
- X. Jiang, X. Sha, H. Xin, X. Xu, J. Gu, W. Xia, S. Chen, Y. Xie, L. Chen, Y. Chen and X. Fang, Biomaterials, 2013, 34, 2969–2979 CrossRef CAS PubMed.
- X. Jiang, H. Xin, J. Gu, X. Xu, W. Xia, S. Chen, Y. Xie, L. Chen, Y. Chen, X. Sha and X. Fang, Biomaterials, 2013, 34, 1739–1746 CrossRef CAS PubMed.
- X. Wang, Y. Yang, H. Jia, W. Jia, S. Miller, B. Bowman, J. Feng and F. Zhan, Biomater. Sci., 2014, 2, 961–971 RSC.
- X. Jiang, H. Xin, J. Gu, F. Du, C. Feng, Y. Xie and X. Fang, J. Pharm. Sci., 2014, 103, 1487–1496 CrossRef CAS PubMed.
- X. Jiang, H. Xin, Q. Ren, J. Gu, L. Zhu, F. Du, C. Feng, Y. Xie, X. Sha and X. Fang, Biomaterials, 2014, 35, 518–529 CrossRef CAS PubMed.
- J. Mindemark, Y. Tabata and T. Bowden, Macromol. Biosci., 2012, 12, 840–848 CrossRef CAS PubMed.
- M. Yamagata, T. Kawano, K. Shiba, T. Mori, Y. Katayama and T. Niidome, Bioorg. Med. Chem., 2007, 15, 526–532 CrossRef CAS PubMed.
- M. Bertschinger, G. Backliwal, A. Schertenleib, M. Jordan, D. L. Hacker and F. M. Wurm, J. Controlled Release, 2006, 116, 96–104 CrossRef CAS PubMed.
- Z. Y. Ong, K. Fukushima, D. J. Coady, Y.-Y. Yang, P. L. R. Ee and J. L. Hedrick, J. Controlled Release, 2011, 152, 120–126 CrossRef CAS PubMed.
- J. P. K. Tan, S. H. Kim, F. Nederberg, E. A. Appel, R. M. Waymouth, Y. Zhang, J. L. Hedrick and Y. Y. Yang, Small, 2009, 5, 1504–1507 CrossRef CAS PubMed.
- K. Fukushima, R. C. Pratt, F. Nederberg, J. P. K. Tan, Y. Y. Yang, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2008, 9, 3051–3056 CrossRef CAS PubMed.
- F. Nederberg, E. Appel, J. P. K. Tan, S. H. Kim, K. Fukushima, J. Sly, R. D. Miller, R. M. Waymouth, Y. Y. Yang and J. L. Hedrick, Biomacromolecules, 2009, 10, 1460–1468 CrossRef CAS PubMed.
- C. Zhang, D. Liu, X. Zhang, P. Wang, Z. Zhen, J. Li, D. Yi, Y. Jin and D. Yang, J. Appl. Polym. Sci., 2015, 132 Search PubMed , 41815.
- L. Yang, J. Li, S. Meng, Y. Jin, J. Zhang, M. Li, J. Guo and Z. Gu, Polymer, 2014, 55, 5111–5124 CrossRef CAS.
- J. P. K. Tan, S. H. Kim, F. Nederberg, K. Fukushima, D. J. Coady, A. Nelson, Y. Y. Yang and J. L. Hedrick, Macromol. Rapid Commun., 2010, 31, 1187–1192 CrossRef CAS PubMed.
- S. H. Kim, J. P. K. Tan, F. Nederberg, K. Fukushima, J. Colson, C. Yang, A. Nelson, Y.-Y. Yang and J. L. Hedrick, Biomaterials, 2010, 31, 8063–8071 CrossRef CAS PubMed.
- X.-Y. Ke, V. W. Lin Ng, S.-J. Gao, Y. W. Tong, J. L. Hedrick and Y. Y. Yang, Biomaterials, 2014, 35, 1096–1108 CrossRef CAS PubMed.
- A. B. Ebrahim Attia, C. Yang, J. P. K. Tan, S. Gao, D. F. Williams, J. L. Hedrick and Y.-Y. Yang, Biomaterials, 2013, 34, 3132–3140 CrossRef CAS PubMed.
- C. Yang, A. B. Ebrahim Attia, J. P. K. Tan, X. Ke, S. Gao, J. L. Hedrick and Y.-Y. Yang, Biomaterials, 2012, 33, 2971–2979 CrossRef CAS PubMed.
- Y. Zhao, Macromolecules, 2012, 45, 3647–3657 CrossRef CAS.
- A. Matsumoto, K. Yamamoto, R. Yoshida, K. Kataoka, T. Aoyagi and Y. Miyahara, Chem. Commun., 2010, 46, 2203–2205 RSC.
- I. Dasgupta, E. A. Tanifum, M. Srivastava, S. S. Phatak, C. N. Cavasotto, M. Analoui and A. Annapragada, PLoS One, 2012, 7, e29585 CAS.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- C. Hiemstra, W. Zhou, Z. Zhong, M. Wouters and J. Feijen, J. Am. Chem. Soc., 2007, 129, 9918–9926 CrossRef CAS PubMed.
- M. K. Joo, M. H. Park, B. G. Choi and B. Jeong, J. Mater. Chem., 2009, 19, 5891–5905 RSC.
- S. Q. Liu, C. Yang, Y. Huang, X. Ding, Y. Li, W. M. Fan, J. L. Hedrick and Y.-Y. Yang, Adv. Mater., 2012, 24, 6484–6489 CrossRef CAS PubMed.
- J. Jansen, G. Mihov, J. Feijen and D. W. Grijpma, Macromol. Biosci., 2012, 12, 692–702 CrossRef CAS PubMed.
- H. J. Moon, D. Y. Ko, M. H. Park, M. K. Joo and B. Jeong, Chem. Soc. Rev., 2012, 41, 4860–4883 RSC.
- L. Xi, T. Wang, F. Zhao, Q. Zheng, X. Li, J. Luo, J. Liu, D. Quan and J. Ge, PLoS One, 2014, 9, e100632 Search PubMed.
- S. H. Kim, J. P. K. Tan, K. Fukushima, F. Nederberg, Y. Y. Yang, R. M. Waymouth and J. L. Hedrick, Biomaterials, 2011, 32, 5505–5514 CrossRef CAS PubMed.
- H. Ajiro, Y. Takahashi and M. Akashi, Macromolecules, 2012, 45, 2668–2674 CrossRef CAS.
- H. Ajiro, Y. Takahashi, M. Akashi and T. Fujiwara, Macromol. Biosci., 2012, 12, 1315–1320 CrossRef CAS PubMed.
- H. Nikaido, Annu. Rev. Biochem., 2009, 78, 119–146 CrossRef CAS PubMed.
- F. Nederberg, Y. Zhang, J. P. K. Tan, K. Xu, H. Wang, C. Yang, S. Gao, X. D. Guo, K. Fukushima, L. Li, J. L. Hedrick and Y.-Y. Yang, Nat. Chem., 2011, 3, 409–414 CrossRef CAS PubMed.
- E. F. Palermo and K. Kuroda, Biomacromolecules, 2009, 10, 1416–1428 CrossRef CAS PubMed.
- E. F. Palermo, S. Vemparala and K. Kuroda, Biomacromolecules, 2012, 13, 1632–1641 CrossRef CAS PubMed.
- K. Kuroda and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 4128–4129 CrossRef CAS PubMed.
- Y. Oda, S. Kanaoka, T. Sato, S. Aoshima and K. Kuroda, Biomacromolecules, 2011, 12, 3581–3591 CrossRef CAS PubMed.
- E. Palermo and K. Kuroda, Appl. Microbiol. Biotechnol., 2010, 87, 1605–1615 CrossRef CAS PubMed.
- A. C. Engler, N. Wiradharma, Z. Y. Ong, D. J. Coady, J. L. Hedrick and Y.-Y. Yang, Nano Today, 2012, 7, 201–222 CrossRef CAS.
- Y. Qiao, C. Yang, D. J. Coady, Z. Y. Ong, J. L. Hedrick and Y.-Y. Yang, Biomaterials, 2012, 33, 1146–1153 CrossRef CAS PubMed.
- V. W. L. Ng, J. P. K. Tan, J. Leong, Z. X. Voo, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2014, 47, 1285–1291 CrossRef CAS.
- A. C. Engler, J. P. K. Tan, Z. Y. Ong, D. J. Coady, V. W. L. Ng, Y. Y. Yang and J. L. Hedrick, Biomacromolecules, 2013, 14, 4331–4339 CrossRef CAS PubMed.
- V. W. L. Ng, X. Ke, A. L. Z. Lee, J. L. Hedrick and Y. Y. Yang, Adv. Mater., 2013, 25, 6730–6736 CrossRef CAS PubMed.
- W. Chin, C. Yang, V. W. L. Ng, Y. Huang, J. Cheng, Y. W. Tong, D. J. Coady, W. Fan, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2013, 46, 8797–8807 CrossRef CAS.
- S. Venkataraman, A. L. Lee, H. T. Maune, J. L. Hedrick, V. M. Prabhu and Y. Y. Yang, Macromolecules, 2013, 46, 4839–4846 CrossRef CAS.
- K. Fukushima, J. P. K. Tan, P. A. Korevaar, Y. Y. Yang, J. Pitera, A. Nelson, H. Maune, D. J. Coady, J. E. Frommer, A. C. Engler, Y. Huang, K. Xu, Z. Ji, Y. Qiao, W. Fan, L. Li, N. Wiradharma, E. W. Meijer and J. L. Hedrick, ACS Nano, 2012, 6, 9191–9199 CrossRef CAS PubMed.
- Y. Li, K. Fukushima, D. J. Coady, A. C. Engler, S. Liu, Y. Huang, J. S. Cho, Y. Guo, L. S. Miller, J. P. K. Tan, P. L. R. Ee, W. Fan, Y. Y. Yang and J. L. Hedrick, Angew. Chem., Int. Ed., 2013, 52, 674–678 CrossRef CAS PubMed.
- K. Fukushima, S. Liu, H. Wu, A. C. Engler, D. J. Coady, H. Maune, J. Pitera, A. Nelson, N. Wiradharma, S. Venkataraman, Y. Huang, W. Fan, J. Y. Ying, Y. Y. Yang and J. L. Hedrick, Nat. Commun., 2013, 4 Search PubMed.
- C. J. Newcomb, S. Sur, J. H. Ortony, O.-S. Lee, J. B. Matson, J. Boekhoven, J. M. Yu, G. C. Schatz and S. I. Stupp, Nat. Commun., 2014, 5 Search PubMed.
- M. Tanaka, K. Sato, E. Kitakami, S. Kobayashi, T. Hoshiba and K. Fukushima, Polym. J., 2015, 47, 114–121 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |