
Benchmarking nitrous oxide adsorption and activation in metal–organic frameworks bearing coordinatively unsaturated metal centers†
Tristan A.
Pitt
a,
Haojun
Jia
 bc,
Tyler J.
Azbell
a,
Mary E.
Zick
a,
Aditya
Nandy
bc,
Heather J.
Kulik
bc and
Phillip J.
Milner
*a
bc,
Tyler J.
Azbell
a,
Mary E.
Zick
a,
Aditya
Nandy
bc,
Heather J.
Kulik
bc and
Phillip J.
Milner
*a
aDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14850, USA. E-mail: pjm347@cornell.edu
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 23rd January 2024
Abstract
Anthropogenic emissions of N2O, the third most abundant greenhouse gas after CO2 and CH4, are contributing to global climate change. Although metal–organic frameworks (MOFs) have been widely studied as adsorbents for CO2 and CH4, less effort has focused on the use of MOFs to remove N2O from emission streams or from air. Further, N2O activation would enable its use as an inexpensive oxidant for fine chemical synthesis. Herein, we identify features that contribute to strong binding and high uptake of N2O at coordinatively unsaturated metal sites in the M2Cl2(btdd) (M = Mn, Co, Ni, Cu; btdd2− = bis(1,2,3-triazolo[4,5-b],[4′,5′-i])dibenzo[1,4]dioxin) and M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn; dobdc4− = 2,5-dioxido-1,4-benzenedicarboxylate) series of MOFs. Combined experimental and computational studies suggest that N2O adsorption at open-metal-sites is primarily based on electrostatic interactions, rather than π-backbonding, causing MOFs with more Lewis acidic metal centers to be superior N2O adsorbents. As a result, Mg2(dobdc) demonstrates strong binding and record-setting N2O uptake (8.75 mmol g−1 at 1 bar and 298 K). Using density functional theory (DFT) to characterize reactive intermediates and transition states, we demonstrate that N2O activation to form a M(IV)–oxo species and N2 is thermodynamically favorable in Mn2(dobdc) and Fe2(dobdc) but appears to be kinetically limited in Mn2(dobdc). Our work lays a foundation for understanding N2O adsorption and activation in MOFs, paving the way for the design of promising next-generation materials for N2O capture and utilization.
Introduction
N2O is the third most prevalent anthropogenic greenhouse gas after CO2 and CH4, accounting for 6% of the effective radiative forcing from 1960 to 2019.1 Although CO2 and CH4 are present in higher concentrations in the atmosphere, the global warming potential of N2O (265) is far greater (1 and 28 for CO2 and CH4, respectively) and its atmospheric lifetime (116 year) is far longer (1 and 12 year for CO2 and CH4, respectively).2 Over the last four decades, global anthropogenic emissions of N2O have increased by 30%.3 Up to 87% of this increase derives from agricultural practices such as nitrogen additions to soils. By nature, the sources of these emissions (i.e., farmlands) are diffuse, in contrast to point sources of N2O emissions such as adipic and nitric acid manufacturing.4 Diffuse emissions are currently largely uncontrolled, except for preventative measures such as the use of more efficient fertilizers.5,6 At point sources, N2O can be catalytically destroyed, but regulations requiring this practice have not been globally adopted.7 Thus, the majority of human-caused N2O emissions are currently unabated, leading to a current estimated rate of increase in atmospheric N2O concentration of 2% per decade.3 In addition to its global warming potential, N2O was found to be the dominant ozone-depleting substance emitted in the 21st century,4 underscoring the urgency of curtailing N2O emissions. The environmental effects of anthropogenic N2O emissions and its long atmospheric lifetime necessitate the development of new materials for N2O capture.Selectively capturing gases from diffuse sources such as air is a unique challenge that requires specially designed sorbents. Metal–organic frameworks (MOFs) are an emerging class of materials that have drawn significant interest for their potential applications in greenhouse gas capture,8 separations,9 catalysis,10 and beyond.11 They are crystalline, highly porous materials formed by connecting metal secondary building units (SBU) with multitopic organic linkers. MOFs have been designed to selectively bind CO2 and CH4 based on electrostatic interactions,12,13 chemical reactivity,14–16 hydrogen bonding,17 and more. In contrast, only a small number of largely unrelated MOFs have been studied for N2O capture to date.18–25 The presence of Lewis acidic open-metal-sites has been shown to enhance N2O binding;19 however, a general lack of structure–property trends informing the design of new materials hinders improvements in N2O capture in porous materials.
Herein, we present a structure–activity study of N2O adsorption in MOFs, with the purpose of identifying features that lead to strong binding and high uptake. Through a combined experimental and computational analysis, we clarify the effects of the ligand field and metal identity on N2O adsorption at Lewis acidic open-metal-sites in the M2Cl2(btdd) (M = Mn, Co, Ni, Cu; btdd2− = bis(1,2,3-triazolo[4,5-b],[4′,5′-i])dibenzo[1,4]dioxin)26–28 and M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn; dobdc4− = 2,5-dioxido-1,4-benzenedicarboxylate) series of MOFs.12,29,30 From this analysis, we identify the M2(dobdc) series, particularly Mg2(dobdc) and Ni2(dobdc), as promising adsorbents that display strong binding and record-setting N2O adsorption capacities.
Beyond reducing its environmental impact, N2O capture is incentivized by the opportunity to utilize it as a cheap, abundant, and potent oxidant in organic synthesis.31–33 As a kinetically inert molecule, transition metal catalysts and/or high temperatures and pressures are required to facilitate N2O activation. Thus, MOFs and zeolites containing Fe(II) sites have been explored as catalysts for N2O utilization.34–39 In particular, Fe2(dobdc) has been demonstrated to catalyze C–H oxidation of hydrocarbons using N2O as an oxidant.37,40–42 Bearing strong similarities to enzymatic iron-based catalysts,43–45 this occurs through a 2 e− transfer from high spin Fe(II) to the oxygen of N2O, cleaving the N–O bond and forming a high spin Fe(IV)–oxo intermediate. This process is calculated to be followed by σ-attack and H atom abstraction by the Fe(IV)–oxo and radical rebound to generate the oxidized product.40 Herein, DFT calculations support that N2O activation is also thermodynamically favorable in Mn2(dobdc), and we map out an approximate reaction coordinate for this process using the climbing-image nudged elastic band method (CI-NEB). While combined analysis of computation and experiment suggests that this reaction is kinetically limited in Mn2(dobdc), our work motivates further study of N2O activation in related Mn-based MOFs. Overall, our findings stimulate the development of new framework materials for selective N2O capture and conversion to mitigate its environmentally destructive impact.
Results and discussion
N2O adsorption analysis
N2O is a weakly σ-donating and π-accepting ligand, which has limited the number of well-characterized transition metal adducts of N2O to only a small handful, typically bound as η1-N or η2-N,N coordinated N2O through π-backbonding interactions.46–52 Although N2O has a relatively weak dipole moment (0.161 D),53 sorbents could compensate by incorporating Lewis acidic sites to strengthen electrostatic interactions between N2O and the sorbent material. MOFs containing highly Lewis acidic coordinatively unsaturated metal centers are effective adsorbents for a wide variety of gases through strong electrostatic interactions.54 To identify robust structure–property trends regarding N2O binding at open-metal-sites, we targeted isostructural frameworks accommodating a range of metal ions in similar ligand fields. These criteria should effectively yield insights into the interplay of the metal identity and ligand field on the binding strength of N2O at open-metal-sites in MOFs.Two groups of MOFs that fulfil these requirements are the M2Cl2(btdd) and M2(dobdc) series. These MOFs are composed of hexagonal-pored structures formed by connecting one-dimensional, rod-like metal SBUs with ditopic organic linkers (Fig. 1). Porous channels extend through the materials and, after activation, become lined with a high density of coordinatively unsaturated metal sites confined in square pyramidal geometries. While the overall structures of the M2Cl2(btdd) and M2(dobdc) series are similar, their SBUs lead to distinctive ligation of the metal sites. In M2Cl2(btdd), metal ions are coordinated to three individual triazolate groups through one nitrogen atom per triazolate. Each triazolate coordinates to three distinct metal ions, and two chlorides (μ2) bridge neighboring metals, forming M–Cl chains. For this study, the isostructural Mn, Co, Ni, and Cu MOFs of this series were synthesized according to reported procedures (Fig. 1a and Sections S3–S6, ESI†).26,28,55 In the M2(dobdc) series, the secondary building units are composed of metal-salicylate chains, forming an oxygen-based ligand field. The Mg, Mn, Fe, Co, Ni, Cu, and Zn variants of M2(dobdc) were synthesized in accordance with the literature (Fig. 1b and Sections S7–S13, ESI†).12,30,56–59 All MOFs analyzed in this study were confirmed to be highly crystalline and match the expected structures via powder X-ray diffraction (PXRD). The porosity of all MOFs was confirmed using 77 K N2 adsorption/desorption measurements. The calculated surface areas are similar to those reported in the literature in every case.
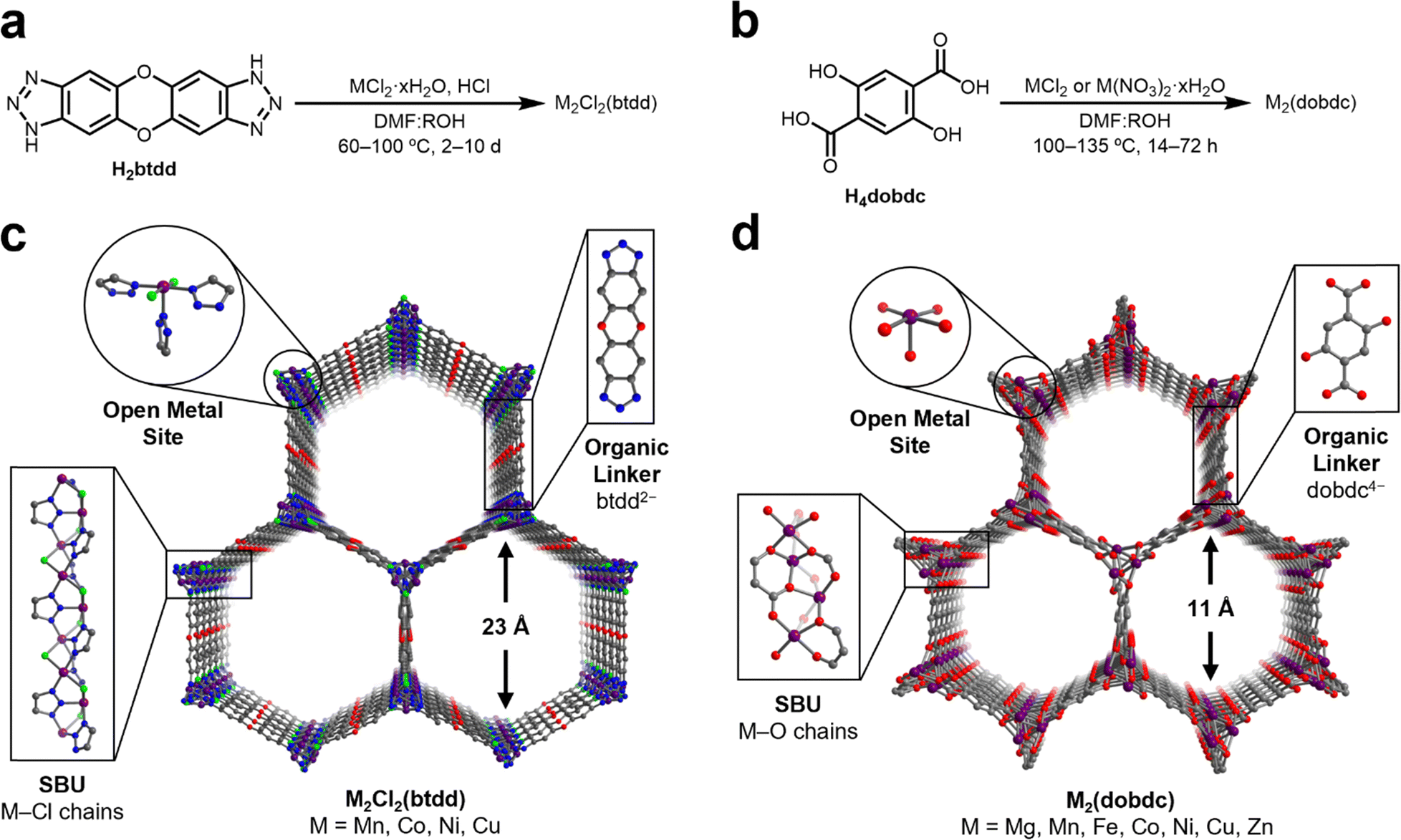 | ||
| Fig. 1 General synthesis conditions of (a) M2Cl2(btdd) (M = Mn, Co, Ni, Cu) and (b) M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn) MOFs (R = Et, Me, iPr, H). Structures of (c) M2Cl2(btdd) and (d) M2(dobdc). Purple, light green, blue, grey, and red spheres represent metal, chlorine, nitrogen, carbon, and oxygen atoms, respectively. Hydrogen atoms are omitted for clarity. | ||
After synthesis, the MOFs were each evaluated as N2O sorbents by measuring N2O adsorption and desorption isotherms at 25 °C, 35 °C, and 45 °C (Sections S3–S13, ESI†). Adsorption data were fit using dual-site Langmuir–Freundlich models (eqn (S1), ESI†). These fits were subsequently used to calculate enthalpies of N2O adsorption (−ΔHads) in each MOF using the Clausius–Clapeyron equation (eqn (S2), ESI†). The resulting −ΔHads values as a function of N2O uptake in both series of MOFs are summarized in Fig. 2 and Table 1. Critically, PXRD and surface area measurements confirm the stability of every MOF towards N2O except for Fe2(dobdc), which has been previously shown to react irreversibly with this gas.37,40–42 As a result, the adsorption data for this MOF were excluded from the analysis below.
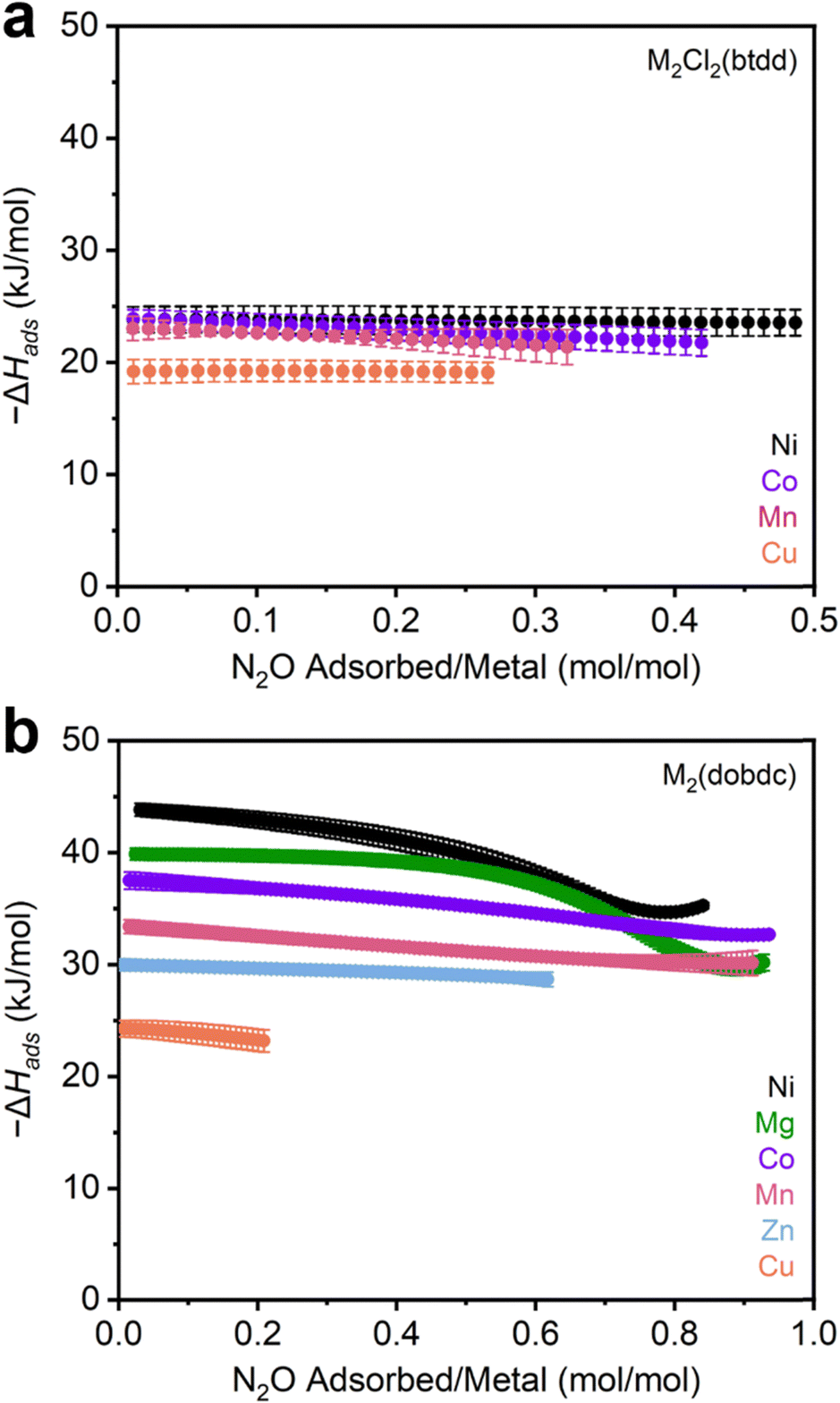 | ||
| Fig. 2 (a) –ΔHadsvs. N2O uptake calculated from N2O adsorption isotherms in (a) M2Cl2(btdd) (M = Ni, Co, Cu, Mn) and (b) M2(dobdc) (M = Ni, Mg, Co, Mn, Zn, Cu) MOFs. | ||
| MOF | −ΔHads N2O (kJ mol−1) | Maximum uptake at 298 K (mmol g−1) |
|---|---|---|
| Mn2Cl2(btdd) | 23.0 ± 1.1 | 2.37 |
| Co2Cl2(btdd) | 23.9 ± 0.8 | 3.10 |
| Ni2Cl2(btdd) | 23.8 ± 1.1 | 3.49 |
| Cu2Cl2(btdd) | 19.2 ± 1.1 | 1.86 |
| Mg2(dobdc) | 39.9 ± 0.5 | 8.75 |
| Mn2(dobdc) | 33.4 ± 0.6 | 7.77 |
| Co2(dobdc) | 37.5 ± 0.8 | 7.26 |
| Ni2(dobdc) | 43.8 ± 0.6 | 6.44 |
| Cu2(dobdc) | 24.3 ± 0.7 | 2.21 |
| Zn2(dobdc) | 30.0 ± 0.5 | 5.37 |
The M2Cl2(btdd) series were first evaluated as N2O sorbents. Despite their high density of open-metal-sites, all MOFs of this series only weakly interact with N2O (−ΔHads < 25 kJ mol−1). The difference in binding strength between the variants is minor: Mn, Co, and Ni analogues exhibit similar binding strengths, followed by Cu (Table 1). MOFs bearing coordinatively unsaturated Cu sites are commonly poor adsorbents due to Jahn–Teller axial distortion at the Cu sites.12,60 Lacking substantial differences in binding strength between variants, the influence of the metal identity appears to be overshadowed by that of the ligand field in the M2Cl2(btdd) series. The Lewis acidities of the metals are likely tempered by the electron-donating coordination environment of triazolate and chloride ligands. These ligands produce electron-rich metal centers that are less Lewis acidic than those found in other materials. Although these MOFs are capable of binding polar gases such as NH3,26 the absence of a significant dipole moment on N2O makes the M2Cl2(btdd) MOFs ineffective N2O adsorbents. Lacking suitable binding sites, the M2Cl2(btdd) series show very limited uptakes; the quantity of N2O adsorbed under equilibrium conditions, even at 1000 mbar N2O and 298 K (Table 1), does not come close to saturating the available open-metal-sites.
Decreasing the ligand field strength surrounding the open-metal-sites should produce more Lewis acidic metal centers and thus lead to stronger electrostatic interactions with N2O. Switching from M2Cl2(btdd) to M2(dobdc) preserves the coordination geometry of the metal centers, but the oxygen-based SBU provides a weaker ligand field overall. Indeed, every member of the M2(dobdc) series binds N2O more strongly at low pressures than the M2Cl2(btdd) MOFs (Fig. 2b). Moreover, the comparatively electron-deficient coordination environments in this series emphasize the influence of the metal identity on N2O binding strengths; enthalpies of adsorption vary by as much as 20 kJ mol−1 among M2(dobdc) variants (Table 1). N2O binding strengths in these series mirror the empirical Irving–Williams series: Cu < Zn < Mn < Co < Mg < Ni.61 Like Cu2Cl2(btdd), Cu2(dobdc) likely exhibits weak N2O adsorption because of axial distortion.12 In contrast, Mg2(dobdc) is an especially effective N2O adsorbent due to the hard nature of its Lewis acidic Mg(II) cations. As the effective charge of the transition metal centers increases from Mn to Ni,62 the N2O binding strengths increase as well. As a result, Ni2(dobdc) is the strongest N2O adsorbent assessed in this study (−ΔHads = 43.8 ± 0.6 kJ mol−1). Compared to other commonly studied gases (CO2, O2, N2), N2O is generally more strongly bound by the M2(dobdc) series, which is likely due to its modest dipole moment. In particular, N2O binding is approximately 2–6 kJ mol−1 stronger than CO2 in all cases except for in Mg2(dobdc), in which CO2 binds more strongly by approximately 3 kJ mol−1.12 O2 and N2 binding enthalpies are consistently 10–20 kJ lower than those of N2O as well.63 These findings highlight the discrepancy between the comparatively strong binding of N2O at metal centers compared to CO2, O2, and N2 and the lack of well-characterized N2O-bound metal complexes.46–50
Strong N2O binding is accompanied by relatively high N2O capacities in the M2(dobdc) series of frameworks. In particular, the second-strongest adsorbent of N2O identified in this work, Mg2(dobdc), adsorbs 8.75 mmol g−1 at 1000 mbar of N2O and 298 K, surpassing the previous record-holder, MIL-100 (Cr) (5.78 mmol g−1, MIL = Materials Institute Lavoisier), by a significant margin.20 The Mn, Co, and Ni M2(dobdc) variants also exceed the previous record for N2O uptake in a MOF. Overall, this comparison underlines the M2(dobdc) series, especially Mg2(dobdc), as promising adsorbents that exhibit high gravimetric N2O capacities coupled with strong and tuneable binding strengths.
Computational survey of N2O adsorption.
After establishing the M2(dobdc) series as effective N2O adsorbents, we further evaluated N2O binding in this series using DFT calculations (Section S14, ESI†). For these calculations, trimetallic cluster models were generated to approximately study the one-dimensional chain SBU (Fig. 3a). Similar cluster models have been used to evaluate N2O reduction in Fe2(dobdc).40,41 For each member of the M2(dobdc) series, excluding Mg2(dobdc) for redundancy, two types of cluster models were simulated: a trimetallic system and a Mg-diluted system. In the Mg-diluted models, both edge metal ions were replaced with Mg(II) ions. The purpose of including the Mg-diluted systems is to simulate only one open-shell metal center and decouple adsorption energetics from the potential influence of metal–metal coupling on N2O adsorption. All systems are neutral with all M(II) ions in the high-spin state where applicable (Table S13, ESI†).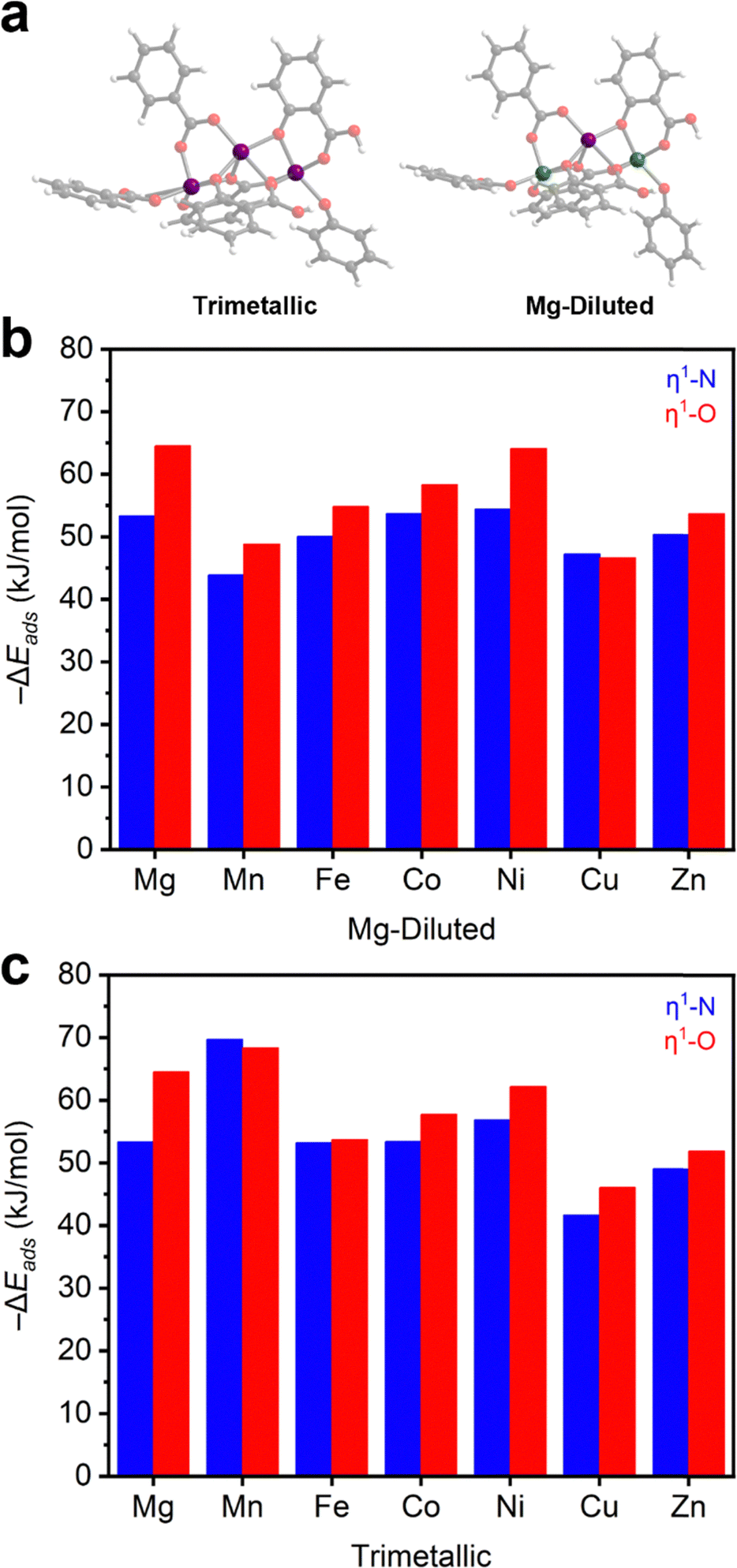 | ||
| Fig. 3 (a) Trimetallic (left) and Mg-diluted (right) cluster models of M2(dobdc). Purple, green, grey, red, and white spheres represent metal, magnesium, carbon, oxygen, and hydrogen atoms, respectively. DFT-calculated negative adsorption energies (−ΔEads) of η1-N (blue) and η1-O (red) coordinated N2O in the (b) Mg-diluted and (c) trimetallic systems. | ||
The energies of adsorption (−ΔEads) for both η1-N and η1-O coordinated N2O adducts in the model clusters were first calculated (Fig. 3b–c and Table S11, ESI†). In both the trimetallic and Mg-diluted models, η1-O coordinated N2O is slightly more stabilized (2–10 kJ mol−1) than η1-N coordinated N2O. This aligns with the approximately 60%/40% η1-O/η1-N population split determined from neutron diffraction data collected on N2O-dosed Fe2(dobdc).37 The calculated formation energies of N2O adducts in the Mg-diluted series (Fig. 3b) approximately match the trend observed experimentally: Cu ≈ Mn < Fe ≈ Zn < Co < Ni ≈ Mg. However, some divergence is noted in the trimetallic systems (Fig. 3c): the Mn-based trimetallic system binds N2O considerably more strongly than the equivalent Mg-diluted model, suggesting multiple metal effects that require consideration of metal–metal coupling (see below). Additionally, η1-N coordination to Mn2(dobdc) is slightly more favorable than η1-O in the trimetallic model.
Across the series, the model systems reveal that η1-O and η1-N adducts of N2O are bent, with bond angles and M–N2O distances in agreement with those solved from the neutron diffraction data in N2O–Fe2(dobdc) (Table S12, ESI†), the only definitively characterized structure of N2O bound to a metal center within a MOF reported to date.37 In the DFT-calculated structure, η1-N coordinated N2O exhibits a typical bond angle of 115–123° (122° in N2O–Fe2(dobdc)), whereas η1-O coordinated N2O exhibits slightly smaller bond angles ranging from 105–120° (117° in N2O–Fe2(dobdc)). Bond lengths of η1-N and η1-O coordinated N2O (2.25–2.57 Å and 2.27–2.54 Å, respectively) vary over only a narrow range. These models reinforce that N2O binding across the M2(dobdc) series is primarily based on electrostatic interactions rather than π-backbonding, which would be expected to lead to linear η1-N (V, Cu, Ru, Rh) or side-on η2-N,N (Co, Ni) interactions with N2O.46–52
It should be noted that N2O adsorption has been modelled previously in a small variety of other open-metal-site MOFs. The Fe(II)- and Cu(II)-based paddlewheel nodes in M3(btc)2 (btc3− = benzene-1,3,5-tricarboxylate) MOFs show bent η1-N and η1-O coordination modes for N2O.54 Likewise, N2O adducts have been modelled for the trinuclear carboxylate-bridged, oxygen-centered nodes (M3(μ3-O)(RCOO)6, M = V, Cr, Mn, Fe, Co, Ni) common among MOFs such as MIL-100, MIL-101, and MIL-127.64 The calculated binding of η1-N coordinated N2O at V centers is linear, but other adducts are bent, with similar bond angles and bond lengths as calculated in the M2(dobdc) series herein. In the Kuratowski-type SBU of Cu-MFU-4l (MFU = Metal–Organic Framework Ulm-University), DFT calculations support an approximately linear η1-N coordinated N2O molecule, indicating possible π-backbonding from the Cu(I) centers.19 Overall, these findings support that N2O is predicted to bind in a bent fashion at most metal centers in MOFs.
A notable exception to the trends outlined above is the trimetallic Mn cluster, in which η1-N coordinated N2O is nearly linear (172°), suggesting that π-backbonding occurs from the Mn d orbitals into the π* orbital of N2O, which has previously been invoked in linear η1-N V, Cu, Ru, and Rh adducts of N2O to justify the stability of those complexes.46,48–50,52 Consistently, the Mn–N bond length (1.95 Å) is considerably shorter than in other models (Fig. S68, ESI†). These characteristics are distinct from the equivalent Mg-diluted cluster, suggesting that metal–metal coupling may affect the binding mode of N2O in these calculations. We thus evaluated N2O binding in the open-shell trimetallic systems (Mn through Cu) using broken-symmetry density functional theory (BS-DFT) with two spin flip configurations (i.e., in the central metal or in one edge metal) to quantify the metal–metal coupling and to extract coupling constants (Fig. S69 and Section S14, ESI†). To ensure that the BS-DFT calculations converged to the desired states, the spin density was visually inspected (Fig. S70, ESI†). In particular, the energetic difference in the trimetallic Mn system in comparison to the dilute case indicates especially strong metal–metal coupling (Table S14, ESI†). From this data, we calculated strong magnetic coupling along the SBU chains in the trimetallic Mn system (J = 314.27 cm−1), which becomes even stronger after N2O binding regardless of the coordination mode (J = 857.47 cm−1 for η1-N, J = 857.47 cm−1 for η1-O). Consistently, magnetic susceptibility measurements (Fig. S77, ESI†) and prior first principles studies support that Mn2(dobdc) exhibits antiferromagnetic coupling below approximately 27 K.62,65 Adsorbate-induced changes in magnetic coupling are also precedented in the M2(dobdc) series; for example, in Fe2(dobdc), the ferromagnetic exchange strength along the SBU chains is attenuated by interaction with weak adsorbates (e.g., CH4, −ΔHads = 20 kJ mol−1), and the coupling becomes antiferromagnetic upon interaction with strong adsorbates (e.g., C2H2, −ΔHads = 47 kJ mol−1).66 Our results point to an additional stabilization of the N2O adducts of Mn2(dobdc) related to enhanced metal coupling, possibly leading to greater π-backbonding from Mn to N2O and favoring linear η1-N coordination. Above 27 K, however, this effect is no longer expected to significantly contribute to the adsorption interactions as the spins become randomly oriented. Consistently, the experimental binding enthalpies do not show enhanced adsorption in Mn2(dobdc) relative to other MOFs in the series. Nevertheless, if a suitable Mn-based open-metal-site MOF with a higher Néel temperature were to be identified, this feature could potentially be leveraged to enhance N2O binding.
Evaluating N2O activation in the M2(dobdc) series
N2O activation to form M(IV)–oxo species and N2 in MOFs has been principally studied in Fe2(dobdc) and other Fe(II)-based frameworks.37–41,67,68 Unfortunately, Fe(II)-based systems are prohibitively air sensitive for practical applications. Identifying more air-stable materials capable of N2O activation would be a significant step towards utilizing N2O as a green oxidant. In order to determine whether this reactivity is unique to Fe2(dobdc), the same cluster models were used to calculate the favorability of M(IV)–oxo formation in the remainder of the M2(dobdc) series (Table S15 and Section 14, ESI†). DFT calculations with both the trimetallic and Mg-diluted cluster model series show that, in addition to Fe2(dobdc), M(IV)–oxo formation is thermodynamically favorable (ΔEf < 0 kJ mol−1) in Mn2(dobdc) (Fig. 4a and Section 14, ESI†). Indeed, M(IV)–oxo formation is calculated to be significantly more thermodynamically favorable in Mn2(dobdc) (−183.1 and −176.1 kJ mol−1 for trimetallic and Mg-diluted systems, respectively) than in Fe2(dobdc) (−55.5 and −52.7 kJ mol−1 for trimetallic and Mg-diluted systems, respectively). In contrast, M(IV)–oxo formation is endothermic in the Mg, Co, Ni, Cu, and Zn analogues, ruling out these materials as potential catalysts for N2O activation. This is fairly expected due to the electronic instability of terminal M(IV)–oxo complexes of octahedral symmetry possessing greater than five d electrons.69 Precedent for N2O activation in Mn-based systems is found in manganese oxides, which catalyze the decomposition of N2O70 and the oxidation of 1-butene at high temperatures.71 Likewise, Mn-substituted polyoxometalates have been shown to activate N2O and catalyze the epoxidation of alkenes,72,73 and Mn-substituted zeolites catalyze N2O decomposition as well.74,75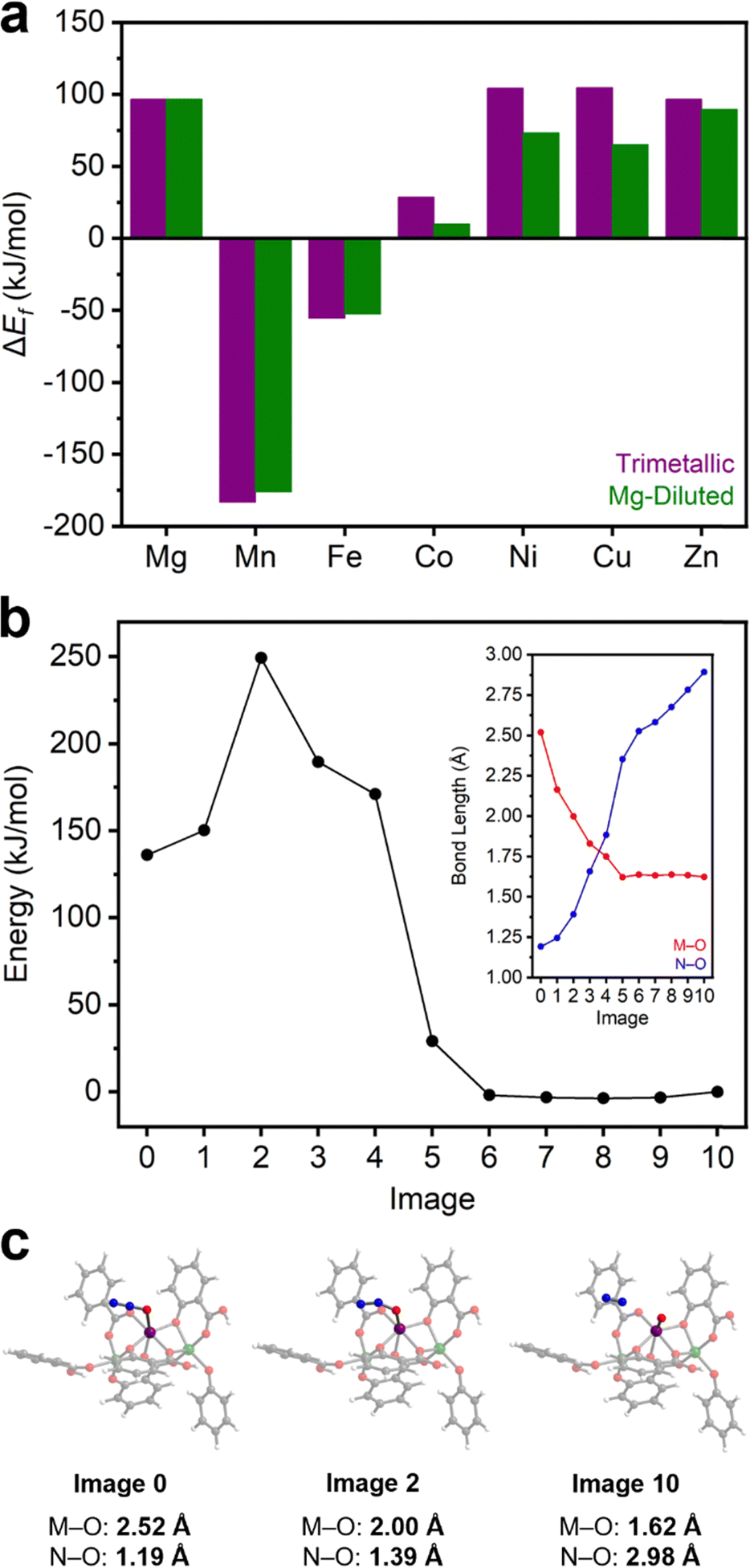 | ||
| Fig. 4 (a) DFT-calculated energies of M–oxo formation (ΔEf) in the trimetallic (purple) and Mg-diluted (green) models of M2(dobdc). (b) CI-NEB-calculated approximate reaction coordinate of Mn–oxo formation from η1-O coordinated N2O in the Mg-diluted system. Inset: M–O (red) and N–O (blue) bond lengths vs. image number. (c) Structures of the initial (left), transition (center), and final (right) states. Purple, green, blue, grey, red, and white spheres represent manganese, magnesium, nitrogen, carbon, oxygen, and hydrogen atoms, respectively. | ||
To determine the potential viability of N2O activation by Mn2(dobdc), an approximate reaction coordinate for this process was modelled using the CI-NEB method (Fig. 4b and Text S1, ESI†). The η1-O coordinated N2O adduct of the Mg-diluted Mn2(dobdc) cluster model was used as the initial state, and the Mn(IV)–oxo-containing Mg-diluted cluster was used as the final state (Section S14, ESI†). During N2O activation, the Mn(IV)–oxo bond is formed and the O–N bond is broken, forming N2. From the initial state, the M–O bond length shrinks significantly and is matched by a substantial lengthening of the O–N bond. During this transition, the energy of the system rises sharply early on, after which it falls to roughly the energy of the final state. Likewise, a constant M–O bond length, consistent with Mn(IV)–oxo formation, was observed in the second half of the reaction coordinate, as the rest of the pathway is characterized by O–N bond elongation as unbound N2 moves away from the cluster. The approximate transition state of the reaction is rather early, with Mn–O and N–O bond lengths of 2.00 Å and 1.39 Å, respectively (Fig. 4c). From the difference in energy between the initial state and this approximate transition state, the kinetic barrier to N2O activation in this model cluster was calculated to be approximately 113 kJ mol−1. This barrier is comparable to the calculated activation barriers of 167 kJ mol−1 in the Mn-based trinuclear MOF nodes and 109 kJ mol−1 in the Mn-substituted polyoxometalate discussed above.64,73
In previous studies, heating N2O-dosed Fe2(dobdc) at only 35 °C was sufficient to partially oxidize the material, and its complete oxidation was accomplished at 60 °C after prolonged heating.37 The activation barrier for Fe(IV)–oxo formation in N2O-bound Fe2(dobdc) was calculated to be 94 kJ mol−1 (enthalpy of activation = 82 kJ mol−1).40 While we note that the method used to calculate the barrier in Fe2(dobdc) differs from this work, it is nonetheless a useful comparison. Although the calculated activation barrier for Mn(IV)–oxo formation in Mn2(dobdc) is notably larger, the conditions that lead to Fe(IV)–oxo formation in Fe2(dobdc) are mild. As such, we evaluated whether more forceful conditions (i.e., higher temperatures) could enable Mn(IV)–oxo formation in Mn2(dobdc). To probe this possibility, N2O adsorption measurements in Mn2(dobdc) at 180 °C, 250 °C, and 300 °C were collected to identify potential N2O activation through changes in the adsorption properties of the material (Fig. S72, ESI†). Fully reversible N2O adsorption was measured at 180 °C; however, measurements at 250 °C and 300 °C consistently yielded anomalous negative adsorption, possibly indicating reactivity with the MOF. For further analysis, a bulk sample of Mn2(dobdc) was prepared by dosing the MOF with N2O at 300 °C (Section S15, ESI,†). The BET surface area of Mn2(dobdc) after heating at 300 °C (1285 ± 3 m2 g−1) under vacuum for 24 h is comparable to that of the pristine MOF (1344 ± 3 m2 g−1), supporting that this MOF is stable at elevated temperatures. Notably, the BET surface area is significantly attenuated after N2O dosing at 300 °C (896 ± 2 m2 g−1) (Fig. S73, ESI†). The reduction in surface area is accompanied by a color change from orange to brown. PXRD measurements confirm that Mn2(dobdc) retains its crystallinity after this process, although some peak-broadening was observed, indicative of partial decomposition (Fig. S74, ESI†).
Magnetic susceptibility measurements were used to characterize the product(s) resulting from high-temperature treatment of Mn2(dobdc) with N2O. The magnetic moments in Mn2(dobdc) calculated from susceptibility measurements before (μeff = 5.94μB) and after (μeff = 5.89μB) (Fig. S80 and S81, ESI†) N2O treatment at 300 °C closely match the value expected for Mn(II) with a spin of 5/2 (5.92μB), indicating a lack of oxidation at the metal center after N2O treatment (Fig. 5). Despite this, increased magnetic susceptibility relative to unreacted Mn2(dobdc) was consistently noted in moment vs. field measurements collected at 5 K after N2O treatment at 300 °C (Fig. S82, ESI†). This finding may point to reactivity between N2O and the redox-active linker instead,76 as oxidation of the high-spin Mn(II) sites to Mn(IV) should result in a decrease in the magnetic susceptibility.
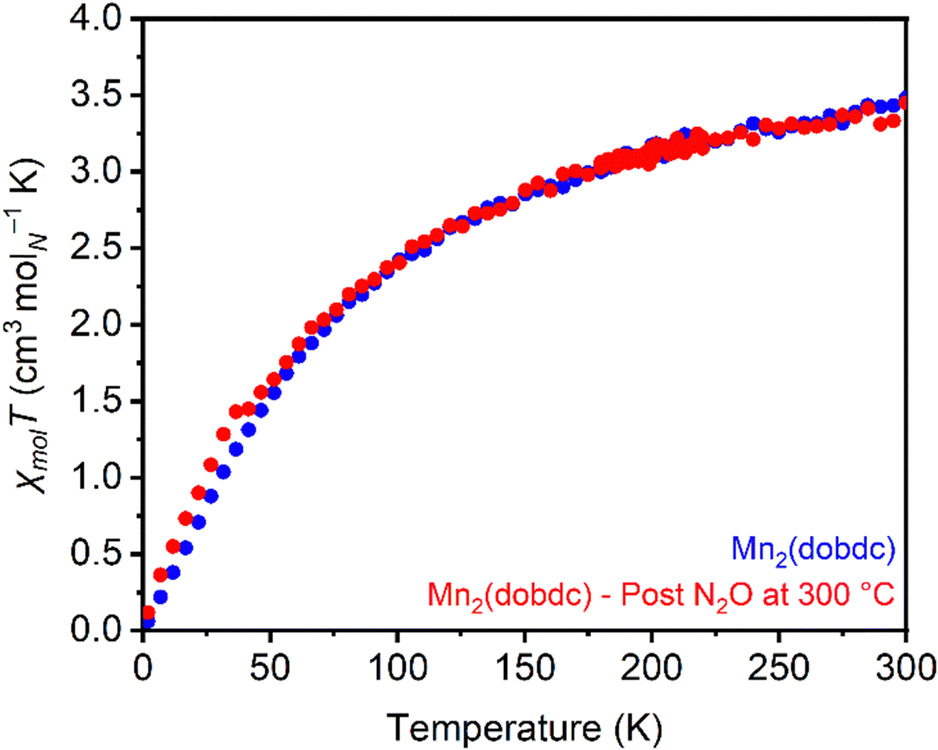 | ||
| Fig. 5 Variable-temperature magnetic susceptibility (χmolT) measurements of Mn2(dobdc) under an applied field of 1000 Oe before (blue) and after (red) N2O treatment at 300 °C. The minor increase in χmolT of the N2O-treated sample from approximately 2–40 K is attributed to slight O2 contamination. | ||
To further characterize the reaction of Mn2(dobdc) with N2O, we utilized variable-temperature diffuse reflectance Fourier transform (DRIFTS) spectroscopy (Fig. S75 and S76, ESI†). A sample of Mn2(dobdc) was heated under an atmosphere of N2O (approx. 1 bar) from 25 °C to 300 °C and held at 300 °C for 15 h. Spectra were collected periodically throughout the duration of the measurement. New Mn–O stretches corresponding to Mn(IV)–oxo (approx. 845 cm−1)77 or Mn(III)–OH species (600–700 cm−1)78 were not observed (Fig. S76, ESI†). Likewise, stretches corresponding to quinone formation due to linker oxidation were not observed (1657 cm−1).76 However, a weak O–H stretching frequency at 3673 cm−1 emerged over time, and the prominent stretch at 1406 cm−1 reduced in intensity after prolonged treatment with N2O (Fig. S75, ESI†). Overall, while the MOF appears to react with N2O at high temperatures, the DRIFTS and magnetic data indicate a lack of oxidation at the metal centers. This finding suggests that the high kinetic barrier to Mn(IV)–oxo formation in Mn2(dobdc) is likely prohibitive, despite its thermodynamic favorability. Elucidating the products of this MOF's reaction with N2O is the focus of ongoing work.
Conclusions
Herein, we identify features that contribute to strong binding and high uptake of N2O at Lewis acidic, coordinatively unsaturated metal sites in MOFs. We utilize the M2Cl2(btdd) and M2(dobdc) series as model systems to facilitate comparisons, as their general structures are related but distinguished by the ligation of the open-metal-sites. In the M2Cl2(btdd) series, the binding enthalpies of N2O are generally low (<25 kJ mol−1) and within error, despite variations in the metal identity. In contrast, the salicylate-based SBU in the M2(dobdc) series provides a weaker, oxygen-based ligand field. N2O binding strengths in these MOFs mostly mirror the Irving–Williams series, with Ni2(dobdc) exhibiting the strongest adsorption of N2O among all tested MOFs (−ΔHads = 43.8 ± 0.6). Notably, Mg2(dobdc) exhibits a record-breaking N2O gravimetric capacity (8.75 mmol g−1 at 1000 mbar of N2O and 298 K) among MOFs. While these Lewis acidic sites are effective for N2O capture, they are not necessarily selective for N2O among other polar(izable) gases, such as H2O.79 Achieving selective adsorption of N2O at open-metal-sites will be the focus of future work.Using the cluster systems to computationally model the oxidation of the M2(dobdc) series by N2O, forming M(IV)–oxo species, we find that M–oxo formation is also thermodynamically favorable in Mn2(dobdc) according to DFT calculations. We thus used the CI-NEB method to map a reaction coordinate for this process and determined an approximate activation barrier of 113 kJ mol−1, which is higher than that calculated for Fe2(dobdc) (94 kJ mol−1). Although experiments indicate that this kinetic barrier is too high to be overcome in Mn2(dobdc), these findings suggest that Mn-based MOFs may be promising alternatives to traditionally studied Fe-based materials for N2O activation.
Overall, this work adds to the growing body of research seeking to utilize N2O as a green oxidant, in which Mn-based solid-state catalysts remain relatively understudied. The results reported herein will help to drive the identification and development of other effective MOF-based sorbents to mitigate environmentally destructive N2O emissions.
Author contributions
P. J. M and H. J. K. conceived the project. T. A. P. carried out the synthesis, characterization, and gas sorption measurements of MOF samples under the supervision of P. J. M. P. J. M. and M. E. Z. also synthesized MOF samples. H. J. and A. N. carried out computational studies under the supervision of H. J. K. T. J. A. carried out magnetic measurements. T. A. P. and H. J. prepared the first draft of the manuscript, which was edited and approved by all co-authors.Conflicts of interest
P. J. M. is listed as a co-inventor on several patents related to metal–organic frameworks.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0021000 (T. A. P., T. J. A., M. E. Z., P. J. M.). Computational modeling was supported by the National Science Foundation under Award Number CBET-1846426 (H. J., A. N., H. J. K.) as well as a National Science Foundation Graduate Research Fellowship under Grant 1122374 (to A. N.). T. J. A. thanks Cornell University for financial support through a recruiting fellowship. We acknowledge the support of a Camille Dreyfus Teacher-Scholar Award to P. J. M. (TC-23-048). This work made use of a Bruker 500 MHz spectrometer, the purchase of which was supported by the National Science Foundation (CHE-1531632). This work made use of the Cornell Center for Materials Research Shared Facilities, which are supported through the NSF MRSEC program (DMR-1719875). This work used Expanse at the San Diego Supercomputing Center through allocation CHE140073 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants 2138259, 2138286, 2138307, 2137603, and 2138296. We thank Prof. Joseph Zadrozny (the Ohio State University) for helpful discussions.Notes and references
- E. A. Davidson and W. Winiwarter, Urgent Abatement of Industrial Sources of Nitrous Oxide, Nat. Clim. Change, 2023, 13(7), 599–601, DOI:10.1038/s41558-023-01723-3.
- Intergovernmental Panel On Climate Change, Climate Change 2021 – The Physical Science Basis: Working Group I Contribution to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, 1st edn, Cambridge University Press, 2023 DOI:10.1017/9781009157896.
- H. Tian, R. Xu, J. G. Canadell, R. L. Thompson, W. Winiwarter, P. Suntharalingam, E. A. Davidson, P. Ciais, R. B. Jackson, G. Janssens-Maenhout, M. J. Prather, P. Regnier, N. Pan, S. Pan, G. P. Peters, H. Shi, F. N. Tubiello, S. Zaehle, F. Zhou, A. Arneth, G. Battaglia, S. Berthet, L. Bopp, A. F. Bouwman, E. T. Buitenhuis, J. Chang, M. P. Chipperfield, S. R. S. Dangal, E. Dlugokencky, J. W. Elkins, B. D. Eyre, B. Fu, B. Hall, A. Ito, F. Joos, P. B. Krummel, A. Landolfi, G. G. Laruelle, R. Lauerwald, W. Li, S. Lienert, T. Maavara, M. MacLeod, D. B. Millet, S. Olin, P. K. Patra, R. G. Prinn, P. A. Raymond, D. J. Ruiz, G. R. Van Der Werf, N. Vuichard, J. Wang, R. F. Weiss, K. C. Wells, C. Wilson, J. Yang and Y. Yao, A Comprehensive Quantification of Global Nitrous Oxide Sources and Sinks, Nature, 2020, 586(7828), 248–256, DOI:10.1038/s41586-020-2780-0.
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Nitrous Oxide (N2O): The Dominant Ozone-Depleting Substance Emitted in the 21st Century, Science, 2009, 326(5949), 123–125, DOI:10.1126/science.1176985.
- M. U. Hassan, M. Aamer, A. Mahmood, M. I. Awan, L. Barbanti, M. F. Seleiman, G. Bakhsh, H. M. Alkharabsheh, E. Babur, J. Shao, A. Rasheed and G. Huang, Management Strategies to Mitigate N2O Emissions in Agriculture, Life, 2022, 12(3), 439, DOI:10.3390/life12030439.
- B. Gu, X. Zhang, S. K. Lam, Y. Yu, H. J. M. Van Grinsven, S. Zhang, X. Wang, B. L. Bodirsky, S. Wang, J. Duan, C. Ren, L. Bouwman, W. De Vries, J. Xu, M. A. Sutton and D. Chen, Cost-Effective Mitigation of Nitrogen Pollution from Global Croplands, Nature, 2023, 613(7942), 77–84, DOI:10.1038/s41586-022-05481-8.
- L. Li, J. Xu, J. Hu and J. Han, Reducing Nitrous Oxide Emissions to Mitigate Climate Change and Protect the Ozone Layer, Environ. Sci. Technol., 2014, 48(9), 5290–5297, DOI:10.1021/es404728s.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Carbon Capture and Conversion Using Metal–Organic Frameworks and MOF-Based Materials, Chem. Soc. Rev., 2019, 48(10), 2783–2828, 10.1039/C8CS00829A.
- S. K. Firooz and D. W. Armstrong, Metal-Organic Frameworks in Separations: A Review, Anal. Chim. Acta, 2022, 1234, 340208, DOI:10.1016/j.aca.2022.340208.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Metal–Organic Frameworks in Heterogeneous Catalysis: Recent Progress, New Trends, and Future Perspectives, Chem. Rev., 2020, 120(16), 8468–8535, DOI:10.1021/acs.chemrev.9b00685.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, The Chemistry and Applications of Metal-Organic Frameworks, Science, 2013, 341(6149), 1230444, DOI:10.1126/science.1230444.
- W. L. Queen, M. R. Hudson, E. D. Bloch, J. A. Mason, M. I. Gonzalez, J. S. Lee, D. Gygi, J. D. Howe, K. Lee, T. A. Darwish, M. James, V. K. Peterson, S. J. Teat, B. Smit, J. B. Neaton, J. R. Long and C. M. Brown, Comprehensive Study of Carbon Dioxide Adsorption in the Metal–Organic Frameworks M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn), Chem. Sci., 2014, 5(12), 4569–4581, 10.1039/C4SC02064B.
- Y. He, W. Zhou, G. Qian and B. Chen, Methane Storage in Metal–Organic Frameworks, Chem. Soc. Rev., 2014, 43(16), 5657–5678, 10.1039/C4CS00032C.
- Y. Lin, C. Kong and L. Chen, Amine-Functionalized Metal–Organic Frameworks: Structure, Synthesis and Applications, RSC Adv., 2016, 6(39), 32598–32614, 10.1039/C6RA01536K.
- Z. Sharifzadeh and A. Morsali, Amine-Functionalized Metal-Organic Frameworks: From Synthetic Design to Scrutiny in Application, Coord. Chem. Rev., 2022, 459, 214445, DOI:10.1016/j.ccr.2022.214445.
- M. E. Zick, D. Cho, J. Ling and P. J. Milner, Carbon Capture Beyond Amines: CO2 Sorption at Nucleophilic Oxygen Sites in Materials, ChemNanoMat, 2023, 9(1), e202200436, DOI:10.1002/cnma.202200436.
- J. Li, X. Han, X. Zhang, A. M. Sheveleva, Y. Cheng, F. Tuna, E. J. L. McInnes, L. J. McCormick McPherson, S. J. Teat, L. L. Daemen, A. J. Ramirez-Cuesta, M. Schröder and S. Yang, Capture of Nitrogen Dioxide and Conversion to Nitric Acid in a Porous Metal–Organic Framework, Nat. Chem., 2019, 11(12), 1085–1090, DOI:10.1038/s41557-019-0356-0.
- X. Zhang, W. Chen, W. Shi and P. Cheng, Highly Selective Sorption of CO2 and N2O and Strong Gas-Framework Interactions in a Nickel(II) Organic Material, J. Mater. Chem. A, 2016, 4(41), 16198–16204, 10.1039/C6TA06572D.
- D. Denysenko, J. Jelic, O. V. Magdysyuk, K. Reuter and D. Volkmer, Elucidating Lewis Acidity of Metal Sites in MFU-4l Metal-Organic Frameworks: N2O and CO2 Adsorption in MFU-4l, CuI-MFU-4l and Li-MFU-4l, Microporous Mesoporous Mater., 2015, 216, 146–150, DOI:10.1016/j.micromeso.2015.03.014.
- J. Yang, B. Du, J. Liu, R. Krishna, F. Zhang, W. Zhou, Y. Wang, J. Li and B. Chen, MIL-100Cr with Open Cr Sites for a Record N2O Capture, Chem. Commun., 2018, 54(100), 14061–14064, 10.1039/C8CC07679K.
- L. Ma, F. Zhang, K. Li, Y. Zhang, Z. Song, L. Wang, J. Yang and J. Li, Improved N2O Capture Performance of Chromium Terephthalate MIL-101 via Substituent Engineering, J. Solid State Chem., 2022, 309, 122951, DOI:10.1016/j.jssc.2022.122951.
- L. Wang, F. Zhang, J. Yang, L. Li and J. Li, The Efficient Separation of N2O/CO2 Using Unsaturated Fe2+ Sites in MIL-100Fe, Chem. Commun., 2021, 57(54), 6636–6639, 10.1039/D1CC01659H.
- D. Saha, Z. Bao, F. Jia and S. Deng, Adsorption of CO2, CH4, N2O, and N2 on MOF-5, MOF-177, and Zeolite 5A, Environ. Sci. Technol., 2010, 44(5), 1820–1826, DOI:10.1021/es9032309.
- L. Wang, Y. Li, Y. Wang, J. Yang, L. Li and J. Li, Research on CO2-N2O Separation Using Flexible Metal Organic Frameworks, Sep. Purif. Technol., 2020, 251, 117311, DOI:10.1016/j.seppur.2020.117311.
- G. Mercuri, M. Moroni, S. Galli, G. Tuci, G. Giambastiani, T. Yan, D. Liu and A. Rossin, Temperature-Dependent Nitrous Oxide/Carbon Dioxide Preferential Adsorption in a Thiazolium-Functionalized NU-1000 Metal–Organic Framework, ACS Appl. Mater. Interfaces, 2021, 13(49), 58982–58993, DOI:10.1021/acsami.1c21437.
- A. J. Rieth, Y. Tulchinsky and M. Dincă, High and Reversible Ammonia Uptake in Mesoporous Azolate Metal–Organic Frameworks with Open Mn, Co, and Ni Sites, J. Am. Chem. Soc., 2016, 138(30), 9401–9404, DOI:10.1021/jacs.6b05723.
- T. J. Azbell, T. A. Pitt, M. M. Bollmeyer, C. Cong, K. M. Lancaster and P. J. Milner, Ionothermal Synthesis of Metal-Organic Frameworks Using Low-Melting Metal Salt Precursors, Angew. Chem., Int. Ed., 2023, 62(17), e2022182, DOI:10.1002/anie.202218252.
- S. S. Park, Y. Tulchinsky and M. Dincă, Single-Ion Li+, Na+, and Mg2+ Solid Electrolytes Supported by a Mesoporous Anionic Cu–Azolate Metal–Organic Framework, J. Am. Chem. Soc., 2017, 139(38), 13260–13263, DOI:10.1021/jacs.7b06197.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, Dramatic Tuning of Carbon Dioxide Uptake via Metal Substitution in a Coordination Polymer with Cylindrical Pores, J. Am. Chem. Soc., 2008, 130(33), 10870–10871, DOI:10.1021/ja8036096.
- E. D. Bloch, L. J. Murray, W. L. Queen, S. Chavan, S. N. Maximoff, J. P. Bigi, R. Krishna, V. K. Peterson, F. Grandjean, G. J. Long, B. Smit, S. Bordiga, C. M. Brown and J. R. Long, Selective Binding of O2 over N2 in a Redox–Active Metal–Organic Framework with Open Iron(II) Coordination Sites, J. Am. Chem. Soc., 2011, 133(37), 14814–14822, DOI:10.1021/ja205976v.
- K. Severin, Synthetic Chemistry with Nitrous Oxide, Chem. Soc. Rev., 2015, 44(17), 6375–6386, 10.1039/C5CS00339C.
- V. N. Parmon, G. I. Panov, A. Uriarte and A. S. Noskov, Nitrous Oxide in Oxidation Chemistry and Catalysis: Application and Production, Catal. Today, 2005, 100(1–2), 115–131, DOI:10.1016/j.cattod.2004.12.012.
- F. Le Vaillant, A. Mateos Calbet, S. González-Pelayo, E. J. Reijerse, S. Ni, J. Busch and J. Cornella, Catalytic Synthesis of Phenols with Nitrous Oxide, Nature, 2022, 604(7907), 677–683, DOI:10.1038/s41586-022-04516-4.
- M. L. Bols, B. E. R. Snyder, H. M. Rhoda, P. Cnudde, G. Fayad, R. A. Schoonheydt, V. Van Speybroeck, E. I. Solomon and B. F. Sels, Coordination and Activation of Nitrous Oxide by Iron Zeolites, Nat. Catal., 2021, 4(4), 332–340, DOI:10.1038/s41929-021-00602-4.
- M. C. Simons, S. D. Prinslow, M. Babucci, A. S. Hoffman, J. Hong, J. G. Vitillo, S. R. Bare, B. C. Gates, C. C. Lu, L. Gagliardi and A. Bhan, Beyond Radical Rebound: Methane Oxidation to Methanol Catalyzed by Iron Species in Metal–Organic Framework Nodes, J. Am. Chem. Soc., 2021, 143(31), 12165–12174, DOI:10.1021/jacs.1c04766.
- M. Barona, S. Ahn, W. Morris, W. Hoover, J. M. Notestein, O. K. Farha and R. Q. Snurr, Computational Predictions and Experimental Validation of Alkane Oxidative Dehydrogenation by Fe2M MOF Nodes, ACS Catal., 2020, 10(2), 1460–1469, DOI:10.1021/acscatal.9b03932.
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocellà, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Oxidation of Ethane to Ethanol by N2O in a Metal–Organic Framework with Coordinatively Unsaturated Iron(II) Sites, Nat. Chem., 2014, 6(7), 590–595, DOI:10.1038/nchem.1956.
- A. Tofoni, F. Tavani, M. Vandone, L. Braglia, E. Borfecchia, P. Ghigna, D. C. Stoian, T. Grell, S. Stolfi, V. Colombo and P. D’Angelo, Full Spectroscopic Characterization of the Molecular Oxygen-Based Methane to Methanol Conversion over Open Fe(II) Sites in a Metal–Organic Framework, J. Am. Chem. Soc., 2023, 145(38), 21040–21052, DOI:10.1021/jacs.3c07216.
- J. G. Vitillo, M. Choudhary, M. C. Simons, L. Gagliardi and A. Bhan, Mechanism of Benzene Hydroxylation on Tri-Iron Oxo-Centered Cluster-Based Metal–Organic Frameworks, J. Phys. Chem. C, 2023, 127(48), 23246–23257, DOI:10.1021/acs.jpcc.3c06423.
- P. Verma, K. D. Vogiatzis, N. Planas, J. Borycz, D. J. Xiao, J. R. Long, L. Gagliardi and D. G. Truhlar, Mechanism of Oxidation of Ethane to Ethanol at Iron(IV)–Oxo Sites in Magnesium-Diluted Fe2(dobdc), J. Am. Chem. Soc., 2015, 137(17), 5770–5781, DOI:10.1021/jacs.5b00382.
- J. Borycz, J. Paier, P. Verma, L. E. Darago, D. J. Xiao, D. G. Truhlar, J. R. Long and L. Gagliardi, Structural and Electronic Effects on the Properties of Fe2(dobdc) upon Oxidation with N2O, Inorg. Chem., 2016, 55(10), 4924–4934, DOI:10.1021/acs.inorgchem.6b00467.
- B. L. Suh and J. Kim, Ligand Insertion in MOF-74 as Effective Design for Oxidation of Ethane to Ethanol, J. Phys. Chem. C, 2018, 122(40), 23078–23083 CrossRef CAS.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates, Chem. Rev., 2004, 104(2), 939–986, DOI:10.1021/cr020628n.
- B. J. Wallar and J. D. Lipscomb, Dioxygen Activation by Enzymes Containing Binuclear Non-Heme Iron Clusters, Chem. Rev., 1996, 96(7), 2625–2658, DOI:10.1021/cr9500489.
- B. Meunier, S. P. De Visser and S. Shaik, Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes, Chem. Rev., 2004, 104(9), 3947–3980, DOI:10.1021/cr020443g.
- V. Zhuravlev and P. J. Malinowski, A Stable Crystalline Copper(I)–N2O Complex Stabilized as the Salt of a Weakly Coordinating Anion, Angew. Chem., Int. Ed., 2018, 57(36), 11697–11700, DOI:10.1002/anie.201806836.
- C. C. Mokhtarzadeh, C. Chan, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Side-On Coordination of Nitrous Oxide to a Mononuclear Cobalt Center, J. Am. Chem. Soc., 2019, 141(38), 15003–15007, DOI:10.1021/jacs.9b08241.
- N. A. Piro, M. F. Lichterman, W. H. Harman and C. J. Chang, A Structurally Characterized Nitrous Oxide Complex of Vanadium, J. Am. Chem. Soc., 2011, 133(7), 2108–2111, DOI:10.1021/ja110798w.
- C. B. Pamplin, E. S. F. Ma, N. Safari, S. J. Rettig and B. R. James, The Nitrous Oxide Complex RuCl2(η1-N2O)(P-N)(PPh3) (P-N = [o-(N,N-Dimethylamino)phenyl]diphenylphosphine); Low Temperature Conversion of N2O to N2 and O2, J. Am. Chem. Soc., 2001, 135(35), 8596–8597, DOI:10.1021/ja0106319.
- F. Paulat, T. Kuschel, C. Näther, V. K. K. Praneeth, O. Sander and N. Lehnert, Spectroscopic Properties and Electronic Structure of Pentammineruthenium(II) Dinitrogen Oxide and Corresponding Nitrosyl Complexes: Binding Mode of N2O and Reactivity, Inorg. Chem., 2004, 43(22), 6979–6994, DOI:10.1021/ic049302i.
- B. Puerta Lombardi, C. Gendy, B. S. Gelfand, G. M. Bertrand, R. E. Wasylishen, H. M. Tuononen and R. Roesler, Side-on Coordination in Isostructural Nitrous Oxide and Carbon Dioxide Complexes of Nickel, Angew. Chem., Int. Ed., 2020, 60(13), 7077–7081, DOI:10.1002/anie.202011301.
- M. R. Gyton, B. Leforestier and A. B. Chaplin, Rhodium(I) Pincer Complexes of Nitrous Oxide, Angew. Chem., 2019, 131(43), 15439–15442, DOI:10.1002/ange.201908333.
- W. B. Tolman, Binding and Activation of N2O at Transition-Metal Centers: Recent Mechanistic Insights, Angew. Chem., Int. Ed., 2010, 49(6), 1018–1024, DOI:10.1002/anie.200905364.
- S. Ketrat, T. Maihom, S. Wannakao, M. Probst, S. Nokbin and J. Limtrakul, Coordinatively Unsaturated Metal–Organic Frameworks M3(btc)2 (M = Cr, Fe, Co, Ni, Cu, and Zn) Catalyzing the Oxidation of CO by N2O: Insight from DFT Calculations, Inorg. Chem., 2017, 56(22), 14005–14012, DOI:10.1021/acs.inorgchem.7b02143.
- A. J. Rieth, A. M. Wright, G. Skorupskii, J. L. Mancuso, C. H. Hendon and M. Dincă, Record-Setting Sorbents for Reversible Water Uptake by Systematic Anion Exchanges in Metal–Organic Frameworks, J. Am. Chem. Soc., 2019, 141(35), 13858–13866, DOI:10.1021/jacs.9b06246.
- M. E. Zick, J.-H. Lee, M. I. Gonzalez, E. O. Velasquez, A. A. Uliana, J. Kim, J. R. Long and P. J. Milner, Fluoroarene Separations in Metal–Organic Frameworks with Two Proximal Mg2+ Coordination Sites, J. Am. Chem. Soc., 2021, 143(4), 1948–1958, DOI:10.1021/jacs.0c11530.
- S. J. Geier, J. A. Mason, E. D. Bloch, W. L. Queen, M. R. Hudson, C. M. Brown and J. R. Long, Selective Adsorption of Ethylene over Ethane and Propylene over Propane in the Metal–Organic Frameworks M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Zn), Chem. Sci., 2013, 4(5), 2054–2061, 10.1039/c3sc00032j.
- M. I. Gonzalez, M. T. Kapelewski, E. D. Bloch, P. J. Milner, D. A. Reed, M. R. Hudson, J. A. Mason, G. Barin, C. M. Brown and J. R. Long, Separation of Xylene Isomers through Multiple Metal Site Interactions in Metal–Organic Frameworks, J. Am. Chem. Soc., 2018, 140(9), 3412–3422, DOI:10.1021/jacs.7b13825.
- J. A. Mason, M. Veenstra and J. R. Long, Evaluating Metal–Organic Frameworks for Natural Gas Storage, Chem. Sci., 2014, 5(1), 32–51, 10.1039/C3SC52633J.
- K. Sumida, D. Stück, L. Mino, J.-D. Chai, E. D. Bloch, O. Zavorotynska, L. J. Murray, M. Dincă, S. Chavan, S. Bordiga, M. Head-Gordon and J. R. Long, Impact of Metal and Anion Substitutions on the Hydrogen Storage Properties of M-BTT Metal–Organic Frameworks, J. Am. Chem. Soc., 2013, 135(3), 1083–1091, DOI:10.1021/ja310173e.
- H. Irving and R. J. P. Williams, Order of Stability of Metal Complexes, Nature, 1948, 162, 746–747, DOI:10.1038/162746a0.
- D. Yu, A. O. Yazaydin, J. R. Lane, P. D. C. Dietzel and R. Q. Snurr, A Combined Experimental and Quantum Chemical Study of CO2 Adsorption in the Metal–Organic Framework CPO-27 with Different Metals, Chem. Sci., 2013, 4(9), 3544–3556, 10.1039/c3sc51319j.
- M. H. Rosnes, D. Sheptyakov, A. Franz, M. Frontzek, P. D. C. Dietzel and P. A. Georgiev, On the Elusive Nature of Oxygen Binding at Coordinatively Unsaturated 3d Transition Metal Centers in Metal–Organic Frameworks, Phys. Chem. Chem. Phys., 2017, 19(38), 26346–26357, 10.1039/C7CP05119K.
- M. Barona and R. Q. Snurr, Exploring the Tunability of Trimetallic MOF Nodes for Partial Oxidation of Methane to Methanol, ACS Appl. Mater. Interfaces, 2020, 12(25), 28217–28231, DOI:10.1021/acsami.0c06241.
- Q. Zhang, B. Li and L. Chen, First-Principles Study of Microporous Magnets M-MOF-74 (M = Ni, Co, Fe, Mn): The Role of Metal Centers, Inorg. Chem., 2013, 52(16), 9356–9362, DOI:10.1021/ic400927m.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Hydrocarbon Separations in a Metal-Organic Framework with Open Iron(II) Coordination Sites, Science, 2012, 335(6076), 1606–1610, DOI:10.1126/science.1217544.
- K. D. Vogiatzis, E. Haldoupis, D. J. Xiao, J. R. Long, J. I. Siepmann and L. Gagliardi, Accelerated Computational Analysis of Metal–Organic Frameworks for Oxidation Catalysis, J. Phys. Chem. C, 2016, 120(33), 18707–18712, DOI:10.1021/acs.jpcc.6b07115.
- J. G. Vitillo, C. C. Lu, C. J. Cramer, A. Bhan and L. Gagliardi, Influence of First and Second Coordination Environment on Structural Fe(II) Sites in MIL-101 for C–H Bond Activation in Methane, ACS Catal., 2021, 11(2), 579–589, DOI:10.1021/acscatal.0c03906.
- V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Iron and Manganese Oxo Complexes, Oxo Wall and Beyond, Nat. Rev. Chem., 2020, 4(8), 404–419, DOI:10.1038/s41570-020-0197-9.
- T. Yamashita and A. Vannice, N2O Decomposition over Manganese Oxides, J. Catal., 1996, 161(1), 254–262, DOI:10.1006/jcat.1996.0183.
- R. A. Ross and C. Fairbridge, Oxidation of 1-Butene by Nitrous Oxide over Manganese(III) and Related Transition Metal Oxides, Can. J. Chem., 1984, 62(8), 1483–1486, DOI:10.1139/v84-252.
- R. Ben-Daniel, L. Weiner and R. Neumann, Activation of Nitrous Oxide and Selective Epoxidation of Alkenes Catalyzed by the Manganese-Substituted Polyoxometalate, [MnIII2ZnW(Zn2W9O34)2]10, J. Am. Chem. Soc., 2002, 124(30), 8788–8789, DOI:10.1021/ja0259077.
- M.-X. Jiang and C.-G. Liu, New Insight into the Catalytic Cycle about Epoxidation of Alkenes by N2O over a Mn–Substituted Keggin-Type Polyoxometalate, J. Mol. Graphics Modell., 2017, 73, 8–17, DOI:10.1016/j.jmgm.2016.12.012.
- Y. Li and J. N. Armor, Catalytic Decomposition of Nitrous Oxide on Metal Exchanged Zeolites, Appl. Catal., B, 1992, 1(3), L21–L29, DOI:10.1016/0926-3373(92)80019-V.
- M. C. Campa, V. Indovina and D. Pietrogiacomi, The Selective Catalytic Reduction of N2O with CH4 on Na-MOR and Na-MFI Exchanged with Copper, Cobalt or Manganese, Appl. Catal., B, 2012, 111–112, 90–95, DOI:10.1016/j.apcatb.2011.09.021.
- A. F. Cozzolino, C. K. Brozek, R. D. Palmer, J. Yano, M. Li and M. Dincă, Ligand Redox Non-Innocence in the Stoichiometric Oxidation of Mn2(2,5-dioxidoterephthalate) (Mn-MOF-74), J. Am. Chem. Soc., 2014, 136(9), 3334–3337, DOI:10.1021/ja411808r.
- R. L. Halbach, D. Gygi, E. D. Bloch, B. L. Anderson and D. G. Nocera, Structurally Characterized Terminal Manganese(iv) Oxo Tris(Alkoxide) Complex, Chem. Sci., 2018, 9(19), 4524–4528, 10.1039/C8SC01164H.
- X. Wang and L. Andrews, Infrared Spectra of M(OH)1,2,3 (M = Mn, Fe, Co, Ni) Molecules in Solid Argon and the Character of First Row Transition Metal Hydroxide Bonding, J. Phys. Chem. A, 2006, 110(33), 10035–10045, DOI:10.1021/jp0624698.
- J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Water Adsorption in MOFs: Fundamentals and Applications, Chem. Soc. Rev., 2014, 43(16), 5594–5617, 10.1039/C4CS00078A.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, and gas sorption measurements of all MOFs, additional computational details, and high-temperature N2O experiments. See DOI: https://doi.org/10.1039/d3tc04492k |
| This journal is © The Royal Society of Chemistry 2024 |