Spent-coffee grounds-derived biochar-supported heterogeneous photocatalyst: a performance evaluation and mechanistic approach for the degradation of pentachlorophenol†
Received
16th May 2023
, Accepted 26th July 2023
First published on 26th July 2023
Abstract
The study presents a facile and green approach to synthesizing crystalline TiO2 nanoparticles on a surface of biochar derived from abundantly available biomass waste, i.e., spent-coffee grounds (SCGs), via a simple sol–gel route. The biochar–TiO2 nanocomposite was used as an effective heterogeneous photocatalyst for the degradation and mineralization of priority pollutants, i.e., pentachlorophenol (PCP) in the aqueous phase. The physiochemical assessment of nanocomposite supports the efficient attachment of TiO2 to biochar, lowers the bandgap energy and particle size, and increases light absorption and durability for easy material separation after use. The radical scavenging experiments results revealed the dominance of superoxide radical anions (O2˙−) followed by hydroxyl radicals (HO˙) and photogenerated holes (h+) for the PCP degradation. Possible degradation mechanism and degradation pathways of PCP was also proposed. Besides, the nanocomposite material exhibited high stability for up to five cycles with negligible activity loss, further confirmed by the FT-IR analysis. Thus, biochar–TiO2 hybrid nanocomposite shows excellent photocatalytic activity for the degradation of organic pollutants in terms of performance metrics, which was far superior to the reported biomass-derived biochar/TiO2 materials and photocatalysts for PCP removal.
Sustainability spotlight
The study provides insightful information on using coffee waste biomass as a sustainable adsorbent and carbon-based material for the photocatalytic treatment of the U.S. Environmental Protection Agency (EPA) listed priority pollutant, i.e., pentachlorophenol. We followed an efficient, low-cost, environmentally friendly, and sustainable approach to synthesizing biomass waste-derived nanocomposites for the photocatalytic treatment of organic pollutants from wastewater. Using such waste biomass for materials synthesis is significant from the perception of green chemistry as it reduces energy consumption and promotes further waste remediation from the perception of green engineering, providing an economical and sustainable approach to treating waste with waste. This research contributes towards water treatment and aligns with Goal 6: Clean Water and Sanitation of the UN SDGs.
|
1. Introduction
Pentachlorophenol (PCP) is a highly toxic halogenated aromatic compound used as a wood preservative, herbicide, and fungicide in various industrial, agricultural, and domestic applications.1,2 As a result, it has been released into the environment, where it can persist for a long time and potentially contaminate soil, water, and air.3 Owing to its toxicity, the U.S. Environmental Protection Agency (EPA) listed it as a priority pollutant in 1979, and the International Agency for Research on Cancer classified it as a potential human carcinogen in 1999.4 The biological degradation of PCP is a slow and complex process, particularly at high concentrations, accompanied by the transformation of PCP to more toxic products like polychlorinated dibenzofurans and polychlorinated dibenzo-p-dioxins.5 Due to its high carcinogenicity and stable structure, it is vital to develop cost-efficient and reliable methods to completely degrade the PCP.
Photocatalytic degradation of organic contaminants using titanium dioxide (TiO2) has been extensively studied due its low-cost, strong oxidizing capabilities, chemical stability, and resistance to photocorrosion.3,6 When illuminated with photons that have energy equal to or greater than its bandgap, TiO2 generates e−−h+ pairs that participate in the formation of reactive oxygen species (ROS), such as hydroxyl radicals (HO˙) and superoxide radical anion (O2˙−).6 However, TiO2 has a relatively large bandgap (∼3.0 and ∼3.2 eV for rutile and anatase TiO2, respectively)7 and absorbs a small portion of the solar spectrum.8 The photogenerated e−−h+ pair undergo fast recombination which limits the reaction rates9 and it has low reactivity towards pollutants with lower concentrations.10 Combining TiO2 with carbon-rich material that is low cost and non-toxic and derived from the valorization of waste biomass could provide an optimal solution for designing stable and versatile heterogenous TiO2 photocatalysts.11,12 Biochar, a carbonaceous material from the pyrolysis of waste biomass, offers an advantage over conventional activated carbon owing to abundant surface functional groups (e.g., carbonyl, phenolic hydroxyl, and carboxyl) and high affinity to immobilize TiO2 semiconductor photocatalysts.13 The synergistic effects of a biochar-supported TiO2 hybrid structure could promote the photocatalytic efficiency of TiO2, in which biochar can act as an electron acceptor and binding site for the organic pollutants to bind, thereby promoting the separation of photoinduced e−−h+.14,15
Coffee is one of the most widely cultivated agricultural products and second only to petroleum as the most traded commodity in the world.16 As a result of the prominent market, the coffee industry is responsible for generating significant quantities of waste, including spent coffee grounds (SCGs) and coffee silverskin. SCGs are lignocellulosic with ∼51% and ∼40% w/w cellulose and hemicellulose, respectively, along with lignin and protein components.17 The valorization of waste SCG for biochar synthesis could solve the problem of recovery of sustainable materials from the waste and reduce the environmental impacts of SCG if discarded in the environment. Although studies have been reported to utilize coffee grounds-derived biochar in diverse applications, including adsorption,16,18 and soil amendments;19 the research addressing the coffee grounds-derived biochar–TiO2 hybrid materials and its performance evaluation for photocatalytic degradation of high priority pollutant, like PCP, have not been reported yet. Herein, we report the degradation of PCP in the aqueous stream by the photocatalytic process utilizing the SCG-derived biochar and TiO2 hybrid material. To the best of our knowledge, this is one of its kinds of study that provides insightful and complete knowledge on the photocatalytic treatment of PCP by using spent coffee grounds-biochar modified with TiO2 prepared by a simple sol–gel methodology. Using such waste biomass for materials synthesis is significant from the perception of green chemistry as it reduces energy consumption, promotes waste minimization and is economical.
2. Experimental
2.1 Chemicals and reagents
Titanium(IV) isopropoxide (TTIP, 97%), ethylenediaminetetraacetic acid (EDTA, 99%), and HPLC grade acetonitrile (≥99.9%) were procured from Sigma-Aldrich. Hydrochloric acid, nitric acid, and sodium chloride were purchased from Fisher Chemicals. AEROXIDE TiO2 (Degussa P25) was procured from Evonik Industries, USA. Polyethylene glycol (PEG, 50%), p-benzoquinone (BQ, 98%), sodium chloride, and isopropyl alcohol (IPA, 99.7%) were purchased from Thermo Scientific. Deionized (DI) water used throughout the study was collected from the Direct-Q® UV water purification system (Milli-Q, Millipore Sigma, Germany).
2.2 Synthesis of biochar
The synthesis of biochar from spent coffee grounds (SCG) was carried out through slow pyrolysis experiments in a lab-scale pyrolyzer system. Briefly, dried SCG was placed inside the reactor and then heated to 500 °C, at a heating rate of 10 °C min−1. The residence time for slow pyrolysis was maintained at 1 h. Nitrogen gas was continuously purged into the reactor to create an inert condition and to sweep the pyrolysis vapor away from the reactor. The condensable vapor (liquid) was collected by a flask connected to the condenser, and the non-condensable vapor (gas) was vented through the fume hood. At the end of the reaction, the reactor was naturally cooled down to room temperature and then opened the reactor to collect the solid product labelled as coffee-ground biochar (CGB).
2.3 Synthesis of CGB/TiO2 composites
Before the synthesis process, the as-prepared CGB was washed with deionized water twice to remove ash and impurities, then soaked in 250 mL of 0.1 M HCl solution for 30 min under ultrasonic cleaner. After that, the mixture was aged for 12 h under static conditions. The CGB was separated from HCl by filtration, followed by washing it with deionized water several times until the filtrate was of neutral pH. The washed biochar was dried at 105 °C overnight and stored for further use. The CGB/TiO2 composites were prepared using a sol–gel method by following these steps: (1) 50 mL of ethanol was mixed with 100 mL of ultrapure water for 30 min to obtain hydrolysate solution; (2) 50 mL of ethanol, 20 mL of TTIP, 2 mL of nitric acid, and 2 mL of PEG were mixed together for 1 h to obtain TiO2 precursor solution; (3) after 1 h, 200 mg of CGB was added to the TiO2 precursor solution and mixed for 30 min; (4) hydrolysate solution was added dropwise to the TiO2 solution while stirring for 4 h followed by ageing; (5) after ageing for 24 h, the homogeneous sol–gel was dried overnight at 110 °C; (6) the dried product was ground to fine powder followed by washing with ultrapure water and ethanol; (7) finally, the powered was calcinated for 4 h at 380 °C and labelled as CGBT. For comparison, TiO2 was prepared through the same procedure without adding biochar and stored for further analysis and application.
2.4 Characterization
The crystalline phases of the as-prepared materials were determined using powder X-ray diffraction (XRD, Siemens D500 XRD) with Cu Kα radiation and operating parameters of 35 kV and 30 mA. The average crystalline size of synthesized materials was determined using the Debye–Scherrer equation, D = Kλ/(β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ), where λ is the wavelength of X-ray (0.154 nm), D is the average diameter of crystal, K is a dimensionless shape factor (K = 0.94), β is the corrected peak width (full width at half-maximum), and θ is the diffraction angle. The morphology and composition of synthesized materials was assessed using field emission scanning electron microscopy-energy dispersive X-ray spectroscopy (Hitachi S-4700 FE Scanning Electron Microscope, Japan) and transmission electron microscopy (FEI, Tecnai 12, 120 kV, Netherland). Brunauer–Emmett–Teller (BET) analysis was performed on the N2 adsorption–desorption isotherms (NOVA 1200e, Quantachrome, USA) to determine the specific surface area. The pore-size distribution was obtained using the Barrett–Joyner–Halenda (BJH) method by analyzing the adsorption branch of the isotherm. The surface characteristics were measured using X-ray photoelectron spectroscopy (XPS, VG Microtech MultiLab ESCA 2000 instruments, Thermo VG Scientific, UK) with an Al Kα radiation source (1486.6 eV), pass energy = 100.0 eV (survey scan) and 30 eV (high resolution scan), current 20 mA and voltage 10 kV). All XPS data analysis was performed using CasaXPS software (Version 2.3.23). The background subtraction and peak fitting was performed using a Shirley Background and symmetric Gaussian/Lorentzian peak shapes, respectively. Fourier Transform Infrared (FT-IR) data was collected using a PerkinElmer, UATR Spectrum 2 spectrometer in the range 450−4000 cm−1 and spectral resolution of 4 cm−1. Ultraviolet-vis diffuse reflectance spectroscopy (UV-vis DRS) was performed on a Cary 5 Series, Agilent Technologies, spectrophotometer. Photoluminescence (PL) spectra were recorded using PerkinElmer LS-55, USA spectrometer with an excitation wavelength of 325 nm.
cosθ), where λ is the wavelength of X-ray (0.154 nm), D is the average diameter of crystal, K is a dimensionless shape factor (K = 0.94), β is the corrected peak width (full width at half-maximum), and θ is the diffraction angle. The morphology and composition of synthesized materials was assessed using field emission scanning electron microscopy-energy dispersive X-ray spectroscopy (Hitachi S-4700 FE Scanning Electron Microscope, Japan) and transmission electron microscopy (FEI, Tecnai 12, 120 kV, Netherland). Brunauer–Emmett–Teller (BET) analysis was performed on the N2 adsorption–desorption isotherms (NOVA 1200e, Quantachrome, USA) to determine the specific surface area. The pore-size distribution was obtained using the Barrett–Joyner–Halenda (BJH) method by analyzing the adsorption branch of the isotherm. The surface characteristics were measured using X-ray photoelectron spectroscopy (XPS, VG Microtech MultiLab ESCA 2000 instruments, Thermo VG Scientific, UK) with an Al Kα radiation source (1486.6 eV), pass energy = 100.0 eV (survey scan) and 30 eV (high resolution scan), current 20 mA and voltage 10 kV). All XPS data analysis was performed using CasaXPS software (Version 2.3.23). The background subtraction and peak fitting was performed using a Shirley Background and symmetric Gaussian/Lorentzian peak shapes, respectively. Fourier Transform Infrared (FT-IR) data was collected using a PerkinElmer, UATR Spectrum 2 spectrometer in the range 450−4000 cm−1 and spectral resolution of 4 cm−1. Ultraviolet-vis diffuse reflectance spectroscopy (UV-vis DRS) was performed on a Cary 5 Series, Agilent Technologies, spectrophotometer. Photoluminescence (PL) spectra were recorded using PerkinElmer LS-55, USA spectrometer with an excitation wavelength of 325 nm.
2.5 Photocatalytic experiments
Batch mode experiments were conducted to assess the photocatalytic activity of as prepared photocatalysts for the degradation of pentachlorophenol (PCP). The properties and chemical structure of PCP are shown in Table S1 (in ESI†). Briefly, 80 mL glass vial was loaded with 50 mL of PCP's solution (10 mg L−1) was placed under the light irradiation sources (UV/visible/solar). The pH of PCP's solution was maintained using 1 M HCl and 1 M NaOH solutions. Prior to the reaction, the PCP solutions were magnetically stirred in the presence of photocatalysts in the dark for 30 min to establish an adsorption–desorption isotherm. The photocatalytic reaction was initiated by placing the reaction solution under the light source. At fixed time intervals, an aliquot of the sample was withdrawn from the reaction vials to determine the residual concentrations of PCP. All the photocatalytic experiments were performed in triplicates, and the mean values were reported. The intensity of light sources was measured at regular intervals with a Dual-Range Traceable Light Meter (Model 3251, Cole-Parmer, USA). The UV and visible light sources were a UV lamp (Eiko F15T8/BL, 14 W nominal power, λmax of 365 nm, measured intensity of 9.77 mW cm−2) and LED bulb (Philips A19, 8 W nominal power, λmax of 450 nm, measured intensity of 4.23 mW cm−2) mounted at the top of the reactor. The solar irradiation experiments were performed between 11:00 AM to 1:00 PM in July–August at Dalhousie Agriculture Campus. The radical trapping experiments were performed to identify the role of reactive species responsible for the degradation of PCP during the photocatalytic process. The degradation experiments were conducted with the addition of 1 mM concentration of isopropyl alcohol (HO˙ scavenger), p-benzoquinone (O2˙− scavenger) and ethylenediaminetetraacetic acid (h+ scavenger) in the PCP solution.
2.6 Analytical methods
The PCP degradation was confirmed using UV-visible spectrophotometer (Hach DRS 6000, USA) with λmax at 219 nm. The residual concentration of PCP in the reaction vials was also confirmed by an HPLC system equipped with a reversed-phase column (Inertsil-ODS-3, 150 × 4.6 mm; 3 μm) and diode array detector (Flexar PDA Plus Detector, PerkinElmer). Elution was made using a mixture of acetonitrile and water (85:15, v/v) with pH adjusted to 3 using acetic acid. The flow rate and column temperature were maintained at 1 mL min−1 and 30 °C, respectively. The detection of PCP was done at 219 nm. During the reaction, the generation of Cl− was analyzed using the Hach test kit (Hach 19600, USA) and according to the APHA (APHA Manual, 4500-Cl−1). The chemical oxygen demand (COD) was also assessed using the dichromate method (Hach, USEPA Reactor Digestion Method 8000, USA). The quantification of degraded products of PCP was done using a gas chromatograph (Clarus 680, PerkinElmer, USA) with a capillary column (Restek, Rxi-5MS, 30 m × 0.25 mm, 0.25 μm) and a mass spectrometer (Clarus 6Q 8T, PerkinElmer, USA), which were programmed through a TurboMass software (Turbomass 6.1.2.). Before analysis, 10 mL of the PCP samples were extracted with 25 mL of dichloromethane along with 0.2 mL of 30% HCl (to protonate phenolates in the solution) with constant shaking for 15 min. The column temperature began at 50 °C (1 min) to 270 °C at a ramp rate of 20 °C min−1 and kept at 270 °C for 2 min. The injection used helium as a carrier gas at a rate of 5.0 mL min−1 (50 mL min−1 including makeup gas).
3. Results and discussion
3.1 Physicochemical properties of the samples
The morphological characteristics of the synthesized materials were analyzed using SEM and TEM techniques. Fig. 1a–j shows the surface morphology and elemental distribution for biochar (CGB), TiO2, and biochar–TiO2 composite (CGBT). It can be observed from Fig. 1a that the biochar has a ridge-like structure with small pores on the structure, making it an ideal skeleton for anchoring TiO2 particles. The enlarged SEM image (Fig. S1†) shows the porosity of biochar and weight percentage of elements present in it. The elemental mapping images of biochar indicate high percentage of C, which was distributed uniformly, along with the low percentages of O, Mg, and K species. The presence of Mg and K was assigned to spent coffee grounds as this biomass contains these minerals.20 The TEM image (Fig. 1b) shows a sheet like structure at the edge of biochar. Fig. 1d–f shows the SEM, EDS mapping and particle size distribution of TiO2. The SEM image of TiO2 (Fig. 1e) shows agglomerates of nanoparticles with an average particle size of 23 ± 6 nm. Fig. 1g–j shows the SEM, EDS maps, particle size distribution and TEM analysis of CGBT composites. The SEM image (Fig. 1g) showed TiO2 coated onto the biochar surface which was further confirmed from the EDS maps (Fig. 1h) which showed uniform distribution of Ti element on the surface of the biochar. The average particle size for TiO2 in the CGBT composites was 11 ± 3 nm which is smaller than the pure TiO2 samples (Fig. 1i). TEM analysis also showed that the TiO2 was polyhedral and attached to the carbon sheets.
 |
| | Fig. 1 (a) SEM image, (b) TEM image, and (c) EDS maps of spent-coffee ground biochar (CGB); (d) SEM image, (e) particle size distribution and (f) EDS maps of TiO2; and (g) SEM image, (h) EDS mapping, (i) particle size distribution and (j) TEM image of CGBT nanocomposites. | |
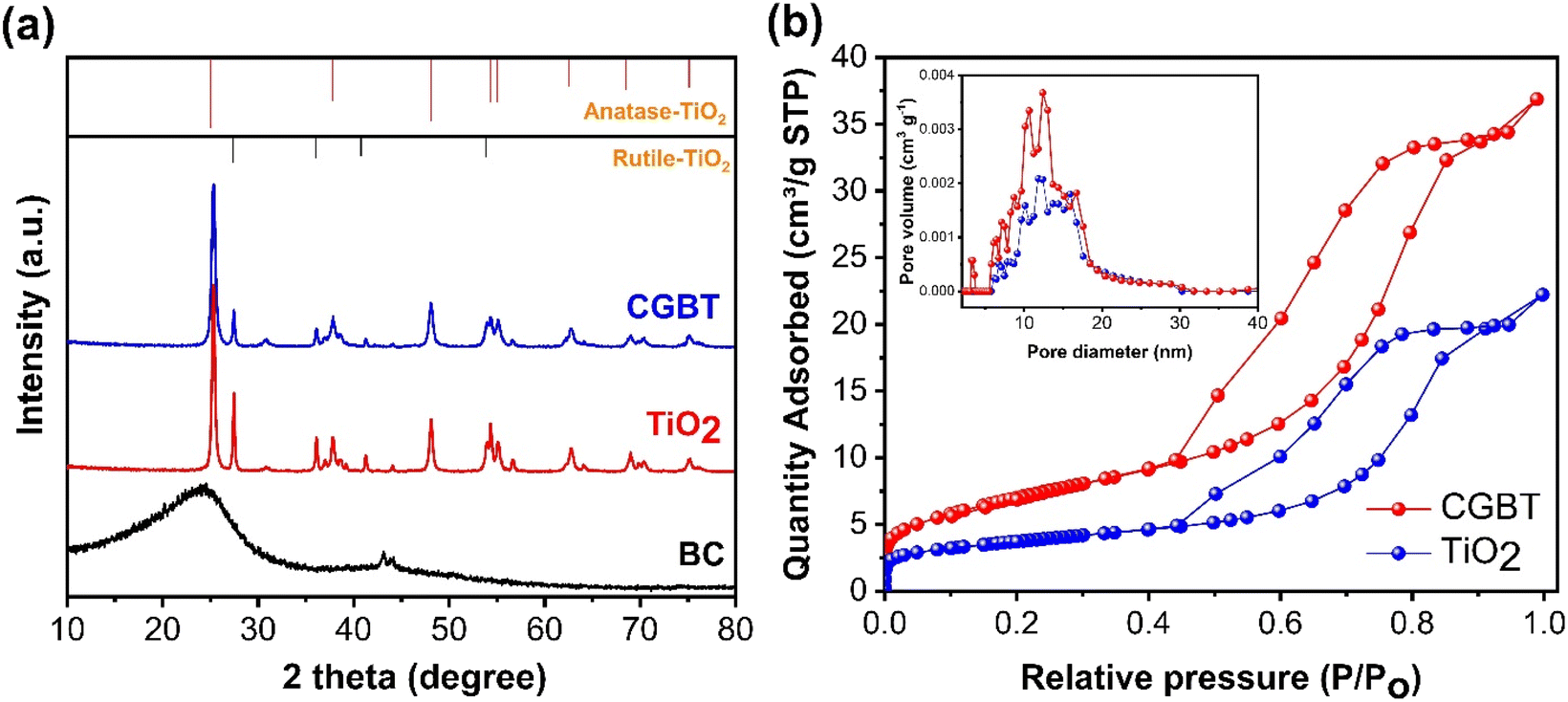
The powder XRD patterns of CGBT, TiO2 and CGB are shown in Fig. 2a. The as-prepared CGB shows two peaks of graphitic carbon and the long-range disordered patterns at 2θ at 23.8° and 43.1°, assigned to the (002) and (100) planes, respectively.21,22 The XRD pattern of TiO2 nanoparticles showed characteristics diffraction peaks centered at 25.36°, 37.88°, 48.12°, 55.16°, 62.68°, 69.0°, and 75.20° corresponding to (101), (004), (200), (211), (204), (116), and (215) planes of anatase TiO2, respectively (JCPDS card no. 21-1272). Besides, some peaks of rutile planes at 27.4°, 36.1°, 41.2°, and 54.3° were also observed in TiO2 diffractogram (JCPDS card no. 21-1276). The anatase TiO2 crystal form has the highest catalytic activity, low e−−h+ pairs recombination, and low mass density compared to other forms of TiO2, i.e., rutile and brookite.3,15 The high intensity of the diffraction peak (101) of TiO2 at 25.36°, signifying an interplanar distance of 0.35 nm,23 is consistent with the TEM results. Compared to CGB and TiO2, the as-prepared CGBT complex shows all the major diffraction peaks for the anatase TiO2, suggesting that the cross-linked structure formed by TiO2 nanoparticles and biochar has no significant effect on the main crystalline structure of TiO2. Based on the relative intensity of peaks, the rutile fraction in TiO2 was reduced in CGBT.24 Besides, the characteristic peaks of graphitic carbon could not be observed in the composite catalyst in XRD patterns due to the stronger crystallinity of TiO2 than CGB. The Scherrer formula25 was used to calculate the grain size of TiO2 and CGBT structure and the data collected are shown in Table 1. As listed in Table 1, the calculated average crystallite size of TiO2 in pure and CGBT complex is 19.5 nm and 13.2 nm, respectively, which agrees with the TEM analysis and indicates formation of single crystalline particles.
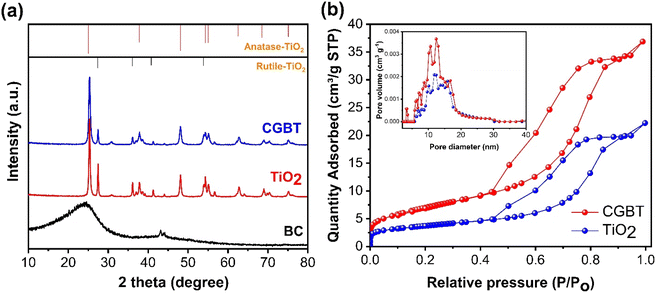 |
| | Fig. 2 (a) XRD diffraction pattern and (b) nitrogen adsorption–desorption isotherms (inset: pore size distribution curves) of synthesized materials. | |
Table 1 Results of XRD, BET, and UV-vis DRS analysis of coffee-ground biochar (CGB), pure TiO2, and coffee ground biochar–TiO2 nanocomposites (CGBT). [-nd-: not determined]
| Sample |
XRD |
BET |
UV-vis DRS |
| Crystalline size (nm) |
Phase |
Surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Pore size (nm) |
Optical gap (eV) |
Absorption threshold (nm) |
| CGB |
-nd- |
-nd- |
3.36 |
2.32 × 10−3 |
8.88 |
-nd- |
-nd- |
| TiO2 |
19.5 |
Anatase/rutile |
12.37 |
3.25 × 10−2 |
9.44 |
3.01 |
422 |
| CGBT |
13.2 |
Anatase/rutile |
21.51 |
5.51 × 10−2 |
7.52 |
2.90 |
439 |
The pore characteristics of CBG, TiO2 and CGBT catalysts were analyzed using N2 adsorption–desorption isotherms and Barrett–Joyner–Halenda (BJH) method (Fig. 2b). All the synthesized materials exhibited a type IV isotherm with H3 hysteresis loop (Fig. 2b), which generally signifies mesoporous structure.9 The pore-size distributions (Fig. 2b, inset) confirmed the presence of meso/microporous structures for all the prepared materials. The specific surface area, average pore volume and pore size of CGB, pure TiO2, and CGBT complex are listed in Table 1. From Table 1, the average pore size of CGBT was 7.52 nm, indicating that it could take advantage of the mesoporous structure and promote the photodegradation process.26 In addition, the specific surface area increased from 12.37 m2 g−1 (TiO2) to 21.51 m2 g−1 (CGBT) with the introduction of spent-coffee grounds-biochar. Typically, the surface area of spent-coffee grounds biochar was the smallest, with only 3.36 m2 g−1. Several studies reported that biochar prepared from food waste generally have a low specific surface area, mainly within 0.01 to 2.2 m2 g−1.27,28
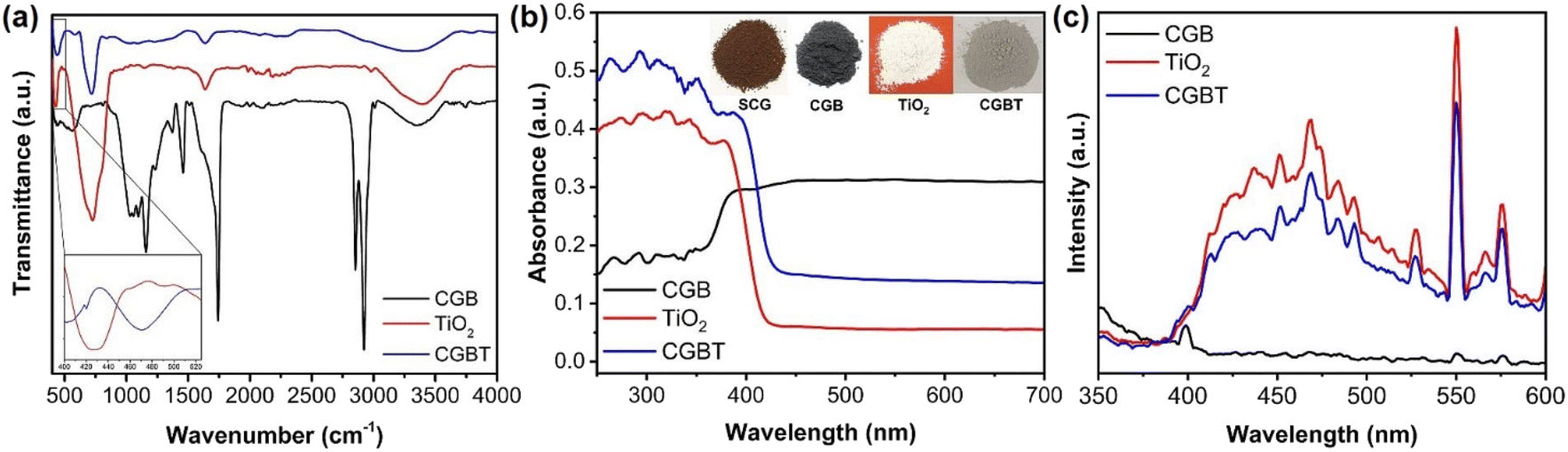
Fourier transform infrared spectra (FT-IR) of CGB, TiO2, and CGBT are depicted in Fig. 3a. In the FT-IR spectra of CGB, a wide band around 3250–3500 cm−1 is ascribed to –OH stretching.21 The sharp peaks at ca. 2923, 1742, and 1158 cm−1 are due to saturated C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and C–O bonds, respectively.29,30 Other small peaks in the range 1646–1202 cm−1 likely due to C–C and CC bond vibrations. In TiO2, the absorption band between 410–900 cm−1 is due to stretching vibrations of Ti–O–Ti bond.31,32 In the FT-IR spectrum of CGBT, the characteristic peaks of TiO2 are preserved well, while the absorption bands for carbon bonds are remarkably weakened. The peak appearing at 1635 cm−1 in all the materials corresponds to deformation vibration for H–O–H bonds of physisorbed water molecules.33 The CGBT composite has a small absorption peak at 826 cm−1, indicating the presence of some peroxy bonds on the surface after the TiO2 loading.21 Besides, the characteristic absorption band at 3250–3500 cm−1 in CGBT composite was less intense when compared to TiO2 and CGB, indicating that TiO2 binding on the biochar surface occurs via condensation of surface hydroxyl groups. The absorption band at around 410–500 cm−1 (confined to Ti–O–Ti bond) shifted to higher wavenumbers likely due to the formation of Ti–O–C linkages in the CGBT (inset of Fig. 3a).34,35 UV-vis DRS was employed to assess the optical properties of synthesized materials and results are shown in Fig. 3b and Table 1. Pure TiO2 shows strong absorption in the UV range (200–380 nm), whereas coffee-ground biochar exhibits a distinctive absorbance in the visible range (380–700 nm). In contrast, the CGBT nanocomposites show enhanced absorption intensity in UV region, along with the noticeable absorbance in the visible range. The enhanced absorption of CGBT nanocomposites in visible range is ascribed to the presence of biochar whereas the absorption in UV range confirms the existence of TiO2 nanoparticles. Similar optical properties have been reported in the literature for biochar-based nanocomposites.15,36,37 The optical gaps of pure TiO2 and CGBT nanocomposites are calculated using the Kubelka–Munk function,32 and the results are depicted in Fig. S2 (in ESI†). The absorption edge is shifted towards the longer wavelength as can be seen for the CGBT nanocomposites (Table 1). It can be observed that the bandgap for TiO2 was 3.01 eV, whereas the CGBT had an optical gap of 2.90 eV. Photoluminescence (PL) spectra was measured to analyze the separation efficiency of e−−h+pairs. Generally, a higher e−−h+ separation efficiency results in a lower photoluminescence intensity, indicating higher photocatalytic activity.38 As shown in Fig. 3c, the position of emission peaks of TiO2 and CGBT was similar, but the peaks intensity decreased in CGBT. This suggests that CGBT nanocomposite has a lower recombination rate of e−−h+ pairs compared with TiO2, indicating that more of the photo-generated charge carriers are available for photocatalysis. The better charge separation is likely due to the heterojunction between TiO2 and the biochar.
O, and C–O bonds, respectively.29,30 Other small peaks in the range 1646–1202 cm−1 likely due to C–C and CC bond vibrations. In TiO2, the absorption band between 410–900 cm−1 is due to stretching vibrations of Ti–O–Ti bond.31,32 In the FT-IR spectrum of CGBT, the characteristic peaks of TiO2 are preserved well, while the absorption bands for carbon bonds are remarkably weakened. The peak appearing at 1635 cm−1 in all the materials corresponds to deformation vibration for H–O–H bonds of physisorbed water molecules.33 The CGBT composite has a small absorption peak at 826 cm−1, indicating the presence of some peroxy bonds on the surface after the TiO2 loading.21 Besides, the characteristic absorption band at 3250–3500 cm−1 in CGBT composite was less intense when compared to TiO2 and CGB, indicating that TiO2 binding on the biochar surface occurs via condensation of surface hydroxyl groups. The absorption band at around 410–500 cm−1 (confined to Ti–O–Ti bond) shifted to higher wavenumbers likely due to the formation of Ti–O–C linkages in the CGBT (inset of Fig. 3a).34,35 UV-vis DRS was employed to assess the optical properties of synthesized materials and results are shown in Fig. 3b and Table 1. Pure TiO2 shows strong absorption in the UV range (200–380 nm), whereas coffee-ground biochar exhibits a distinctive absorbance in the visible range (380–700 nm). In contrast, the CGBT nanocomposites show enhanced absorption intensity in UV region, along with the noticeable absorbance in the visible range. The enhanced absorption of CGBT nanocomposites in visible range is ascribed to the presence of biochar whereas the absorption in UV range confirms the existence of TiO2 nanoparticles. Similar optical properties have been reported in the literature for biochar-based nanocomposites.15,36,37 The optical gaps of pure TiO2 and CGBT nanocomposites are calculated using the Kubelka–Munk function,32 and the results are depicted in Fig. S2 (in ESI†). The absorption edge is shifted towards the longer wavelength as can be seen for the CGBT nanocomposites (Table 1). It can be observed that the bandgap for TiO2 was 3.01 eV, whereas the CGBT had an optical gap of 2.90 eV. Photoluminescence (PL) spectra was measured to analyze the separation efficiency of e−−h+pairs. Generally, a higher e−−h+ separation efficiency results in a lower photoluminescence intensity, indicating higher photocatalytic activity.38 As shown in Fig. 3c, the position of emission peaks of TiO2 and CGBT was similar, but the peaks intensity decreased in CGBT. This suggests that CGBT nanocomposite has a lower recombination rate of e−−h+ pairs compared with TiO2, indicating that more of the photo-generated charge carriers are available for photocatalysis. The better charge separation is likely due to the heterojunction between TiO2 and the biochar.
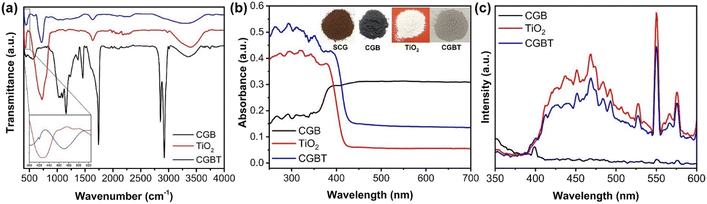 |
| | Fig. 3 (a) FT-IR spectra, (b) UV-vis DRS analysis (inset: picture of powder form of spent-coffee grounds (SCG), coffee-grounds biochar (CGB), TiO2, and coffee-ground biochar-supported TiO2 nanocomposites (CGBT)), and (c) photoluminescence spectra of the synthesized materials. | |
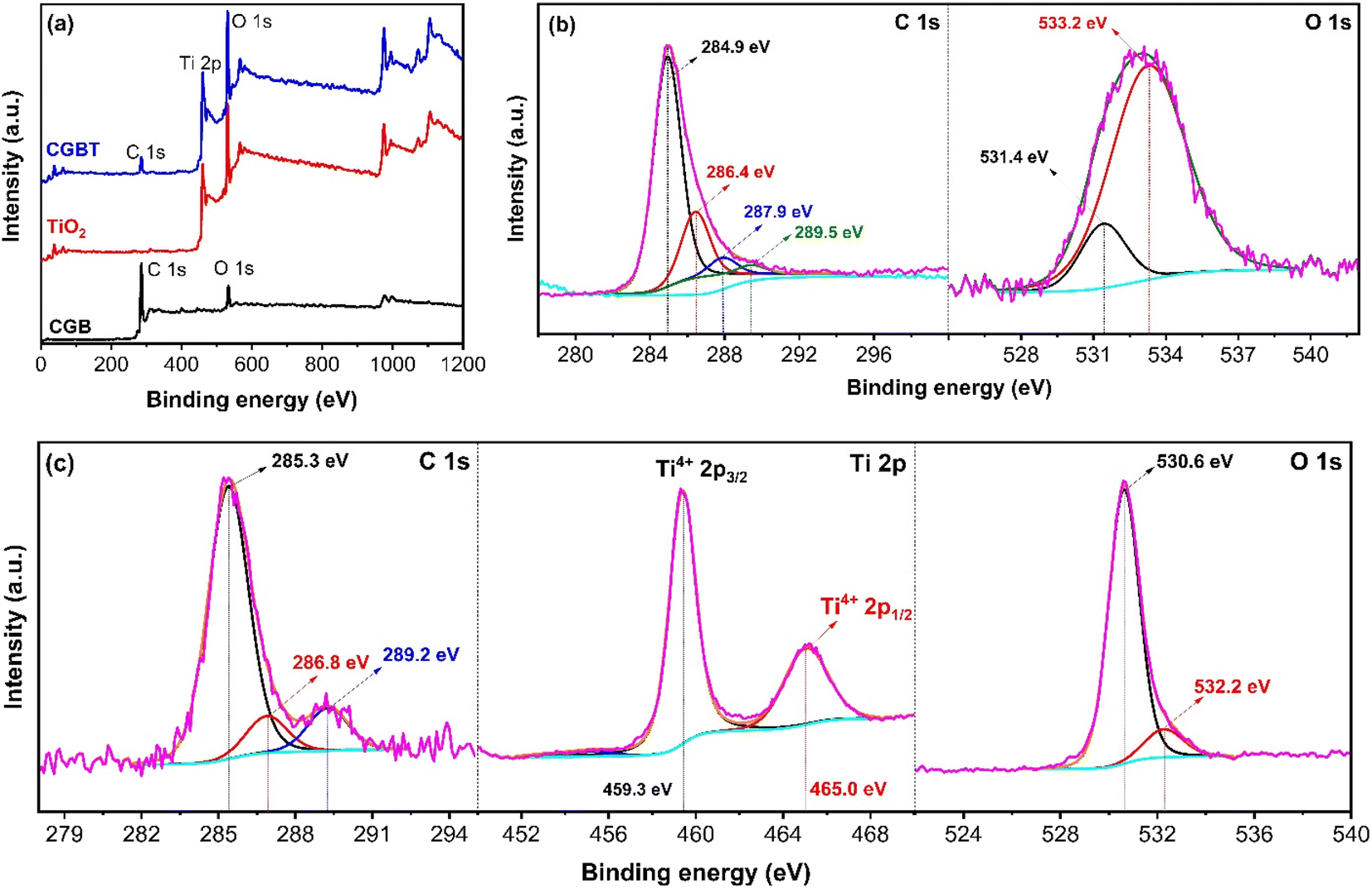
Fig. 4 shows the chemical state and the surface environment of the CGB, TiO2 and CGBT composite as recorded by the XPS technique. The survey spectra of all the synthesized materials are shown in Fig. 4a. In CGB, only C 1s and O 1s peaks can be observed (Fig. 4b), whereas typical C 1s, Ti 2p, and O 1s peaks could be seen in biochar–TiO2 composite (Fig. 4c), confirming that TiO2 nanoparticles were supported on coffee-ground biochar surface. The C 1s high-resolution spectra of CGB and CGBT are deconvoluted and are depicted in Fig. 4b and c, respectively. In the CGB, four peaks can be assigned to Cn (∼284.9 eV),9,39 C–OH (∼286.4 eV), CO (∼287.9 eV), and O–CO (∼289.5 eV).40 Similar peaks were observed for the CGBT composite, except the O–CO (∼289.5 eV) peak was missing, and other characteristic peaks C–O (from 286.4 to 286.8 eV) and CO (from 287.9 to 289 eV) shifted towards higher binding energies. Besides, there is a drop in the concentration area of the C–O peak and increase in concentration area of the CO peak. With the loading of TiO2 nanoparticles, Ti–O–C bond was formed on the surface of CGB, and the chemical environment on the surface changed. Thus, carbon in biochar material and TiO2 nanoparticles were linked via oxygen atom (Ti–O–C) rather than the Ti–C bond under mild conditions.41 The O 1s high-resolution spectra of the CGB and CGBT composite were deconvoluted and are depicted in Fig. 4b and c, respectively. In CGB, the two deconvoluted peak at ∼531.4 eV and ∼533.2 eV are assigned to –COOH and C–O bond.9 Compared with CGB, a new peak appeared at ∼532.6 eV in the CGBT composite, which is assigned to Ti–O bond from the TiO2 lattice.39,40 The existence of this peak in the composite material further demonstrated the successful deposition of TiO2 nanoparticles onto the coffee-ground biochar surface via an oxygen atom, which is also consistent with the FT-IR and TEM results. Fig. 4c represents the Ti 2p high-resolution spectra of the CGBT composite. The Ti 2p high-resolution spectra of TiO2 nanoparticle were also deconvoluted and is shown in Fig. S3 (in ESI†). Both the TiO2 and CGBT composite show two typical deconvoluted peaks at ∼459.3 eV and ∼465.0 eV ascribed to Ti2p3/24+ and Ti2p1/24+.9 The Ti 2p results are close to the theoretical values 458.8 eV (Ti2p3/2) and 462.2 eV (Ti2p1/2), indicating that the Ti valence state in TiO2 and CGBT composite is Ti4+ and the material exists as TiO2 in the synthesized material.32,42 Therefore, the XPS results, in alignment with other physicochemical analysis, confirmed that the CGBT composite material consists of TiO2 and graphene-like C in biochar with a strong interaction between them, both of which provide the beneficial physicochemical properties to composite for improving the overall catalytic performance.
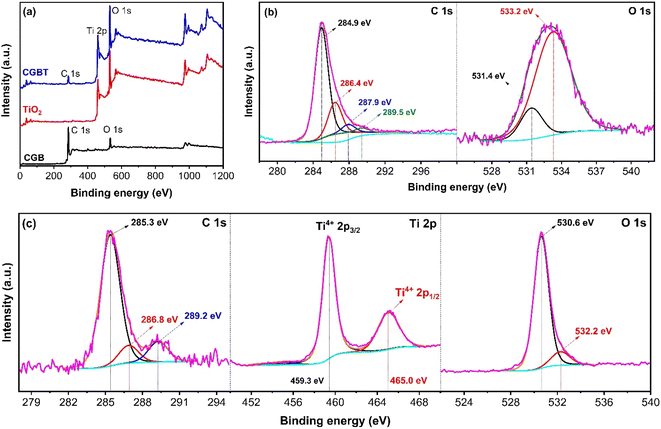 |
| | Fig. 4 (a) XPS full-scan spectra of the synthesized materials. (b) High-resolution XPS C 1s and O 1s spectra of coffee ground-biochar (CGB). (c) High-resolution XPS C 1s, Ti 2p and O 1s spectra of CGBT nanocomposites. | |
3.2 Photocatalytic activity of as-prepared CGBT composite
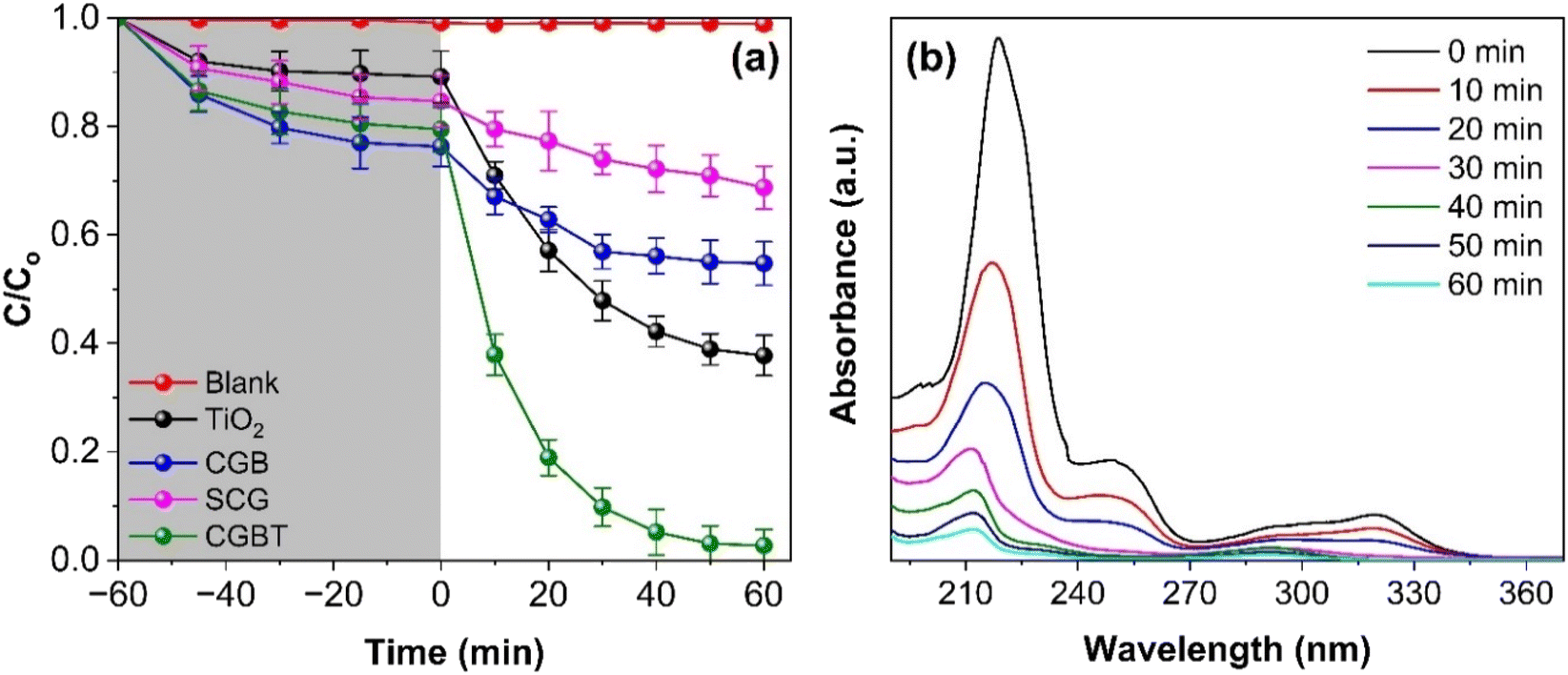
Photocatalytic activity of as-prepared materials was tested against the degradation of pentachlorophenol (PCP) under a heterogeneous photocatalytic oxidation process. As depicted in Fig. 5a, PCP degradation was insignificant under dark and direct UV illumination. Fig. 5a suggested that for all the synthesized materials, the PCP adsorption reached equilibrium within 60 min. The PCP adsorbing capacity of ∼23% coincides with the high surface area of CGBT composite, whereas TiO2 and CBG alone can adsorb 13 and 17% of PCP, respectively. Thus, the degradation process can be divided into two processes, i.e., adsorption and degradation. Fig. 5b shows representative data for PCP degradation where the characteristic absorbance peak 219 nm decreased with the increase in photocatalytic treatment time. The photocatalytic activity of CGBT composite was higher than TiO2, CGB, and spent-coffee grounds (SCG) alone, where only 49, 26 and 13% degradation of PCP was achieved in 1 h under UV light illumination. However, 97% of PCP was degraded in the presence of CGBT nanocomposite (with an initial CGBT concentration of 1 g L−1), suggesting a superior photocatalytic activity of composite material that could be ascribed to the synergistic effect of TiO2 and biochar under light irradiation. Fig. S4 (in ESI†) depicts the degradation kinetics of PCP which can be fit to the first-order rate kinetics model [ln(C/Co) = k1t]. In the rate-kinetics model, C and Co are the concentrations of PCP at a given time interval and in the corresponding dark control, respectively. Table 2 presents the data on the k1 constant, degradation (%), pollutant destruction rate (μmol g−1 h−1), and determination coefficient (R2), as obtained for photocatalytic degradation of PCP under different reaction setups. In the case of CGBT, an accelerated degradation of PCP was observed with k1 value of 0.0602 min−1 compared to CGB and TiO2 which has values of 0.053 and 0.0145 min−1, respectively. Further, the higher destruction rate of 34.84 μmol g−1 h−1 for PCP in the presence of CGBT demonstrated that CGBT composite exhibited rapid and better photocatalytic activity than original SCG, coffee-ground biochar, and TiO2 alone. The improved performance of CGBT nanocomposite can be attributed to increased light absorption and PCP adsorption, presence of a heterojunction which promotes the rapid transfer of photogenerated e− s and inhibits the e−−h+ recombination process,43,44 thereby leading to the enhanced degradation of the PCP pollutant.
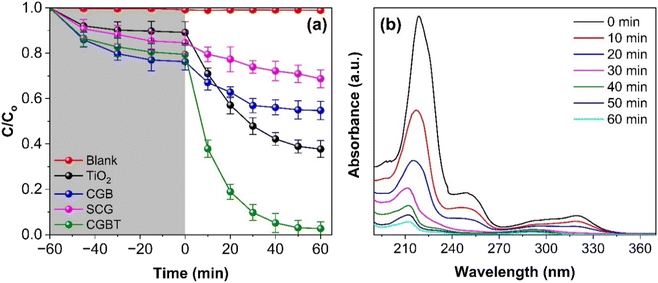 |
| | Fig. 5 (a) Photocatalytic degradation of PCP under photolysis and in the presence of catalysts: TiO2, SCG, CGB, and CGBT nanocomposite, and (b) temporal band changes in absorbance spectra of PCP during photocatalytic degradation by CGBT nanocomposites. [(PCP)o = 10 mg L−1, catalyst dosage of 1 g L−1, pH 5, and UV light irradiation). | |
Table 2 Data on rate constant (k1), degradation (%), destruction rate (μmol g−1 h−1), and determination coefficient (R2) obtained using photocatalytic degradation of PCP in the presence of as-prepared materials [(PCP)o = 10 mg L−1]
| Sample |
k (min−1) |
R
2
|
Degradation (%) |
Destruction rate (μmol g−1 h−1) |
| PCP alone |
1.97 × 10−5 |
0.9789 |
Negligible |
Negligible |
| SCG |
0.0032 |
0.9876 |
13 ± 4 |
0.9 |
| CGB |
0.0053 |
0.9914 |
26 ± 3 |
2.49 |
| TiO2 |
0.0145 |
0.9877 |
49 ± 3 |
9.13 |
| CGBT |
0.0602 |
0.9902 |
96 ± 3 |
34.84 |
The ideal concentration of CGBT and optimal pH of PCP solution were investigated for achieving the enhanced degradation of PCP. Fig. S5a and b (in ESI†) show the effect of varying concentrations of CGBT nanocomposites on the degradation of PCP and its corresponding rate kinetics, respectively. The results demonstrated that with the increase of CGBT concentration from 0.4 g L−1 to 1 g L−1, the degradation of PCP increased from 87% to 96% with the increase in corresponding rate k1 from 0.0294 min−1 to 0.0619 min−1. With the further increase in CGBT concentration, the degradation rate of PCP reduces with the photocatalytic treatment time. It is possible that further increase in CGBT concentration can lead to increased light scattering or agglomeration of the nanocomposites which would reduce the active sites on CGBT to adsorb PCP molecules, thereby reducing the degradation of PCP. Both the surface binding sites on the photocatalyst and aqueous chemistry are greatly influenced by the pH of the solution. Therefore, prior to the pH optimization, the point of zero charges (pHPZC) of CGB, TiO2, and CGBT composites was determined by the procedure described in the ESI (Text S1†). The pHPZC of CGB, TiO2, and CGBT composites was determined to be 4.17, 6.02, and 6.47 (Fig. S6, in ESI†). The initial pHPZC of CGB was calculated to be 5.76, but after the acid-treatment (as described in the Section 2.3), the pHPZC of CGB reduced to 4.17. For the solution pH closer to pHPZC, the surface of CGBT is neutral, while for the higher pH, the surface is negatively charged. Besides, PCP is a weak acid and becomes negatively charged (PCP−) at pH above 4.7 (pKa of PCP is 4.7 at 25 °C).45 Thus, with the increase in solution pH above the pHPZC value, the surface of CGBT composite and PCP will acquire a negative charge, leading to increased electrostatic repulsion and decreased degradation at higher pH (as evident from Fig. S5c, in ESI†). In Fig. S5d,† the rate k1 increased to 0.0629 min−1 at solution pH 5 from the k1 value of 0.0526 min−1 at pH 3, and it decreased to 0.0101 min−1 at pH 11. Therefore, pH 5 is favorable condition to adsorb the negatively charged PCP molecules on the positively charged CGBT composite, leading to a higher degradation of PCP. Besides, at pH 5, biochar and TiO2 alone showed lower degradation of PCP (Fig. 5a), signifying the synergistic effect of biochar and TiO2 on the adsorption and photocatalytic oxidation of PCP.
3.3 Effect of light sources and water matrices
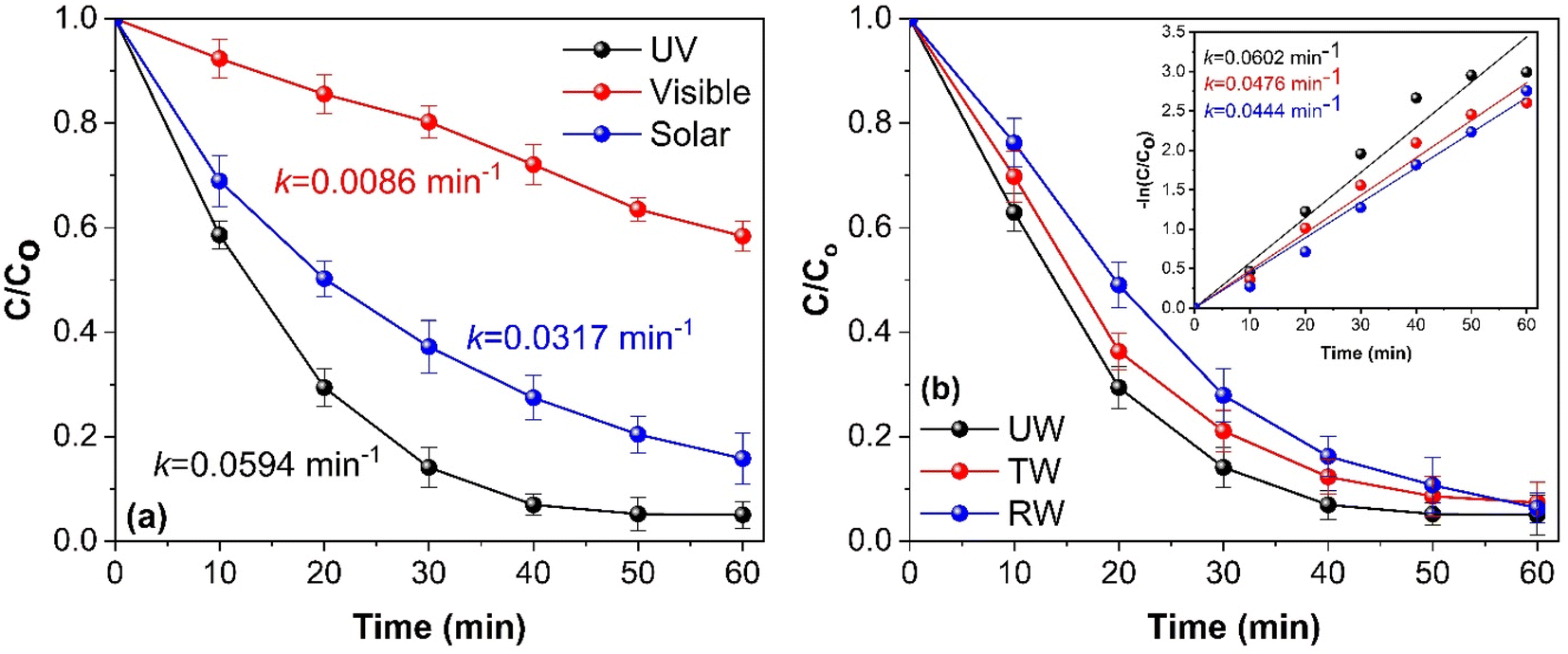
To evaluate the photocatalytic activity of synthesized CGBT nanocomposites, the degradation of PCP was performed under UV, visible, and direct solar light. Fig. 6a shows that the synthesized photocatalyst showed high activity under UV light (PCP degradation of 96%) compared to solar light (86%) or visible light (48%). The synthesized materials showed lower activity under visible light illumination, likely due to lower light absorption by the photocatalyst in that range. The natural sunlight showed decent activity due to the presence of UV-component in the solar spectrum. This is promising as solar light irradiation offers possible pathways for harvesting renewable energy sources for degradation of pollutants. Fig. 6b shows the effect of different water matrices, i.e., deionized water (DI), river water (RW), and tap water (TW), on the photocatalytic degradation of PCP using CGBT nanocomposites. The river water was collected from the Salmon River near the Dalhousie Agriculture Campus, Truro. Tap water was collected from the local drinking water supply. The results indicated that the synthesized nanocomposites efficiently degraded PCP in all the different water bodies in 1 h of photocatalytic treatment. However, the degradation rate of PCP reduced from 0.0602 min−1 with deionized water to 0.0476 and 0.044 min−1 with tap and river water, respectively. Furthermore, it can be speculated that the interaction of reactive radical species (O2˙−, HO˙ and h+) with the ions and dissolved solids in water matrices was insignificant in inhibiting the degradation of PCP in 60 min. Thus, irrespective of the chemical composition of the water matrices, CGBT nanocomposites were able to degrade PCP and hence validated the potential real-world application of as-prepared materials derived from the strong linkage of waste-derived biochar and TiO2 nanomaterials.
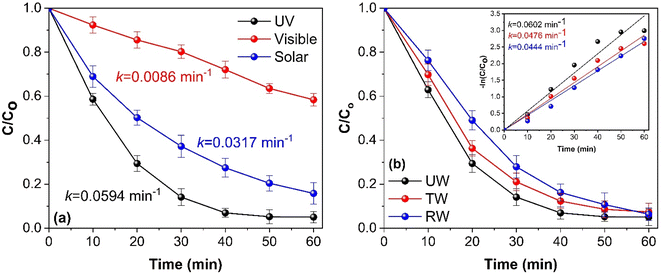 |
| | Fig. 6 (a) Effect of different light sources and (b) water matrix on the photocatalytic degradation of PCP (inset: rate kinetics). [(PCP)o = 10 mg L−1, CGBT dosage of 1 g L−1, pH 5, and UV light irradiation). | |
3.4 Possible reaction mechanism
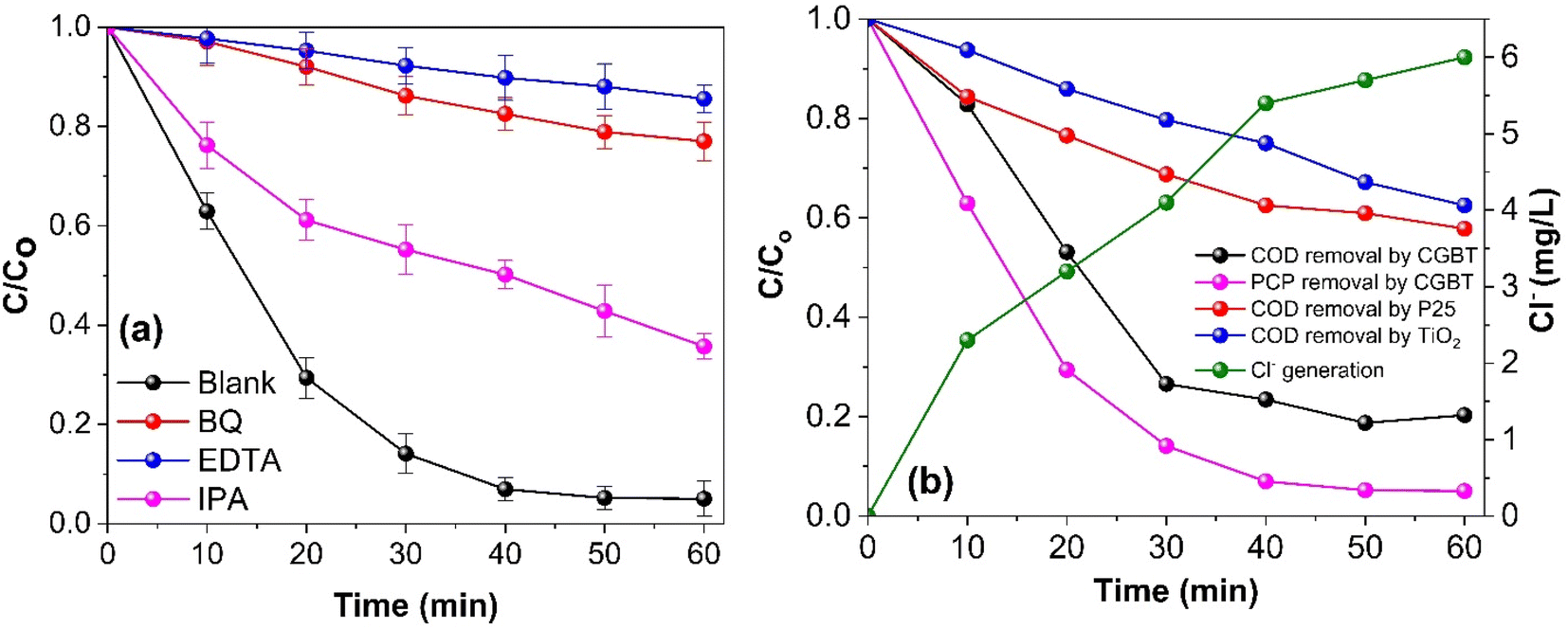
The photocatalytic degradation of the organic pollutant PCP is intimately linked to the nature of the reactive radical species. Thus, free radical trapping experiments were performed by employing a certain concentration (1 mM) of isopropyl alcohol (IPA), ethylenediaminetetraacetic acid (EDTA), and p-benzoquinone (BQ) as a scavenger of HO˙, h+, and O2˙−, respectively to elucidate their contribution to the photocatalytic degradation of PCP. As shown in Fig. 7a, compared with the blank reaction without any scavenger, PCP degradation using CGBT nanocomposite showed different inhibition degrees after the addition of EDTA (PCP degradation efficiency of 24%), BQ (31%), and IPA (68%) with the corresponding k1 values of 0.0026, 0.0046, and 0.0178 min−1, respectively, after 60 min under light irradiation. The results revealed the participation of all the reactive species, where h+, and O2˙− were the predominant ones in the reaction system, followed by the HO˙. The photogenerated h+ can directly mineralize the PCP or generate HO˙ to promote the degradation.46 Similar trends have been observed for radical trapping experiments with biochar/hydrochar-derived catalysts used in the photocatalytic degradation of organic pollutants.9,44,47 To further support the influence of CGBT nanocomposite on the mineralization of PCP, the COD removal efficiency of PCP was assessed during the photocatalytic reaction process. It can be observed from Fig. 7b that the CGBT nanocomposite resulted in effective oxidation and high mineralization of PCP (∼79% of COD removal) in 1 h of photocatalytic reaction. The commercial TiO2 (Degussa P25, average particle size of 21 nm) and synthesized TiO2 led to lower mineralization of PCP (42% and 37.5% by P25 and TiO2, respectively) than the CGBT nanocomposite. The higher mineralization efficiency of CGBT nanocomposite than commercial TiO2 demonstrates the advantage of a lower recombination rate of photoinduced charged species and better light harvesting ability.34,48
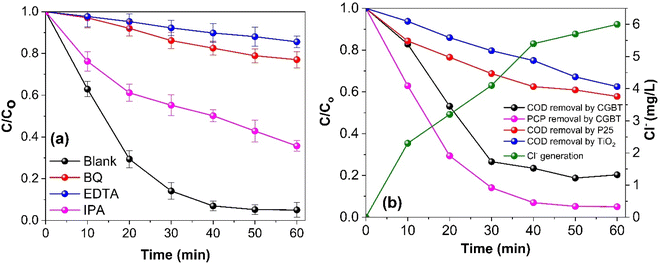 |
| | Fig. 7 (a) Effect of radical trapping experiments on the photocatalytic degradation of PCP, and (b) the removal of COD and the generation of Cl− obtained during the photocatalytic degradation of PCP. [(PCP)o = 10 mg L−1, CGBT and TiO2 dosage of 1 g L−1, pH 5, and UV light irradiation). | |
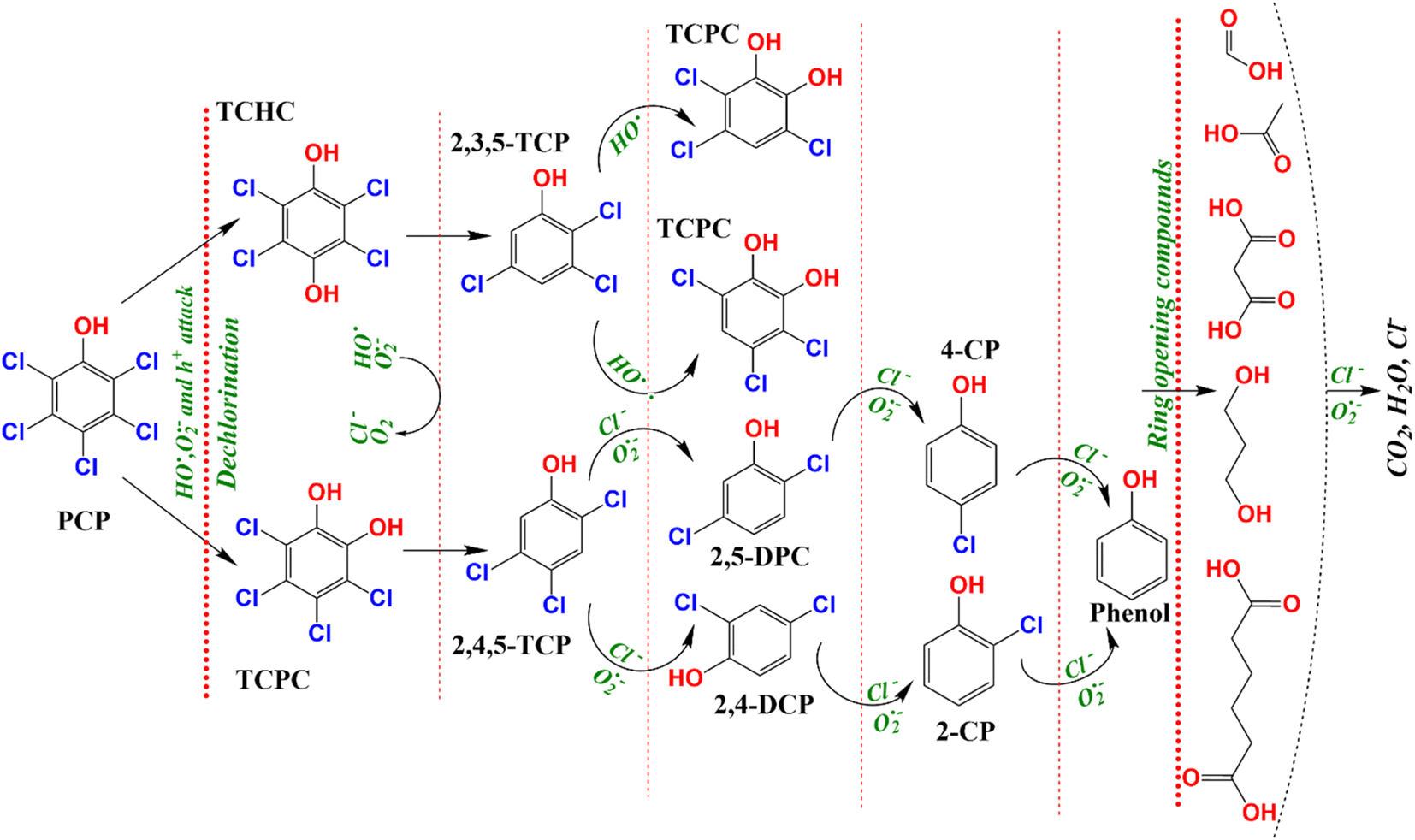
PCP is a chlorinated molecule which can generate other aromatic products and some free chloride ions upon degradation.49,50Fig. 7b depicts the time course of PCP degradation and generation of chloride ions (Cl−) with the CBGT nanocomposites. An increase in Cl− ions indicates that photocatalytic treatment of PCP with CGBT nanocomposite facilitates dichlorination and breakdown of PCP molecules to some aromatic intermediate or transformed products (TPs). To further understand the degradation, gas chromatography-mass spectroscopy (GC-MS) analysis was conducted to identify the intermediates or products generated during the reaction. GC-MS analysis identified six major TPs of PCP based on the comparison with the mass spectra provided by the NIST library, the presence of chlorine isotopic patterns, and EI fragmentation patterns. The details of identified TPs, including the retention time (tR) and their spectral characteristics, are appended in Table S2 (in ESI†). The GC chromatogram and mass spectra of identified products have been shown in Fig. S7, S8 and S9 (in ESI†). Based on the TPs, the tentative photocatalytic degradation pathways for PCP were proposed (Fig. 8). PCP is a chlorinated compound with an electron-withdrawn chlorine group, and the C atoms of the benzene ring are electrophilic in nature.51 Meanwhile, the O2˙−generated during the reaction were strongly nucleophilic and can cleave the bond between C and chlorine atoms. Eventually, in our experiments, tetrachlorobenzenediol, trichlorophenol, dichlorophenol and phenol were identified as major transformed products. Considering the electron negativity and resonance effect, the reactive species could attack the aromatic ring of PCP at the ortho and para-positions,49 thereby generating tetrachloro-pyrocatechol (TCPC) or tetrachloro-hydroquinone (TCHC) with m/z ratio of 247.88. The later TPs have been reported in previous studies as major TPs of PCP during ozonation,52 Fenton's process,53 and heterogeneous photocatalysis.49 The generation of hydroxybenzene also indicated that oxidative dichlorination of PCP indeed occurred in our experiments. As a result, the aliphatic carboxylic acids could undergo further oxidation to generate CO2 and H2O. Fortunately, highly toxic polychlorinated dibenzofurans and polychlorinated dibenzo-p-dioxins intermediates were not found during the experiments, indicating the photocatalytic degradation of PCP by CGBT is an effective and green method to degrade the highly toxic aromatic compounds. Overall, the mechanism of photocatalytic oxidation of PCP has been demonstrated to occur primarily by O2˙− and HO˙ attack and dichlorination process, as evidenced by the presence of residual hydroxylated intermediates and released chloride ions. Control studies performed in the presence of CGBT and absence of PCP did not show formation of these molecules indicating that they are not originating from the biochar itself.
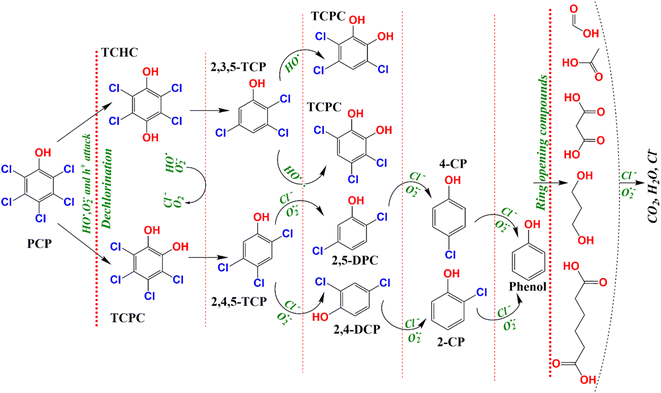 |
| | Fig. 8 The tentative degradation pathways of PCP over CGBT nanocomposites under UV light irradiation. | |
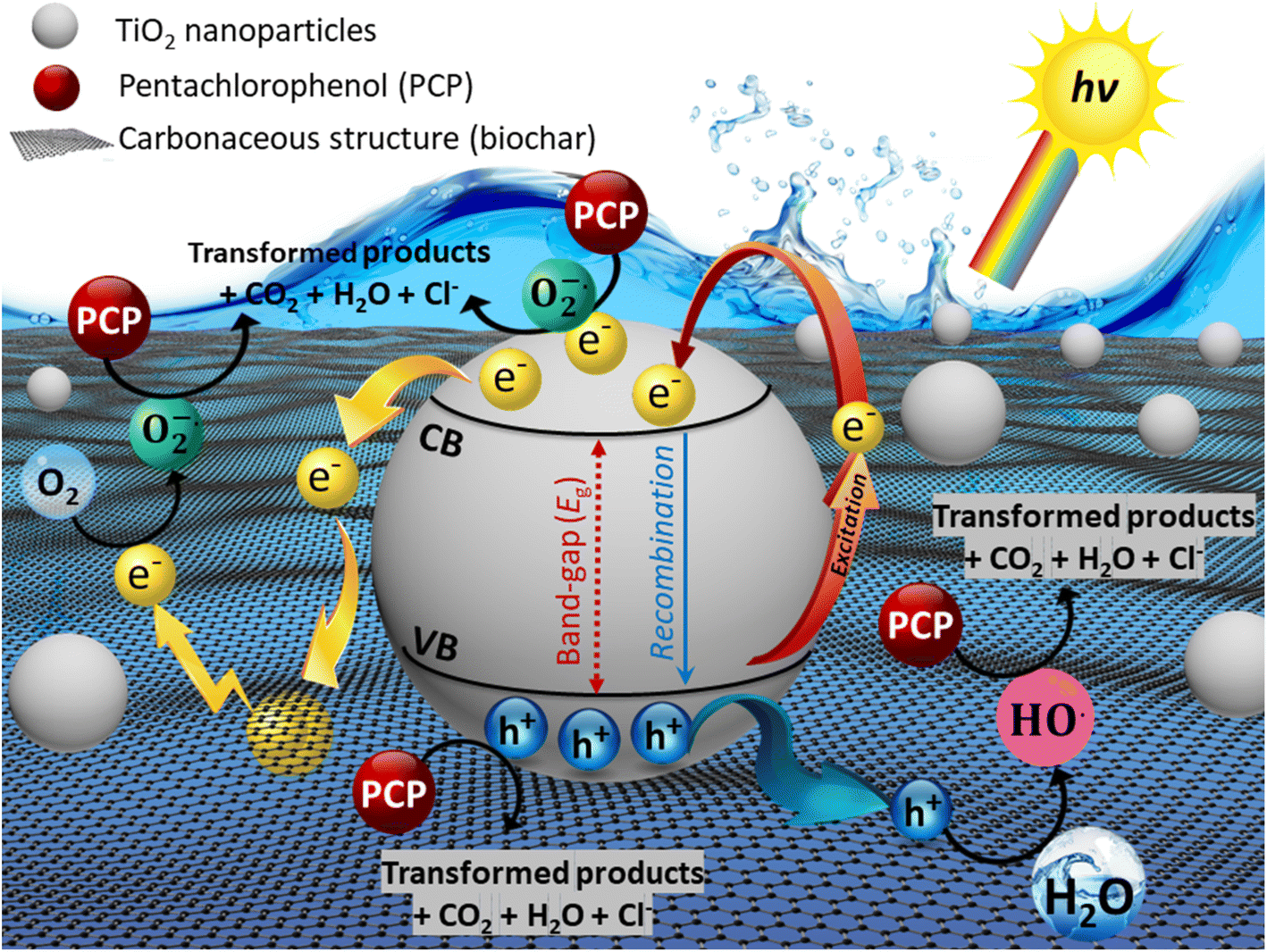
The possible degradation mechanism of PCP over as-prepared CGBT nanocomposite is shown in Fig. 9. Firstly, the PCP molecules could be adsorbed on the composite due to the rich oxygen-functional groups and good porosity of the biochar. Under light irradiation, e− are excited from the valence band (VB) to the conduction band (CB) of the TiO2, leaving the photogenerated h+ behind in the VB. Meanwhile, the biochar could act as an e− trapping layer that could prevent the recombination of e−−h+ pairs, thereby enhancing the e− transfer rate. Secondly, photoexcited e− reacts directly with the dissolved and/or adsorbed O2 to generate superoxide radical anions (O2˙−), while the HO˙ were formed due to the action of h+ with the water molecules. Therefore, the efficient breakdown of PCP molecules could be achieved by the co-functions of O2˙−, HO˙, and h+ and their interaction with the PCP molecules, subsequently generating CO2 and releasing Cl− ions. The high surface area of the CGBT composite also provides numerous adsorption sites for PCP molecules. The ROS generated does not have to diffuse far into the solution for the degradation to occur which further improves the reaction rates.
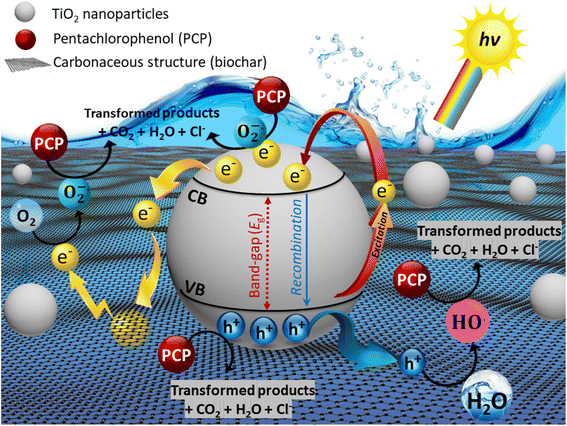 |
| | Fig. 9 Possible reaction mechanisms of CGBT for the photocatalytic degradation. | |
3.5 Comparison with other nanocomposites
Metrics based on incident irradiation and photon flux are vital for evaluating photocatalytic wastewater treatment systems. However, a catalyst performing well under given experimental conditions may not necessarily provide optimal performance under variable conditions. Consequently, there is an imperative need to assess multiple metrics such as the quantum yield (QY), figure-of-merits (FOM), and space-time yield (STY) to evaluate the performance of photocatalytic systems on a quantitative basis. The details of QY, STY, and FoM have been described in Text S2 (in ESI†). Accordingly, we compared our reaction system employing CGBT nanocomposite for wastewater treatment with the other reported studies by considering all associated variables in the reaction system and the results are presented in Table S3 (in ESI†). Based on the performance metrics tools, the as-prepared biochar–TiO2 nanocomposites in this study had shown superior activity with high QY, STY, and FoM of 3.07 × 10−7 molecule per photon, 6.14 × 10−6 molecule per photon per g and 3.24 × 10−9 L g−1 mol−1 J−1 h−1, respectively among the other synthesized biochar–TiO2 composites and photocatalysts employed for the degradation of PCP molecules. In addition, our CGBT-employed photocatalytic system outperformed in terms of QY, STY and FoM in comparison to the photocatalytic system using commercial P25 Degussa TiO2 nanoparticles (QY, STY, and FoM of 1.58 × 10−7 molecules per photon, 3.15 × 10−6 molecules per photon per g and 8.53 × 10−10 L g−1 mol−1 J−1 h−1, respectively).
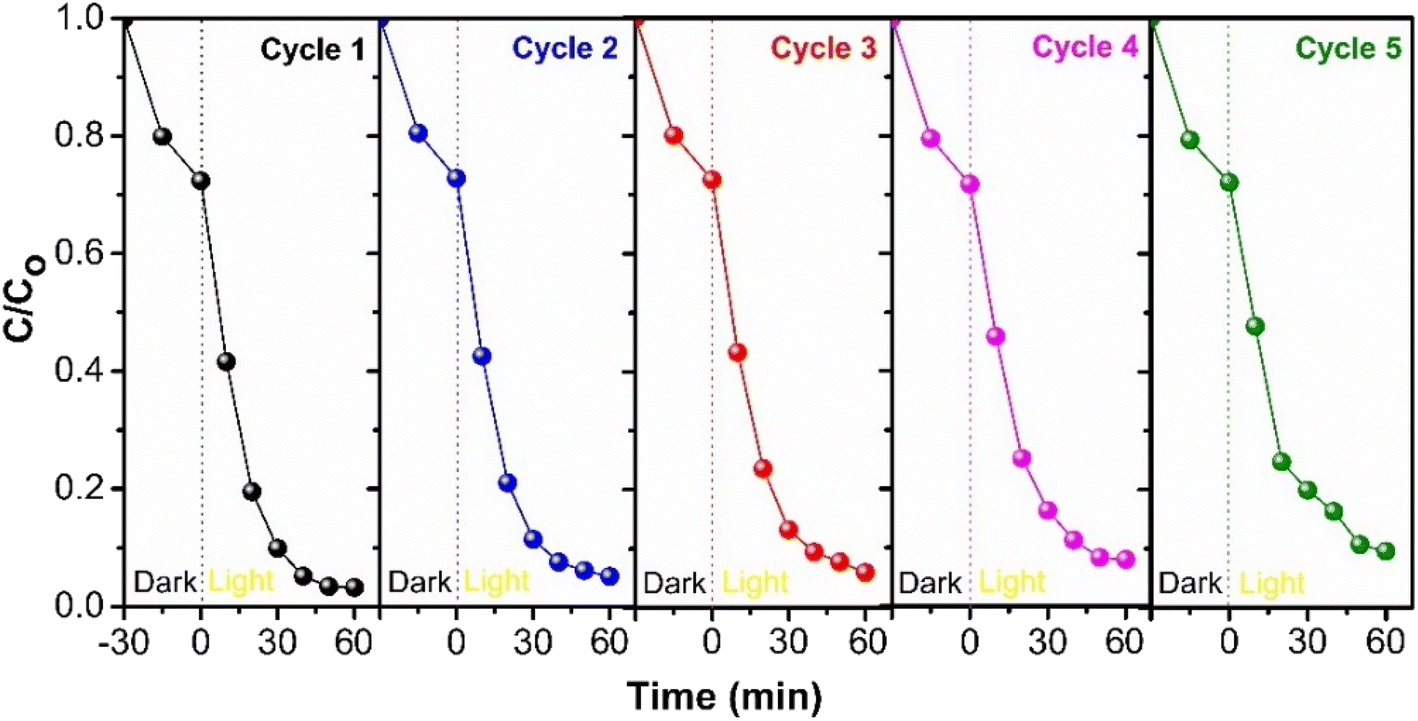
3.6 Stability of CGBT nanocomposites
To assess the long-term stability of the synthesized CGBT nanocomposites, consecutive recycling tests were performed under identical conditions for the degradation of PCP. After each cycle, the photocatalyst was filtered and washed with deionized water, followed by oven drying at 80 °C overnight before use for the next cycle. Fig. 10 depicts the results of successive cycles of photocatalytic degradation with the loss of about ∼9% of PCP degradation activity after five cycles. The regeneration method of photocatalyst could affect the recycling efficiency,54 and besides, there could be a loss of TiO2 film in composite materials during washing.55 However, the results revealed promising stability and recyclability of synthesized CGBT nanocomposites for photocatalytic treatment applications. To further assess the structural stability, the CGBT photocatalyst after successive usage for 5 cycles was subjected to characterization by FT-IR analysis. As shown in Fig. S10 (in ESI†), the FT-IR spectra of recovered CGBT was almost similar to the fresh CGBT nanocomposite. The broad peak at around 3250−3500 cm−1 suggests that the surface of CGBT nanocomposite is rich in –OH groups after 5 consecutive cycles, which could be evidence for the presence of Ti–OH.7,15 The absorption band in the region 450−520 cm−1 is intact and is confined to Ti–O–C bond,34 whereas the sharp band in the range 580−830 cm−1 is assigned to Ti–O–Ti bond in the TiO2.32 Combined with the FT-IR results and cycling experiments, the prepared nanocomposite material exhibited structural and application stability.
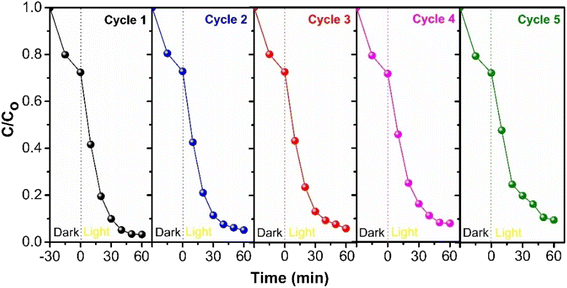 |
| | Fig. 10 The reusability potential of as-prepared CGBT nanocomposites for the photocatalytic degradation of PCP. | |
4. Conclusion
The study aimed to demonstrate the low-cost and environmentally beneficial approach to synthesizing and improving the photoactivity of TiO2-based spent coffee grounds (CGBT) nanocomposites. This was achieved by preparing biochar from spent coffee grounds via pyrolysis and, subsequently, synthesizing biochar–TiO2 hybrid nanocomposites using a simple sol–gel method. Such interfacial chemistry of waste-derived materials with a semiconductor photocatalyst not only optimizes the transfer pathways of photogenerated charged carriers across the interface of TiO2 and biochar but also promotes the low recombination rates of charged carriers in the hybrid structure as well as it might significantly affect the physicochemical properties of the hybrid nanocomposites materials. The superior performance of CGBT nanocomposites exhibited several favourable properties, including combination of anatase and rutile TiO2 nanoparticles, the increased surface area of hybrid materials, better light absorption and enhanced photocatalytic activity to remove organic pollutants. Overall, the CGBT nanocomposite was the most photocatalytic active in PCP degradation (∼97% degradation and ∼79% mineralization in 60 min) compared to other reported photocatalysts for PCP degradation. This study also emphasizes addressing the potential of our photocatalytic system with reported literature by using various performance metrics tools that are rarely encountered in the literature. Obtained results revealed the existence of intimate contact between coffee-ground biochar and TiO2 in the hybrid structure capable of acquiring easy electron transferability and excellent electron–hole separation efficiency on the resulting nanocomposite. This works offers an easy, economical, and efficient route to synthesize biochar/TiO2 photocatalysts with superior activity and durability for applications in water and wastewater treatment, along with the possible contribution towards the high-value application of abundantly available waste biomass, i.e., spent coffee grounds.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the financial support from the Killam Trusts (Killam post-doctoral fellowship) and Natural Science and Engineering Research Council (NSERC), Canada, Discovery Program (grant number RGPIN-2020-05695).
References
- W. Zheng, X. Wang, H. Yu, X. Tao, Y. Zhou and W. Qu, Environ. Sci. Technol., 2011, 45, 4668–4675 CrossRef CAS PubMed.
- H.-Y. Ma, L. Zhao, L.-H. Guo, H. Zhang, F.-J. Chen and W.-C. Yu, J. Hazard. Mater., 2019, 369, 719–726 CrossRef CAS.
- K. Su, Z. Ai and L. Zhang, J. Phys. Chem. C, 2012, 116, 17118–17123 CrossRef CAS.
- G. S. Cooper and S. Jones, Environ. Health Perspect., 2008, 116, 1001–1008 CrossRef CAS PubMed.
- M. E. Harnly, M. X. Petreas, J. Flattery and L. R. Goldman, Environ. Sci. Technol., 2000, 34, 1143–1149 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- B. Han, W. Wei, L. Chang, P. Cheng and Y. H. Hu, ACS Catal., 2016, 6, 494–497 CrossRef CAS.
- J. Shi, W. Huang, H. Zhu, J. Xiong, H. Bei and S. Wang, ACS Omega, 2022, 7, 12158–12170 CrossRef CAS PubMed.
- M. Abellán, B. Bayarri, J. Giménez and J. Costa, Appl. Catal., B, 2007, 74, 233–241 CrossRef.
- Y. H. Ng, S. Ikeda, M. Matsumura and R. Amal, Energy Environ. Sci., 2012, 5, 9307–9318 RSC.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- W.-J. Liu, H. Jiang and H.-Q. Yu, Chem. Rev., 2015, 115, 12251–12285 CrossRef CAS PubMed.
- X. Xiong, K. Iris, L. Cao, D. C. Tsang, S. Zhang and Y. S. Ok, Bioresour. Technol., 2017, 246, 254–270 CrossRef CAS PubMed.
- P. Lisowski, J. C. Colmenares, O. Masek, W. Lisowski, D. Lisovytskiy, A. Kaminska and D. Łomot, ACS Sustainable Chem. Eng., 2017, 5, 6274–6287 CrossRef CAS.
- M.-S. Kim, H.-G. Min, N. Koo, J. Park, S.-H. Lee, G.-I. Bak and J.-G. Kim, J. Environ. Manage., 2014, 146, 124–130 CrossRef CAS PubMed.
- J. McNutt, J. Ind. Eng. Chem., 2019, 71, 78–88 CrossRef CAS.
- V.-T. Nguyen, T.-B. Nguyen, C.-W. Chen, C.-M. Hung, J.-H. Chang and C.-D. Dong, Bioresour. Technol., 2019, 284, 197–203 CrossRef CAS PubMed.
- H.-B. Kim, S.-H. Kim, E.-K. Jeon, D.-H. Kim, D. C. Tsang, D. S. Alessi, E. E. Kwon and K. Baek, Sci. Total Environ., 2018, 636, 1241–1248 CrossRef CAS PubMed.
- L. F. Ballesteros, J. A. Teixeira and S. I. Mussatto, Food Bioprocess Technol., 2014, 7, 3493–3503 CrossRef CAS.
- A. M. Abodif, L. Meng, S. Ma, A. S. Ahmed, N. Belvett, Z. Z. Wei and D. Ning, ACS Omega, 2020, 5, 13630–13640 CrossRef CAS PubMed.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- M. Shi, W. Wei, Z. Jiang, H. Han, J. Gao and J. Xie, RSC Adv., 2016, 6, 25255–25266 RSC.
- H. Zhang and J. F. Banfield, J. Phys. Chem. B, 2000, 104, 3481–3487 CrossRef CAS.
- D. Guo, D. Feng, Y. Zhang, Z. Zhang, J. Wu, Y. Zhao and S. Sun, Fuel Process. Technol., 2022, 229, 107200 CrossRef CAS.
- X. H. Lin, Y. Wu, J. Xiang, D. He and S. F. Y. Li, Appl. Catal., B, 2016, 199, 64–74 CrossRef CAS.
- M.-M. Fu, C.-H. Mo, H. Li, Y.-N. Zhang, W.-X. Huang and M. H. Wong, J. Cleaner Prod., 2019, 236, 117637 CrossRef CAS.
- Y. Shao, H. Tan, D. Shen, Y. Zhou, Z. Jin, D. Zhou, W. Lu and Y. Long, Fuel, 2020, 266, 117146 CrossRef CAS.
- H. Luo, S. Yu, M. Zhong, Y. Han, B. Su and Z. Lei, J. Alloys Compd., 2022, 899, 163287 CrossRef CAS.
- A. Mukherjee, J. A. Okolie, C. Niu and A. K. Dalai, ACS Omega, 2021, 7, 638–653 CrossRef PubMed.
- W. Liu, J. Cai, Z. Ding and Z. Li, Appl. Catal., B, 2015, 174, 421–426 CrossRef.
- W. Wang, J. Zhang, T. Chen, J. Sun, X. Ma, Y. Wang, J. Wang and Z. Xie, Sci. Rep., 2020, 10, 6588 CrossRef CAS PubMed.
- L. Qian, Z.-S. Jin, S.-Y. Yang, Z.-L. Du and X.-R. Xu, Chem. Mater., 2005, 17, 5334–5338 CrossRef CAS.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed.
- S. Umrao, S. Abraham, F. Theil, S. Pandey, V. Ciobota, P. Shukla, C. J. Rupp, S. Chakraborty, R. Ahuja and J. Popp, RSC Adv., 2014, 4, 59890–59901 RSC.
- X. Xie, S. Li, H. Zhang, Z. Wang and H. Huang, Sci. Total Environ., 2019, 659, 529–539 CrossRef CAS PubMed.
- T. Fazal, A. Razzaq, F. Javed, A. Hafeez, N. Rashid, U. S. Amjad, M. S. U. Rehman, A. Faisal and F. Rehman, J. Hazard. Mater., 2020, 390, 121623 CrossRef CAS PubMed.
- R. Shan, L. Lu, J. Gu, Y. Zhang, H. Yuan, Y. Chen and B. Luo, Mater. Sci. Semicond. Process., 2020, 114, 105088 CrossRef CAS.
- J. Zhong, F. Chen and J. Zhang, J. Phys. Chem. C, 2010, 114, 933–939 CrossRef CAS.
- J. Liu, L. Han, N. An, L. Xing, H. Ma, L. Cheng, J. Yang and Q. Zhang, Appl. Catal., B, 2017, 202, 642–652 CrossRef CAS.
- L. Zhao, X. Chen, X. Wang, Y. Zhang, W. Wei, Y. Sun, M. Antonietti and M.-M. Titirici, Adv. Mater., 2010, 22, 3317 CrossRef CAS PubMed.
- J. Li, W. Chen, H. Yu, B. Wu, W. Huang, M. Wang, S. Huang, W. Lin, L. Zhang and S. Li, J. Phys. Chem. Solids, 2014, 75, 505–511 CrossRef CAS.
- Z. Jiang, X. Zhang, Z. Yuan, J. Chen, B. Huang, D. D. Dionysiou and G. Yang, Chem. Eng. J., 2018, 348, 592–598 CrossRef CAS.
- J. Leichtweis, S. Silvestri, N. Stefanello and E. Carissimi, Chemosphere, 2021, 281, 130987 CrossRef CAS PubMed.
- S. Yang, Y. Liu and C. Sun, Appl. Catal., A, 2006, 301, 284–291 CrossRef CAS.
- S. Li, Q. Ma, L. Chen, Z. Yang, M. A. Kamran and B. Chen, Chem. Eng. J., 2022, 433, 134492 CrossRef CAS.
- S. Wang and J. Wang, J. Hazard. Mater., 2022, 423, 127207 CrossRef CAS PubMed.
- S. D. Perera, R. G. Mariano, K. Vu, N. Nour, O. Seitz, Y. Chabal and K. J. Balkus Jr, ACS Catal., 2012, 2, 949–956 CrossRef CAS.
- G. Mills and M. R. Hoffmann, Environ. Sci. Technol., 1993, 27, 1681–1689 CrossRef CAS.
- J. P. Wilcoxon, J. Phys. Chem. B, 2000, 104, 7334–7343 CrossRef CAS.
- L. K. Weavers, N. Malmstadt and M. R. Hoffmann, Environ. Sci. Technol., 2000, 34, 1280–1285 CrossRef CAS.
- P. A. Hong and Y. Zeng, Water Res., 2002, 36, 4243–4254 CrossRef CAS PubMed.
- J. A. Zimbron and K. F. Reardon, Water Res., 2009, 43, 1831–1840 CrossRef CAS PubMed.
- S. Zhang and X. Lu, Chemosphere, 2018, 206, 777–783 CrossRef CAS.
- D. T. Sponza and R. Oztekin, Environ. Technol., 2016, 37, 865–879 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c and
Quan (Sophia)
He
*c and
Quan (Sophia)
He