
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of DP and MMD of the pulps used in the Ioncell® process on processability and fiber properties†
Yibo
Ma
 *,
Xiang
You
,
Kaarlo
Nieminen
,
Daisuke
Sawada
and
Herbert
Sixta
*
*,
Xiang
You
,
Kaarlo
Nieminen
,
Daisuke
Sawada
and
Herbert
Sixta
*
Department of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, P.O. Box 16300, 00076 Aalto, Finland. E-mail: angeless1115@gmail.com; Herbert.sixta@aalto.fi
First published on 11th July 2023
Abstract
In this study, we have demonstrated for the first time the ability of the Ioncell fiber spinning process to produce high-quality regenerated cellulose fibers from dissolving pulps with a wide range of intrinsic viscosity from 140 to 1300 mL g−1 and different molecular mass distributions prepared by the applied acid, enzyme and sequential acid–enzyme pretreatments. To minimize the influence of the hemicellulose content, only dissolving pulps with a hemicellulose content of less than 4% were used. The rheological properties exhibited certain threshold values beyond which the spinnability of the prepared dope can be severely affected. A spinning dope with a dynamic modulus higher than 6000 Pa s or less than 1600 Pa s is not spinnable or results in an acceptable draw ratio. The best spinnability can be obtained at a crossover point for the dynamic moduli between 2800 and 3500 Pa and for the angular frequency between 0.4 and 1.2 rad s−1. In these ranges, the highest draw ratios of the spun filaments can be obtained for pulps with a viscosity of 420 to 500 mL g−1 and the lowest linear density (fiber titer) for pulps with viscosity from 500 to 730 mL g−1. The strength of the spun fibers is strongly DP dependent. The fiber tenacity gradually decreases with reducing pulp viscosity until non-spinnable dopes are prepared at a pulp viscosity below 300 mL g−1. Compared to acid-treated pulp, fibres made from enzyme-treated pulp generally have lower strength, which is due to a high polydispersity expressed in a high proportion of short-chain cellulose. At a suitable pulp viscosity between 360 and 820 mL g−1 and a cellulose concentration in the spinning dope of 11 to 15 wt%, Ioncell fibres with excellent tensile strength and toughness properties can be produced.
Sustainability spotlightThe textile industry is known to be one of the biggest polluters of environmental emissions. However, spinning staple or continuous fibres from sustainable raw materials such as dissolving wood pulp or recycled cellulosic textile waste makes a decisive contribution to the shift towards sustainable closed-loop production of textiles, provided that the entire spinning process can be carried out in a closed loop. In the present work, we demonstrate the production of sustainable man-made cellulosic fibres from a solution of dissolving pulp in a green direct solvent, 1,5-diazabicyclo[4.3.0]non-5-enium acetate. By selectively adjusting the initial degree of polymerization and the molecular weight distribution of the dissolving wood pulps, regenerated cellulose fibers with extremely high mechanical strengths approaching those of polyester can be produced. The current work at hand aligns with the UN sustainability development goals 12 for sustainable consumption and production and 13, 14, 15 for climate and life protection on the earth. |
1. Introduction
Man-made cellulose fibers (MMCF) represent a class of fiber that is regenerated from the cellulose solution form in a solvent. The development of regenerated cellulose fibers can be traced back to 1855 when technology was invented to produce the cellulose fiber by regeneration from cellulose nitrate.1 By far the most successful MMCF technology is the viscose process. It utilizes an indirect dissolution method to shape the cellulose into fibers by wet spinning. In the viscose process, cellulose is first converted to an alkali-cellulose by steeping in an 18% NaOH solution, which is then derivatized to cellulose xanthate by gas phase treatment with CS2 before being dissolved in dilute NaOH to form a spinning solution, which is wet-spun in an acidic aqueous coagulation bath and finally regenerated to a viscose fiber.2 The production of viscose fibres reached its first peak in the 1970s. Since then, the market share has been pushed back considerably by the strong development of petroleum-based synthetic fibers. Due to their high mechanical performance, excellent processability and applicability, and usually lower cost, synthetic fibers immediately penetrated the textile market and are still by far the dominant raw material for textile applications.1 However, the fast-paced development of mankind's society severely impacts environmental sustainability. The extensive production, utilization and inadequate recycling of petroleum-based materials caused serious problems to the ecosystem. In view of these challenges, the major governments have set specific targets to achieve sustainability goals by 2030. Within this framework, the production of cellulose-based regenerated fibers could play an important role in the transition from a linear, unsustainable economy, to a sustainable circular economy.3In addition to the sustainability approach, the stagnant cotton production since the beginning of the century also urged regeneration of cellulose fibers to close the gap between production and demand. It was predicted that, by 2030, the anticipated need for an additional twenty million tons of cellulose fiber is expected in order to fulfil the market demand.4 Against this background, the research and development of regenerated cellulose fibers has experienced an upturn in the last ten years. In addition to the viscose process, NMMO lyocell technology has established itself as another successful MMCF technology in the last 20 to 30 years, even though the annual production of NMMO lyocell fibers at 400 kt/a only accounts for about 6% of viscose fiber production. However, due to the many lyocell capacities under construction, this share will increase significantly in the coming years (The Fiber Year, 2022). Lyocell technology uses a direct cellulose dissolution strategy before spinning, which simplifies the processing technology and avoids the use of harmful chemicals.5,6
Ioncell technology is an MMCF process that has not yet been commercialized and belongs to the category of lyocell technology. In this process, as in the NMMO lyocell spinning process, regenerated cellulose fibers are produced by dry-jet wet spinning (or so-called air gap spinning).7–9 After a decade of development, the process entered the pilot phase in 2021 and is expected to be commercialized in the coming years after proof of concept. The Ioncell technology utilizes superbase-based ILs jointly developed by the University of Helsinki and Aalto University in Finland. The standard Ioncell fiber is produced by first direct dissolution of dissolving wood pulp in 1,5-diaza-bicyclo[4.3.0]non-5-enium acetate ([DBNH][OAc]) and then spun into a water bath via an air gap.10,12 The IL dissolves the cellulose by forming hydrogen bonds between the anion of the IL and the polar hydroxyl groups of the cellulose, with the cation of the IL interacting with the cellulose via hydrophobic interactions.11 During the spinning process, the extruded solution filaments can be stretched in the air gap, causing the randomly oriented molecular chains in the solution to align parallel to the molecular axis. Following coagulation and crystallization of the cellulose in the antisolvent water, a high-performance regenerated cellulose fiber can be obtained.9 As a result of the Ioncell spinning process using a commercial hardwood prehydrolysis kraft pulp (PHK) without further purification measures, a standard Ioncell fiber exhibits a tenacity of ∼50 cN per tex, an elongation at break of ∼12% and a modulus of elasticity of 20–30 GPa under dry conditions.7,8
The mechanical performance of regenerated cellulose fibers is governed by several key factors, including IL properties, spinning process conditions, and most importantly, pulp properties (chemical composition, DP and molar mass distribution etc.), to name a few.10
Both the spinning processes themselves and the spinnability of the spinning solutions determine the properties of the resulting fibers. The wet-spun standard viscose fiber generally exhibits relatively low strength properties compared to the lyocell fiber, which is mainly due to the lower stretchability which, after all, takes place entirely in the spinning bath and results from the only slightly decoupled coagulation and regeneration compared to the modal fiber or supercord fiber process.14 After extrusion into the coagulation bath, the cellulose viscose gel immediately regenerated with a stretch-ratio of 50–60%, resulting in a weak fiber with a poorly orientated, fringed micellar structure of crystallite sections linked with amorphous regions. To improve the stretchability of the viscose gel and thus the fiber strength, additives such as ZnSO4 and/or organic modifiers are added to the coagulation bath to retard regeneration and allow greater stretch of the filament prior to crystallization. Under these conditions, high tenacity fibers such as modal and supercord fibers can be produced.15
When spinning lyocell fibers, the process conditions, such as temperature and cellulose concentration, must be carefully controlled so that the spinning dope exhibits suitable rheological properties that allow the extensive stretching of the filaments in the air gap.13,16 It has been suggested that fibers produced with increasing stretch ratio (up to a certain value) exhibit fringed fibril structures with increasing crystallite orientation and long period (length of the successive crystalline and amorphous zone) and thus potentially improved strength.1 In addition, the air gap conditions, such as a suitable air gap distance and appropriate conditioning in the air gap, also promote the high stretchability of the dope and the high strength of the spun fibers.17,18
The characteristics of cellulosic raw material are also very crucial for the properties of the regenerated fibers. To ensure a uniform dissolution of the raw material in a solvent (as a prerequisite for the spinning), the lignocellulose must be separated into its polymeric constituents by a chemical pulping process, firstly to break up the recalcitrant structure and secondly to remove the non-cellulose components allowing the solvent to penetrate into the cellulose structures.19–23 Under given process conditions, the use of a pulp with increasing cellulose content correlates with the production of regenerated cellulose fibers with increasing mechanical strengths, especially if the length of the cellulose molecules is evenly distributed. In the development of the Ioncell technology, this was demonstrated by using a high purity dissolving pulp with a hemicellulose content of only 0.7% and an intrinsic viscosity of 594 mL g−1, producing an Ioncell fiber with a dry strength of 61.5 cN per tex.24 Thus, by simply replacing a standard dissolving pulp (6.8% hemicellulose and 482 mL g−1 viscosity) with a high-purity dissolving pulp, the fiber strength could be increased by about 12 cN per tex.25
Several studies done by Ma et al. demonstrated the negative influences of hemicellulose and lignin on the dope spinnability and fiber properties. In extreme cases, a raw material consisting of 50% lignin and 50% cellulose or 22% hemicellulose, 24% lignin and 54% cellulose may lead to an Ioncell fiber that is still easily spinnable but only has a tenacity similar to viscose fibers.21,26
As shown above with an example, DP and molecular weight distribution are other critical pulp properties that control the spinning performance and fiber properties. Already during the development of viscose fibers in the course of the 20th century, numerous studies were carried out on the effects of DP on fiber properties. The DP is known to affect the strength of fibers and in particular regenerated cellulose fibers.1
As early as the 1930s, Hermann Staudinger observed that a minimum DP, measured as DPn = 60–100 and DPw = 100–150, was required for the production of regenerated cellulose fibers with useable strengths.27,28. Flory was the first to assume a linear relationship between tensile strength and reciprocal DPn.29 This finding is based on the results of Sooke and Harris, who have shown that the tensile strength of cellulose acetate fibers depends explicitly on DPn.30 When plotting the tensile strength (T) values against the reciprocal DPn following the equation, T = a0 + a1/![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n, a straight-line relationship was obtained, where the DPn was measured by the osmotic pressure method. This explicit correlation with the DPn (or n) was interpreted by Spurlin such that the breakage starts from the ends of the molecules and therefore the fiber strength should depend on the number of (reducing) ends.31
n, a straight-line relationship was obtained, where the DPn was measured by the osmotic pressure method. This explicit correlation with the DPn (or n) was interpreted by Spurlin such that the breakage starts from the ends of the molecules and therefore the fiber strength should depend on the number of (reducing) ends.31
In an extensive study, regenerated cellulose fibers with different molecular masses in the range of DP 786-111 were spun from fractionated cellulose diacetates dissolved in concentrated acetic acid and spun under the same conditions in an aqueous potassium acetate–acetic acid solution. The filaments were stretched in boiling water to form filaments with different degrees of orientation and then saponified in aqueous alkaline solution.32 The results confirm that when strength data are selected for a narrow birefringence range of 0.030 ± 0.006, there is a good linear correlation between the dry as well as wet tensile strengths with the reciprocal DPn, where the intersection of the straight lines with a tensile strength of zero can be calculated at a DP in the range of 80 to 100, indicating a theoretical DP minimum. At a low degree of polymerization, the cellulose molecules are not long enough to span at least two crystallites, causing the tensile strength to decrease sharply and eventually drop to zero. These results are consistent with the fringe-micellar theory, which assumes that crystalline domains are separated by amorphous domains in which molecules can span two or more crystalline domains. This explains that as the average length of molecules increases, they become more likely to connect two or more crystallites. Thus, the more molecules that connect the crystalline domains, the greater the strength of the filament. It has been shown that above a certain chain length, the DP no longer has a measurable effect on strength.33
From the above considerations, it can be summarized that in the case of a continuous structure, the proportion of intact bonds at a given number-average degree of polymerization is proportional to the expression 1 − 1/DPn. In the presence of elementary fibrils, as in cellulose fibers, morphological units exist. In this case, there is proportionality between the proportion of intact bonds and the expression (1/DPnl − 1/DPn), which gives the length of the fiber-forming cellulose molecules relative to the length of the elementary crystallites, where DPnl corresponds to the crystallite length, which can be approximated by the level-off-DP.34
In addition to the degree of polymerization of the cellulose molecules, the crystallinity and the degree of orientation of the morphological units and their interlinks affect the tensile strength of MMCFs. Krässig and Kitchen have proposed an empirical model according to which the conditioned tensile strengths of different MMCFs depend linearly on the product of the difference in the reciprocal number-average degree of polymerization of the crystallites and cellulose molecules, according to (1/DPnl − 1/DPn), the crystallinity CrI, and the square of the degree of crystalline orientation, fr2.34
More recent studies on the strength of lyocell and ionic liquid-based fibers have confirmed that the tensile strength of the fibers rises with increasing DP of the dissolving pulp.35–38 However, there are no systematic studies on the influence of the DP of the pulp on fiber strength, and moreover, the previous studies only used pulps with a limited DP range.
Therefore, in our current study, we have prepared a series of high purity pulps with intrinsic viscosities (represents the DPv) ranging from ∼1300 down to 140 mL g−1. The scope is to determine the DP limitation of the cellulose–IL dope spinnability and the development of the fiber strength at this pulp viscosity range. In addition, pulps with similar viscosities but different MMDs were prepared by acid and enzyme treatment to assess the role of MMDs in spinning and fiber performance.
2. Experimental methods
2.1 Material
A softwood acid sulfite (SWS) dissolving pulp (Rayonier HV+) was acquired from Rayonier Inc., Florida, USA. A hardwood pre-hydrolysed kraft (HWPHK) pulp (Bracell Crystal LD HV) was kindly provided by Bracell limited, Bahia, Brazil. The initial properties of the pulps are listed in Table 1. The ionic liquid (IL) 1,5-diazabicyclo[4.3.0]non-5-enium acetate ([DBNH][OAc]) was used as a cellulose solvent. [DBNH][OAc] was synthesized from 1,5-diazabicyclo[4.3.0]non-5-ene (DBN, supplied by Fluorochem, UK, 99% purity) and acetic acid (glacial, supplied by Merck, Germany, 100% purity). An equimolar amount of acetic acid was slowly added to DBN. The temperature was controlled and allowed to rise to 70 °C in order to avoid crystallization of the IL. The IL, 1-ethyl-3-methylimidazolium acetate ([EMIM][OAc], ≥98%), was acquired from PROIONIC GmbH, Austria. Sulfuric acid (purity 95–97%) was supplied by Merck, Germany. The enzyme ECOPULP® R (endoglucanase) was provided by AB Enzymes Oy, Finland.| Intrinsic viscosity (mL g−1) | Cellulose (%) | Hemicellulosea (%) | Lignin (%) | M w (Da) | DP > 2000 | DP < 100 | PDI | |
|---|---|---|---|---|---|---|---|---|
| a Sum of glucuronoxylan and glucomannan in the case of hardwoods and arabinoxylan and galactoglucomannan in the case of softwood. | ||||||||
| SWS | 1456 | 93.5 | 5.7 | 0.8 | 427![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
1.7 | 56.6 | 3.2 |
| SWS-IP | 1474 | 96.8 | 2.0 | 1.2 | 421000 |
0.2 | 58.4 | 2.2 |
| HWPHK | 653 | 95.6 | 4.0 | 0.4 | 231300 |
24.1 | 1.8 | 2.4 |
2.2 Pulp purification and adjustment of the degree of polymerization (DP)
The SWS pulp was treated using Ioncell-P technology, causing partial extraction of hemicelluloses. Hemicellulose removal for SWS pulp was performed using [EMIM][OAc] water mixture in a vertical kneader system at 60 °C under ambient pressure for 3 hours. The pulp to solution ratio is 5% pulp, 81% IL and 14% H2O. The mixture was subsequently filtered in a hydraulic press filter (metal filter mesh with 5 μm absolute fineness, Gebr. Kufferath AG, Germany) to separate the solid residue and the liquid phase in which the hemicelluloses are dissolved. The solid residue, which contained mainly cellulose, was washed twice with warm water (60 °C) and twice with tap water to remove the residual IL and extracted hemicellulose in the solid residue. The wash water was then added to the separated liquid phase to precipitate the hemicelluloses. The precipitate was separated from the IL–water mixture by centrifugation, and the remaining IL–water mixture was recovered by evaporation using a thin film evaporator as described by Elsayed et al.39 (Elsayed et al. 2020). The recycled IL was again utilized for the hemicellulose extraction of the SWS pulp.The washed solid cellulose residue (SWS – IP) and the HWPHK pulp were subjected to acidic and enzymatic hydrolysis for DP adjustment in a glass vessel reactor. The acidic treatment was carried out at an H2SO4 concentration of 0.1 mol L−1 at 80 °C and 5% pulp consistency. By adjusting the treatment duration, different DP targets of the pulps could be achieved. The enzymatic hydrolysis was performed at 50 °C for 180 min with a pulp consistency of 5%. Various enzyme dosages were applied to achieve pulps with different DPs. Experiments were also conducted in which the DP was adjusted by successive treatments with acid and enzymes.
2.3 Pulp dissolution and spinning trials
[DBNH][OAc] was first melted at 80 °C, then blended with the air-dried pulp (ground with a Willey mill with 1 mm mesh sieves), stirred for 2 h at 80 °C and 30 rpm at reduced pressure (1–10 mbar) using a vertical kneader system. The polymer concentration of the dope was adjusted according to the intrinsic viscosity of the pulps. The solutions were filtered through a hydraulic press filter device (metal filter mesh with 5 μm absolute fineness, Gebr. Kufferath AG, Germany) at 2 MPa and 80 °C to remove the undissolved substrate, which would lead to unstable spinning. The prepared dope was finally shaped into the dimensions of the spinning cylinder and solidified upon cooling overnight to ensure filling without the inclusion of air bubbles.Multi-filaments were spun with a customized laboratory piston spinning system (Fourné Polymertechnik, Germany). The solidified spinning dope was heated to 70–90 °C (depending on dope rheology) in the spinning cylinder to form a highly viscous, air-bubble-free spinning dope. The molten solution was then extruded through a 200-hole spinneret with a capillary diameter of 100 μm and a length-to-diameter ratio (L/D) of 0.2. After the fluid filaments had passed an air gap of 10 mm, they were coagulated in a water bath (10 to 15 °C) in which they were guided by Teflon rollers to the godet couple. The extrusion velocity (Ve) was set to 5.5 mL min−1 (3.5 m min−1), while the take-up velocity (Vt) of the godet was varied until the maximum draw ratio (DR = Vt/Ve) was reached. The fibers were then washed off-line in hot water (60 °C) and air-dried.
2.4 Rheology measurement
The rheological behaviour of the dope was measured using an Anton Paar MCr 300 rheometer having parallel plate geometry (plate diameter = 25 mm; gap size = 1 mm). The dynamic strain sweep tests were performed with a 0.5% strain in the temperature range 60–100° over an angular velocity range 0.1–100 s−1. Complex viscosity and the dynamic moduli were recorded. The zero-shear viscosity was calculated using a cross-viscosity model, considering that the Cox–Merz rule is valid.2.5 Pulp properties
The molar mass distribution (MMD) of the pulps was measured using a Dionex Ultimate 3000 HPLC system set-up equipped with a Viscotek/Malvern SEC/MALS 20 multi-angle light-scattering (MALS) detector. After solvent exchange (water/acetone/DMAc), the samples were dissolved in a saturated LiCl/DMAc solution followed by the dilution to reach an eluent concentration of 0.9% LiCl/DMAc. 100 μl of each sample solution was placed into the four-column system (PLgel MIXED-A) operating at a flow-rate of 0.75 mL min−1. A narrow polystyrene standard (Mw = 96000 g mol−1, Đ = 1.04) was used to obtain the detector constants for MALS and DRI, whereas a broad polystyrene sample (Mw = 248000 g mol−1, Đ = 1.73) was applied to test the calibration of the detectors. According to Potthast et al., a ∂n/∂c value of 0.136 mL g−1 was reported for cellulose dissolved LiCl/DMAc (0.9%).40 In addition to the differential molar mass distribution, the distribution of the cumulative weight fractions over the entire molar mass range was also evaluated in order to be able to calculate the weight fractions DP < 100 and DP > 2000.
The intrinsic viscosity of the pulp was determined in cupriethylenediamine (CED) according to the standard method SCAN-CM 15:99.
The carbohydrate and hemicellulose contents were determined according to the NREL/TP-510-42618 measured by high-performance anion exchange chromatography with pulse amperometric detection (HPAEC-PAD) using a Dionex ICS-3000 system. Janson formula was utilized to interpret the actual composition of cellulose and hemicellulose in the materials.41
2.6 Mechanical properties of the spun fibers
The tensile properties of the fibers were measured using an automatic single-fiber tester Favigraph instrument (Textechno H. Stein GmbH & Co, Germany) with at least 0.06 cN per tex pretension. Tenacity and elongation of the fibers were measured both in the conditioned and wet states. 20 fibers from each sample were tested at 23 °C and 60 ± 2% RH. The gauge length was 20 mm and speed 20 mm min−1.2.7 WAXS measurements
WAXS experiments were performed in the transmission mode of a Xeuss 3.0 (Xenocs) CuK X-ray machine at 50 kV and 0.6 mA. Selected fiber samples, in the form of fiber bundles, were placed vertically on the sample holder and irradiated with X-rays. The sample chamber was kept under vacuum (=0.16 mbar) during the measurement. The scattering intensity was recorded with a 2D detector Eiger2 R 1M (Detris) with a distance of 56 mm between the sample and detector and corrected for cosmic background.The 2D scattering patterns were processed using pyFAI software (the fast azimuthal integration Python library: pyFAI, Journal of Applied Crystallography). For oriented cellulose fiber samples, the 2D scattering patterns were processed to obtain structural parameters of the cellulose crystal as previously described for cellulose fibers.42
3. Results and discussion
3.1 Viscosity adjustment
Scheme 1 shows the basic flow diagram for the pretreatment of the SWS and HWPHK pulps. Prior to viscosity adjustment, an Ioncell-P stage was applied to SWS pulp to yield a pulp with significantly lower hemicellulose content (SWS-IP). In the Ioncell-P process, an IL–water mixture is used to reduce the dissolving power so that only the low molecular weight hemicelluloses can be extracted.43,44 As shown in Table 1, the IL–water mixture was able to extract hemicellulose from the SWS without degrading the cellulose, which is due to the lower dissolving power of IL in the presence of water, so that only polysaccharide fractions with a molecular weight typical of hemicellulose could be dissolved. The resulting SWS-IP has a high cellulose content with only 2% hemicellulose in the pulp.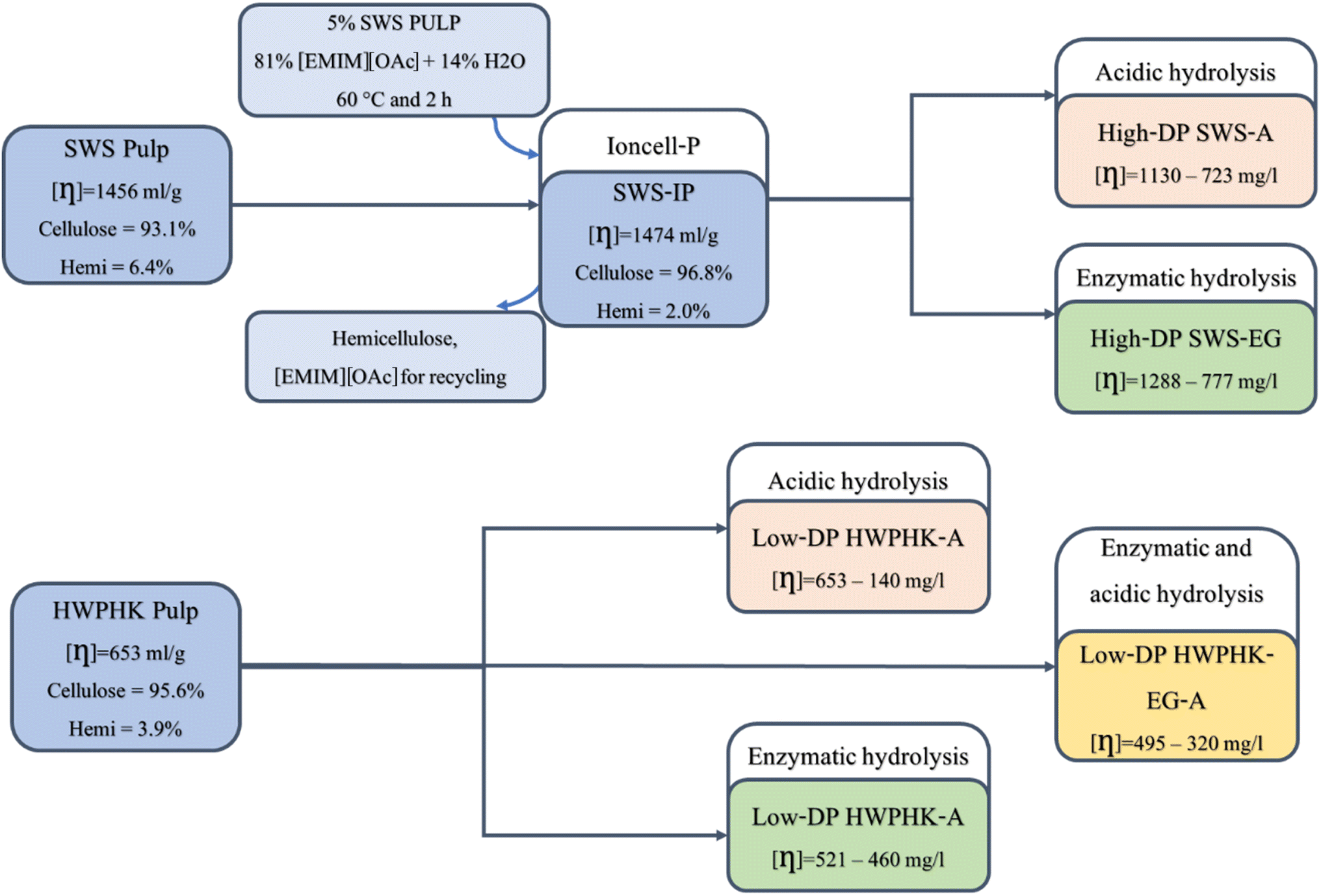 | ||
| Scheme 1 Schematic flow diagram of the pretreatment of SWS and HWPHK pulps for the production of dissolving pulps with a broad DP spectrum and different molecular weight distributions (MWDs). | ||
To obtain pulps with an extensive range of viscosity, the SWS-IP and HWPHK pulps were subjected to acid, enzyme and acid–enzyme combined treatments. A series of pulps were prepared by each pre-treatment method to assess the influence of pulp degradation methods on the pulp properties and their spinnability. Table 2 lists the properties of the degraded pulps utilizing the three mentioned degradation methods.
| Intrinsic viscosity (mL g−1) | M w (Da) | PDI | DP > 2000 | DP < 100 | Cellulose (%) | Hemi (%) | Lignin (%) | |
|---|---|---|---|---|---|---|---|---|
| Acid treated SWS-IP | 1130 | 405700 |
2.9 | 52.4 | 1.2 | 96.0 | 2.9 | 1.1 |
| 948 | 343600 |
2.1 | 44.2 | 0.2 | 97.5 | 1.9 | 0.6 | |
| 726 | 271000 |
2.0 | 29.3 | 0.2 | 97.9 | 1.9 | 0.2 | |
| Acid treated HWPHK | 653 (untreated) | 231300 |
2.4 | 24.1 | 1.8 | 95.6 | 4.0 | 0.4 |
| 525 | 189600 |
2.2 | 16.4 | 1.5 | 95.6 | 4.1 | 0.3 | |
| 476 | 173700 |
2.6 | 13.9 | 4.1 | 95.7 | 4.0 | 0.3 | |
| 420 | 136400 |
2.3 | 7.8 | 4.0 | 96.0 | 3.9 | 0.1 | |
| 364 | 115800 |
2.2 | 4.8 | 5.0 | 96.0 | 3.9 | 0.1 | |
| 294 | 98100 |
2.5 | 2.9 | 8.6 | 95.9 | 4.1 | 0.0 | |
| 140 | 37300 |
2.4 | 0.2 | 26.5 | 95.7 | 4.0 | 0.3 | |
| Enzyme treated SWS-IP | 1288 | 308700 |
2.0 | 46.0 | 0.0 | 96.8 | 1.9 | 1.3 |
| 1098 | 302800 |
2.3 | 46.2 | 1.2 | 97.2 | 2.1 | 0.7 | |
| 886 | 251900 |
3.2 | 34.9 | 3.8 | 97.4 | 1.8 | 0.8 | |
| 777 | 213100 |
5.0 | 30.3 | 9.7 | 97.2 | 2.4 | 0.4 | |
| Enzyme treated HWPHK | 521 | 170500 |
3.5 | 15.7 | 7.5 | 94.3 | 3.7 | 2.0 |
| 460 | 161300 |
3.9 | 14.7 | 8.4 | 94.7 | 3.8 | 1.5 | |
| Enzyme + acid treated HWPHK | 495 | 175500 |
2.8 | 15.3 | 4.6 | 95.1 | 4.3 | 0.6 |
| 422 | 148400 |
3.1 | 10.9 | 7.1 | 95.3 | 4.3 | 0.4 | |
| 378 | 134300 |
4.1 | 9.6 | 10.9 | 94.3 | 3.7 | 2.0 | |
| 320 | 107800 |
4.8 | 5.6 | 14.4 | 95.4 | 4.2 | 0.4 |
The DP adjustments made by acid treatment were described as the chain scission rate (DP0/DP − 1 as a function of treatment duration) where DP is the pulp DP after treatment and DP0 is the DP of the pulp before treatment (DP = Mw/162, where 162 is the molar mass of an anhydrous glucose unit). The kinetics of chain scission was described with a simple model as shown in Fig. 1. An extended description of the kinetics of the DP adjustment and the chain scission is depicted in ESI Section 1.†
 | ||
| Fig. 1 The chain scission rate of cellulose during the acid (left) and endoglucanase treatment (right). | ||
It was found that the acidic softwood sulphite pulps degraded significantly faster under identical conditions, which was due to better accessibility, a higher proportion of carbonyl groups and lower hemicellulose content.45,46 In addition, the high intrinsic initial viscosity also contributed to the rapid degradation. Acid-catalysed degradation of cellulose occurs through the random cleavage of glycosidic bonds. A cellulose with high DP provides more reaction sites, which promotes the hydrolysis reaction rate.47 Acidic hydrolytic degradation of HWPHK pulp was relatively slow compared to SWS-IP, due to the pulping process, its conditions and the resulting higher hemicellulose content and lower carbonyl group content, as well as the lower intrinsic initial viscosity (Tables 3 and 4).
| Pulp | Conditions | Rate constants | ||||||
|---|---|---|---|---|---|---|---|---|
| H2SO4 (mol L−1) | Temperature (°C) | Consistency (%) | Time (min) | M 0 | k 1 | W 0 | k 2 | |
| SWS-IP | 0.10 | 80 | 5 | 0–15 | 1.88 | 0.1130 | — | — |
| HWPHK | 0.10 | 80 | 5 | 0–240 | 0.42 | 0.0700 | 54.3 | 8.8 × 10−5 |
| 0.10 | 80 | 5 | 0–240 | 1.81 | 0.0078 | — | — | |
| Pulp | Conditions | Rate constants | ||||
|---|---|---|---|---|---|---|
| EG dose (mg g−1) | Temperature (°C) | Consistency (%) | Time (min) | CSv,max (per chain) | K M (mg g−1) | |
| SWS-IP | 0–2 | 50 | 5 | 180 | 1.32 | 0.80 |
| HWPHK | 0–5 | 50 | 5 | 180 | 0.64 | 0.41 |
Degradation by endoglucanase was controlled by the endoglucanase dosage. Chain degradation can be modelled using Michaelis–Menten kinetics (ESI eqn (4)†). Here, the SWS-IP pulps also show significantly higher chain scission under the same conditions due to the lower hemicellulose content.48 Unlike acid-catalyzed hydrolysis, the major hydrolysis reaction of the enzyme takes place at the surface of the cellulose, therefore, the hydrolysis rate tends to level off as the reaction progresses.49 This phenomenon can be clearly observed in Fig. 1 as the degradation rate of the enzymatic treatment followed well the Michaelis–Menten kinetics. For this reason, the lowest intrinsic viscosity that the pulp can reach with the EG treatment is 460 mL g−1.
Even though both acidic and enzymatic hydrolysis could produce pulps with a similar intrinsic viscosity, their properties differed, which was clearly reflected in their molecular mass distributions (MMDs). The effects of the degradation process on the MMD are shown in Fig. 2. The acid-treated pulps exhibited a more uniform and narrower MMD, whereas a broader distribution was observed for the enzyme-treated pulps. The acid and EG treatments did not differ in terms of the weight fraction of the long chains (DP > 2000). However, the EG treatment increased the formation of the short-chain fraction due to the extensive chain scissions of the surface layers of the cellulose. Likewise, the extreme heterogeneous hydrolysis caused by the EG treatment was the reason for this non-uniform MMD. As shown in Fig. 2, the MMD pattern of EG-treated pulp demonstrated a distinct hump at the low molar mass domain indicating the formation of short chain cellulose. As a result, the polydispersity of the endoglucanase-treated pulps at a given DP is greater than that of the acid-treated pulps.
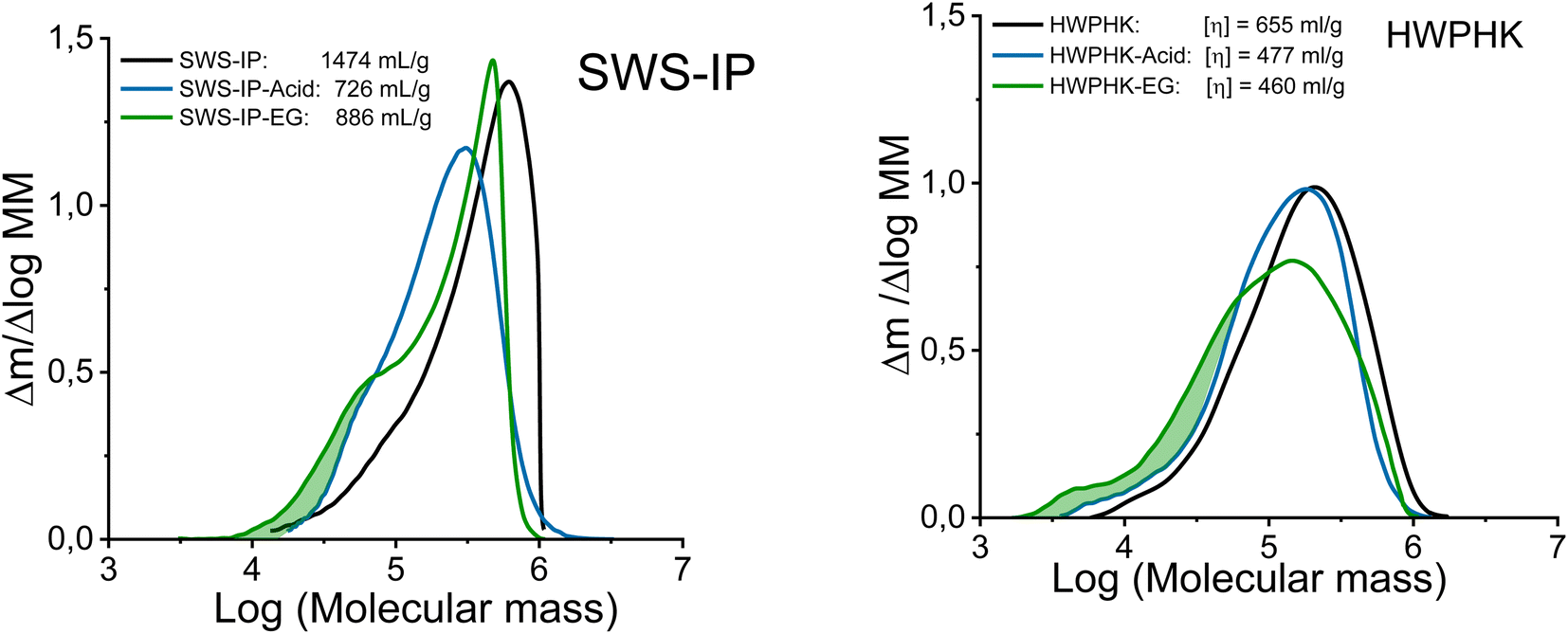 | ||
| Fig. 2 Molecular mass distribution of the acid and enzyme-treated SWS-IP (left) and HWPHK pulps (right). | ||
To minimize the effect of the short-chain fraction on spinnability and fiber properties, and to further decrease the pulp viscosity of the enzyme treatment-derived pulps, a combined enzyme–acid treatment was also applied. Although these pulps had lower percentage of low molecular weight fraction than neat enzyme-treated pulps, the PDI values are still slightly higher than those of acid-treated pulp (Table 2).
The combined endoglucanase–acid treatment is dominated by the acid treatment after 120 minutes, as shown in Fig. S1.† The previous endoglucanase treatment contributed little to further chain scission of cellulose, even at a relatively high dosage of 2 mg g−1. The differences are evident only at short treatment durations of the acid hydrolysis step.
Fig. 3 illustrates the development of the weight fraction of long-chain (DP > 2000) and short-chain (DP < 100) cellulose as a function of the number of chain scissions. There is no doubt that the long-chain polymer was reduced by acid and EG hydrolysis, and for both SWS-IP and HWPHK pulps the trend is similar. However, as mentioned earlier, the amount of low molecular weight fraction was different at a given intrinsic viscosity. The short chain cellulose polymer remained at a similar level (<1%) for acid-treated SWS-IP pulps (from intrinsic viscosity 1474 to 726 mL g−1) owing to its high initial DP and more uniform degradation reaction. In contrast to the acid treatment, the EG treatment had already produced 1% short-chain polymer when the viscosity of the SWS-IP pulp was lowered to 1098 mL g−1 and reached almost 4% when the intrinsic viscosity was adjusted to 886 mL g−1. When the HWPHK was subjected to acid and EG treatment, the short-chain molecules also gradually increased. Similar to SWS-IP pulp, HWPHK pulp also contained a higher proportion of short-chain polymers after EG treatment, which increased with rising chain scission rate.
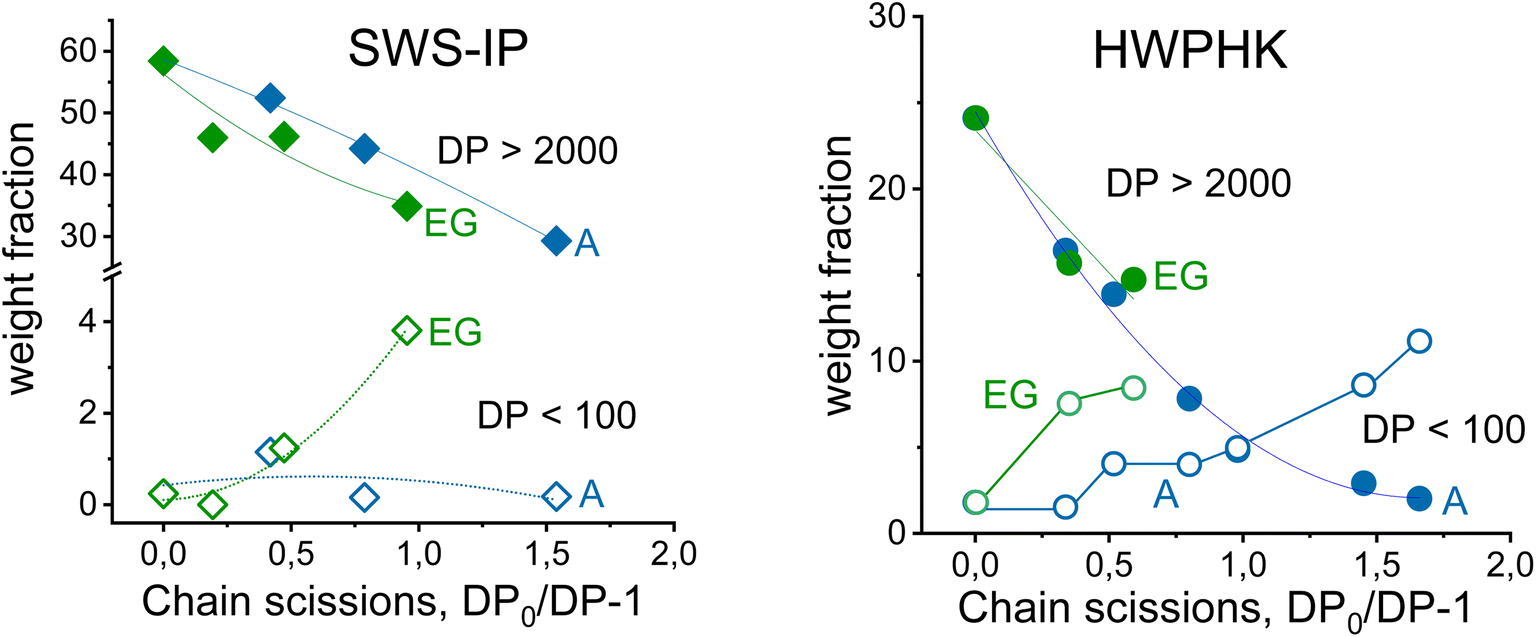 | ||
| Fig. 3 The cellulose chain scission rate as a function of molecular weight fractions: left: SWS-IP pulp, right: HW-PHK pulp. Green symbols: endoglucanase (EG), blue symbols: acid (A) treatments. Full symbols, circles and diamonds: DP > 2000 fractions; open symbols, circles and diamonds: DP < 100 fractions. | ||
3.2 Rheological properties of spinning dopes
All pulps produced with the degradation processes are easily dissolved in IL. Acid and enzyme hydrolyses are reported to improve the accessibility of the pulp and thus increase the dissolution rate.50 Before spinning, the rheological properties were determined. The rheological behaviour is crucial for the spinnability of the spun pulp and the production of high tenacity fibres.51 The rheological properties of the dopes were determined by oscillatory shear measurements to obtain the complex viscosity and dynamic moduli as a function of angular frequency as a master curve. The zero shear viscosity and the dynamic moduli such as storage modulus (G′) and loss modulus (G′′) were extrapolated to the cross-over point (COP) to investigate the relationship between these rheological parameters and the spinnability of the dope. The cellulose concentration in the dope was determined according to the intrinsic viscosity of the pulp and the optimal rheological properties for the IL spinning dopes reported previously (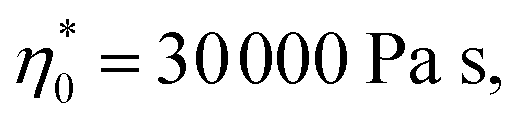 G′ = G′′ at COP = 3000–5000 Pa at 0.8–1.5 1 per s).8,37 For each pulp, two or three dopes were prepared to ensure the best possible spinnability of the pulp. A comprehensive table of the rheological properties of the corresponding dopes, spinning conditions and the resulting fiber properties is listed in ESI Table S1.†
G′ = G′′ at COP = 3000–5000 Pa at 0.8–1.5 1 per s).8,37 For each pulp, two or three dopes were prepared to ensure the best possible spinnability of the pulp. A comprehensive table of the rheological properties of the corresponding dopes, spinning conditions and the resulting fiber properties is listed in ESI Table S1.†
Theoretically, the long chain polymer contributes most to the rheological properties of the solution.52,53 The intrinsic viscosity of the pulps prepared in this study demonstrated a positive linear relationship with their high molar mass fractions (Fig. S2†). Therefore, it was justified to relate the rheological properties to the intrinsic viscosity of the pulp. The complex viscosity and zero shear viscosity of the spinning dopes are closely related to the pulp viscosity and the polymer concentration, as shown in the example of the 10, 13, and 14 wt% pulp solution in the dope after acid and endoglucanase treatment, respectively (Fig. 4). It can be seen that the complex viscosity of the spinning dope increases sharply with increasing pulp viscosity. A change in pulp viscosity can be compensated to some extent by adjusting the pulp concentration in the dope.
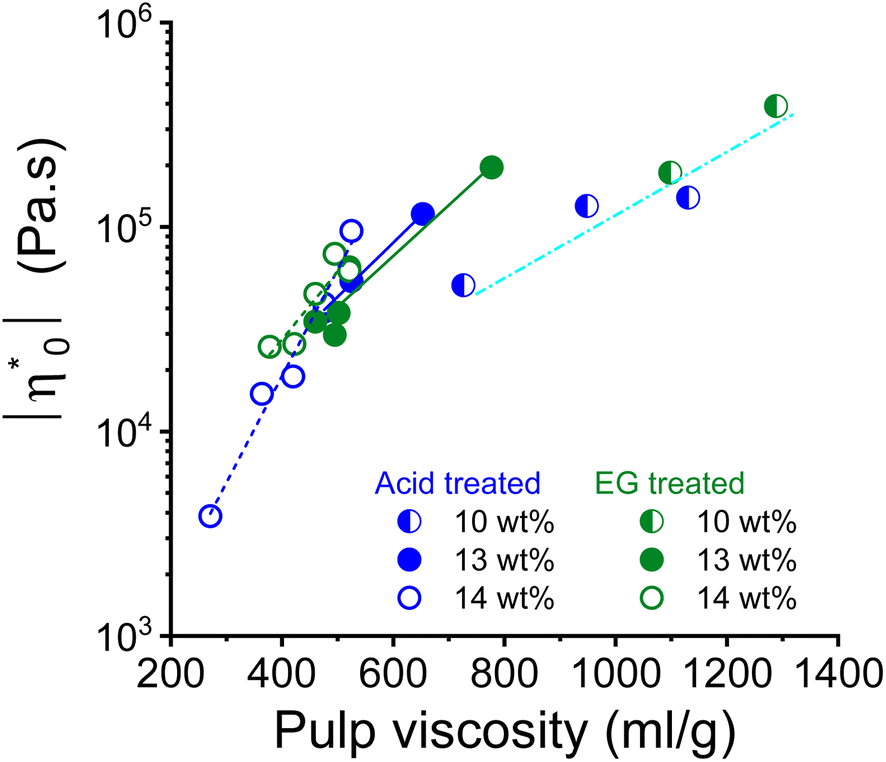 | ||
| Fig. 4 Relationship between dope zero shear viscosity and pulp viscosity for the dope concentrations 10, 13 and 14 wt%. | ||
As shown in Fig. 5, the complex moduli at COP decreased with increasing pulp viscosity and increasing polydispersity, while high polymer concentration increased the moduli at the COP. The same applies to the angular frequency at COP (Fig. 6). However, a higher pulp viscosity significantly enhanced the chain entanglement which suppressed the relaxation of the polymer networks. Eventually, the dynamic moduli of the cellulose solutions at COP decreased (Fig. 5). A broader molar mass distribution in the EG-treated pulp reduces the dynamic moduli of the spinning solution at comparable pulp viscosity and polymer concentration. The dopes prepared from the broadly distributed EG-pulps (which contain a higher weight fraction of short cellulose chains) exhibited a shift to lower angular frequencies at COP compared to the spinning solutions prepared from the acid-treated pulps, indicating a longer relaxation time which in turn leads to lower dynamic moduli (Fig. 6).
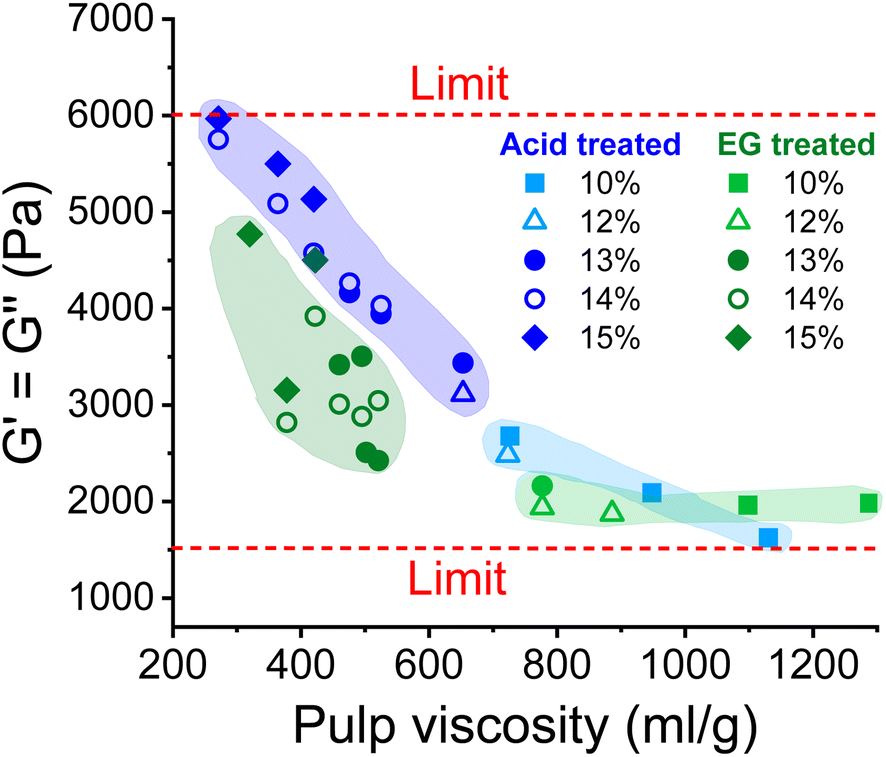 | ||
| Fig. 5 The effect of pulp viscosity and dope concentration on the dynamic moduli at the COP. The dynamic moduli were selected at the temperature at which the dope demonstrated best spinnability. | ||
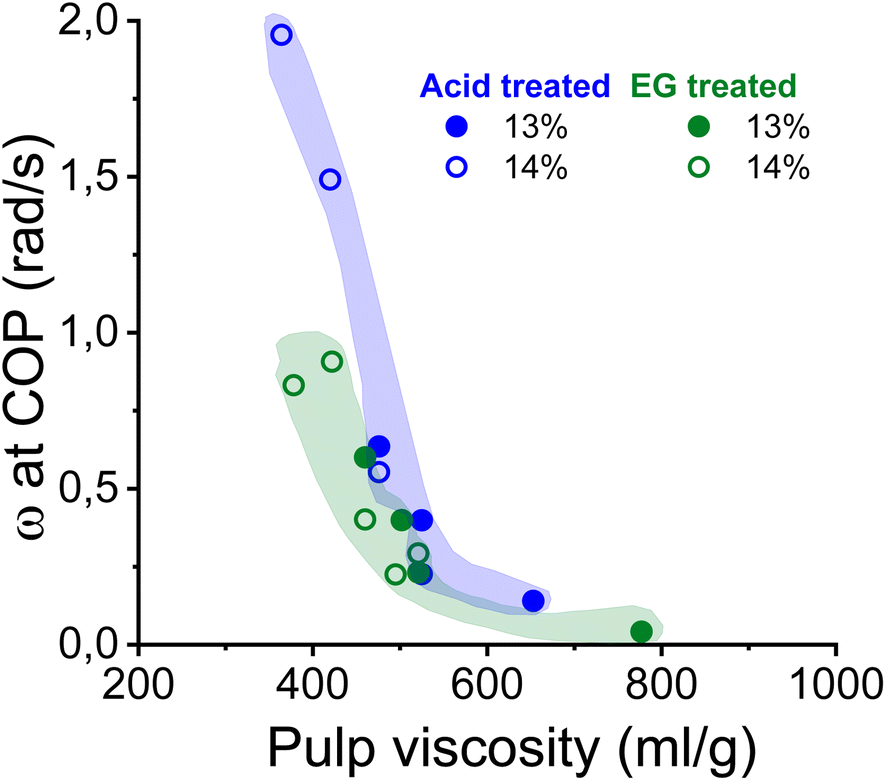 | ||
| Fig. 6 The effect of pulp viscosity and dope concentration on the angular frequency at the COP. The angular frequency was selected at the temperature at which the dope demonstrated best spinnability. | ||
3.3 Dope spinnability
Spinnability is defined as the ability to withstand the applied forces to adjust the fiber thickness.7,17 The spinnability of dopes is usually determined by the draw ratio of extruded filaments in the air gap during spinning. A practical value for spinnability is the minimum fiber titre or linear density, i.e., the thinnest cellulose fibers that can be achieved with the respective air-jet spinning process. Good spinnability is achieved when fibers with a titer of 1.0–1.3 dtex can be produced by applying a certain draw ratio at a reasonably fast extrusion velocity.Maximum draw ratios result in fibers with a titer below or above the optimal titer, which are then associated with excellent or poor spinnability, respectively. Spinning dopes that cannot be drawn are referred to as non-spinnable. Spinnability can be governed by several factors, including the chemical and physical properties, the chemical composition of the starting raw material, the process conditions and, above all, the rheological properties of the dopes, which are reflected, for example, in pulp viscosity and cellulose concentration in the dope.10
Fig. 7 shows the spinnability of the dopes prepared from pulps pretreated with acid, EG and acid–EG, measured as maximum draw ratio, as a function of pulp viscosity and pulp concentration in the dope. The contour plot reveals a clear picture of the influence of pulp viscosity and dope concentration on spinnability. The highest DR can be achieved at a dope concentration of 12 to 14 wt% with pulp viscosity from ∼420 to 500 mL g−1. Spinnability decreased rapidly when using a high concentration dope with a pulp viscosity less than 320 mL g−1. For a pulp with a viscosity of 140 mL g−1, high dope concentrations were designated to maintain the reasonable viscoelastic properties of the dope. However, the short cellulose chains formed a fragile fibrillar network resulting in a less elastic dope. With increasing pulp viscosity, the pulp concentration in the dope was lowered to avoid extreme viscosity compromising filterability. The draw ratio during dry-jet wet spinning of the dope had to be gradually reduced as the pulp viscosity increased. If the cellulose concentration in the spinning solution becomes less than 8–9 wt%, the dope loses its spinnability. This means that despite the high molecular weight of the cellulose, at low polymer concentrations the spinning solution suffers a loss of the elastic properties required for air gap spinning.
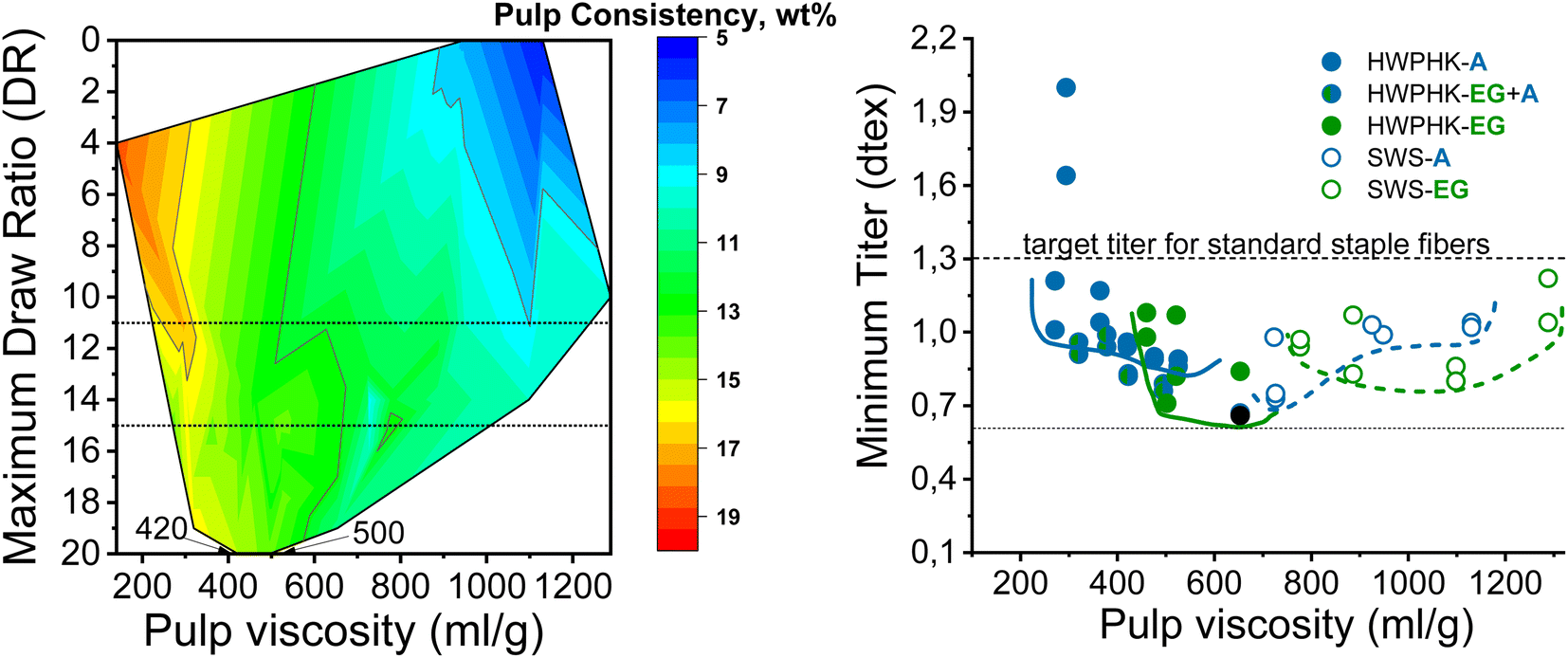 | ||
| Fig. 7 Effect of pulp viscosity and dope concentration on the maximum filament draw ratios (left) and the effect of pulp viscosity on the minimum titer of the spun fiber (right). | ||
When the dope spinnability is related to the rheological properties, it is easy to find that it is hardly related to the complex viscosity. The dope can be spun in a wide range of zero shear viscosity from 4000 Pa s to nearly 130000 Pa s as shown in Fig. 4. However, there are obvious thresholds for the dynamic moduli outside of which the dope is not spinnable (Fig. 5). The spinning performance deteriorates for dopes with dynamic moduli at COP below 1600 Pa (concentration > 8 wt% with pulp viscosity < 1100 mL g−1) or higher than 6000 Pa (concentration < 15 wt% with pulp viscosity > 280 mL g−1).
In this study, the minimum titer is achieved at different draw ratios depending on the concentration of the pulp in the spinning solution (Fig. 7). Therefore, the draw ratio is not really a good indicator of spinnability when the hole diameters of the nozzle and the polymer concentration differ in the experiments. As a consequence, we use the minimum titer as an indicator of spinnability instead of the draw ratio. Fig. 8 illustrates the relationship between pulp viscosity, dynamic moduli and the minimum titer of the spun fibers. The minimum titer of all the spun fibers is reached at 550 to nearly 700 mL g−1 with a spinning solution that has a complex modulus at COP of 2700–3500 Pa, or at lower pulp viscosity with a correspondingly higher complex modulus. The spinning solution with a high viscosity pulp is characterized by a relatively low draw ratio, as shown in Fig. 7, but due to the simultaneous lower pulp concentration in the dope, it leads to a low fiber titer of 1.1–1.3 dtex, which is acceptable for textile processing. A similar picture emerges for the angular frequency at COP with respect to optimal spinnability (Fig. 9). Thus, values around 0.4–1.2 rad s−1 in the above-mentioned viscosity range of the pulp determine the optimum spinnability. The range of the COP can be extended to 0.1 to 2.0 rad s−1 for acceptable dope spinnability. An extended analysis of pulp viscosity and dope concentration selection for the optimum dope spinnability is demonstrated in ESI Section 4.†
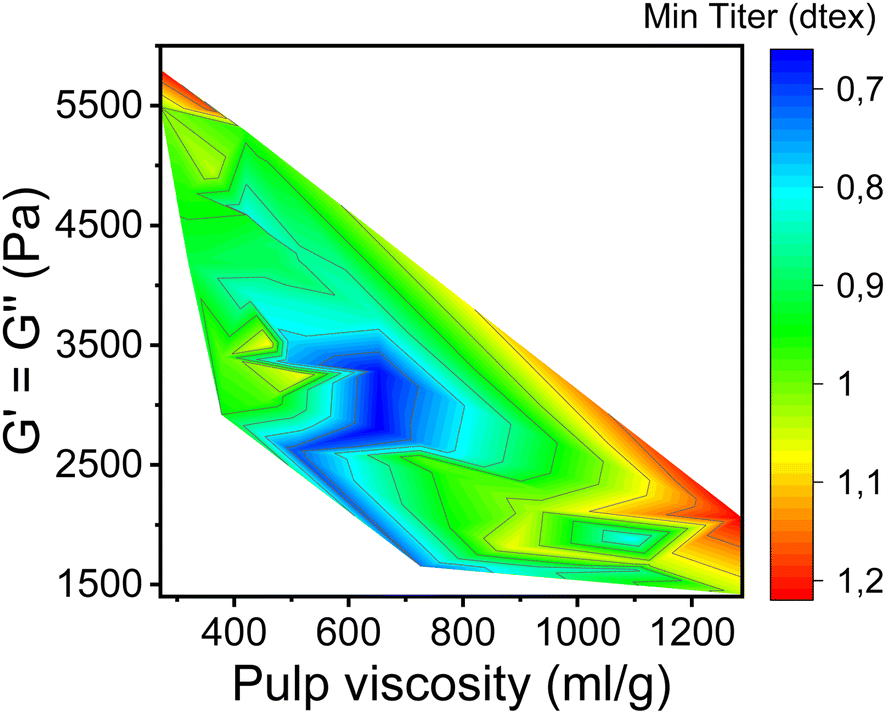 | ||
| Fig. 8 Contour plot showing the dependence of the minimum fiber titer on the complex moduli at COP and the pulp viscosity. | ||
 | ||
| Fig. 9 Contour plot showing the dependence of the minimum fiber titer on the angular frequencies at COP and pulp viscosity. | ||
3.4 Fiber properties
The mechanical properties of the spun fibers depend on several factors, most of which are related to DP and molecular weight distributions in this study. When the overall tenacity of the obtained spun fibers is plotted as a function of the pulp viscosity (Fig. 10 on the left side), there is a clear trend that the fiber strength increases with increasing pulp viscosity. The results again confirmed the theory that the strength of regenerated cellulose fibers is DP-dependent.35–38 The DP of the cellulose chain is linearly related to the crystallite length of the cellulose molecules. A higher DP forms longer crystallites in the regenerated cellulose fibers, resulting in stronger fibers due to more pronounced internal cohesion of the cellulose molecules. The hemicellulose-lean SWS-IP pulp exhibited slightly higher fiber tenacity compared to the HWPHK pulp with intrinsic viscosity ranging from 350 to 650 mL g−1. The fiber strength breaks down when the pulp viscosity reaches values around 300 mL g−1 and continues to decrease further as the DP of the pulp decreases. Furthermore, if one wants to produce Ioncell fibers with the highest possible strength, spinning can be conducted with a 9–10 wt% solution of high-purity dissolving pulp with an intrinsic viscosity of about 730 mL g−1 (Fig. S4†). Simple dry-jet wet spinning systems can produce fibers with a strength of about 61 cN per tex without air gap conditioning, despite suboptimal spinneret geometry, while simultaneously producing microfibers.24 This combination has not been described before and can therefore be noted as another unexpected result.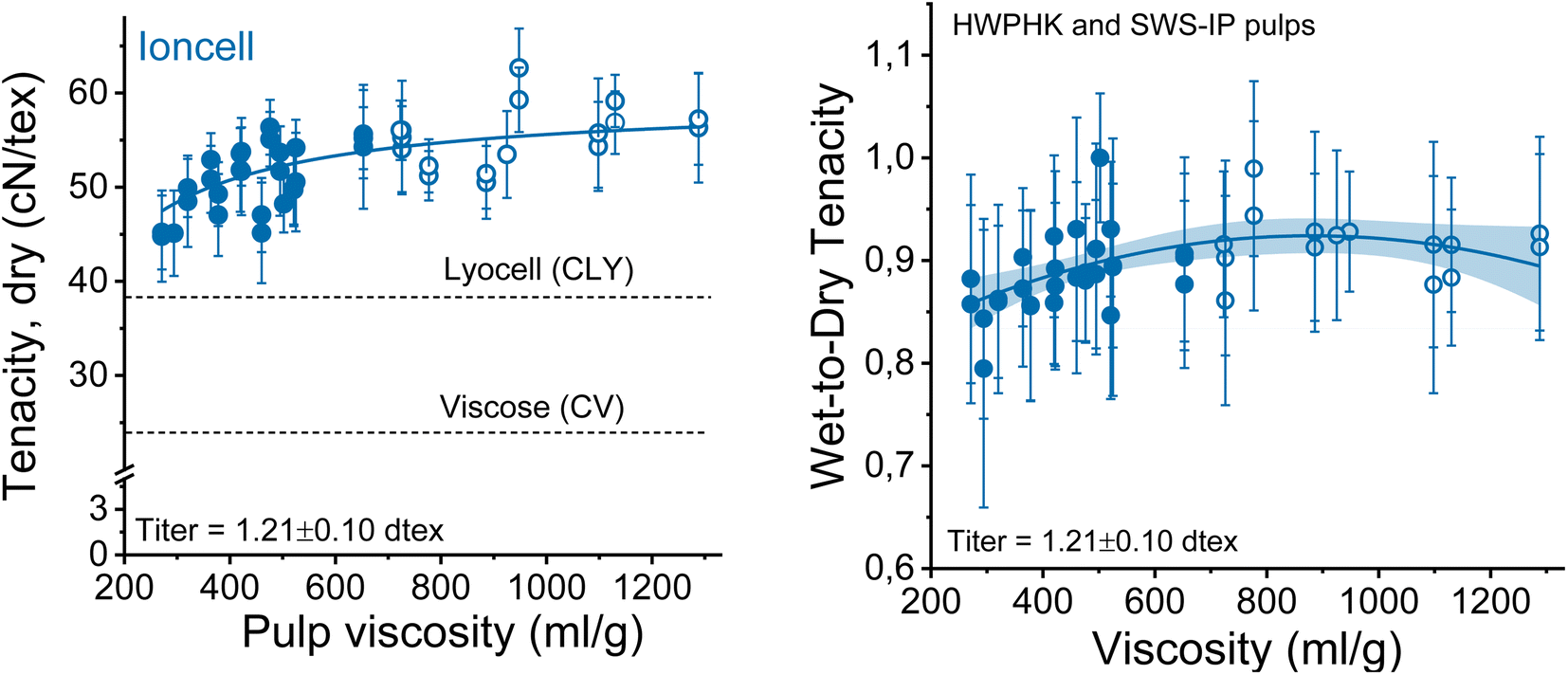 | ||
| Fig. 10 Strength properties of the spun fiber as a function of pulp viscosity. Left: conditioned tenacity, right: ratio between conditioned and wet tenacities (closed circles: HWPHK pulps; open circles: SWS-IP pulps). | ||
It has to be noted that the relatively high cellulose purity (thanks to the Ioncell-P treatment) also contributed to the strength improvement of the regenerated fibers.
The wet and dry tenacities of the spun fibers were relatively close to each other. As depicted in Fig. 10 on the right, the ratio between wet and dry tenacities ranges from 0.8 to 0.95 with a broad maximum at a pulp viscosity ∼800 mL g−1. In addition to tenacity, the modulus of toughness showed a similar trend. The highest modulus of toughness was observed for fibers produced with a pulp viscosity 500 to 700 mL g−1, and a notable decrease in toughness began at a pulp viscosity of 400 mL g−1 (Fig. 11).
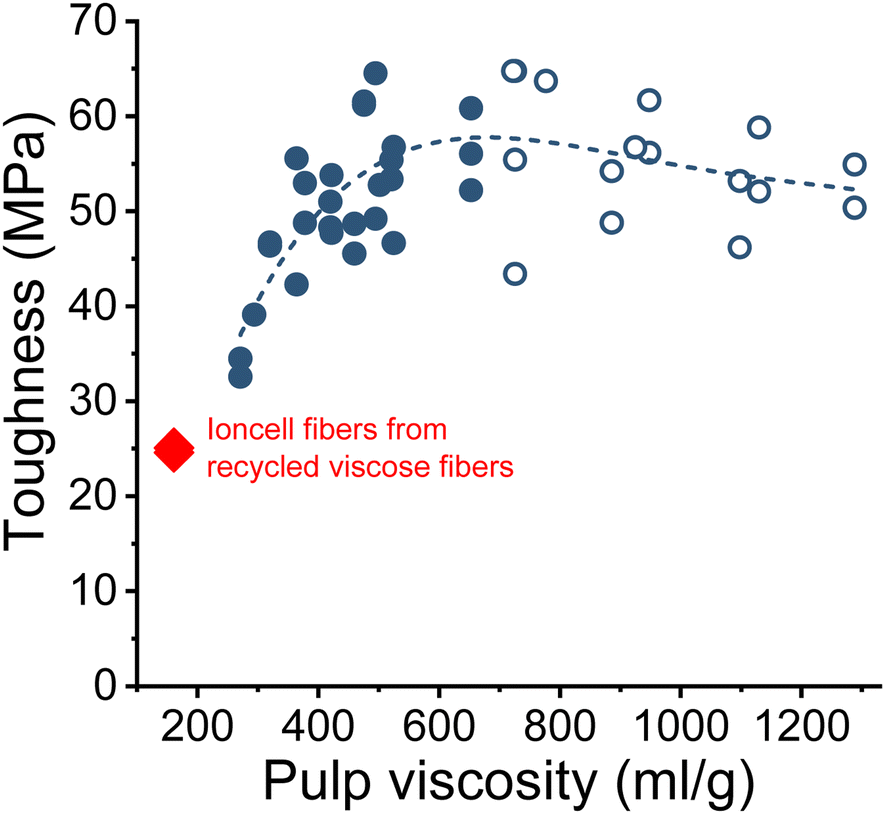 | ||
| Fig. 11 The modulus of toughness of the Ioncell fibers as a function of pulp viscosity (closed circles: HWPHK pulps; open circles: SWS-IP pulps). | ||
The most significant difference between the acid-treated and the enzyme-treated pulps is the fiber strength. In previous studies, Michud et al. claimed that stronger Ioncell fibers require a high proportion of long cellulose chains and a low proportion of short cellulose chains.54 Our current study revealed a similar conclusion. The fibers made from enzyme-treated pulp had a significantly lower dry tenacity than those made from acid-treated pulp, although the viscosity was even slightly lower (Fig. 12). Apparently, the higher proportion of short-chain fractions in the EG-treated pulp deteriorated the mechanical properties of the spun fibers. To produce fibers with improved strength from the EG-treated pulps, the EG treated pulp can subsequently be treated with acid to reduce the proportion of short chain fractions. Fibers spun from pulp treated subsequently with EG and acid exhibited slightly improved tenacity than the fibers spun from pulp treated only with EG at an equivalent viscosity level (Fig. 12). The results are consistent with an earlier study by Haslinger et al. using EG-acid treated cotton.55
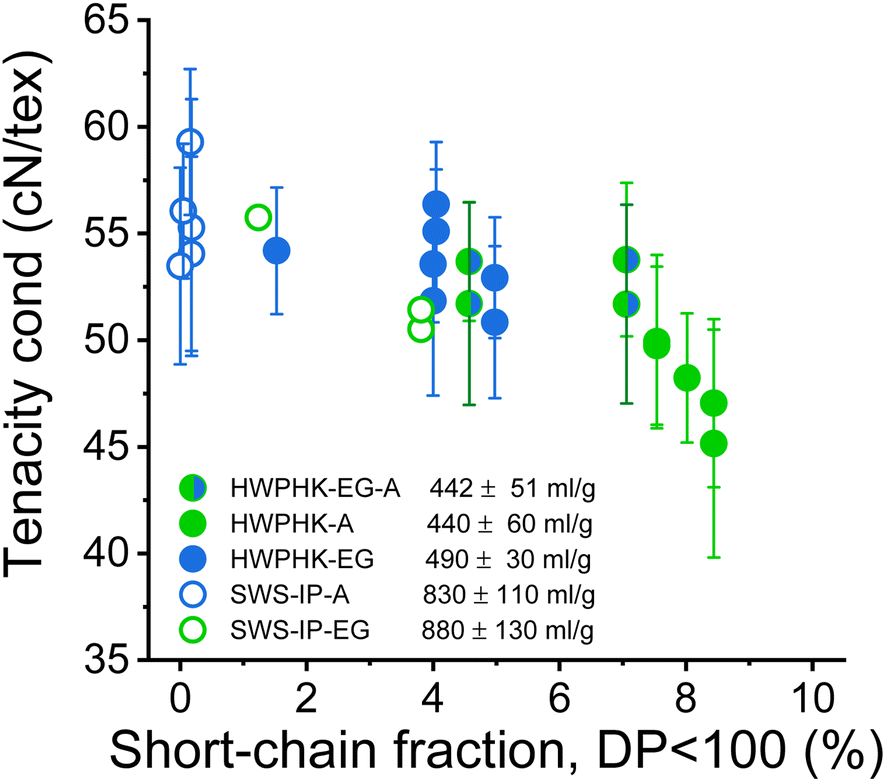 | ||
| Fig. 12 Influence of the short-chain content DP < 100 of the pulps on the conditioned strength of the staple fibers produced from them. | ||
3.5 Structure and properties
The dependence of the fiber strength on the molecular weight becomes apparent if, instead of the intrinsic viscosity, the number average of the degree of polymerization DPn determined by GPC is related to the fiber strength. Thus, the proportion of the short-chain fractions is taken into account. Despite the heterogeneity of the samples due to the use of very different pulps, there is an acceptable correlation between the conditioned tensile strength of fibers with a titer of 1.21 ± 0.10 dtex and the reciprocal DPn with an r2 of 0.70 as shown in Fig. S5.†Surprisingly, the slope of this function is similar to that reported for rayon fibers by Cumberbirch et al.32 but with only half the tensile strength. The significantly lower tensile strength of rayon fibers is mainly due to their much lower orientation in both crystalline and amorphous domains (Hermans and birefringence), as well as their shorter long period (Lm).24,56
In a next step, we investigated whether other measured structural parameters such as crystallinity and Hermans orientation function have an additional influence on the determined fiber strengths. Since crystallinities hardly differ from each other, their consideration does not show an improved relationship to the fiber data (Table S2†). In contrast, the Hermans orientation function shows significant differences in the fibers with a trend towards higher orientation with decreasing DPn as illustrated in Fig. S5.†
Nevertheless, a simple linear combination between the reciprocal DPn and the Hermans orientation function, 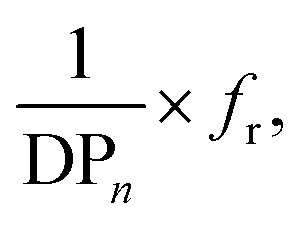 is shown to improve the correlation with fiber strength (Fig. 13). In contrast to Krässig, the square of the orientation function does not lead to an improved correlation.
is shown to improve the correlation with fiber strength (Fig. 13). In contrast to Krässig, the square of the orientation function does not lead to an improved correlation.
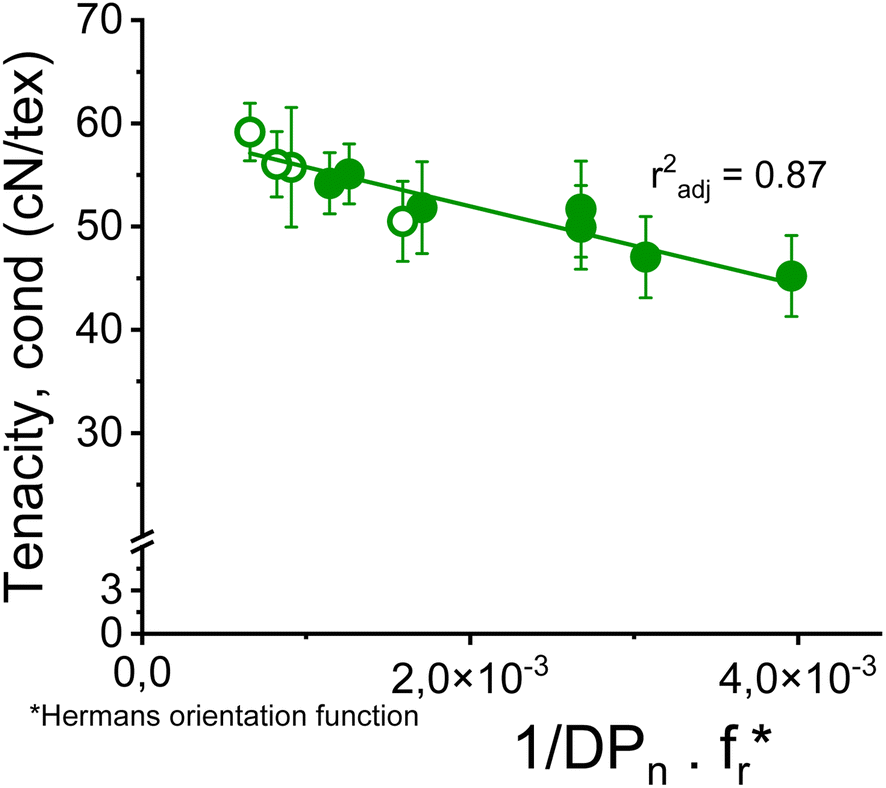 | ||
| Fig. 13 Conditioned tenacity of Ioncell fibers with titers of 1.21 ± 0.10 dtex made from SWS-IP (open circles) and HWPHK pulps (closed circles) as a function of the reciprocal DPn times the Hermans orientation function fr. | ||
At this point it should be mentioned that only a smaller selection of fiber samples was investigated with regard to the structural parameters, which limits the significance of the relationships shown to the smaller number of samples.
4. Conclusion
In this study, we have demonstrated for the first time that the Ioncell process is capable of producing high-quality regenerated cellulose fibers from dissolving pulps with a wide range of intrinsic viscosity from 140 to 1300 mL g−1 and different molecular mass distributions (MMD). The corresponding pulps were produced by aqueous acid (A), enzyme (EG) and sequential enzyme–acid pretreatment. Pulps from enzyme treatment showed a significantly broader MMD than after acid treatment, as the degradation reaction was restricted to the fibre surface. As a result, the proportion of short cellulose chains is remarkably high. To avoid the influence of different hemicellulose contents on fiber properties, a hardwood PHK pulp with a relatively low hemicellulose content of ∼4% was used, and for the softwood acid sulfite pulp (SWS), the hemicellulose content was adjusted from the original of about 6% to 2% by an efficient and mild Ioncell-P treatment without affecting other properties.The spinnability of the prepared dopes demonstrated definite threshold values for the rheological properties outside of which the spinnability is affected. The complex viscosity of the spinning solution allows spinnability in a relatively wide range, which is mainly limited by the encountered pressures and associated pumping and flow properties of the spinning system. However, the dynamic moduli at COP are the determining factors for spinnability. A dope with a dynamic modulus higher than 6000 Pa s, as occurs at high cellulose concentration combined with low pulp viscosity, or less than 1600 Pa s, as occurs at (very) low cellulose concentration combined with high pulp viscosity, is not spinnable or cannot be drawn to an acceptable draw ratio. Due to the increased proportion of short-chain fractions, the dopes from enzyme-treated pulp were less elastic than those from acid-treated pulp, proving that the high molecular weight cellulose contributes most to the rheological behaviour. The best spinnability is obtained at a COP for the dynamic moduli between 2800 and 3500 Pa and for the angular frequency between 0.4 and 1.2 rad s−1. In these ranges, the highest draw ratios of the spun filaments were obtained for pulps with an intrinsic viscosity of 420 to 500 mL g−1 and the lowest titers (linear density) for pulps with an intrinsic viscosity from 500 to 730 mL g−1.
The strength of the spun fibers is strongly DP dependent. The highest correlation with the achieved conditioned fiber tenacity was obtained in the investigated range with the product of the reciprocal DPn and the Hermans orientation factor fr. The fiber tenacity gradually decreases with decreasing pulp viscosity before collapsing at a pulp viscosity below 300 mL g−1. The highest conditioned fiber strength of 61 cN per tex was obtained with a softwood acid sulfite pulp treated with Ioncell-P (SWS-IP) to bring the hemicellulose content to a level comparable to that of other pulps studied, and after the intrinsic viscosity was adjusted to 730 mL g−1 by acid treatment. The cellulose solution in the spinning solution was 10 wt%. The low dope consistency also resulted in microfiber with a titer of only 0.75 dtex. The fibers produced with enzyme-treated pulps generally led to fibers with 10% lower strength compared to fibers from acid treatment with even lower pulp viscosity.
From this study, the suitable viscosity range of the pulp for spinning Ioncell fibers is between 360 and 820 mL g−1 at a dope concentration of 11 to 15 wt%. In this range, fibers with excellent tenacity and toughness properties can be obtained. However, considering the economic aspect of the process, a higher cellulose content in the spinning dope should be achieved. The optimum intrinsic viscosity range of the dissolving pulps for the Ioncell air gap spinning process is between 360 and 500 mL g−1 in conjunction with cellulose concentrations in the spinning dope between 15 and 13 wt%.
However, to avoid misunderstandings, it should be noted that the Ioncell fibre spinning process, like any other lyocell fibre process, can only process pulp or recycled cellulose waste of any kind with a constant DP and a reasonably constant MMD. Varying DPs or MMDs influence the rheological behaviour of the cellulose solution and thus the spinning behaviour. For this reason, for any continuous fibre production, the DP value must be chosen in the above-mentioned range (e.g. 360–500 mL g−1) and fixed together with the polymer concentration (e.g. 13–15 wt%) and kept constant during production.
In terms of sustainability, the aim should be to achieve the highest possible cellulose concentration in the spinning dope in order to keep the amount of water to be evaporated as low as possible. Simply increasing the cellulose concentration from 13 to 15 wt% reduces the amount of water to be evaporated from 15.4 t H2O/odt fiber to 13.1 t H2O/odt fiber, assuming a 30% IL concentration in the combined spinning and washing baths and a residual water content in the IL of 3%.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author would like to acknowledge Sateri International, Singapore for the financial support of the project. We would also like to thank RYAM for providing the high viscosity softwood dissolving pulp and Bracell for providing the high purity hardwood dissolving pulp.References
- J. W. S. Hearle and C. Woodings, in Regenerated Cellulose Fibres, Woodhead, Boca Raton, 2001, pp. 156–173 Search PubMed.
- K. Fischer, H. Sendner, R. Büchner and A. Schlesinger, in Progress and Trends in Rheology II, Steinkopff, Heidelberg, 1988, pp. 388–391 Search PubMed.
- C. Felgueiras, N. G. Azoia, C. Gonçalves, M. Gama and F. Dourado, Front. Bioeng. Biotechnol., 2021, 9, 1–20 CrossRef PubMed.
- F. M. Hämmerle, Lenzinger Ber., 2011, 89, 12–21 Search PubMed.
- T. Rosenau, A. Potthast, H. Sixta and P. Kosma, Prog. Polym. Sci., 2001, 26, 1763–1837 CrossRef CAS.
- D. Eichinger and M. Eibl, Chem. Fibers Int., 1996, 46, 28–30 Search PubMed.
- H. Sixta, A. Michud, L. Hauru, S. Asaadi, Y. Ma, A. W. T. King, I. Kilpeläinen and M. Hummel, Nord. Pulp Pap. Res. J., 2015, 30, 43–57 CrossRef CAS.
- A. Michud, M. Tanttu, S. Asaadi, Y. Ma, E. Netti, P. Kääriainen, A. Persson, A. Berntsson, M. Hummel and H. Sixta, Text. Res. J., 2016, 86, 543–552 CrossRef CAS.
- M. Hummel, A. Michud, M. Tanttu, S. Asaadi, Y. Ma, L. K. J. Hauru, A. Parviainen, A. W. T. King, I. Kilpeläinen and H. Sixta, Adv. Polym. Sci., 2015, 271, 133–168 CrossRef.
- Y. Ma, Fibre spinning from various low refined, recycled lignocelluloses using ionic liquid, Doctoral dissertations 171/2018, Aalto University, School of Chemical Engineering, Department of Bioproducts and Biosystems, 2018.
- H. Liu, K. L. Sale, B. M. Holmes, B. A. Simmons and S. Singh, J. Phys. Chem. B, 2010, 114, 4293–4301 CrossRef CAS PubMed.
- L. K. J. Hauru, M. Hummel, A. Michud and H. Sixta, Cellulose, 2014, 21, 4471–4481 CrossRef CAS.
- H. P. Fink, P. Weigel, H. J. Purz and J. Ganster, Prog. Polym. Sci., 2001, 26, 1473–1524 CrossRef CAS.
- K. Götze, ed. K. Götze, Chemiefasern nach dem Viskoseverfahren, Springer Berlin Heidelberg, Berlin, Heidelberg, 3rd edn, 1967, pp. 525–578 Search PubMed.
- J. Eriksson, Pilot spinning of viscose staple fibres, Masters thesis, Umeå University, 2015.
- X. Xia, M. Gong, C. Wang, B. Wang, Y. Zhang and H. Wang, Cellulose, 2015, 22, 1963–1976 CrossRef CAS.
- L. K. J. Hauru, M. Hummel, K. Nieminen, A. Michud and H. Sixta, Soft Matter, 2016, 12, 1487–1495 RSC.
- C. Guizani, K. Nieminen, M. Rissanen, S. Larkiala, M. Hummel and H. Sixta, Cellulose, 2020, 27, 4931–4948 CrossRef CAS.
- Y. Ma, B. Zeng, X. Wang and N. Byrne, ACS Sustainable Chem. Eng., 2019, 7, 11937–11943 CAS.
- H. Q. Lê, Y. Ma, M. Borrega and H. Sixta, Green Chem., 2016, 18, 5466–5476 RSC.
- Y. Ma, J. Stubb, I. Kontro, K. Nieminen, M. Hummel and H. Sixta, Carbohydr. Polym., 2018, 179, 145–151 CrossRef CAS PubMed.
- Y. Ma, B. Nasri-Nasrabadi, X. You, X. Wang, T. J. Rainey and N. Byrne, J. Nat. Fibers, 2020, 1–13 Search PubMed.
- Y. Ma, M. Hummel, I. Kontro and H. Sixta, Green Chem., 2018, 20, 160–169 RSC.
- K. Moriam, D. Sawada, K. Nieminen, M. Hummel, Y. Ma, M. Rissanen and H. Sixta, Cellulose, 2021, 28, 9547–9566 CrossRef CAS.
- K. Moriam, Aalto University, 2022.
- Y. Ma, S. Asaadi, L.-S. Johansson, P. Ahvenainen, M. Reza, M. Alekhina, L. Rautkari, A. Michud, L. Hauru, M. Hummel and H. Sixta, ChemSusChem, 2015, 8, 4030–4039 CrossRef CAS PubMed.
- H. Staudinger, Melliand Textilber., 1937, 18, 53 CAS.
- H. Staudinger, Text Rundsch., 1949, 4, 1 Search PubMed.
- P. J. Flory, J. Am. Chem. Soc., 1945, 67, 2048–2050 CrossRef CAS.
- A. M. Sookne and M. Hilton, Ind. Eng. Chem., 1945, 37, 478–482 CrossRef CAS.
- M. Spurlin, Cellulose and Cellulose Derivatives, Interscience, New York, 1913 Search PubMed.
- R. J. E. Cumberbirch and W. G. Harland, J. Text. Inst., Trans., 1959, 50, T311–T334 CrossRef CAS.
- R. J. E. Cumberbirch and C. Mack, J. Text. Inst., Trans., 1960, 51, T458–T483 CrossRef.
- H. Krassig and W. Kitchen, J. Polym. Sci., 1961, 51, 123–172 CrossRef CAS.
- R. de Silva and N. Byrne, Carbohydr. Polym., 2017, 174, 89–94 CrossRef CAS PubMed.
- S. Kim and J. Jang, Fibers Polym., 2013, 14, 909–914 CrossRef CAS.
- S. Asaadi, M. Hummel, S. Hellsten, T. Härkäsalmi, Y. Ma, A. Michud and H. Sixta, ChemSusChem, 2016, 9, 3250–3258 CrossRef CAS PubMed.
- M. K. Yoo, M. S. Reza, I. M. Kim and K. J. Kim, Fibers Polym., 2015, 16, 1618–1628 CrossRef CAS.
- S. Elsayed, S. Hellsten, C. Guizani, J. Witos, M. Rissanen, A. H. Rantamäki, P. Varis, S. K. Wiedmer and H. Sixta, ACS Sustainable Chem. Eng., 2020, 8, 14217–14227 CrossRef CAS.
- A. Potthast, S. Radosta, B. Saake, S. Lebioda, T. Heinze, U. Henniges, A. Isogai, A. Koschella, P. Kosma, T. Rosenau, S. Schiehser, H. Sixta, M. Strlič, G. Strobin, W. Vorwerg and H. Wetzel, Cellulose, 2015, 22, 1591–1613 CrossRef CAS.
- J. Janson, Papper Och Trä., 1970, 5, 323–329 Search PubMed.
- M. Trogen, N.-D. Le, D. Sawada, C. Guizani, T. V. Lourençon, L. Pitkänen, H. Sixta, R. Shah, H. O'Neill, M. Balakshin, N. Byrne and M. Hummel, Carbohydr. Polym., 2021, 252, 117133 CrossRef CAS PubMed.
- A. Roselli, M. Hummel, A. Monshizadeh, T. Maloney and H. Sixta, Cellulose, 2014, 21, 3655–3666 CrossRef CAS.
- A. Roselli, S. Asikainen, A. Stepan, A. Monshizadeh, N. von Weymarn, K. Kovasin, Y. Wang, H. Xiong, O. Turunen, M. Hummel and H. Sixta, Holzforschung, 2016, 70, 291–296 CrossRef CAS.
- N. Kvarnlöf and U. Germgård, Bioresources, 2015, 10, 3934–3947, DOI:10.15376/biores.10.3.3934-3947.
- S. Y. Yoon, S. H. Han and S. J. Shin, Energy, 2014, 77, 19–24 CrossRef CAS.
- K. M. Vanhatalo and O. P. Dahl, Bioresources, 2014, 9, 4729–4740 Search PubMed.
- S. I. Mussatto, M. Fernandes, A. M. F. Milagres and I. C. Roberto, Enzyme Microb. Technol., 2008, 43, 124–129 CrossRef CAS.
- L. T. Fan, Y.-H. Lee and D. H. Beardmore, Biotechnol. Bioeng., 1980, 22, 177–199 CrossRef CAS.
- S. Ceccherini, M. Ståhl, D. Sawada, M. Hummel and T. Maloney, Biomacromolecules, 2021, 22, 4805–4813 CrossRef CAS.
- M. Hummel, A. Michud, S. Asaadi, Y. Ma, L. K. J. Hauru, E. Hartikainen and H. Sixta, Annu. Trans.–Nord. Rheol. Soc., 2015, 23, 13–20 CAS.
- R. J. Sammons, J. R. Collier, T. G. Rials and S. Petrovan, J. Appl. Polym. Sci., 2008, 110, 1175–1181 CrossRef CAS.
- S. J. Haward, V. Sharma, C. P. Butts, G. H. McKinley and S. S. Rahatekar, Biomacromolecules, 2012, 13, 1688–1699 CrossRef CAS PubMed.
- A. Michud, M. Hummel and H. Sixta, Polymer, 2015, 75, 1–9 CrossRef CAS.
- S. Haslinger, M. Hummel, A. Anghelescu-Hakala, M. Määttänen and H. Sixta, Waste Manage., 2019, 97, 88–96 CrossRef CAS.
- D. Sawada, Y. Nishiyama, T. Röder, L. Porcar, H. Zahra, M. Trogen, H. Sixta and M. Hummel, Polymer, 2021, 218, 123510 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00013c |
| This journal is © The Royal Society of Chemistry 2023 |