
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel synthesis of semiconductor chalcohalide anti-perovskites by low-temperature molecular precursor ink deposition methodologies†
Ivan
Caño
 *ab,
Jonathan W.
Turnley
c,
Pol
Benítez
ab,
Cibrán
López-Álvarez
ab,
José-Miguel
Asensi
d,
David
Payno
e,
Joaquim
Puigdollers
ab,
Marcel
Placidi
ab,
Claudio
Cazorla
a,
Rakesh
Agrawal
c and
Edgardo
Saucedo
ab
*ab,
Jonathan W.
Turnley
c,
Pol
Benítez
ab,
Cibrán
López-Álvarez
ab,
José-Miguel
Asensi
d,
David
Payno
e,
Joaquim
Puigdollers
ab,
Marcel
Placidi
ab,
Claudio
Cazorla
a,
Rakesh
Agrawal
c and
Edgardo
Saucedo
ab
aUniversitat Politècnica de Catalunya (UPC), EEBE, Av. Eduard Maristany 10-14, Barcelona 08019, Catalonia, Spain. E-mail: ivan.cano.prades@upc.edu
bUniversitat Politècnica de Catalunya (UPC), Barcelona Centre for Multiscale Science & Engineering, Av. Eduard Maristany 10-14, Barcelona 08019, Catalonia, Spain
cPurdue University, Davidson School of Chemical Engineering, Forney Hall of Chemical Engineering, 480 Stadium Mall Drive, West Lafayette, IN 47907-2100, USA
dUniversitat de Barcelona (UB), Departament de Física Aplicada, Av. Martí i Franquès 1, Barcelona 08028, Spain
eCatalonia Institute for Energy Research (IREC), Jardins de les Dones de Negre 1, Sant Adrià del Besòs 08930, Spain
First published on 1st February 2024
Abstract
In recent years, a growing interest in the development of new energy harvesting technologies based on earth-abundant, environmentally-friendly semiconductors, has led to the re-discovery of hitherto overlooked materials. Among them, Ag-based chalcohalides stand out for their abundancy and low-toxicity, as well as the crystal structure analogous to perovskite, albeit with cations in place of anions and vice-versa (i.e. anti-perovskite). Until now, inorganic anti-perovskites have generally been studied as solid-state electrolytes. Indeed, Ag3SI was identified in the 1960s as a superionic conductor. On the other hand, theorical calculations have demonstrated bandgaps in the visible range, suggesting that they could be suitable for PV applications. However, there is little published information on their potential as energy harvesting materials and so far, thin films have been prepared by solid-state reactions or physical vapor deposition techniques at high temperature and/or vacuum conditions, which limits their commercial viability owing to costly, non-scalable processes. In this work, we present a new procedure to synthesize Ag-based chalcohalides by a low-temperature solution-based methodology, using an thiol-amine reactive solvent system to dissolve Ag2S and AgX (X = Br, I) precursors, followed by spin coating deposition to obtain polycrystalline films. Through this process, it has been possible to synthesize Ag3S(IxBr1−x) (x = 0–1) films for the first time, which have been characterized, demonstrating the formation of the anti-perovskite phase and a linear correlation between structural parameters and composition. Optical characterization shows bandgap ranging from 0.9 eV (Ag3SI) to 1.0 eV (Ag3SBr), with a bowing effect for the intermediate solid solutions. First solar cells prototypes demonstrate photo-response and promising electrical characteristics.
Introduction
Metal chalcohalides constitute an extended family of semiconductor materials encompassing different compositions, structural and optoelectronic properties. Since the 1960, several chalcohalide materials have been reported to possess photoconductive, electro-optical and ferroelectric effects, and their wide heterogeneity of compositions and structures lead to a broad bandgap range, potentially suitable for photovoltaics (PV).1 Nevertheless, these semiconductors have been largely overlooked among the PV community, likely overshadowed by the prevalence of high performance Si in the solar cell market, and the success of other thin film technologies such as CdTe, Cu(In,Ga)Se2 and lead halide perovskites.2,3 Interestingly, recent studies have reported an excellent performance of several chalcohalide materials including SbSI and SbSeI, reaching efficiencies up to a 4% in a very short period of time, benefited by their quasi-1D structure which favors enhanced transport properties.4–7 Likewise, transition metal chalcohalides such as Ag3SX (X = Br, I) stand out for their crystalline structure analogous to that of the successful halide perovskites, suggesting that they might possess similar defect-tolerant properties. Indeed, their structure presents a perovskite-like arrangement, albeit switching cation sites by anions, and anion sites by cations; thus, the denomination anti-perovskite. In fact, very recently, the possible relevance of this type of compounds has been highlighted in several review papers, which also note the lack of methodologies to synthesize suitable thin films for optoelectronic applications.1,8Ag3SI and Ag3SBr were first reported by Reuter and Hardel in 1960,9 and after a short time they were identified as superionic conductors by Takahashi and Yamamoto.10 Consequently, they have been mainly investigated for use as solid electrolytes. However, recent research by Yin, Lapidus et al. have shown that there are at least four polymorphs of Ag3SI stable at different temperatures – which diverge upon the ordering of S2−/I− anions and the location of Ag+ cations –, of whom only the high-temperature phase can be genuinely regarded as superionic (S2−/I− disordered cubic phase), with ionic conductivity around 1.5 S cm−1.11 Interestingly, theoretical calculations have shown that Ag anti-perovskites have bandgaps in the 0.9–2.0 eV range, with no consensus for the exact value, but suggesting that they could be suitable for single-junction solar cells as well as for tandem configurations, hence opening a new horizon of possibilities for this family of materials.12,13 Despite these promising properties, there is an unresolved gap regarding their implementation into PV and other energy applications, most likely due to predominant costly and time-consuming synthetic approaches, involving high temperatures as well as vacuum processing, which have hindered their development. Therefore, anti-perovskites have been largely overlooked as potential semiconductors for energy applications.
In this work, Ag3S(Br1−xIx) thin films have been prepared for the first time using a novel methodology based on the deposition of thiol-amine molecular precursor inks at very low temperatures. This is the first process based on chemical deposition reported for this family of materials. Until now, Ag3SX films have been produced by pulsed laser deposition (which requires high vacuum),14 and laser ablation (from powders prepared at temperature above 600 °C).15 Furthermore, in most studies on chalcohalide anti-perovskites, powders and pellets have been fabricated by a high temperature annealing (typically around 600 °C) of the solid-state precursors (Ag2S and AgX),16,17 whereas we propose a chemically versatile and cost-effective method that allows synthesis at low temperatures (220 °C). More recently, a revived interest in anti-perovskites has resulted in new works reporting the synthesis of high-purity Ag3SBr and Ag3SI powders at temperatures below 300 °C by mechanochemical synthesis.14 However, vacuum processes are still required for layer deposition. Also, to the best of our knowledge, there are no published reports on Ag3S(BrxI1−x) solid solution preparation in thin film form from low temperature chemical methods, and some of their properties remain largely unknown, such as the bandgap.
In addition to the ecologically benign nature of their constituents (earth-abundancy, non-toxicity) and suitable bandgaps, Ag3SX anti-perovskites also stand out for their chemical adaptability. They tolerate different cation and anion-substitutions such as Ag3S(BrxI1−x) and (CuxAg1−x)3SX,18 which offers a viable approach for bandgap tuning and for adjusting specific properties towards the development of tailored materials for advanced energy applications, including photovoltaics and solid-state batteries. In this regard, the methodology proposed in this work is highly convenient, since it entails great versatility by allowing the preparation of precursor solutions with virtually any possible composition contained within the compositional space of solid solutions, through adjusting the relative molar amounts of precursors.
Following this procedure, polycrystalline anti-perovskite films have been synthesized at temperatures in the 200–300 °C range, covering the whole Br–I compositional spectrum (x = 0 to 1). Composition, structure, morphology and optical properties of the whole family of compounds have been extensively characterized for the first time, demonstrating the formation of the anti-perovskite phase, the impact of anionic substitution to the structural parameters, and a very high compositional uniformity of the films. Also, first simple solar cell prototypes have been manufactured, demonstrating diode behavior and promising optoelectronic properties. Importantly, our method does not require any vacuum or high-temperature treatment at furnace, thus opening up new possibilities for the fabrication and development of anti-perovskites, including their implementation into energy harvesting and power devices. Moreover, solution-based processes are simple, cost-effective and easily scalable, making research and development of these materials for the first time accessible to a larger number of laboratories. Finally, first-principles calculations based on density functional theory (DFT) have been carried out, which are in excellent agreement with the experimental observations after taking into consideration thermal effects (e.g., electron–phonon coupling and ionic diffusion) thus proving that anti-perovskites are highly anharmonic materials with ostensible ionic conductivity above room temperature.
Materials and methods
Materials
The following materials have been used to fabricate the Ag3S(BrxI1−x) samples: AgBr (99.998% metal basis) and AgI (99.99% metal basis) were purchased from Thermo-Scientific. Ag2S (powder, 99.99% metal basis), 1,2-ethanedithiol (EDT; >98%), propylamine (PA; >99%), titanium diisopropoxide bis(acetyl-acetonate) (75% in isopropyl alcohol), poly(3-hexylthiophene-2,5-diyl) (P3HT), thiourea (TU; >99%), and dimethylformamide (DMF) were purchased from Sigma-Aldrich. All chemicals have been used as received.Synthesis of glass/FTO/Ag3SX (X = Br, I) films is based on the following sequential steps.
Preparation of precursor solutions
For a successful solution-based synthesis, it is necessary to prepare Ag2S and AgX (X = Br, I) precursor solutions separately, using the thiol-amine solvent mixture (EDT + PA). Importantly, solution preparation is performed under inert atmosphere in a glovebox, using high-purity grade N2 gas with O2 amount below 0.1 ppm. Also, all these manipulations take place in darkroom conditions, avoiding exposure to sunlight, to prevent decomposition of AgX into silver metal and Br2 or I2. First, AgX (X = Br, I) solutions are prepared dissolving 0.2 mmol of AgX powders in 75 μl EDT and 225 μl PA (1 EDT: 3 PA) under continuous stirring at 35 °C. Similarly, Ag2S solutions have been prepared by dissolving 0.8 mmols of Ag2S powder in 790 μl PA and 410 μl EDT with continuous stirring at 35 °C for 12 to 24 hours. Due to the lower solubility of Ag2S (this is the main reason for using thiol-amine mixture in this process), the EDT + PA ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in this case. Nevertheless, it is important to use the minimum EDT to avoid the solutions becoming very viscous, or the precursor solution being able to dissolve the Ag3SX layer upon performing multiple deposits for stacks. Note that both solutions have a concentration of 0.67 M.
:2 in this case. Nevertheless, it is important to use the minimum EDT to avoid the solutions becoming very viscous, or the precursor solution being able to dissolve the Ag3SX layer upon performing multiple deposits for stacks. Note that both solutions have a concentration of 0.67 M.
Ag3SX molecular ink
When both AgX and Ag2S mixtures are fully dissolved (in the case of Ag2S it can take 12–24 hours), 250 μl from the Ag2S solution are added to the halide solution using a pipette. Then, the Ag3SX precursor solution (0.36 M) is left under continuous stirring at 35 °C for 2–4 hours.Film deposition
The 0.36 M Ag3SX thiol-amine precursor solution is deposited by spin-coating onto 2 × 2 cm2 FTO-coated glass substrates, using a SPIN150i-NPP single substrate spin processor from SPS Polos. The deposition is performed in N2 glovebox at 2000 rpm for 30 s, spinning 100 ul precursor solution per layer. After each coating, the films are immediately annealed on a preheated hot-plate at 220 °C for 2 min, see Fig. 1 for a schematic layout of the synthesis methodology. The whole process is performed in darkroom conditions to avoid the decomposition of precursors into Ag(0).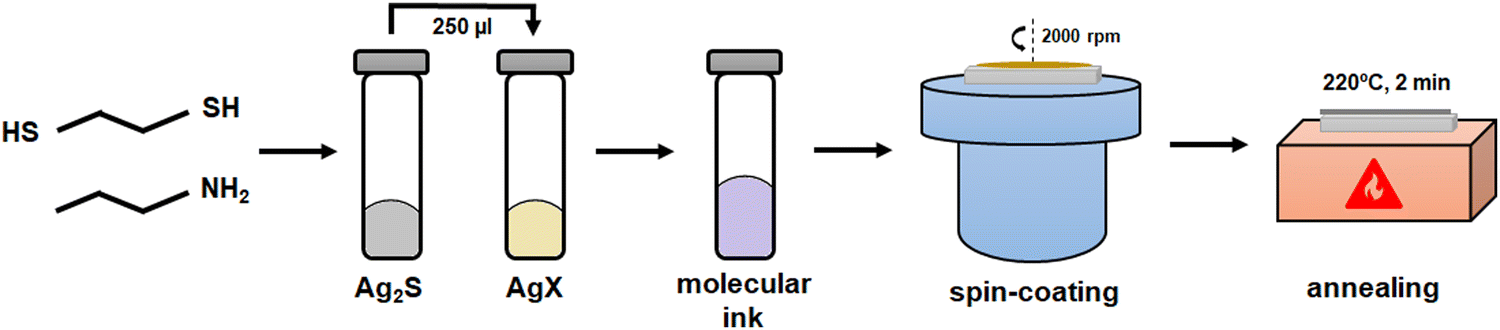 | ||
| Fig. 1 Schematic outlining the synthesis process of Ag3SX by thiol-amine route, beginning with the preparation of Ag2S and AgX (X = Br, I) solutions, and the molecular ink. Then, the material is deposited by spin-coating and crystallized by heating treatment on a hot-plate. | ||
Additionally, prototype photovoltaic devices have also been prepared using a superstrate architecture based on the following structure: FTO/TiO2/Ag3SX/P3HT/Au, where TiO2 acts as electron transport layer (ETL), and P3HT is the hole transport layer (HTL). Preparation of PV devices is based on the following step-by-step process.
Compact TiO2 layer deposition
The TiO2 solution is prepared diluting 1 ml of titanium diisopropoxide bis[acetylacetonate] commercial solution with 19 ml ethanol. The 30 nm-thick TiO2 layer is deposited onto FTO-coated glass substrate by spray-pyrolysis; placing the sample on a hot plate at 500 °C, while the atomized diluted TiO2 is sprayed using a hand-sprayer.Ag3SX absorber deposition
Following the procedure described above.P3HT and Au deposition
The P3HT solution is prepared dissolving 11 mg of P3HT in 1 ml chlorobenzene (1 wt%). Subsequently, 100 ul of the HTL solution are deposited onto the SLG/FTO/TiO2/Ag3SX sample using a two-step spin-coating process: 350 rpm for 4 s + 3000 rpm for 20 s, followed by a single annealing at 170 °C for 5 min. Finally, 200 nm of Au are thermally evaporated as the top conductive contact using a mask with an active area of 9 mm2.19,20The bare absorbers (glass/FTO/Ag3SX samples) and PV devices have been studied using a number of techniques for structural, optical and electronic characterization, offering a first general overview of the properties of chalcohalide anti-perovskites as synthesized using molecular precursor inks, as well as a first clear insight on their optical bandgap.
Characterization
Structural characterization by X-ray diffraction (PXRD) has been performed with a Bruker D8 Advance diffractometer equipped with a Cu X-ray tube (40 kV, 40 mA) and a Sol-X detector, with discriminator for the Kβ line and fluorescence effect reduction. The measurements are done using grazing incidence configuration (GIXRD), and data treatment and analysis have been performed with the FullProf suite.21Absorbance and optical bandgap measurements have been performed by Photothermal Deflection Spectroscopy (PDS). The setup used here consists of 100 W tungsten halogen lamp, monochromator (spectral range 400–2000 nm), an optical chopper, position-sensitive detector connected to a lock-in amplifier to measure the deflection of the laser probe beam, and a quartz cuvette filled with Fluorinert liquid (TM FC-40) to place the samples.22,23 Bandgap and Urbach front properties have been acquired using the Tauc-Urbach model (see Annex 1 in ESI† for further details on this model).
Scanning Electron Macroscopy (SEM) images have been obtained with a Neon40 Crossbeam FIB-SEM workstation from Carl Zeiss, equipped with a GEMINI-SEM column and an INCAPentaFETx3 EDX detector for chemical analysis. Finally, a Sun 3000 AAA class simulator from Abet technologies has been used to acquire current density vs voltage (JV) curves from photovoltaic devices (under AM 1.5 spectrum illumination).
DFT calculations
First-principles calculations based on density functional theory (DFT) were carried out with the PBEsol exchange–correlation energy functional,24,25 as it is implemented in the VASP software.26 The projector-augmented wave method (PAW) was employed to represent the ionic cores by considering the following electronic states as valence: Ag 1s 10d; S 2s 4p; I 2s 5p; Br 2s 5p.27 The Grimme's et al. D3 scheme was employed for a better treatment of the dispersion interactions in the system.28 An energy cutoff of 650 eV and a regular k-point grid center at Gamma of 8 × 8 × 8 were used for integration within the Brillouin zone, leading to total energies converging to within 1 meV per formula unit. Atomic relaxations were concluded when the forces in the atoms were all below 0.005 eV Å−1. The electronic properties of the different materials were estimated with the accurate screened hybrid exchange–correlation functional HSEsol by considering relativistic spin–orbit coupling effects and adopting the geometries previously determined at the PBEsol + D3 level.29,30 Finite-temperature ab initio molecular dynamics simulations were carried out for 2 × 2 × 2 and 4 × 4 × 4 supercells containing a total of 40 and 320 atoms, respectively. These were performed at T = 200 and 600 K and lasted for 100 and 50 ps, respectively. From all the configurations generated during the dynamical simulations, a total of 10 equally spaced geometries were selected to perform the statistical averages.Results and discussion
The main challenge in the synthesis of Ag-based anti-perovskites by chemical route lies in the difficulty of forming a stable molecular precursor ink, given the poor solubility of silver precursors such as Ag halides and Ag2S. Interestingly, it has been reported that the solubility of Ag halides increases in polar solvents when 2 TU per halide are present in the solution.31 However, the mixture of AgX and TU (1:2) in DMF does not lead to successful formation of the anti-perovskite phase. Indeed, structural and compositional characterization revealed that the dominant crystalline phase corresponds to AgBr regardless the annealing conditions, with very small intensity signals compatible, albeit not univocally indexable to Ag3SBr (see the XRD patterns in Fig. S1, ESI†). This observation is similar to that of Murria et al. where it was revealed that chloride ligands could not be completely displaced by thiolates upon dissolution of CuCl or CuCl2 in an thiol-amine mixture, resulting in the persistent presence of chlorine in the thin films prepared from these solutions.32 In order to produce an effective molecular precursor ink with the precise stoichiometric amounts of Ag, S and halide, a new approach was devised involving the use of Ag2S and AgX (X = I, Br) as chemical precursors dissolved in an thiol-amine solvent system. In this case, the very low solubility of Ag2S constitutes a major limitation when it comes to finding the right solvent. However, it has been shown that thiol-amine mixtures can easily dissolve a wide range of bulk solids which have been generally thought to be insoluble, such as transition metals (i.e., Cu, In, Zn) and chalcogenide compounds, resulting in the designation of the solvent as an alkahest.33–37
A series of anti-perovskite samples have been prepared, which Br–I content ranges from pure bromine up to pure iodine – i.e. Ag3S(BrxI1−x) with x = 1 to x = 0 – including three intermediate solid solutions (with x = 0.7, 0.5, 0.3). Structural characterization has been performed by GIXRD, see the patterns in Fig. 2(a). They have been indexed, showing phases corresponding to FTO (the substrate – ICDD file 01-077-0449) and Ag3S(BrxI1−x), with no apparent secondary phases, suggesting that pure anti-perovskite phases were obtained in all the cases. Interestingly, all the solid solutions containing bromine exhibit a Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m cubic structure (indexed from ICDD file 01-074-0117). Conversely, the pure iodine compound also presents cubic structure, but Imm symmetry (01-084-1172). As discussed elsewhere, the Ag3SI system is characterized by having multiple polymorphs which are stabilized at different temperatures. In particular, theoretical analyses have shown that Ag3SI has two polymorphs with space group Imm (α-Ag3SI and α*-Ag3SI), which are characterized by the absence of long-range order of the anion species, i.e. anion sites are randomly occupied by either S2− or I−. On the other hand, the cubic Pmm structure is characterized by the presence of long-range order of the anion species, e.g. S2− and I− occupy vertices and center of the anti-perovskite unit cell respectively, see structures in Fig. 2(c). Also note that depending on the structure, the three Ag+ per unit cell are randomly distributed over octahedral, tetrahedral or trigonal positions.38 Significantly, it has been reported that the α phase is stable at higher temperatures (∼569 K), with ionic conductivity around 1.5 S cm−1, while the α* phase appears by quenching the α-Ag3SI polymorph, but has a lower ionic conductivity at 0.13 S cm−1. Finally, the Pmm structure (β-Ag3SI) is the most stable at room temperature and has the lowest ionic conductivity (approximately two orders of magnitude lower than α*-Ag3SI).11,39 Based on its higher stability at room temperature and the lower ionic conductivity, we consider the β-Ag3S(BrxI1−x) structure to present the greatest interest for PV applications.
m cubic structure (indexed from ICDD file 01-074-0117). Conversely, the pure iodine compound also presents cubic structure, but Imm symmetry (01-084-1172). As discussed elsewhere, the Ag3SI system is characterized by having multiple polymorphs which are stabilized at different temperatures. In particular, theoretical analyses have shown that Ag3SI has two polymorphs with space group Imm (α-Ag3SI and α*-Ag3SI), which are characterized by the absence of long-range order of the anion species, i.e. anion sites are randomly occupied by either S2− or I−. On the other hand, the cubic Pmm structure is characterized by the presence of long-range order of the anion species, e.g. S2− and I− occupy vertices and center of the anti-perovskite unit cell respectively, see structures in Fig. 2(c). Also note that depending on the structure, the three Ag+ per unit cell are randomly distributed over octahedral, tetrahedral or trigonal positions.38 Significantly, it has been reported that the α phase is stable at higher temperatures (∼569 K), with ionic conductivity around 1.5 S cm−1, while the α* phase appears by quenching the α-Ag3SI polymorph, but has a lower ionic conductivity at 0.13 S cm−1. Finally, the Pmm structure (β-Ag3SI) is the most stable at room temperature and has the lowest ionic conductivity (approximately two orders of magnitude lower than α*-Ag3SI).11,39 Based on its higher stability at room temperature and the lower ionic conductivity, we consider the β-Ag3S(BrxI1−x) structure to present the greatest interest for PV applications.
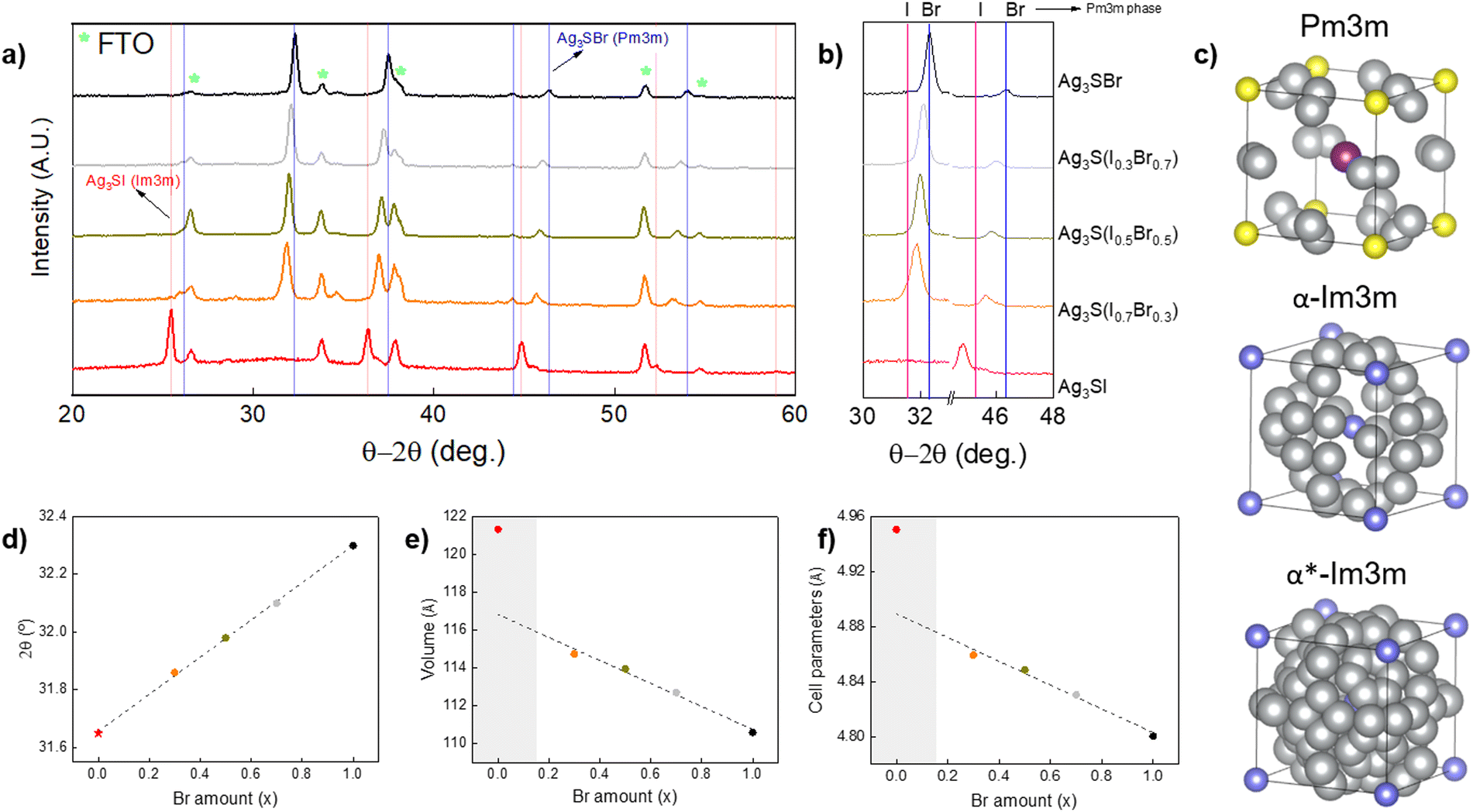 | ||
| Fig. 2 (a) GIXRD patterns of a series of Ag3S(BrxI1−x) samples, with x = 1, 0.7, 0.5, 0.3 and 0. (b) Enlarged detail of the patterns in Figure a (blue and pink lines indicate Pmm Ag3SBr and Pmm Ag3SI, respectively). (c) Ag3SI unit cell structures obtained from S. Hull et al.,38 represented with VESTA software: grey – Ag, Purple – I, yellow – S, blue – I/S (anionic disorder)40 (d) 2θ position of the diffraction peak that appears around 32° as a function of x. Value for x = 0 (star) has been extracted from the Pmm Ag3SI ICDD file: 01-084-1167. (e) Cell volume as a function of x. (f) Lattice parameter as a function of x. | ||
Besides these cases of polymorphism, the GIXRD patterns also suggest that the solid solutions have been successfully formed. Indeed, note that the Ag3S(BrxI1−x) diffraction peaks shift to lower 2θ values by decreasing x (the Br amount) with respect to the precisely indexed Ag3SBr Pmm phase, while the pure iodine structure separates from the trend presenting a completely different pattern (owing to its Imm symmetry, as discussed above), see Fig. 2(b). A linear correlation is observed between the bromine amount x and the 2θ reflections, which extrapolates consistently to the values for Ag3SI with Pmm cubic symmetry, which has been inserted from tabulated values (01-084-1167 ICDD file). This confirms the existence of a solid solution in the whole Br–I compositional range, and the full miscibility of the Ag3SBr and Ag3SI compounds, see Fig. 2(d). Also, analysis and pattern refinement have been performed with FullProf suite, applying the Le Bail method for all the crystalline phases observed. As a result, it is demonstrated that the cell volume and lattice parameters increase linearly along with the iodine content, which can be explained by the bigger volume of I− anions with respect to Br−, resulting in lattice expansion, see Fig. 2(e) and (f). The linear trend of these figures gives validity to our hypothesis of the formation of stable Ag3S(BrxI1−x) solid solutions with a cubic structure. On the other hand, a structural transition is observed between x = 0.3 and x = 0 (from Pmm to Imm space groups). This transition may be due to the larger size of the I− anion, triggering a phase change under the synthesis conditions used for this work. However, it is necessary to further investigate the formation mechanism of the solid solutions to fully understand the cause of this phenomenon. In addition, the nature of the molecular precursors and their impact on the formation of materials will be studied through solution analysis techniques by mass spectroscopy and nuclear magnetic resonance. See the calculated cell parameters and cell volume in Table S1 (ESI†).
To investigate the morphology and composition of the anti-perovskite samples, top-view SEM images have been acquired, along with energy dispersive spectroscopy analysis (EDX) measurements, see Fig. 3. At a microscopic level, all the samples present a homogeneous appearance, being constituted by small grains which form a compact layer. Also, the composition is remarkably constant. Notice in the EDX scan mappings that the distributions of Ag, S, and halide are highly uniform in every sample. Likewise, the incorporation of the halide in different proportions is confirmed, as can be noted by the chemical density gradation of the Br/I EDX colour maps as the halide amount (x) changes. Importantly, for the solid solutions, no Br or I-rich areas have been observed, indicating the successful formation of the mixed Ag3S(BrxI1−x) compounds instead of segregated phases. Nevertheless, despite the local homogeneity of these films, the 2 × 2 cm2 samples are not constituted by an entirely uniform layer, but rather interconnected islands of material, separated by regions where the substrate remains exposed, see Fig. S3 (ESI†). This might be caused by a poor adhesion between coating and substrate, whereby a different choice of solvents, or different proportion of the components in the solvent mixture could improve the deposition by modifying its surface tension. Alternately, these non-uniformities may result from a nucleation and crystallization process favoring the aggregation of the material, in which case using chelating agents, surfactants or polymer assisted methods could offer viable alternatives.41 All in all, SEM imaging characterization shows a remarkably high compositional homogeneity which, particularly in the case of samples containing both Br and I, holds up our hypothesis that this material supports the formation of solid solutions. However, it is necessary to improve the overall uniformity of the layers to achieve continuous thin films with a stable thickness.
 | ||
| Fig. 3 SEM images and EDX top-view scan maps of a series of Ag3S(BrxI1−x) samples, with x = 1, 0.7, 0.5, 0.3 and 0. | ||
The absorption spectra of Ag3S(BrxI1−x) samples have been acquired by photothermal deflection spectroscopy (PDS), see Fig. 4(a). With these measurements, the optical absorbance was obtained with a high precision (despite the small thickness and non-uniformity of the layers).42,43 Then, bandgap was calculated by applying a combined Tauc-Urbach model for the absorption front (see Annex 1 in ESI†), resulting in values of 0.88 eV and 0.97 eV for Ag3SI and Ag3SBr respectively. These bandgaps are in the appropriate range to absorb most of the solar spectra, making Ag3SI and Ag3SBr compatible for single junction solar cells or tandem solar cells as the bottom absorber material. The solid solution samples were also measured, presenting bandgap values around 1.0 eV (see Fig. S4, ESI†). Interestingly, unlike what can be observed with their structural properties, the bandgaps of Ag3S(BrxI1−x) anti-perovskites do not show a linear trend between the extreme cases (x = 1 and x = 0). Instead, we observe that the bandgap increases, saturates after a certain value of x (between 0.5 and 0.7), and then decreases again, see Fig. 4(b). The highest bandgap of this series of samples is 1.029 eV for Ag3S(Br0.5I0.5). This bandgap nonlinearity, also denominated “bandgap bowing effect”, is a common feature of both lead halide and oxide perovskite solid solutions, which has been attributed to the mismatch between atomic orbitals of the elements that constitute the solid solution.44–46 Overall, here it is shown that changing the halide (Br/I) does not have a significant impact on the optical properties, especially bandgap and absorption coefficient. Hence, we believe that it may be interesting to explore other areas of the anti-perovskite compositional map, including (AgyCu1−y)3(SzSe1−z)Br, to discover effective strategies for tuning the bandgap. Lastly, the large Urbach energy and sub-bandgap absorption indicate the need to optimize the material growth conditions, improving its morphology and crystallinity, and increasing the thickness (see Urbach energies in Table S2, ESI†). Nevertheless, previous optical analyses did not show a clear onset in the PDS spectra, which would allow for a straightforward determination of the bandgap, but rather a shallow slope ranging between 0.8 and 1.6 eV for Ag3SBr.14 Furthermore, first principle calculations performed at different levels of theory have typically yielded a broad dispersion of bandgap values, leading to some uncertainty concerning the optical properties of anti-perovskites, see Table 1.14,47 On the other hand, the measurements shown here present a well-defined absorption front, and bandgap values have been obtained through a model that correctly fits the experimental spectrum, suggesting that the synthetic approach holds promise to develop good quality crystalline materials. Furthermore, the DFT calculations presented below show good agreement with the measurements, thus advancing towards a better understanding of the properties of anti-perovskites and the development of a more appropriate method for its characterization.
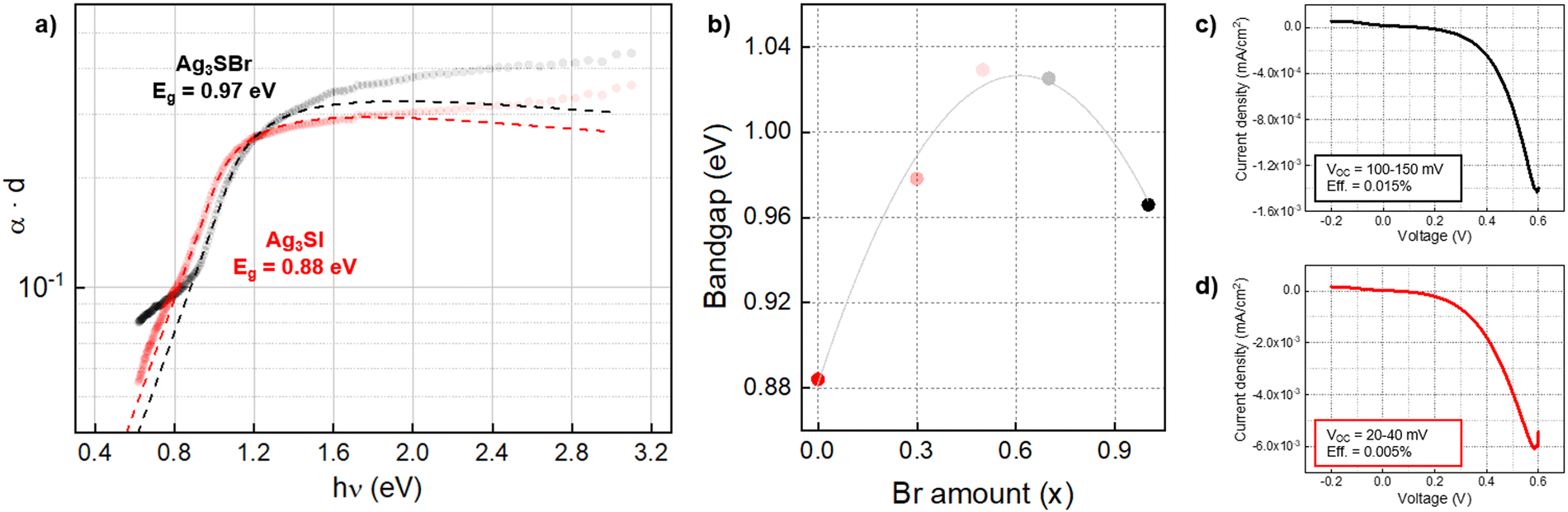 | ||
| Fig. 4 (a) Absorption spectra of Ag3SBr and Ag3SI films by PDS. (b) Bandgap as a function of x (Br amount). (c) Current–voltage characteristics of a Ag3SBr prototype solar cell. (d) Current–voltage characteristics of a Ag3SI prototype solar cell. | ||
Additionally, prototype solar cells have been prepared with the following superstrate architecture: FTO/TiO2/Ag3SX/P3HT/Au, where TiO2 works as electron transport layer (ETL) and P3HT is the hole transport layer (HTL). Ag3SBr and Ag3SI devices have both shown diode behavior with VOC values up to 150 mV, see Fig. 4(b) and (c). We highlight that these devices are not optimized, and that the anti-perovskite absorber presents a highly irregular, non-uniform morphology, with large and numerous pores which can clearly be responsible for significant shunt losses. Nevertheless, the fact that despite these faults we still obtain a diode current–voltage curve and a photoactive response indicates that these materials have a tangible potential as solar absorbers, and that by improving the uniformity and architecture of the device, their photovoltaic performance can also be enhanced.
The electronic and optical properties of the ternary compounds Ag3SBr and Ag3SI have been analyzed using theoretical first-principles methods based on density functional theory (DFT),24 considering a cubic Pmm structure for both materials. The lattice parameters and atomic positions were first optimized at the semi-local PBEsol level and the bandgap and optical absorption coefficients were subsequently estimated using a hybrid exchange–correlation functional and considering spin–orbit coupling effects.25,29,30 It is worth noting that semi-local DFT functionals are not able to provide reliable estimations of the bandgap due to their limitations related to self-interaction errors, however, the use of hybrid DFT functionals alleviates these technical problems to some extent.29,30 For the perfect equilibrium geometries determined at 0 K, bandgaps of 1.93 eV and 1.41 eV were obtained for the Ag3SBr and Ag3SI compounds, respectively. Remarkably, these theoretical values present significant discrepancies with the values measured by PDS in this work (see Fig. 4(a)). Indeed, they are approximately two times larger than the experimental results, indicating a non-satisfactory agreement between empirical evidence and calculations.
However, Ag-based anti-perovskites have been reported to be extremely anharmonic materials,48 suggesting that thermal effects such as electron–phonon coupling could play a relevant role on the estimation of the bandgap. In order to consider this possibility, finite-temperature ab initio molecular dynamics simulations at 200 and 600 K were carried out. Then, the bandgap was recalculated by performing statistical averages over a number of uncorrelated configurations obtained at these temperatures. It is worth noting that simulations performed at 600 K show the existence of ionic diffusion through mobility of the Ag+ cation. Considering these effects, the recalculated bandgap amounted to 1.37 and 1.15 eV for Ag3SBr (at 200 and 600 K, respectively), and 1.15 and 0.92 eV for Ag3SI; see Table 1, where the bandgap results from this work are presented along with previous DFT calculations and measurements. Therefore, after considering thermal and ionic diffusion effects, the agreement between theoretical and experimental optical properties (bandgap) is excellent within the statistical uncertainties. Consequently, the wide bandgap variation driven by temperature (roughly amounting to 1 eV from 0 to 600 K) may be the reason for the poor consensus among the existing DFT data on the optical properties of chalcohalide anti-perovskites.
Our experiments have shown that changing the relative Br/I content in Ag3SX (X = Br, I) anti-perovskites does not have a significant impact on the bandgap. Aimed at unravelling the causes for this experimentally observed behavior, we calculated the total and projected densities of electronic states for Ag3SBr and Ag3SI (see Fig. S5, ESI†). Our first-principles DFT results show that irrespective of the considered halide species, the Ag orbitals contribute the most to the top of the valence band and bottom of the conduction band, the two energy levels that define the band gap. Consequently, changing the relative content of Br/I has an almost negligible effect on Eg while performing Ag substitutions probably may induce more substantial bandgap variations.
Finally, the optical properties of Ag3SBr and Ag3SI were also theoretically analyzed. Fig. S6 (ESI†) shows the refractive index (n), extinction coefficient (κ), absorption coefficient (α), and reflectivity (R) estimated as a function of the incident photon energy. It is appreciated that Ag3SBr and Ag3SI present similar optical properties, with Ag3SI exhibiting overall larger values for all the investigated magnitudes. For instance, the refractive index of Ag3SI is about 10% larger than that of Ag3SBr within the whole visible spectrum range. Importantly, from the absorption coefficient results it can be concluded that both materials possess suitable properties for light harvesting since they exhibit α values of the order of 103–105 cm−1 within the whole visible spectrum range. Thus, our first-principles DFT calculations prove that Ag3SX (X = Br, I) anti-perovskites may be good candidates for photovoltaic applications.
In this work, the potential of Ag3SX (X = Br, I) anti-perovskites for PV or other energy applications has been discussed, based on their bandgap, perovskite-analogous structure and evidence of photoactive effect. Likewise, the proposed synthesis methodology allows to obtain the material at low temperature with tuneable composition, through a low-cost and easily scalable solution-based technique that does not require vacuum or high power. However, there are still some limitations that need to be overcome to achieve the maximum potential of the material and method. First, the morphological non-uniformities are a major obstacle to developing electronic devices, since the presence of layer discontinuities can be a source of resistance losses and carrier recombination. This can be addressed by optimizing the molecular precursor inks using other solvent mixtures or chelating agents. Also, by developing uniform layers it will be possible to determine their thickness accurately. Secondly, a thorough stability analysis is required to evaluate the degradation (or lack thereof) of the material upon exposure to moisture and sunlight. Third, morphology and crystallinity (i.e. grain size) need to be improved to reduce the impact of a large Urbach energy. Finally, to achieve a better understanding of the material it is necessary to perform a deeper characterization of its electronic, defect and phase-transition properties.
Conclusion
In this work we have demonstrated a solution-based low-temperature methodology to fabricate Ag3SX (X = Br, I) anti-perovskite materials, based on the deposition of thiol-amine molecular precursor inks. It has been shown that a solvent mixture of ethanedithiol and propylamine allows Ag3S(BrxI1−x) layers to be obtained without secondary phases at very low temperature (around 220 °C), with a cubic Pmm structure for the samples containing Br. Likewise, solid solutions throughout the whole Br–I range have been successfully synthesized, their formation confirmed by a remarkable compositional homogeneity (no Br or I-rich segregated phases), a linearly increasing trend in lattice parameters as the Br amount decreases, and bandgap bowing effect (lower bandgap for pure Br/I compounds than the intermediate compositions), similar to that reported for well-documented perovskite solutions. The optical bandgap in the 0.9–1.0 eV range and photo-active diode response also indicate that Ag chalcohalide anti-perovskites have a clear potential as semiconductors for energy applications, hence opening the door to a new family of highly versatile earth-abundant materials for thin-film photovoltaics. Finally, DFT calculations shed new light on the controversial anti-perovskite bandgap determination, showing that thermal effects such as electron–phonon coupling and ionic diffusion have a clear impact on the optical properties of the material, which might explain the wide dispersion of results reported so far.
Author contributions
I. C. Conceptualization, formal analysis, investigation, methodology, writing – original draft. J. W. T. Conceptualization, methodology, writing – review & editing. P. B. Data curation, investigation, software, writing – original draft. C. L. Investigation, software, methodology. J. M. A. Formal analysis, methodology. D. P. Methodology. J. P. Funding acquisition, resources, supervision. M. P. Funding acquisition, resources, supervision. C.C. Funding acquisition, resources, software, supervision. R. A. Conceptualization, funding acquisition, methodology, resources, supervision, writing – review & editing. E. S. Conceptualization, funding acquisition, methodology, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research has received funding from the European Union H2020 Framework Program under through the SENSATE project: low dimensional semiconductors for optically tunable solar harvesters (grant agreement number 866018). Other funding from the Ministry of science and innovation of the Government of Spain under the MATER-ONE (PID2020-116719RB-C41) and MIRACLE projects. I. C. acknowledges UPC and Banco Santander for his FPI-UPC PhD scholarship. M. P. acknowledges the financial support from Ministry of science and innovation within the Ramon y Cajal program (RYC-2017-23758). C. C. acknowledges support from the Spanish Ministry of Science, Innovation and Universities under the fellowship RYC2018-024947-I and grant TED2021-130265B-C22. P. B., C. L. and C. C. thankfully acknowledge the computer resources at MareNostrum and the technical support provided by Barcelona Supercomputing Center (RES-FI-1-0006, RES-FI-2022-2-0003, RES-FI-2023-1-0002 and RES-FI-2023-2-0004). E. S. acknowledges the ICREA Academia program. J. W. T. and R. A. are appreciative of the funding support from the National Science Foundation through grants 1735282-NRT (SFEWS) and 10001536 (INFEWS).References
- F. Palazon, Metal Chalcohalides: Next Generation Photovoltaic Materials?, Sol. RRL, 2022, 6(2), 2100829, DOI:10.1002/solr.202100829.
- M. Green, E. Dunlop, J. Hohl-Ebinger, M. Yoshita, N. Kopidakis and X. Hao, Solar Cell Efficiency Tables (Version 57), Prog. Photovoltaics Res. Appl., 2021, 29(1), 3–15, DOI:10.1002/pip.3371.
- A. Zakutayev, J. D. Major, X. Hao, A. Walsh, J. Tang, T. K. Todorov, L. H. Wong and E. Saucedo, Emerging Inorganic Solar Cell Efficiency Tables (Version 2), JPhys Energy, 2021, 3(3) DOI:10.1088/2515-7655/abebca.
- R. Nie, M. Hu, A. M. Risqi, Z. Li and S. Seok II, Efficient and Stable Antimony Selenoiodide Solar Cells, Adv. Sci., 2021, 8(8), 2003172, DOI:10.1002/advs.202003172.
- R. Nie, H. S. Yun, M. J. Paik, A. Mehta, B. W. Park, Y. C. Choi and S. Seok, Il. Efficient Solar Cells Based on Light-Harvesting Antimony Sulfoiodide, Adv. Energy Mater., 2018, 8(7), 1–7, DOI:10.1002/aenm.201701901.
- Y. C. Choi and K. W. Jung, One-Step Solution Deposition of Antimony Selenoiodide Films via Precursor Engineering for Lead-Free Solar Cell Applications, Nanomaterials, 2021, 11(12), 3206, DOI:10.3390/nano11123206.
- K. W. Jung and Y. C. Choi, Compositional Engineering of Antimony Chalcoiodides via a Two-Step Solution Process for Solar Cell Applications, ACS Appl. Energy Mater., 2021, 5, 5348–5355, DOI:10.1021/acsaem.1c02676.
- U. V. Ghorpade, M. P. Suryawanshi, M. A. Green, T. Wu, X. Hao and K. M. Ryan, Emerging Chalcohalide Materials for Energy Applications, Chem. Rev., 2023, 123(1), 327–378, DOI:10.1021/acs.chemrev.2c00422.
- B. Reuter and K. Hardel, Silbersulfidbromid Und Silbersulfidjodid, Angew. Chem., Int. Ed. Engl., 1960, 72(4), 138–139, DOI:10.1002/zaac.19653400308.
- T. Takahashi and O. Yamamoto, The Ag/Ag3SI/I2 Solid-Electrolyte Cell, Electrochim. Acta, 1966, 11, 779–789, DOI:10.1016/0013-4686(66)87055-X.
- L. Yin, M. Murphy, K. Kim, L. Hu, J. Cabana, D. J. Siegel and S. H. Lapidus, Synthesis of Antiperovskite Solid Electrolytes: Comparing Li3SI, Na3SI, and Ag3SI, Inorg. Chem., 2020, 59(16), 11244–11247, DOI:10.1021/acs.inorgchem.0c01705.
- Z. Liu, R. Mi, G. Ji, Y. Liu, P. Fu, S. Hu, B. Xia and Z. Xiao, Bandgap Engineering and Thermodynamic Stability of Oxyhalide and Chalcohalide Antiperovskites, Ceram. Int., 2021, 47(23), 32634–32640, DOI:10.1016/j.ceramint.2021.08.159.
- W. Shimosaka, S. Kashida and M. Kobayashi, Electronic Structure of Ag3SI, Solid State Ionics, 2005, 176(3–4), 349–355, DOI:10.1016/j.ssi.2004.08.001.
- P. Sebastiá-Luna, N. Rodkey, A. S. Mirza, S. Mertens, S. Lal, A. M. Gaona Carranza, J. Calbo, M. Righetto, M. Sessolo, L. M. Herz, K. Vandewal, E. Ortí, M. Morales-Masis, H. J. Bolink and F. Palazon, Chalcohalide Antiperovskite Thin Films with Visible Light Absorption and High Charge-Carrier Mobility Processed by Solvent-Free and Low-Temperature Methods, Chem. Mater., 2023, 35(16), 6482–6490, DOI:10.1021/acs.chemmater.3c01349.
- Y. S. Tver’yanovich, M. D. Bal’makov, V. V. Tomaev, E. N. Borisov and O. Volobueva, Ion-Conducting Multilayer Films Based on Alternating Nanolayers Ag3SI, AgI and Ag2S, AgI, Glass Phys. Chem., 2008, 34(2), 150–154, DOI:10.1134/S1087659608020065.
- R. Blachnik and H. A. Dreisbach, The Phase Diagrams of Ag2X-AgY (X = S, Se, Te; Y = Cl, Br, I): Mixtures and the Structure of Ag5Te2CI, 1985, vol. 60 Search PubMed.
- M. V. Moroz, M. V. Prokhorenko, S. V. Prokhorenko and O. V. Reshetnyak, Thermodynamic Study of Phase Equilibrium of Superionic Alloys of Ag3SBr1−xClx System in the Concentration Range 0.0–0.4 and Temperature Range 370–395 K, Arch. Thermodyn., 2017, 38(1), 27–38, DOI:10.1515/aoter-2017-0002.
- R. B. Beeken and E. M. Beeken, Ionic Conductivity in Cu-Substituted Ag3SBr, Solid State Ionics, 2000, 136–137, 463–467, DOI:10.1016/S0167-2738(00)00564-6.
- M. Salado, D. Payno and S. Ahmad, Enhancing Operational Stability in Perovskite Solar Cells by Solvent-Free Encapsulation Method, Sustainable Energy Fuels, 2022, 6(9), 2264–2275, 10.1039/d1se02054d.
- N. Juneja, S. Mandati, A. Katerski, N. Spalatu, S. Daskeviciute-Geguziene, A. Vembris, S. Karazhanov, V. Getautis, M. Krunks and I. Oja Acik, Sb2S3 Solar Cells with a Cost-Effective and Dopant-Free Fluorene-Based Enamine as a Hole Transport Material, Sustainable Energy Fuels, 2022, 6(13), 3220–3229, 10.1039/d2se00356b.
- J. Rodríguez-Carvajal; T. Roisnel and J. Gonzales-Platas FullProf Suite. Laboratoire Léon Brillouin, CEA-CNRS, CEN Saclay, France 2005.
- I. Caño, P. Vidal-Fuentes, L. Calvo-Barrio, X. Alcobé, J. M. Asensi, S. Giraldo, Y. Sánchez, Z. Jehl, M. Placidi, J. Puigdollers, V. Izquierdo-Roca and E. Saucedo, Does Sb2Se3 Admit Nonstoichiometric Conditions? How Modifying the Overall Se Content Affects the Structural, Optical, and Optoelectronic Properties of Sb2Se3 Thin Films, ACS Appl. Mater. Interfaces, 2022, 14(9), 11222–11234, DOI:10.1021/acsami.1c20764.
- J. Serra, J. Andreu, G. Sardin, C. Roch, J. M. Asensi, J. Bertomeu and J. Esteve, Hydrogen Related Effects in A-Si:H Studied by Photothermal Deflection Spectroscopy, Phys. B, 1991, 170(1–4), 269–272, DOI:10.1016/0921-4526(91)90136-3.
- C. Cazorla and J. Boronat, Simulation and Understanding of Atomic and Molecular Quantum Crystals, Rev. Mod. Phys., 2017, 89(3) DOI:10.1103/RevModPhys.89.035003.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces, Phys. Rev. Lett., 2008, 100(13) DOI:10.1103/PhysRevLett.100.136406.
- G. Kresse and J. Furthmü, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15) DOI:10.1063/1.3382344.
- A. M. Ganose, M. Cuff, K. T. Butler, A. Walsh and D. O. Scanlon, Interplay of Orbital and Relativistic Effects in Bismuth Oxyhalides: BiOF, BiOCl, BiOBr, and BiOI, Chem. Mater., 2016, 28(7), 1980–1984, DOI:10.1021/acs.chemmater.6b00349.
- E. N. Brothers, A. F. Izmaylov, J. O. Normand, V. Barone and G. E. Scuseria, Accurate Solid-State Band Gaps via Screened Hybrid Electronic Structure Calculations, J. Chem. Phys., 2008, 129(1) DOI:10.1063/1.2955460.
- J. A. Clark, A. Murray, J. M. Lee, T. S. Autrey, A. D. Collord and H. W. Hillhouse, Complexation Chemistry in N,N-Dimethylformamide-Based Molecular Inks for Chalcogenide Semiconductors and Photovoltaic Devices, J. Am. Chem. Soc., 2019, 141(1), 298–308, DOI:10.1021/jacs.8b09966.
- P. Murria, C. K. Miskin, R. Boyne, L. T. Cain, R. Yerabolu, R. Zhang, E. C. Wegener, J. T. Miller, H. I. Kenttämaa and R. Agrawal, Speciation of CuCl and CuCl2 Thiol-Amine Solutions and Characterization of Resulting Films: Implications for Semiconductor Device Fabrication, Inorg. Chem., 2017, 56(23), 14396–14407, DOI:10.1021/acs.inorgchem.7b01359.
- K. M. Koskela, M. J. Strumolo and R. L. Brutchey, Progress of Thiol-Amine ‘Alkahest’ Solutions for Thin Film Deposition, Trends Chem., 2021, 1, 1061–1073, DOI:10.1016/j.trechm.2021.09.006.
- D. H. Webber and R. L. Brutchey, Alkahest for V2VI3 Chalcogenides: Dissolution of Nine Bulk Semiconductors in a Diamine-Dithiol Solvent Mixture, J. Am. Chem. Soc., 2013, 135(42), 15722–15725, DOI:10.1021/ja4084336.
- J. W. Turnley, K. C. Vincent, A. A. Pradhan, I. Panicker, R. Swope, M. C. Uible, S. C. Bart and R. Agrawal, Solution Deposition for Chalcogenide Perovskites: A Low-Temperature Route to BaMS3 Materials (M = Ti, Zr, Hf), J. Am. Chem. Soc., 2022, 144(40), 18234–18239, DOI:10.1021/jacs.2c06985.
- R. Zhang, S. Cho, D. G. Lim, X. Hu, E. A. Stach, C. A. Handwerker and R. Agrawal, Metal–Metal Chalcogenide Molecular Precursors to Binary, Ternary, and Quaternary Metal Chalcogenide Thin Films for Electronic Devices, Chem. Commun., 2016, 52(28), 5007–5010, 10.1039/c5cc09915c.
- J. Guo, Y. Pei, Z. Zhou, W. Zhou, D. Kou and S. Wu, Solution-Processed Cu2ZnSn(S,Se)4 Thin-Film Solar Cells Using Elemental Cu, Zn, Sn, S, and Se Powders as Source, Nanoscale Res. Lett., 2015, 10(1) DOI:10.1186/s11671-015-1045-6.
- S. Hull, D. A. Keen, J. G. Gardner and W. Hayes, The Crystal Structures of Superionic Ag3SI, J. Phys.: Condens. Matter, 2001, 13, 2295–2316, DOI:10.1088/0953-8984/13/10/321.
- S. Hoshino, H. Fujishita, M. Takashige and T. Sakuma, Phase Transition of Ag3SX (X = I, Br), Solid State Ionics, 1981, 3(4), 35–39, DOI:10.1016/0167-2738(81)90050-3.
- K. Momma and F. Izumi, VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data, J. Appl. Crystallogr., 2011, 44(6), 1272–1276, DOI:10.1107/S0021889811038970.
- J. M. Vila-Fungueiriño, B. Rivas-Murias, J. Rubio-Zuazo, A. Carretero-Genevrier, M. Lazzari and F. Rivadulla, Polymer Assisted Deposition of Epitaxial Oxide Thin Films, J. Mater. Chem. C, 2018, 3834–3844, 10.1039/c8tc00626a.
- W. B. Jackson, N. M. Amer, A. C. Boccara and D. Fournier, Photothermal Deflection Spectroscopy and Detection, Appl. Opt., 1981, 20(8), 1333, DOI:10.1364/ao.20.001333.
- A. J. Lopez-Garcia, C. Voz, J. M. Asensi, J. Puigdollers, V. Izquierdo-Roca and A. Pérez-Rodríguez, Ultrathin A-Si:H/Oxide Transparent Solar Cells Exhibiting UV-Blue Selective-Like Absorption, Sol. RRL, 2023, 7, 2200928, DOI:10.1002/solr.202200928.
- A. Goyal, S. McKechnie, D. Pashov, W. Tumas, M. Schilfgaarde and V. Van Stevanović, Origin of Pronounced Nonlinear Band Gap Behavior in Lead-Tin Hybrid Perovskite Alloys, Chem. Mater., 2018, 30(11), 3920–3928, DOI:10.1021/acs.chemmater.8b01695.
- Q. Gao, H. Sahin, J. Kang and S. H. Wei, Origin of Anomalous Band-Gap Bowing in Two-Dimensional Tin-Lead Mixed Perovskite Alloys, Phys. Rev. B, 2021, 104(6) DOI:10.1103/PhysRevB.104.064204.
- S. Lee, R. D. Levi, W. Qu, S. C. Lee and C. A. Randall, Band-Gap Nonlinearity in Perovskite Structured Solid Solutions, J. Appl. Phys., 2010, 107(2) DOI:10.1063/1.3291102.
- M. Kurita, K. Nakagawa and K. Wakamura, Fundamental Absorption Edge of α-AgI-Type Superionic Conductor Ag3SBr, 2000, 39, 1786, DOI:10.1143/JJAP.39.1786.
- K. Wakamura, F. Miura and T. Kanashiro, Observation of Anomalously Increasing Phonon Damping Constant in the P Phase of the Fast-Ionic Conductor Ag3SI, 1990, 41, 5, DOI:10.1103/PhysRevB.41.2758.
- M. Kurita, K. Nakagawa and F. Akao, Photoacoustic Spectrum in Superionic Conductors Ag3SI and AgI, Jpn. J. Appl. Phys., 1988, 27(10), 1920–1922 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc04410f |
| This journal is © The Royal Society of Chemistry 2024 |